
Advances in deep eutectic solvents and water: applications in metal- and biocatalyzed processes, in the synthesis of APIs, and other biologically active compounds
Luciana
Cicco†
 ,
Giuseppe
Dilauro†
,
Filippo Maria
Perna
*,
Paola
Vitale
* and
Vito
Capriati
*
,
Giuseppe
Dilauro†
,
Filippo Maria
Perna
*,
Paola
Vitale
* and
Vito
Capriati
*
Dipartimento di Farmacia-Scienze del Farmaco, Università di Bari “Aldo Moro”, Consorzio C.I.N.M.P.I.S., Via E. Orabona 4, 70125, Bari, Italy. E-mail: filippo.perna@uniba.it; paola.vitale@uniba.it; vito.capriati@uniba.it
First published on 12th January 2021
Abstract
Owing to a growing awareness towards environmental impact, the search for “greener”, safer, and cost-effective solvents able to replace petroleum-derived solvents has never been greater today. In this context, the use of environmentally responsible solvents like water and the so-called deep eutectic solvents (DESs), constructed from bio-based compounds, has recently experienced important growth in several fields of sciences. This short review highlights the key features of the chemistry of water and (hydrated) DESs when applied to metal- and biocatalyzed transformations as well as to the synthesis of active pharmaceutical ingredients (APIs) and other biologically relevant compounds by providing, through discussion of all relevant literature over the past five years, a comparison of the outcomes of the reactions when carried out in one or the other solvent.
 Luciana Cicco | Luciana Cicco obtained her MSc degree in Pharmacy from the University of Bari (Italy) in 2013, where she received her Ph.D., under the supervision of Professor V. Capriati and Dr F. M. Perna, in 2017. During her Ph.D., she spent some periods abroad, at the University of Rouen in France, and at the University of Oviedo in Spain. Thanks to a fellowship funded by the “Fondazione Puglia”, she then spent six months at the University of Oviedo, working under the supervision of Professor J. García Álvarez. She is currently a fixed-term Researcher in the group of Professor V. Capriati. |
 Giuseppe Dilauro | Giuseppe Dilauro received his MSc degree in Chemistry and Pharmaceutical Technologies (summa cum laude) from the University of Bari (Italy) in 2016, where he received his Ph.D., under the supervision of Professor V. Capriati, in 2020. In 2019 he spent a three-month internship in the research group of Professor K. Koszinowski at the University of Göttingen, Germany. He is currently a Postdoctoral Researcher in Lecce working under the supervision of Professor A. Salomone. His research interests focus on the development of metal-mediated and metal-catalyzed reactions using green and biorenewable reaction media (e.g., deep eutectic solvents, water). |
 Filippo Maria Perna | Filippo M. Perna received his Ph.D. from the University of Bari (Italy) in 2004 under the supervision of Professor S. Florio. He has been a Visiting Researcher at the University of Oviedo in Spain working in the group of Professor J. Barluenga. In 2006 he became Assistant Professor in Organic Chemistry at the University of Bari and joined the research groups of Professors V. Capriati and S. Florio. His current research interests are focused on the use of sustainable solvents in organometallic chemistry, metal-, bio-, and organocatalyzed reactions, and the asymmetric and green synthesis of target molecules with pharmacological importance. |
 Paola Vitale | Paola Vitale graduated in 2000 at the University of Bari (Italy) (summa cum laude) in Chemistry and Pharmaceutical Technologies, and was awarded as the Best Graduate student of her course. She received her Ph.D. at the University of Bari, carrying out a research on new heterocyclic NSAIDs. From 2004 to 2006 she worked as a Postdoctoral Researcher, and then became Researcher at the University of Bari, developing new synthetic methodologies of heterocycles in several interdisciplinary projects. Her research is mainly focused on developing new “green” approaches for the preparation of fine chemicals and APIs by combining biocatalysis and organometallic chemistry. |
 Vito Capriati | Vito Capriati is full Professor of Organic Chemistry at the University of Bari (Italy). He has been Visiting Scientist at the Ohio State University (USA) (2001, with Professor G. Fraenkel) and Visiting Professor at the Göthenburg University (Sweden) (2003). Since 2016 he is the Director of the Interuniversity Consortium CINMPIS. His awards include the 2009 CINMPIS Prize and the 2014 Prize of the Division of Organic Chemistry of the Italian Chemical Society. Current research interests revolve around the chemistry of s-block elements, and the development of novel sustainable chemical transformations. Since 2019 he is Fellow of the Royal Society of Chemistry. |
1. Introduction
The growing environmental concerns in society, which are mainly related to the increase of pollution, the climate change and resource depletion, a combination of factors that are plaguing people's and animal's lives and affecting their health, has impelled chemistry research in developing products and synthetic protocols from renewable resources.1 In this context, the search for cost-effective and more environmentally responsible and safe solvents, in place of toxic and often harmful fossil-generated volatile organic compounds (VOCs), is central as solvents represent the largest source of waste associated with chemical production, accounting for more than 80% of the total mass used in a synthetic process carried out in batch reactors.2It was thus with no surprise that in recent years an explosive growth of publications has focused on the use of bio-based solvents like deep eutectic solvents (DESs) and water, both conforming to the principles of green chemistry.3 DESs and water share several physicochemical properties (e.g., negligible vapour pressure, thermal stability, non-flammability, cheapness), and are both characterized by a strong hydrogen-bond network. DESs are, indeed, usually formed by binary or ternary mixtures comprising at least one hydrogen bond donor (HBD) and at least one hydrogen bond acceptor (HBA). When these components are mixed in a proper molar ratio, the corresponding mixtures have a melting point much lower than that of either of the individual components and, per definition, also to that an ideal liquid mixture.4 DESs are tunable solvents par excellence exhibiting a broad set of polarities because of the possible inclusion of either hydrophilic or hydrophobic components in their structure.5 In addition, those coming from renewable sources also show low toxicity and high biodegradability.6
Intriguingly, water and DESs do have another aspect in common: they are both key players in nature's game. Indeed, if it is true, on the one hand, that water has been (and still is) the “universal solvent” on Earth for several biosynthetic processes for millions of years, on the other hand, DESs formed from primary metabolites (e.g., sugars, natural amino alcohols and carboxylic acids, amino acids, vitamins) (sometimes called natural deep eutectic solvents, NADESs) seem to be the missing link in understanding cellular metabolism and plant physiology. By acting as the third liquid state in the living organisms besides water and lipid, DESs may similarly allow biological processes to take place, while also facilitating the storing and the transportation of poorly water-soluble macromolecules, and the protection of living organisms from cold and drought stresses.7 At the same time, water has also been proven to play a major role in DES formation by modifying the physicochemical properties of the corresponding supramolecular network. This, in turn, affects DES interactions with natural and bioactive molecules (vide infra).8 However, if a small amount of water in hydrated DESs appears to strengthen the hydrogen-bond network, recent studies have disclosed that DES nanostructure is retained up to 42 wt% water. At 51 wt% water, the solution starts exhibiting a behaviour closer to that of solutes in an aqueous solution.9
Several outstanding reviews and book chapters are today already available on the chemistry of both DESs10 and water11 as these are hot topics in many academic workplaces, with new and emerging applications, in particular, in the fields of main group chemistry,12 metal-,13 bio-,14 and organocatalysis,15 electrochemistry,16 photosynthesis,17 and solar technology.18 The reader is referred to these publications for more detailed information. Notwithstanding the similarities existing between DESs and water, these “solvents”, likewise in nature, sometimes offer complementary performances on organic reactivity when tested individually in certain reactions. The reasons are still not clear, and the knowledge of mechanisms of action at a molecular level have become an intense matter of investigation amongst researchers.19
This short review collects and discusses recent progresses over the past five years in (a) cross-coupling processes, (b) biocatalyzed transformations promoted by whole-cell microoorganisms, and (c) the synthesis of active pharmaceutical ingredients (APIs) and other biologically active compounds, showcasing topics/reactions where comparison between water and DESs are available. The aim is to give the reader an overview of the great assortment of reactions feasible, and the potential benefits thereof, when synergistically or singularly using DESs and/or water in (bio)synthetic processes. On the other hand, micelle-promoted transition metal-catalyzed cross-couplings “in water” will not be detailed here, and the interested reader is referred to the comprehensive reviews/monographies already available in the literature.20
2. Cross-coupling reactions in DESs and water
Over the years, transition-metal-catalyzed cross-coupling reactions have become ubiquitous in organic chemistry as they provide an impressive and powerful tool for forging C–C bonds.20e,21 The direct formation of C(sp2)–C(sp2), C(sp)–C(sp2) or C(sp3)–C(sp2) bonds is, indeed, central to natural product synthesis, materials science, biological-, medicinal-, and supramolecular chemistry, and also to catalysis, coordination chemistry and polymer synthesis.22 In this context, solvents are crucial because of the considerable influence they have on the rate and selectivity of such processes as well as on their equilibria.23 The stability of catalysts, in turn, can be influenced by coordinating solvents used in the synthesis, which compete with the ligands (if any), whereas the function of acids and bases could alternatively be played by a complementary hydrogen bond-donor or hydrogen bond acceptor functionality, if present on the solvent. In order to minimize industrial waste and to promote a sustainable environment,24 many efforts have been made over the last decade to accomplish transition-metal catalyzed cross-couplings in more environmentally benign protocols, like those based on nature-inspired reaction media.25 Remarkable developments have been achieved, especially when using green solvents such as water or DESs. The field of cross-coupling reactions in DESs has been recently reviewed by Varma, Ramón and co-workers.26 Thus, in this section, we summarize and discuss only selected examples of metal-catalyzed cross-couplings where a comparison of the reactivity and the outcome of the reaction in water and in DES is available.2.1 The Suzuki–Miyaura reaction
The Suzuki–Miyaura (SM) reaction has been heavily exploited in this decade because of several advantages over other similar reactions, such as mild reaction conditions, the commercial availability of an array of precursors, functional group tolerance, and the deployment of nontoxic and easily synthesized boronic acids and their derivatives (e.g., potassium organotrifluoroborates, boronate esters). A survey of the literature disclosed many examples of Pd, Ni and Au complexes as efficient catalysts for such cross-couplings.27 In 2006, König and co-workers first reported that phenyl boronic acid could be successfully coupled with three aryl bromides at 90 °C in a low-melting sugar–urea–salt mixture made by mannitol–N,N′-dimethyl urea (DMU)–NH4Cl (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40:10 w/w) as an eco-friendly solvent, in the presence of 10 mol% of Pd(OAc)2 as a catalyst and Na2CO3 as a base. A quantitative conversion was achieved after 6 h, and the hydrodeboronation reaction was not observed as a competing process (Scheme 1a).28
:40:10 w/w) as an eco-friendly solvent, in the presence of 10 mol% of Pd(OAc)2 as a catalyst and Na2CO3 as a base. A quantitative conversion was achieved after 6 h, and the hydrodeboronation reaction was not observed as a competing process (Scheme 1a).28
 | ||
| Scheme 1 Examples of Suzuki–Miyaura cross-coupling reactions in (a) low-melting mixtures and (b) in DESs. | ||
In the following years, numerous papers have been published on such a cross-coupling reaction in DESs, mostly using boronic acids as the nucleophilic partners, a large amount of electrophile, and palladium catalysts often associated with structurally engineered phosphine ligands in order to obtain good yields in short reaction times. Ramón and co-workers, in particular, introduced three different protocols: (a) the use of PdCl2 as a catalyst, K2CO3 as a base, and cationic structurally engineered phosphine ligands in a choline chloride (ChCl)-based DES;29 (b) the use of a N-heterocyclic carbene (NHC)-Pd complex as a catalyst, K2CO3 as a base in a ChCl/ethylene glycol (EG) (1:2 mol/mol) eutectic mixture, in the presence of 10 equiv. of water;30 and (c) the employment of a DES-compatible bipyridine palladium complex as a general pre-catalyst, K2CO3 as a base in a ChCl/EG (1:2 mol/mol) mixture at 100 °C.31
In 2018, Capriati and co-workers developed a Pd-catalyzed SM reaction between (hetero)aryl halides (Cl, Br, I) and air- and moisture-stable mono- and bifunctional potassium aryltrifluoroborates (employed in a strict 1:1 stoichiometric ratio). Under these conditions, the coupling was found to proceed efficiently and chemoselectively in air and under generally mild conditions (60 °C), using 1 mol% of Pd(OAc)2 as a catalyst, Na2CO3 as a base in a ChCl/glycerol (Gly) (1:2 mol/mol) eutectic mixture, which is used as a recyclable, sustainable and environmentally responsible medium (Scheme 1b).32 The same ligand-free approach has been then applied to promote smooth SM coupling reactions, under aerobic conditions, between dihalogeno-substituted benzodithiophenes (BDTs) and aryl-, alkenyl- and alkynyltrifluoroborate salts in a ChCl/Gly eutectic mixture to synthesize valuable π-conjugated BDT adducts in up to 79% yield, whose optical and electrochemical properties have been deeply investigated by means of absorption and cyclic voltammetry measurements.33
The use of potassium aryltrifluoroborates in pure water was less effective. In 2015, Schatz and co-workers described an efficient, broadly applicable and sustainable SM protocol in pure water with a Pd loading as low as 0.4 mol%, without any ligand or organic co-solvent, working with a range of activated and nonactivated aryl halides (Scheme 2). Nevertheless, quantitative conversions could not be achieved when using the easy-to-handle and air-stable potassium phenyltrifluoroborate in place of the corresponding phenylboronic acid. The catalytic activity was boosted by using NHC ligand precursors and/or supramolecular additives.34
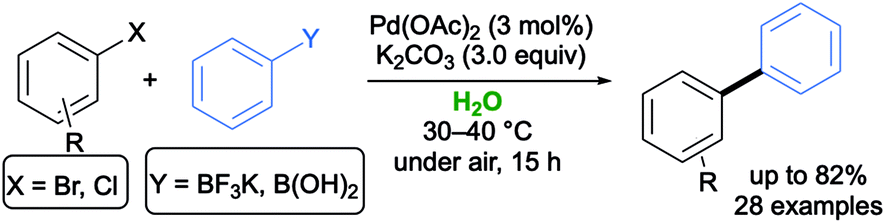 | ||
| Scheme 2 Pd acetate-catalyzed on-water Suzuki Miyaura reactions. | ||
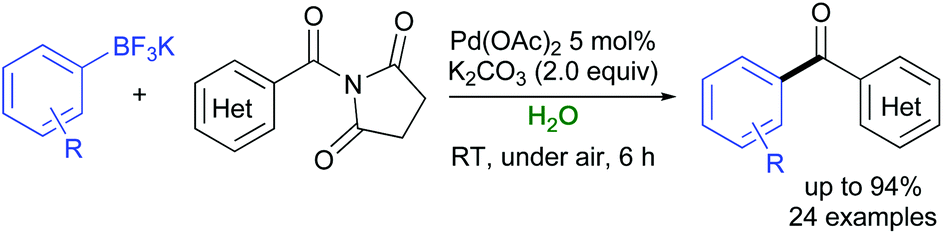
In 2018, an efficient and ligandless catalytic protocol was developed for the coupling between potassium aryltrifluoroborates with various aryl and heteroaryl bromides in water with Na2CO3 as a base. Using tetraethylammonium tetrahydroborate (TEAB) as an additive and Pd(OAc)2 as a catalyst, the desired adducts were isolated in moderate to excellent yields (up to 94%).35 In 2020, a green general strategy for the synthesis of aryl ketones from twisted amides and air- and moisture-stable potassium aryltrifluoroborates has been set up by Liu, Zhu, Zeng and co-workers using Pd(OAc)2 as a catalyst and K2CO3 as a base. The reported protocol showed a high functional group tolerance, with a variety of ketones synthesized in very good yields (up to 94%) under air, and within 6 h reaction time (Scheme 3). Of note, this approach, which relied on the high reactivity of N-acylsuccinimides, also demonstrated the first water phase, room temperature, and ligand-free Suzuki–Miyaura coupling successfully taking place through a C–N bond cleavage.36
 | ||
| Scheme 3 Water phase, room temperature, and ligand-free Suzuki–Miyaura cross-coupling reaction. | ||
2.2 The Sonogashira–Hagihara reaction
The Sonogashira–Hagihara (SH) reaction is the cross-coupling reaction between terminal alkynes and aryl or vinyl halides (pseudohalides) to generate conjugated enynes. This represents one of the most powerful methods for the rapid build up of C(sp2)–C(sp) bonds in synthetic chemistry. This methodology has been widely applied to prepare biologically active molecules, natural products, conducting polymers/engineering materials, and macrocycles with acetylene links.37,38 After pioneering studies by König,39 different copper-free procedures between aryl iodides or bromides and terminal aromatic and/or aliphatic alkynes have been developed in DESs.40,41(Hetero)aryl bromides and iodides were successfully cross-coupled to terminal alkynes in DESs by Ramón, Alonso and co-workers according to the following approaches: (a) use of PdCl2 associated to cationic structurally engineered phosphine ligands and i-Pr2NH as a base in Ph3PMeBr/Gly (1:2 mol/mol) (Scheme 4a);29 (b) use of a NHC-Pd complex and i-PrNH2 in AcChCl/urea (1:2 mol/mol) (Scheme 4b);30 and (c) use of a bipyridine palladium complex as a pre-catalyst in Ph3PMeBr/Gly (1:2 mol/mol) in the presence of i-Pr2NH (Scheme 4c).31
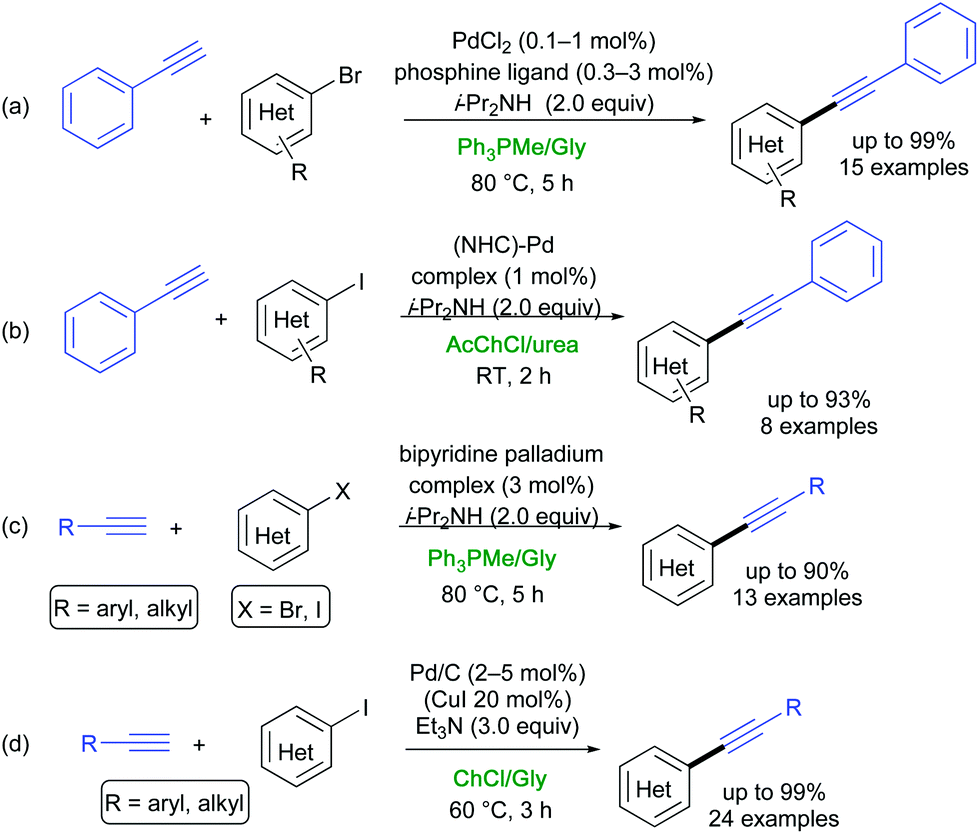 | ||
| Scheme 4 Examples of Sonogashira–Hagihara coupling reactions in DESs (a) in the presence of a phosphine ligand, (b) using a NHC-Pd or (c) a bipyridine Pd complex, or (d) under ligand-free conditions. | ||
A highly efficient, recyclable and ligand-free protocol was developed by Salomone, Capriati and co-workers. Working under heterogeneous conditions, the commercially available Pd/C was found to be an effective catalyst for promoting the coupling between (hetero)aryl iodides (including electron-rich iodides) and aromatic and aliphatic alkynes, within 3 h at 60 °C in yields ranging from 50 to 99%, in the ChCl/Gly eutectic mixture using Et3N as a base (Scheme 4d).42
Notwithstanding the well-known poor solubility of organic substrates in water, Bhattacharya and Sengupta first demonstrated the feasibility of running the alkynylation of aryl iodides and bromides in pure water without any organic co-solvent, phase-transfer catalysts or water-soluble phosphine ligands.43 These couplings, which took place under truly heterogeneous conditions, are remarkably fast (30 min at 70 °C) affording aryl alkynes in 75–92% yield when using 0.5 mol% of Pd(PPh3)4, 1 mol% CuI, and Et3N or N,N-diisopropylethylamine (DIEA) as a base. The reaction showed excellent group tolerance and sensitive functionalities like an aldehyde group were also tolerated. It is worth noting that the addition of an organic solvent like DMF (DMF/H2O 9:1) slowed down the reaction rate and led to the appearance of side-products.
The synthesis of arylpropargylic amines was then achieved by Costa and co-workers via the SH coupling of aryl iodides with propargylic amines,44 working under the so-called “on water” conditions.45 The desired adducts were isolated in up to 85% yield in 24–48 h when using less than 0.2 mol% of Pd(PPh3)4 and 1.5 mol% CuI in the presence of DIEA at 95 °C.44
In 2015, Nasrollahzadeh and colleagues set up an easy and cost-effective synthesis of Pd nanoparticles (NPs) via the reduction of aqueous Pd2+ ions using Piper longum fruit extracts without any surfactant or stabilizer, most probably because the flavonoid present in the extract acted themselves as both reducing and stabilizer agents. The synthesized Pd NPs (stable and storable for months under inert atmosphere) proved to be effective catalysts (loading up to 3 mol%) to perform ligand-, amine- and copper-free SH couplings between aromatic aryl halides and terminal alkynes in neat water under aerobic conditions at 80 °C in the presence of K2CO3 as a base. The expected adducts were isolated in 84–96% yield with the tolerance of a variety of functional groups, and the catalyst was recycled up to five times without any loss of catalytic activity (Scheme 5a).46
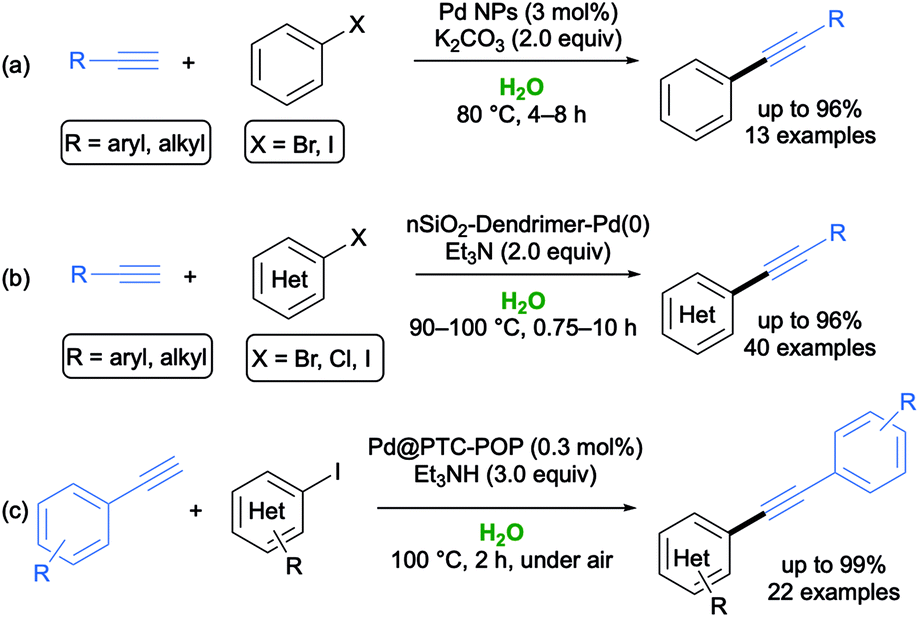 | ||
| Scheme 5 Examples of Sonogashira–Hagihara coupling reactions run in water catalyzed by (a) Pd NPs, or (b) dendrimer-encapsulated Pd(0) nanoparticles immobilized on nanosilica, or (c) a porous organic polymer with in situ generated Pd NPs (Pd@PTC-POP). | ||
In 2016, Sardarian and co-workers reported the synthesis of novel dendrimer-encapsulated Pd(0) nanoparticles immobilized on nanosilica with surface amino groups using natural and waste material from rice husk as the source of the silica components.47 This catalytic system revealed to be an efficient catalyst for promoting copper- and phosphine-free SH couplings between aryl halides and terminal alkynes in water at 90–100 °C in the presence of Et3N, with the desired internal alkynes isolated in 62–97% yield. The robustness of the methodology was further highlighted by the reuse of the catalyst over five cycles without any significant loss in its catalytic activity (Scheme 5b).47
In 2019, a novel highly active and reusable solid phase-transfer catalyst, consisting of a porous organic polymer with in situ generated Pd NPs (Pd@PTC-POP), was fabricated by Dong and co-workers by exploiting a one-pot Suzuki cross-coupling approach. This catalyst promoted SH cross-coupling reactions in aqueous phase, especially from iodo-substituted aromatic substrates and arynes, with the corresponding adducts being synthesized in very good yields (up to 99%) within 2 h at 100 °C in the presence of Et3N and with a broad substrate scope (Scheme 5c).48
2.3 The Murahashi coupling
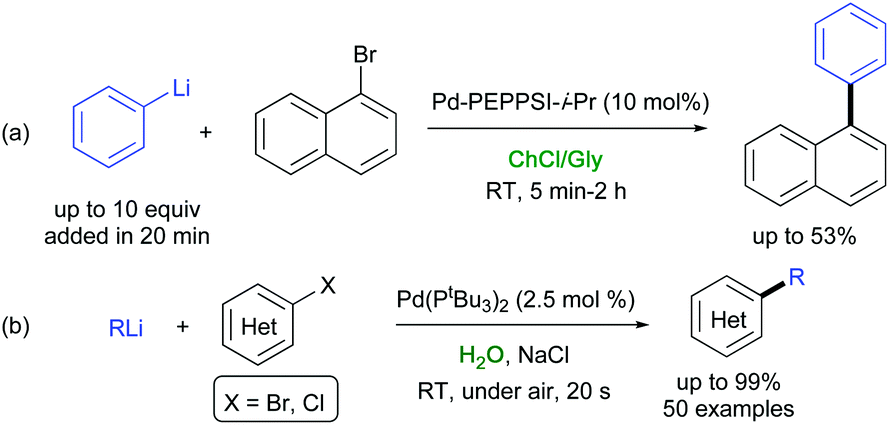
Organolithium compounds are one of the most important classes of organometallic compounds as they are ubiquitous in organic synthesis.49 These cheap, commodity reagents are either commercially available or can be easily accessed by halogen-metal exchange or a direct metalation reaction. Because of their high reactivity, cross-coupling of organolithium species were often proven to suffer from either a fast lithium-halogen exchange, leading to homocoupled products, or a β-hydride elimination reaction, responsible for the formation of dehalogenated products.50 Seminal contributions by Murahashi and co-workers to this field disclosed that Pd(0)-catalyzed cross-couplings between alkenyl halides and organolithium reagents are indeed feasible, though only at high temperature, long reaction time, and at high catalyst loading.51The Murahashi coupling has been recently revised by Feringa and co-workers who disclosed that several alkyl-, alkenyl-, and (hetero)aryllithium reagents could be directly cross-coupled with aryl and alkenyl (pseudo)halides in the presence of palladium or nickel complexes, not experiencing the above drawbacks, when (i) using a nonpolar solvents (toluene), which affects the reactivity of the organolithium reagents, or working with no additional solvents, in conjunction with a Pd catalyst with bulky electron-rich ligands (e.g., Pd/Pt-Bu3), and (ii) performing a slow addition of the organolithium reagent to the reaction mixture.52 Feringa, Hornillos and co-workers also reported that the slow addition (20 min reaction time) of an excess PhLi (2–10 equiv.) to 1-bromonaphthalene using 10 mol% of Pd-PEPSI-i-Pr in a ChCl/Gly eutectic mixture produced the desired adduct, though in low yield (28–53% conversion) (Scheme 6a).53 On the other hand, when alkyl- or aryllithium reagents were added over a mixture of arylbromide and the Pd-catalyst (down to 0.1 mol%) with no additional solvents, cross-coupling reactions were found to proceed fast (10 min) at ambient temperature and also in open air, thereby providing the desired adducts in high yields (81–98%), excellent selectivities, and high scalability.53
 | ||
| Scheme 6 Pd-Catalyzed Murahashi cross-coupling using (a) DES or (b) water as the reaction medium. | ||
In 2019, Capriati and co-workers reported a direct Pd-catalyzed cross-coupling between organolithium reagents and a variety of (hetero)-aryl halides (Br, Cl), taking place at ambient temperature under air, with water as the only reaction medium and in the presence of NaCl as a cheap additive (Scheme 6b).54 A water-accelerated catalysis was the key for achieving C(sp3)–C(sp2), C(sp2)–C(sp2), and C(sp)–C(sp2) cross-coupling reactions, which proceeded fast (20 s reaction time), in yields up to 99%, in competition with protonolysis, and in the absence of dehalogenated/homocoupling side products. Both the catalyst and water could be easily recycled up to 10 times, with an E-factor as low as 7.35. Several eutectic mixtures were also tested, though with both lower yields and selectivity.54
2.4 The Heck–Mirozoki reaction
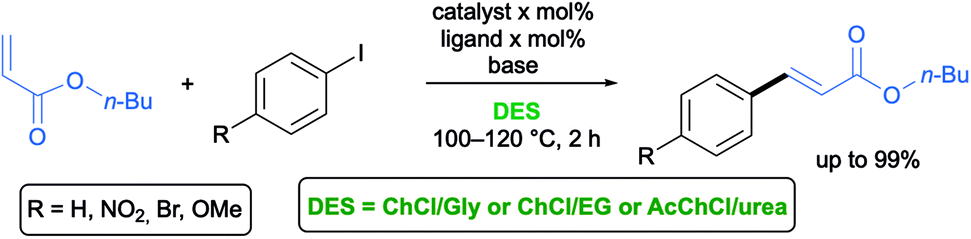
The Heck–Mirozoki (HM) reaction is generally referred to as the Pd-catalyzed C–C coupling between aryl or vinyl (pseudo)halides and activated alkenes in the presence of a base to form a substituted alkene.55 Cross-coupling reactions between aryl iodides with different substitution patterns and methylacrylate have been successfully carried out by Ramón and co-workers in various eutectic mixtures like ChCl/Gly (1:2), ChCl/EG (1:2) and AcChCl/urea (1:2) using cationic pyridiniophospine or bipyridine or strong σ-donor carbene ligands for palladium in the presence of NaOAc as a base at 100–120 °C with good to excellent results (Scheme 7).29–31
 | ||
| Scheme 7 Pd-Catalyzed Heck–Mirozoki cross-coupling reaction between aryl iodides and methyl acrylate in DESs. | ||
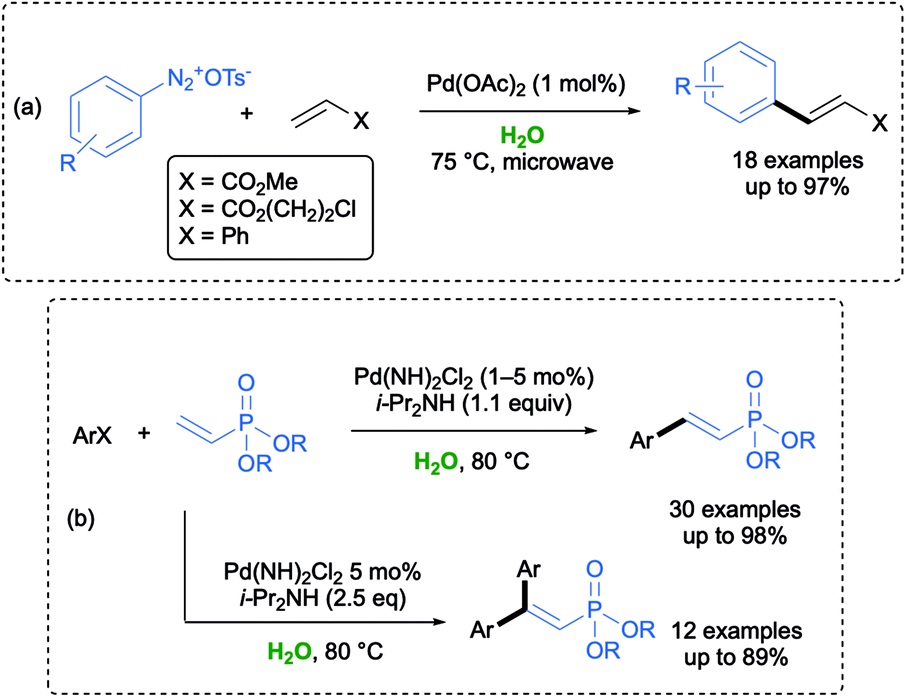
Among Pd-catalyzed cross-coupling reactions, the Heck coupling turned out to be one of the most challenging in an aqueous environment, and several homogeneous and immobilized catalysts, including nanoparticles, have been developed.56 An environmentally friendly Pd-catalyzed Matsuda–Heck reaction between arenediazonium tosylates and different activated alkenes has been first developed by Postnikov and co-workers in water. Under optimized conditions, arylation of aldehydes took place with a broad substrate scope with the desired products isolated with a high purity and in a considerable short reaction time (up to 1 min) when using microwave irradiation (Scheme 8a).57
 | ||
| Scheme 8 Pd-Catalyzed (a) Matsumada–Heck reaction with arendiazonium tosylates, and (b) Heck–Mirozoki reaction with dialkyl vinylphosphonates in water. | ||
In 2017, Tsai and co-workers reported that mono or double Pd-catalyzed MH couplings between dialkyl vinylphosphonates and aryl halides could be carried out in water under air using i-Pr2NH as a base, by simply selecting aryl halides or dialkyl vinylphosphonates as the limiting reagent. A plausible mechanism was also reported. When aryl iodides were used as the electrophiles, the coupling could be run at 80 °C under a low catalyst loading (0.1–1 mol%), whereas aryl bromides required a higher temperature (120 °C), a catalyst loading up to 5 mol%, and a longer reaction time. The greenness of the reaction was showcased by an effective reuse of the residual aqueous solution for both the reported couplings (Scheme 8b).58
2.5 The Ulmmann-type reaction
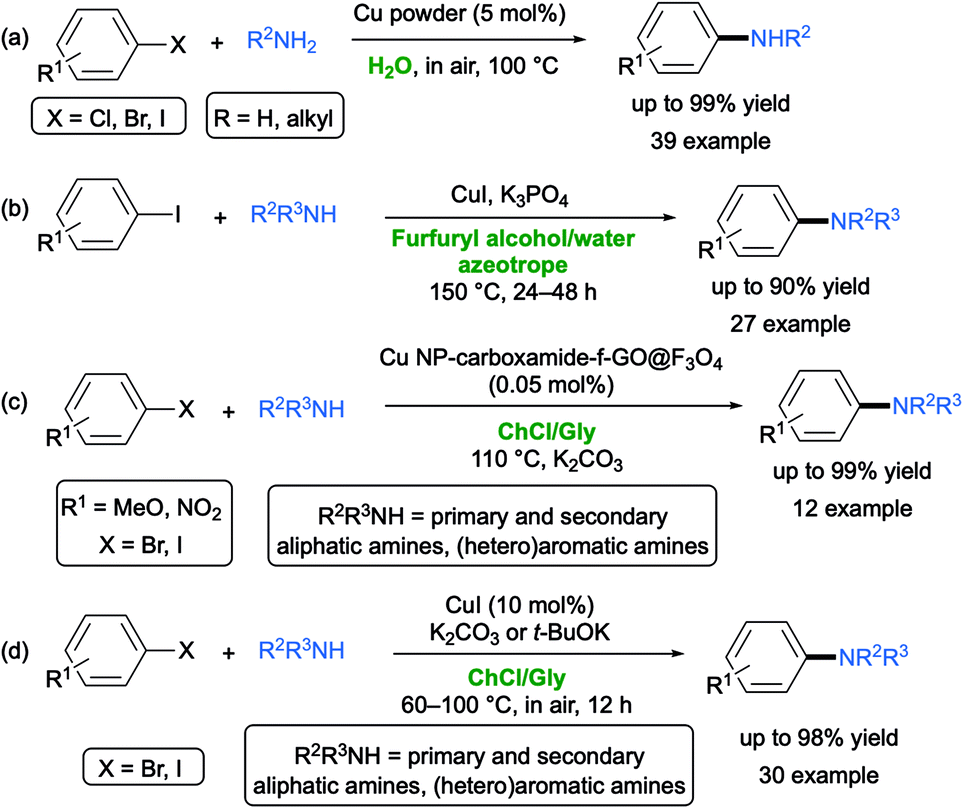
In contrast to the “classical” Ullmann reaction, which is the direct synthesis of symmetric biaryls via copper-catalyzed coupling at elevated temperature (>180 °C),59 the so-called “Ullmann-type” reaction encompasses copper-catalyzed nucleophilic aromatic substitution between aryl halides and various nucleophiles.60 Key aspects of all these reactions are the use of (a) polydentate auxiliary ligands with oxygen- or nitrogen coordination sites (e.g., diols, diamines, oximes) aimed at improving the solubility of the copper precursors and the stability of the active catalyst, and/or (b) toxic VOCs (e.g., DMSO, DMF, dioxane, THF).Copper-catalyzed couplings in aqueous media are known, though they often require harsh conditions (high temperatures up to 100–130 °C) and the presence of phase-transfer catalysts and ligands.61 In 2011, Wei and co-workers reported the copper powder-catalyzed ligand-free amination of aryl halides with aliphatic amines under aerobic conditions in pure water at 100 °C. Under these conditions, however, although sensitive functional groups were tolerated (e.g. CHO, MeCO, CN), secondary amines and aniline derivatives were proven to be unreactive (Scheme 9a).62
 | ||
| Scheme 9 Cu-Catalyzed Ullmann amine cross-coupling between aryl halides and amines (a) in water, (b) in bio-based solvents, (c) in DES, in the presence of Cu NP carboxamide functionalized magnetic graphene oxide as a nanocatalyst, and (d) in DES, with no additional ligands under air. | ||
In 2018, Vaccaro and co-workers developed a waste-minimized protocol to promote the Ullmann-type copper-catalyzed coupling between aryl halides and heteroaromatic or aliphatic amines in the biomass-derived furfuryl alcohol (FA) as an effective bio-based co-solvent/bidentate ligand for Cu(I) catalyst. The reaction showed a broad substrate scope, with the corresponding adducts being isolated in generally good yields. Of note, when the FA was mixed with water, it formed an azeotrope (20 wt% of FA), and thus it could be easily recovered and reused (Scheme 9b).63
In 2018, Shaabani and co-workers reported the Ullmann C–N coupling of aryl halides with N-heterocycles or aniline derivatives in ChCl/Gly (1:2), taking place in high yields (75–99%) and short reaction times, catalyzed by a new copper nanoparticle (Cu NP) modified carboxamide-functionalized magnetic graphene oxide (carboxamide-f-GO@Fe3O4) nanocatalyst, which in turn was synthesized via a one-pot four component UGI reaction. The Cu NP-carboxamide-f-GO@Fe3O4 nanocomposite and DES were easily recovered and reused for five consecutive runs. The authors related the high efficiency of the N-arylation process to the combination of the synthesized nanocatalyst with DES whose extensive hydrogen-bonding network creates a strain inside its cavity so as to trigger the coupling process (Scheme 9c).64
A practical and efficient protocol for performing CuI-catalyzed Ullmann amine synthesis between (hetero)aryl halides (Br, I) and a variety of aromatic or aliphatic primary and secondary amines has been then developed in the same biodegradable ChCl/Gly mixture by Capriati and co-workers. Notable features of this report are (a) a broad substrate scope starting from both electron-deficient and electron-rich (hetero)aryl halides with the tolerance of several functional groups, and with the desired adducts isolated in up to 98% yield; (b) mild conditions (60–100 °C) in air, with K2CO3 or t-BuOK (in the case of aromatic amines) as a base, and a catalyst loading of CuI up to 10 mol%; and (c) an effective recycling of the DES, the catalyst and the base over 6 cycles with an excellent performance of the copper species in the employed eutectic mixture (Scheme 9d).65
3. Sustainable synthesis of active pharmaceutical ingredients (APIS) and biologically active compounds
Recommendations by institutions and organizations, such as The American Chemical Society's (ACS) Green Chemistry Institute Pharmaceutical Roundtable (GCIPR) (whose aim is to catalyze the integration of green chemistry and engineering in the pharmaceutical industry), and the European Union and the United States Environmental Protection Agency (EPA), are concerned with the need to replace conventional hazardous VOCs in favour of safer and bio-based reaction media.2a,66 Indeed, as has been pointed out in the introduction, the appropriate selection of a solvent with a low ecological footprint in a synthetic strategy not only is important for solving global sustainability issues, but also for improving the sustainability of a chemical production process. This is because solvents are heavily used in the pharmaceutical industry as reaction media and in designing purification steps. As a consequence, a number of solvent selection guides have been produced over the years by pharmaceutical industries (e.g., GSK,2d,67 Sanofi,68 Pfizer69), which are useful both for academia and industrial manufacture.In the last two decades, DESs have gained a lot of momentum in the realm of APIs as nanocarriers for increasing the drug delivery and its bioavailability either by creating new formulations or by forming novel eutectic mixtures with APIs themselves.10g,i,70 In this context, the development of sustainable protocols for synthesizing relevant APIs directly in DESs are emerging more and more also for the unique catalytic properties displayed by these mixtures, often resulting in higher selectivities, conversions and yields, shorter reaction times than in conventional VOCs, as well as simpler work-ups and purification procedures.
In the field of transition-metal-catalyzed transformations (see section 2), Pd-catalyzed Suzuki–Miyaura coupling reactions have proven to be effective for synthesizing the analgesic, nonsteroidal, anti-inflammatory drugs Felbinac (3)71 and Diflunisal (7).72 These drugs were prepared in high yields (95–98%) in a ChCl/Gly eutectic mixture and in the absence of additional ligands, by reacting 2-(4-chlorophenyl)acetic acid (1) (Scheme 10a) or 2-hydroxy-5-iodobenzoic acid (4) (Scheme 10b) with potassium phenyltrifluoroborate (2) or potassium 2,4-difluorophenylborate (5), respectively. Of note, the reaction yield of Diflunisal dropped down to 52% starting from the corresponding 2,4-difluoroboronic acid (6).32
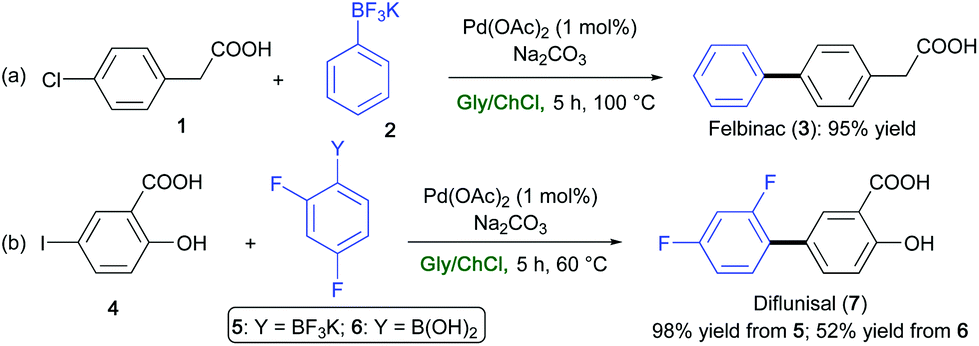 | ||
| Scheme 10 Synthesis of (a) Felbinac (3), and (b) Diflunisal (7) via a Pd-catalyzed Suzuki–Miyaura coupling reaction in ChCl/Gly. | ||
Similarly, the commercially available and cheap Pd/C was found to promote a ligand-free Sonogashira coupling in ChCl/Gly in the presence of CuI (20 mol%) and Et3N between the 6-iodouracil derivative 8 and phenylacetylene (9) to afford the desired alkynylated adduct 10 in 50% yield (Scheme 11).42 Uracil derivatives are privileged structures in drug discovery with a wide array of biological and pharmacological activities.73
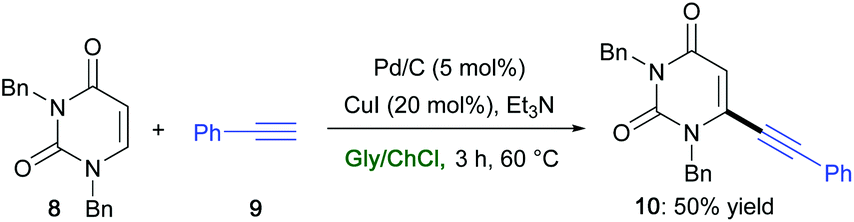 | ||
| Scheme 11 Synthesis of uracil derivative 10via a Pd-catalyzed Sonogashira cross-coupling reaction in ChCl/Gly. | ||
The nucleophilic addition of Grignard and organolithium reagents to chiral non-racemic aromatic and aliphatic N-tert-butanesulfinyl imines was found to proceed smoothly and quickly (2 min reaction time) in the biodegradable eutectic mixture D-sorbitol/ChCl, at ambient temperature and under air, to provide a mixture of diastereomeric sulfinamides in very good yields (74–98%) and with a broad substrate scope.74 Scaling up the process on a gram-scale was also successful. This methodology was applied to the synthesis of the chiral amine side-chain of (R,R)-formoterol (14) (Scheme 12), which is a β2-agonist useful as a bronchodilator in the management of asthma and chronic obstructive pulmonary disease.75
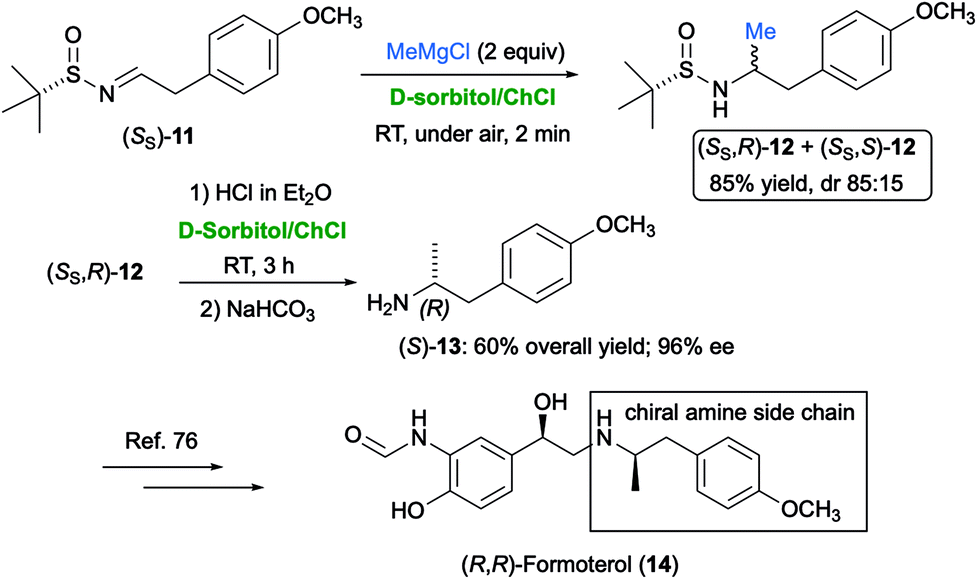 | ||
| Scheme 12 Synthesis of the chiral amine side-chain of (R,R)-formoterol (14) via nucleophilic addition of MeMgCl to sulfinyl imine (SS)-11 in D-sorbitol/ChCl. | ||
To this end, sulfinyl imine (SS)-11 was reacted with MeMgBr to provide a mixture of the two diastereomeric sulfinamides (SS,R)- and (SS,S)-12 [85% overall yield; diastereomeric ratio (dr) = 85:15]. After separation by flash chromatography, the two diastereomers could be deprotected with an acid treatment in the same eutectic mixture, at ambient temperature and under air, to afford the corresponding enantioenriched free amines. Chiral, p-methoxyphenyl-substituted amine (S)-13, which is an important chiral intermediate en route to (R,R)-formoterol (14),76 was isolated in 60% yield and 96% enantiomeric excess (ee) from (SS,R)-12 (Scheme 12).74
The enantiopure (R)-cinacalcet (19) (98% ee), which is a blockbuster drug used as calcimimetic to treat secondary hyperparathyroidism,77 was prepared in only three steps in D-sorbitol/ChCl. First, the enantiopure amine (R)-1-(1-naphthyl)ethylamine (17) was synthesized by reacting MeMgBr with sulfinyl imine (SS)-15, which furnished a mixture of the two diastereomeric sulfinamides (SS,R)- and (SS,R)-16 [86% overall yield; dr = 50:50], with the auxiliary group being removed by acidifying the eutectic mixture with a solution of HCl (2.0 M). Then, the amine (R)-17, obtained from (SS,R)-16, was straightforwardly N-alkylated with alcohol 18 in the presence of the iridium catalyst [Cp*IrCl]2 (1 mol%) to furnish the target drug (R)-19 in 98% yield and 98% ee, after 12 h reaction time at 60 °C working under aerobic conditions (Scheme 13). By performing the above alkylation in toluene at 100 °C for 17 h, in a sealed reactor and under argon, (R)-19 was isolated in an overall yield of only 50%.78 Amine alkylation with alcohols has also been achieved with good results by Williams and co-workers using 1 mol% [Cp*IrI2]2 in water, in the absence of base or other additives.79
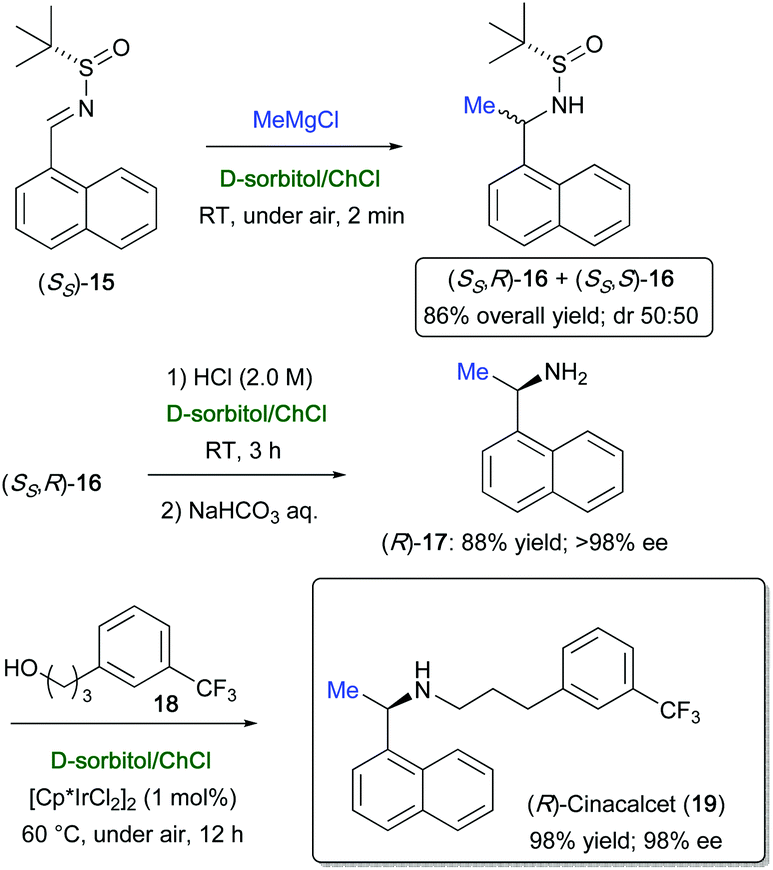 | ||
| Scheme 13 Asymmetric synthesis of (R)-cinacalcet (19) in a D-sorbitol/ChCl eutectic mixture. | ||
Vortioxetine (24) is a multi-modal acting drug with high affinity towards a range of serotoninergic targets, and is now on the market for the treatment of major depressive disorder.80 A transition-metal-free synthesis of thioether 22, which is an important precursor of Vortioxetine,81 has been developed by Gallos and co-workers in ChCl-based eutectic mixtures, like ChCl/Gly or ChCl/urea.82 The reaction occurred between bis(2,4-dimethyl)iodonium bromide (20) (easily accessible from m-xylene) and the commercially available 2-aminothiophenol disulfide (21) using t-BuOK as a base, which was added under stirring at 40–45 °C to the aforementioned mixture in DES in a sealed tube. After 3 h at 80 °C, thioether 22 was isolated in 85–88% yield. The iodoxylene 23, formed as a by-product, could be recycled to 20. The synthesis is also applicable in multi-gram scale (Scheme 14).
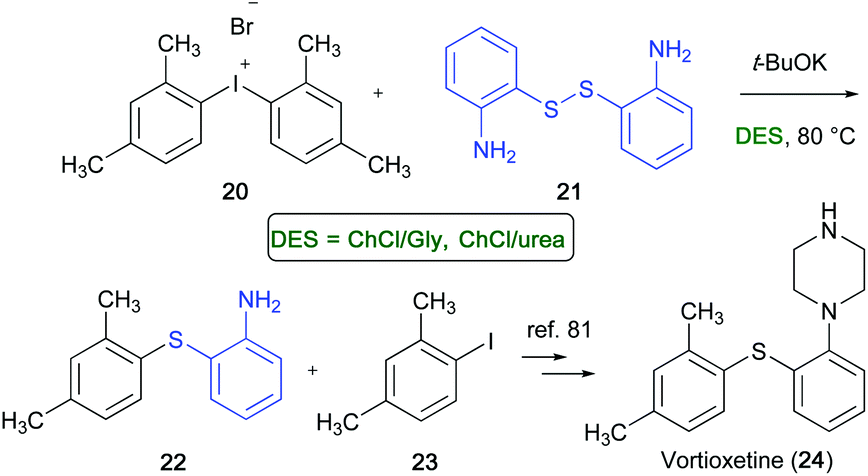 | ||
| Scheme 14 Synthesis of unsymmetrical thioether 22 en route to Vortioxetine (24). | ||
Indole ring and its derivatives have emerged as privileged pharmacophores in thousands of compounds exhibiting a plethora of biological and pharmaceutical activities (e.g., anti-inflammatory, analgesic, antimicrobial, antitumor, and anticonvulsivant).83 An improved synthesis of a key thiazole–indole intermediate (28) en route to selected indolin-2-one derivatives 29, has been set up by synergistically combining the action of a ChCl/urea eutectic mixture with ultrasound (US).84 By reacting 3-bromoacetyl coumarin (27) with hydrazine thioamide (26), in turn prepared from isatin (25), under the aforementioned conditions, compound 28 was isolated in a yield as high as 95% after only 1 h (Scheme 15). On the other hand, by performing the same organic transformation in conventional VOCs (e.g. dioxane), adduct 28 was isolated in 44–68% yield after 3–4 h.84 It was envisaged that the extended network of hydrogen bonds typical of DES may be playing a key role in catalyzing the transformation. This was supported by DFT calculations at the B3LYP/6-31+G(d,p) level of theory. The synthesized indolin-2-one derivatives 29 showed good anti-inflammatory and/or analgesic activity as compared to the standard drugs.
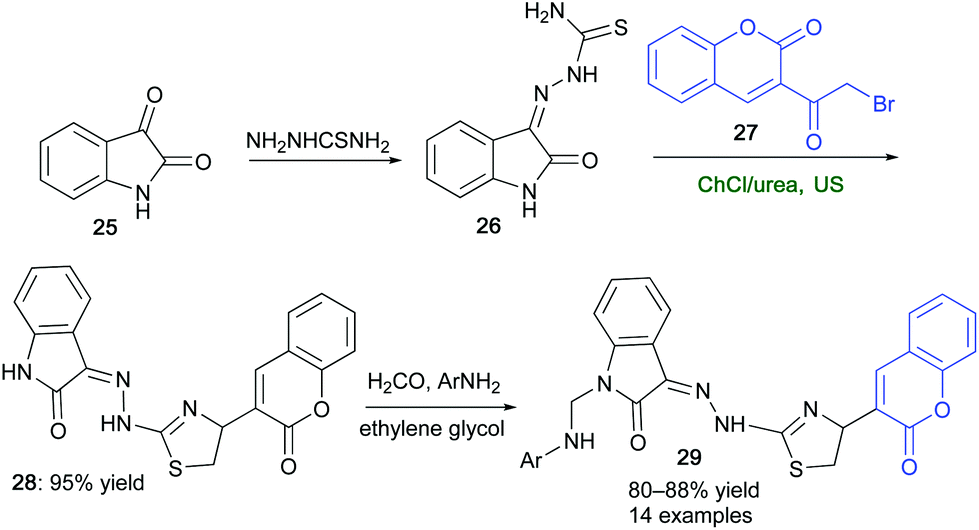 | ||
| Scheme 15 Synthesis of a key thiazole–indole derivative (28) by combining ChCl/urea and US towards indolin-2-ones 29. | ||
Isoxazoline derivatives are biologically important scaffolds with antibacterial, analgesics, anti-inflammatory, antitubercular, and anticancer activity.85 With the aim of synthesizing selected derivatives in an eco-friendly manner, novel DESs formed by mixing benzalkonium chloride (BZK) and urea in different molar ratios were prepared and physically and spectroscopically characterized by Bakht and co-workers.86 By reacting chalcone derivatives 30 with hydroxylamine hydrochloride, it was found that the best performance in the synthesis of isooxazoline derivatives 31, in terms of reaction time (up to 60 min), yield (up to 85%) and energy utilization (up to 88% energy saved), were achieved using the combination BZK:urea (1:2)-US. By applying the nonultrasonic thermal method, lower yields of 31 were obtained (up to 71%) with longer reaction times ranging from 15 to 4–5 h if conventional VOCs or the above DES are used, respectively (Scheme 16).
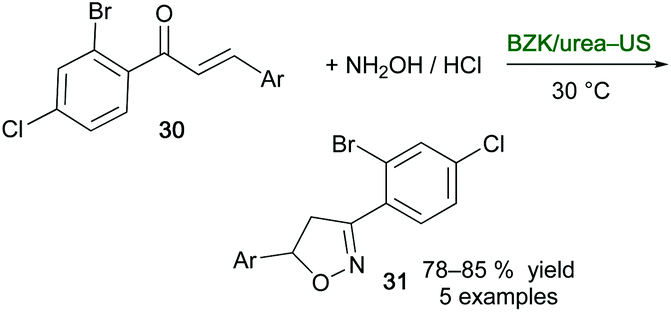 | ||
| Scheme 16 Synthesis of isooxazolines 31 from chalcone derivatives 30 using a combination of benzalkonium chloride (BZK)/urea and US. | ||
Fused pyrazolopyranopyrimidines are interesting compounds displaying a wide spectrum of biological and pharmacological activities.87 A multicomponent domino reaction, aimed at obtaining biologically active tricyclic-fused pyrazolopyranopyrimidines, has been devised by Bhosle and co-worker using DESs as environmentally benign reaction media with the dual role of solvents and catalysts.88 After extensive screening, it was found that the cyclocondensation of various aromatic aldehydes, barbituric acid, hydrazine hydrate, and ethyl acetoacetate took place smoothly in a ChCl/urea (1:2) eutectic mixture, in the presence of EtOH, to provide valuable pyrazolopyranopyrimidine derivatives 32 with a broad substrate scope and very good yields (75–92%) within 1 h at 80 °C (Scheme 17).88 Lower yields (21–34%) were instead obtained after heating the reaction mixture at 80 °C for several hours using VOCs as reaction media. The feasibility of recycling the above eutectic mixture was evaluated, and a plausible reaction mechanism for the DES-catalyzed domino reaction was also proposed.
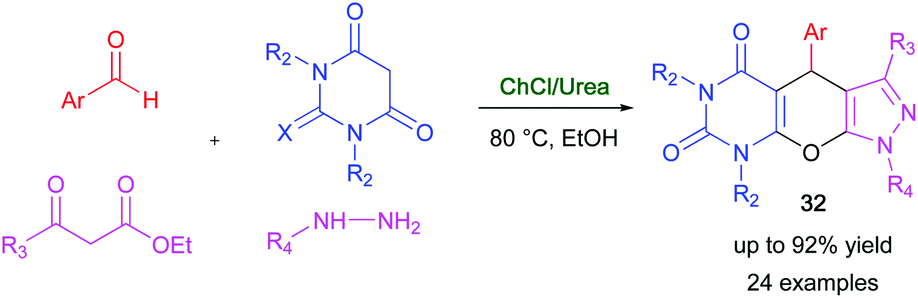 | ||
| Scheme 17 Synthesis of fused pyrazolopyranopyrimidines 32 in ChCl/urea. | ||
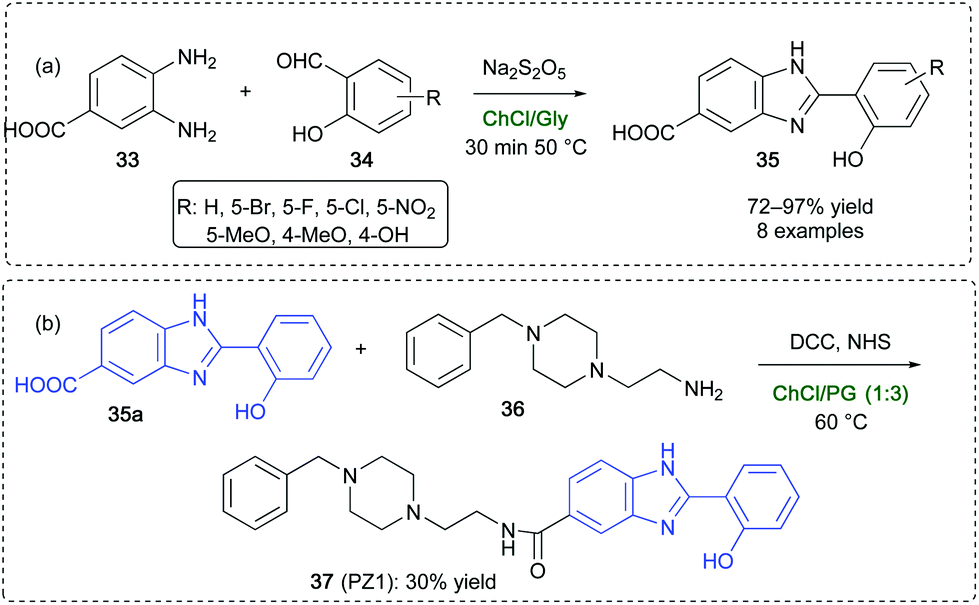
Benzimidazole-based structures are found in many drugs and drug-like candidates89 (like the antihypertensive Telmisartan and the antiviral compound Maribavir), in antiulcer agents (like the blockbuster drug Omeprazole),90 and in several β-secretase inhibitors and Alzheimer's disease targets.91 An efficient cyclodehydration reactions for the synthesis of 2-hydroxybenzimidazoles 35 has been developed by Capriati and coworkers by reacting 3,4-diaminobenzoic acid 33 with a variety of salicylaldehyde derivatives 34 in ChCl/Gly (1:2) using Na2S2O5 as the oxidant.92 Compared to VOCs, the synthesis of 35 in the above DES occurs in better yields (up to 97%), and has the advantage of shorter reaction time (30 min vs. 12 h) and milder reaction conditions (50 vs. 100 °C) (Scheme 18a). In addition, the whole synthesis of the Donepezil-like, hit compound PZ1 (37), which is known to be a new promising multi-target directed compound in Alzheimer's disease treatment,93 has been performed and optimized in DESs taking advantage of shorter reaction time in each single step and/or the avoidance of harsh conditions and toxic and harmful reagents/VOCs.92 The one-pot two-step synthesis of PZ1 has also been achieved in a ChCl/propylene glycol (PG) (1:3) by reacting the N-benzylated adduct 36 with 2-hydroxyphenylbenzimidazole derivative 35a in the presence of N,N′-dicyclohexylcarbodiimide (DCC) and N-hydroxysuccinimide (NHS), with an overall yield of 30% at 60 °C after 60 h reaction time, compared to the 21% yield obtained when working in DMF at 25 °C for 60 h (Scheme 18b).
 | ||
| Scheme 18 Synthesis of (a) 2-hydroxyphenyl benzimidazole derivatives 35 and (b) Donepezil-like compound PZ1 37 in DES. | ||
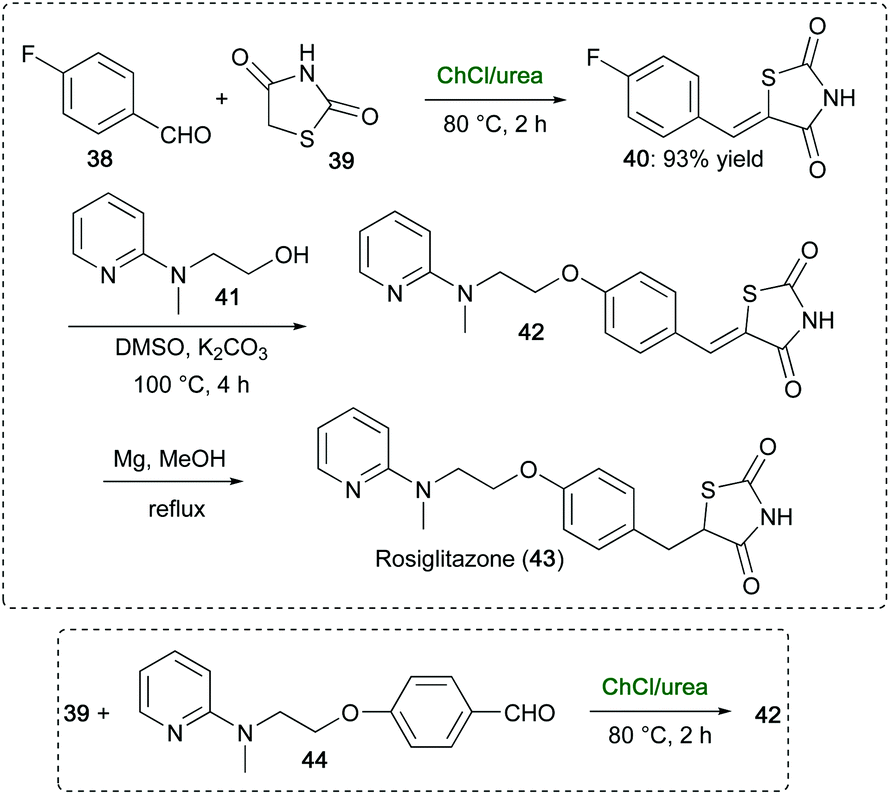
An attractive, scalable route for the synthesis of rosiglitazone (43), a drug used to treat type 2 diabetes mellitus,94 has been developed by Mane and co-workers avoiding toxic/hazardous chemicals/solvents reported for other routes.95 This multistep route made use of ChCl/urea in a key step to promote the Knoevenagel condensation of 4-fluorobenzaldehyde (38) with 2,4-thiazolidinedione (39), which furnished 40 in 93% yield within 2 h at 80 °C (Scheme 19). Compound 40 was then condensed with pyridinylamino alcohol 41 to give adduct 42, which on reduction with Mg-MeOH gave rosiglitazone (43). Intermediate 42 could also be synthesized in ChCl/urea, within 2 h at 80 °C, via condensation of 39 with pyridinylamino benzaldehyde 44, thereby avoiding the use of toluene and piperidine, which are recommended in similar condensation reactions reported by Cantello and co-workers (Scheme 19).94
 | ||
| Scheme 19 Sustainable and improved synthesis of rosiglitazone drug using ChCl/urea in the synthesis of key intermediates 40 and 42. | ||
Quinazolinones are key heterocyclic cores in many naturally occurring alkaloids and marketed drugs [e.g., methaqualone (quaalude), mebroqualone, mecloqualone (Casfen)] with sedative, hypnotic and anxiolytic properties.96 Nagarajan and Ghosh have recently reported on a DES-mediated synthesis of two important natural alkaloids like bouchardatine and schizocommunin,97 the former known to have biological properties as anti-cancer, anti-inflammatory and anti-tubercolar activities,98 the latter showing a cytotoxic activity against murine linphoma cells and human hepatoma cells, and also knowing to be a potent activator of the aryl hydrocarbon receptor.99
The quinazolinone skeleton of bouchardatine (48) was obtained by preliminarily reacting anthranilamide (45) with indole-2-carboxaldehyde (46) in a L-(+)-tartaric acid–DMU (3:7) melt at 90 °C for 8 h to give the quinazolinone intermediate 47 in 81% yield, which was finally formylated at indole C3 position to provide 48, according to a procedure previously reported by the same research group (Scheme 20a).100 Schizocommunin (52), in turn, was prepared by first treating 45 with acetaldehyde (49) in the above green melt at 90 °C for 2 h to give dihydroquinazolinone 50 in 79% yield. Next, 50 was aromatized with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) to furnish quinazolinone 51 in 85% yield. Target drug 52 was finally obtained in one-step from 51, following Nishida's procedure (Scheme 20b).101 In a similar way, drugs like methaqualone (56a), mecloqualone (56b), mebroqualone (56c), and the alkaloid methyl 2-(2-methyl-4-oxoquinazolin-3(4H)-yl) benzoate 56d, all featuring the 2,3-disubstituted quinazolinone skeleton (56), could be prepared in good overall yields (78–89%) by reacting in the aforementioned low melting mixture at 90 °C for 2–6 h the selected 2-amino-N-(substituted)benzamide 53 with the appropriate aldehyde 54, followed by aromatization with DDQ of the corresponding dihydroquinazolinones 55 (Scheme 20c).97
 | ||
| Scheme 20 Synthesis of (a and b) 2-substituted quinazolinone alkaloids bouchardatine (48) and schizocommunin (52), and (c) 2,3-disubstituted quinazolinones (56) (drugs and natural products). | ||
4. Biocatalyzed transformations in aqueous DES mixtures
Biocatalytic reactions carried out in DESs as unconventional reaction media have experienced an accelerated development over the last years as testified by the impressive number of papers published dealing with either biocatalysis promoted by whole-cell microorganisms102 or by isolated enzymes (e.g., alcohol-dehydrogenases,14c,103 lyases,104 lipases,105 laccases,106 hydrolases,107 decarboxylases,108etc.).109 In this section, some important points and updates related to whole-cell biocatalysis in DESs over that run in an aqueous environment will be discussed, paying particular attention (a) to the green aspects and cost-benefits resulting from this catalysis as compared to that of isolated enzymes, and (b) to the biocatalyst performance when using environmentally responsible DES media110 due to higher substrate loadings111 and stability,112 and an improved (or opposite) selectivity displayed by the biocatalyst itself.113The behaviour of lipases and oxidoreductases in DESs and in DES/aqueous mixtures has been thoroughly investigated,105,114 also with the support of molecular dynamics simulations, that have revealed the importance of the role of “water content” in these systems,103,115 thereby confirming that water layer is necessary for achieving an efficient conformational flexibility of the biocatalyst, which is the key to maintain its catalytic activity.116
Water addition to DES mixtures allows a lower viscosity of the medium, while preserving the supramolecular network of DES with percentage up to 42 wt% water,8a,9 with the resulting biotransformations often displaying an increased productivity yield in comparison with water or buffer as the sole reaction medium. Solvent choice in organic reactions is also known to influence green chemistry metrics, which are used to evaluate the real sustainability of biocatalytic processes. These include, among others, productivity (g product per L reactor per h), the E-factor (kg waste per kg product), water intensity (g water used per g product), and solvent intensity (g solvent used per g product).
The ideal medium of enzymes and cells is water, but in many synthetic processes it has been seen that organic reagents and catalysts are much more prone to non-polar or even anhydrous solvents.111a Compared to bio-transformations mediated by isolated enzymes, whole cell biocatalytic processes have major advantages such as low costs, an easier catalyst preparation, intracellular enzymes stability in their natural environment, and an accessible cofactors regeneration in situ.
Biocatalyzed synthesis of steroids, which are worldwide used in clinical therapy as endocrine regulators, allow higher stereo- and regioselectivity and efficiency when compared to other synthetic approaches. Different DESs have been screened to overcome the problem of the poor solubility of steroids in aqueous solutions. The addition of 6% (v/v) of ChCl/urea (1:2) boosted the dehydrogenation of cortisone acetate into prednisone acetate by Arthrobacter simplex,117 whereas the mixture ChCl/Gly (1:2) was found to improve the 15α-hydroxylation of D-ethylgonendione (57) to the corresponding 15α-hydroxy-D-ethylgonendione (58), mediated by Penicillium raistrickii. This is a key biotransformation in the industrial synthesis of Gestodene steroids (Scheme 21).118 In the absence of DESs, 58 was isolated in 33% yield only, after 72 h incubation of Penicillium raistrickii mycelium with 57 (2 g L−1). On the other hand, the suspension of this microorganism in a solution containing 4% (v/v) ChCl/Gly (1:2) improved the conversion up to 82% thanks to a beneficial effect of DES on the substrate solubility, and the mass transfer within mycelium cells.
 | ||
| Scheme 21 Biotransformation of D-ethylgonendione (57) to 15α-hydroxy-D-ethylgonendione (58), mediated by Penicillium raistrickii in ChCl/Gly. | ||
DESs have been recently employed also in the flavonoids synthesis, which is an important process in the production of APIs, flavors and fragrances. The production of L-rhamnose and isoquercitrin from the enzymatic hydrolysis of rutin by recombinant Aspergillus niger α-L-rhamnosidase glycosyl hydrolase was recently reported in a buffer mixture with ChCl–urea. The latter mixture, which was selected as the best co-solvent among other 24 different DESs tested, was found to increase by 1.12 times the synthesis of both L-rhamnose and isoquercitrin from rutin. ChCl/urea was also proven to be an efficient extractive solvent for L-rhamnose and isoquercitrin, which were isolated in 96% and 94% purity, respectively, by an aqueous two-phase systems technology.119
The aforementioned biotransformation of rutin (59) into isoquercitrin (60) was also investigated using whole cells in various eutectic mixtures, which were added to the aqueous medium containing E. coli cells expressing α-L-rhamnosidase. While the observed catalytic activity in ChCl/GIy (1:2), ChCl/EG (1:2) and ChCl/acetamide (1:2) resulted only slightly higher compared to the PBS control (52.9%), the yield of isoquercitrin increased to up to 66.7% when using ChCl/urea (Scheme 22). The latter mixture was also found to increase the cell permeability of the E. coli BL21-pET21a-rhaB1 whole-cell biocatalyst by increasing the contact between the enzyme and the substrate, thereby improving the catalytic performance in the isoquercitrin production.120
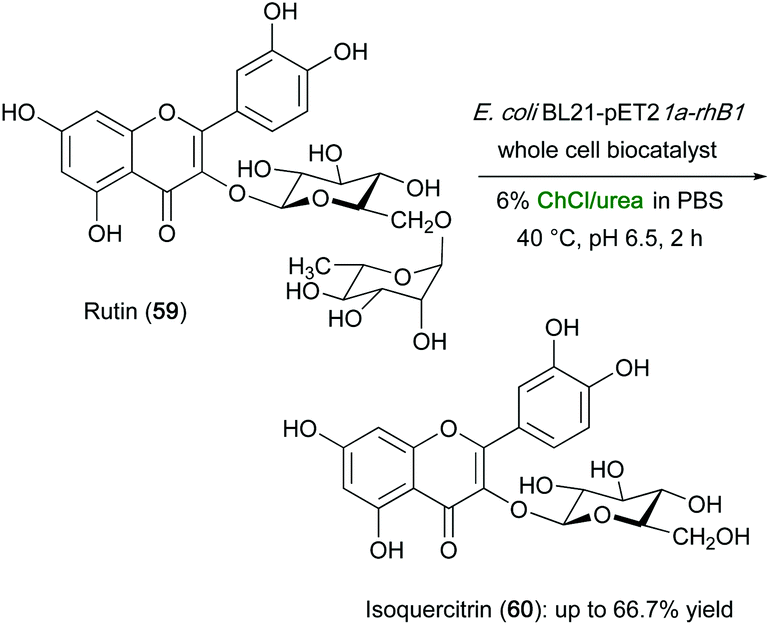 | ||
| Scheme 22 Enzymatic hydrolysis of rutin (59) to isoquercitrin (60) using recombinant E. coli whole cells in aqueous DESs. | ||
Similarly, four DESs and 13 NADESs have been screened in the bioreduction of 4-(trifluoromethyl)acetophenone (61) to (R)-1-[4-(trifluoromethyl)phenyl]ethanol (62) promoted by the recombinant E. coli LXCAR-S154Y and Geotrichum ZJPH1810 biocatalysts (Scheme 23).121 Compound 62 is a key intermediate in the preparation of several pharmaceuticals, including the chemokine CCR5 antagonist, which is widely used for the treatment of AIDS, the DP1 receptor antagonist, and the antifungal econazole.121 The desired secondary alcohol was isolated in high yields (up to 92.4%) and enantioselectivity (up to >99%), also on a preparative scale. The best results were obtained when betaine (B), L-proline (Pro) and L-carnitine were used as HBAs in place of ChCl, with an improved catalytic efficiency due to the preservation of cell membrane integrity by the above DES components and an effective coenzyme regeneration. In addition, lysine (Lys)-based NADES were found to reverse the enantioselectivity of the bioreduction when the reaction was carried out with high water content in the reaction medium. With the B/Lys (1:2) mixture, yields improved up to 88.1% in the presence of 400 mM of substrate, whereas the product was isolated in 85.2% yields when using the B:glucose (Glc):Gly (1:1:2) mixture. A lower yield (77%) was observed when the biotransformation was carried out in an aqueous buffer system, with the same 400 mM substrate concentration.121
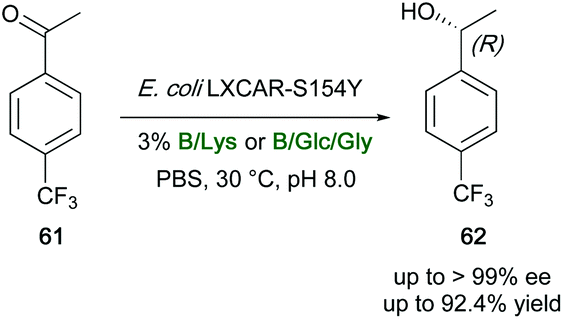 | ||
| Scheme 23 Bioreduction of acetophenone derivative 61 to the secondary alcohol 62 by recombinant E. coli cells in various aqueous eutectic mixtures. | ||
Various NADESs have also been screened in the bioreduction of 2-chloro-1-(3,4-difluorophenyl)ethanone (63) to (S)-2-chloro-1-(3,4-difluorophenyl)ethanol (64), which is a key chiral synthon for the synthesis of P2Y12 receptor antagonist Ticagrelor (Brilinta®), by the recombinant E. coli BL21 (DE3), expressing a carbonyl reductase from Leifsonia xyli HS0904.122 Mixtures of ChCl salts and amino acids [e.g., ChCl/glycine (Gly), ChCl/glutathione (GSH), ChCl/Glc, ChCl/alanine (Ala), choline acetate (ChAc)/GSH, ChAc/glutamic acid (Glu)] were found to affect strongly the pH of the medium when amounts up to 1% w/v were added to cells suspension with respect to buffer solution, in many cases with a decreased reaction yield, probably due to a change of the binding of carbonyl reductase to the substrate. Conversely, E. coli BL21 (DE3) efficiently allowed the asymmetric production (also on a preparative scale) of (S)-64 after incubating cells in a reaction medium containing ChAc/Lys, the substrate loading being 3.3-fold higher than in a pure aqueous mixture. The enantiopure chloridrin was isolated in a yield (87%) higher than that obtained when the biotransformation was run in a buffer solution (68%), and with an enantioselectivity up to 99% ee (Scheme 24). It was also demonstrated that the eutectic mixture ChAc/Lys improved NADH coenzyme regeneration of 1.4–2.0-fold, compared to that resulting by the addition of the single components of the eutectic mixture at the same molar ratio.
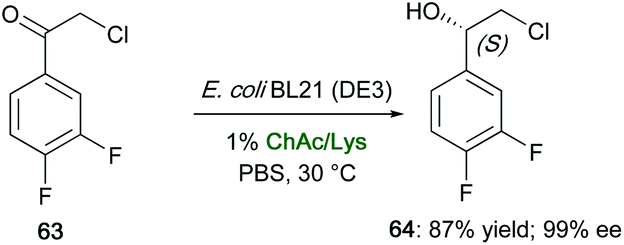 | ||
| Scheme 24 Bioreduction of α-chloro ketone 63 to the chloridrin 64 by the recombinant E. coli BL21 (DE3) whole cells in the presence of ChAc/Lys as a co-solvent. | ||
Similar results have been reported by Lou and co-workers,123 who investigated the bioreduction of 3-chloropropiophenone (65) to (S)-3-chloro-1-phenylpropanol (66) by means of immobilized whole cells of Acetobacter CCTCC M 209061 in the presence of different DESs as co-solvents. The results collected showed that while acidic HBDs strongly inactivated the carbonyl reductase of Acetobacter, secondary alcohol 66 was produced in the highest yield of 82.3% (>99% ee) in the presence of 5% v/v of ChCl/urea, with an increased ketone concentration up to 3-fold with respect to buffer control (Scheme 25).
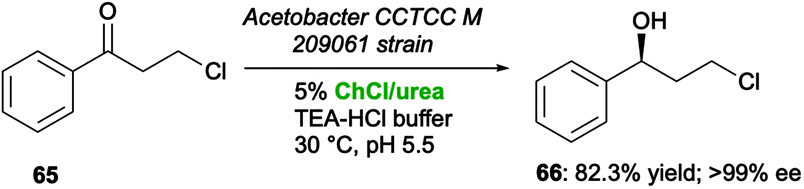 | ||
| Scheme 25 Bioreduction of β-chloroketone 65 to β-chloroalcohol 66 by immobilized whole cells of Acetobacter CCTCC M 209061 in the presence of ChCl/urea as a co-solvent. | ||
The impact of 24 DESs and 21 NADESs as co-solvents (up to 20% v/v) was examined in the biotransformation of isoeugenol (67) to vanillin (68) catalyzed by Lysinibacillus fusiformis CGMCC1347 whole cells (Scheme 26).124 Both types of solvents improved the production yields up to 142% and 132% compared to the ones obtained in (NA)DES-free aqueous systems (Scheme 26). The reaction yields, in particular, were found to depend on the nature of the HBD component of the NADES. The order of efficiency was organic acids < alcohols < sugars, since HBDs affected the pH of the reaction medium. The results showed that a pH = 7.0 was the optimal for the bioconversion of isoeugenol into vanillin. Cell immobilization in polyvinyl alcohol–alginate beads increased even more the production yield than in pure water (up to 181%), with the catalytic activity still maintained for at least 13 cycles. NADESs used as cosolvents also increased both the solubility of isoeugenol up to 1.9–2.7 fold and the production yield with respect to the same transformation run in aqueous systems, thanks to an improved permeability of the cell membrane, resulting by the combination of a surfactant action without decreasing the cell viability.
 | ||
| Scheme 26 Bioconversion of isoeugenol (67) to vanillin (68) catalyzed by Lysinibacillus fusiformis CGMCC1347 whole cells in DES or NADES mixtures. | ||
The exploitation of the biocatalysis in NADES by the Candida tropicalis 104 whole cells was investigated in the asymmetric synthesis of (S)-1-[3,5-bis(trifluoromethyl)phenyl]ethanol (70), which is a key intermediate towards pharmaceutic lipid-lowering agents,125 from the corresponding carbonyl compound 69 (Scheme 27). The reaction yield increased from 74% to 86% when such a bioreduction was carried out in the presence of 1% w/v of ChCl:trehalose (Tre) (1:1) mixture, with respect to the sole PBS buffer medium. ChCl:Tre, like other DES mixtures, enhanced the cell viability and the cell membrane permeability to the substrate, while exerting a protective effect against ketone cytotoxicity. This allowed an higher substrate charge up to 200–300 mM, with a consequent 6-fold increase of the substrate-to-catalyst ratio of the process with respect to that performed in water alone.126
 | ||
| Scheme 27 Bioreduction of acetophenone derivative 69 to the corresponding secondary alcohol 70 by Candida tropicalis104 whole cells in the presence of ChCl/Tre as a co-solvent. | ||
Chemo-enzymatic multi-step processes represent a new challenge in the “green chemistry” approach, since they require the use of chemical and biological catalysts together. Biphasic systems are, in general, the way to carry out such processes,127 using batch conditions and discontinuous systems, although non-continuous processes represent a limitation for semi-preparative or large-scale syntheses.128 Since the first pioneering works of Domínguez de María and co-workers on the tandem combination of proline-based organocatalytic and lipase transesterification reactions in DES mixtures,129 other research groups have reported on the successful combination of various DES/water monophasic systems for organo-, bio-, and metal-catalyzed transformations. Several sustainable bio-reductions promoted by isolated enzymes jointly with metal-catalyzed reactions have been carried out in eutectic mixtures and/or water to obtain enantioenriched chiral products by means of well-projected chemo-enzymatic sequences in which the compatibility of each catalyst with previous catalysts/reagents has been carefully evaluated.14b,d,19d,130
On the other hand, the use of whole-cell biocatalysts within multistep cascade processes is still in its infancy. To date, there are only a few examples in which the addition of bio-sourced eutectic mixtures to the reaction medium was proven to increase the biocatalytic efficiency of a whole-cell-mediated process, without other complicated strategies.131 A low efficiency was often observed especially when heating or monophasic media were required, either because of the incompatibility/toxicity of metal- or chemical catalysts for the cells viability or because of the metal catalyst inactivation/chelation/leaking in the presence of proteins or metabolites.132 Thus, major efforts need to be made towards this direction, in order to benefit of the advantages of whole cells also in chemo-enzymatic processes.133
5. Conclusions
The main purpose of this short review has been to give the reader an overview on the feasibility of performing sustainable metal- and biocatalized reactions as well as the synthesis of relevant APIs and other biologically active compounds when using water or DESs as privileged reaction media. All through this review, we have tried to trace a comparison between the different solvent performances, which sometimes are similar, other times are different. Although water and DESs share several physicochemical properties (see Introduction), the solubility of substrates/catalysts is not always the same in these solvents. Thus, the fact of working under homogeneous/heterogeneous conditions should be, each time, carefully evaluated in order to provide a rationalization of the observed results, and to get an understanding of the mechanisms thereof.Major breakthroughs are expected to come (a) by the use of cheap earth-crust abundant metals in place of noble and expensive metals as catalysts in cross-coupling reactions performed directly in DES mixtures or water, at ambient temperature and in air, and (b) by scaling up DES/water-based processes, which are traditionally carried out in batch reactors, operating under a continuous flow regime. This is important to boost a sustainable industrial production of pharmaceutically relevant drugs. As for this last point, recent papers highlighting the possibility of (a) achieving an efficient continuous synthesis of DESs,134 or (b) combining DESs with the microflow technology,15b,135 are truly inspiring and encouraging towards this goal.
Thanks to the biotechnological progresses over the last decades, a number of recombinant biocatalysts expressing the enzymes necessary for the desired transformations have been engineered, with reduction of side reactions. The benefits arising from this technology could be hopefully expanded in DES media, with potential amazing outcomes. Medium engineering136 and recombinant biocatalysts engineering will certainly allow in the next future more improvements in multistep chemo-enzymatic stereoselective processes with whole cells, through a rationale project of high-tolerant and more efficient whole-cell catalysts, even immobilized on natural supports. If successful, this will reduce the number of reaction steps, thereby avoiding the use of additional VOCs for the purification or extraction of products and intermediates, and will certainly contribute to solve the problem of the environmental impact of many synthetic processes.
We hope that this review can contribute to further raise the interest of researchers and practitioners turning their attention on the possibility (and the advantages) of building a truly sustainable chemistry for the studied transformations based on water or bio-based solvents.
Author contributions
All authors contributed to all aspects of manuscript preparation, revision and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out under the framework of the National PRIN project “Unlocking Sustainable Technologies Through Nature-Inspired Solvents” (Code: 2017A5HXFC_002) and financially supported by MUR, the Interuniversity Consortium C.I.N.M.P.I.S., and the University of Bari.Notes and references
- (a) F. Cavani, G. Centi, S. Perathoner and F. Trifirò, in Sustainable industrial chemistry, Wiley-VCH, Verlag Gmbh & Co. KGaA, 2009 CrossRef; (b) R. Dach, J. J. Song, F. Roschangar, W. Samstag and C. H. Senanayake, Org. Process Res. Dev., 2012, 16, 1697 CrossRef CAS; (c) S. Sharma, J. Das, W. M. Braje, A. K. Dash and S. Handa, ChemSusChem, 2020, 13, 2859 CrossRef CAS; (d) J. L. Tucker, Org. Process Res. Dev., 2010, 14, 328 CrossRef CAS.
- (a) T. Laird, Org. Process Res. Dev., 2012, 16, 1 CrossRef CAS; (b) B. H. Lipshutz, Chemistry, 2018, 4, 2004 CrossRef CAS; (c) C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927 RSC; (d) C. S. Slat, M. J. Savelski and W. A. Carole, in Solvent use and waste issues in green chemistry in the pharmaceutical industry, Wiley-VCH, Verlag Gmbh & Co. KGaA, 2009, p. 49 Search PubMed; (e) R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854 RSC.
- (a) P. T. Anastas and J. C. Warner, in Green chemistry: Theory and practice, Oxford University Press, New York, 1998 Search PubMed; (b) C. S. Slater and M. Savelski, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2007, 42, 1595 CrossRef CAS.
- (a) A. P. Abbott, Chem. Commun., 2003, 1, 70 RSC; (b) M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2018, 1 Search PubMed.
- (a) Y. Liu, J. B. Friesen, J. B. McAlpine, D. C. Lankin, S. N. Chen and G. F. Pauli, J. Nat. Prod., 2018, 81, 679 CrossRef CAS; (b) C. Florindo, L. C. Branco and I. M. Marrucho, ChemSusChem, 2019, 12, 1549 CrossRef CAS; (c) D. J. G. P. van Osch, C. H. J. T. Dietz, J. van Spronsen, M. C. Kroon, F. Gallucci, M. v. S. Annaland and R. Tuinier, ACS Sustainable Chem. Eng., 2019, 7, 2933 CrossRef CAS.
- (a) K. Radosevic, M. C. Bubalo, V. G. Srcek, D. Grgas, T. L. Dragisevic and I. R. Redovnikovic, Ecotoxicol. Environ. Saf., 2015, 51, 4877 Search PubMed; (b) M. Hayyan, Y. P. Mbous, C. Y. Looi, W. F. Wong, A. Hayyan, Z. Salleh and O. Mohd-Ali, SpringerPlus, 2016, 5, 913 CrossRef.
- (a) Y. H. Choi, J. van Spronsen, Y. Dai, M. Verbene, F. Hollmann, I. W. C. E. Arends, G.-J. Witkamp and R. Verpoorte, Plant Physiol., 2011, 156, 1701 CrossRef CAS; (b) Y. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61 CrossRef CAS.
- (a) Y. Dai, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Food Chem., 2015, 187, 14 CrossRef CAS; (b) T. El Achkar, S. Fourmentin and H. Greige-Gerges, J. Mol. Liq., 2019, 288, 111028 CrossRef CAS.
- (a) O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736 RSC; (b) L. Weng and M. Toner, Phys. Chem. Chem. Phys., 2018, 20, 22455 RSC; (c) R. Ahmadi, B. Hemmateenejad, A. Safavi, Z. Shojaeifard, A. Shahsavar, A. Mohajeri, M. Heydari Dokoohaki and A. R. Zolghadr, Phys. Chem. Chem. Phys., 2018, 20, 18463 RSC; (d) O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782 CrossRef CAS.
- Selected reviews/book chapters: (a) E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060 CrossRef CAS; (b) J. García-Álvarez, Eur. J. Inorg. Chem., 2015, 5147 CrossRef; (c) D. Alonso, A. Baeza, R. Chinchilla, G. Guillena, I. M. Pastor and D. Ramón, Eur. J. Org. Chem., 2016, 612 CrossRef CAS; (d) F. M. Perna, P. Vitale and V. Capriati, Curr. Opin. Green Sustain. Chem., 2020, 21, 27 CrossRef; (e) T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202010963; (f) A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063 CrossRef CAS; (g) D. Carriazo, M. C. Serrano, M. C. Gutierrez, M. L. Ferrer and F. D. Monte, Chem. Soc. Rev., 2012, 41, 4996 RSC; (h) Z. Yang, Adv. Biochem. Eng./Biotechnol., 2019, 168, 31 CrossRef CAS; (i) Q. Zhang, K. D. O. Vigier, S. Royer and F. Jerôme, Chem. Soc. Rev., 2012, 41, 7108 RSC; (j) M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074 CrossRef CAS; (k) H. Vanda, Y. Dai, E. G. Wilson, R. Verpoorte and Y. H. Cho, C. R. Chim., 2018, 21, 628 CrossRef CAS; (l) L. Benvenutti, A. A. F. Zielinski and S. R. S. Ferreira, Trends Food Sci. Technol., 2019, 90, 133 CrossRef CAS; (m) A. V. Gómez, A. Biswas, C. C. Tadini, R. F. Furtado, C. R. Alves and H. N. Cheng, J. Braz. Chem. Soc., 2019, 30, 717 Search PubMed; (n) A. Mišan, J. Nađpal, A. Stupar, M. Pojić, A. Mandić, R. Verpoorte and Y. H. Choi, Crit. Rev. Food Sci. Nutr., 2020, 60, 2564 CrossRef; (o) Deep Eutectic Solvents and Their Applications as New Green and Biorenewable Reaction media, in Handbook of Solvents, Vol. 2: Use, Health, and Environment, ed. G. Wypych, ChemTec Publishing, Toronto, Canada, 3rd edn, 2019 Search PubMed; (p) A. Kovács, E. C. Neyts, I. Cornet, M. Wijnants and P. Billen, ChemSusChem, 2020, 13, 3789 CrossRef; (q) Deep Eutectic Solvents: Synthesis, properties, and Applications, ed. D. J. Ramón and G. Guillena, Wiley-VCH, Weinheim, 2019, pp. 1–384 Search PubMed; (r) B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangor, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00385.
- (a) R. Breslow, The principles of and reasons for using water as a solvent for green chemistry, in Handbook of green chemistry, WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed; (b) C.-J. Li and T.-H. Chan, Organic reactions in aqueous media, John Wiley & Sons, New York, 1997 Search PubMed; (c) C.-J. Li and T.-H. Chan, Comprehensive organic reactions in aqueous media, Wiley-interscience, 2007 CrossRef; (d) C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68 RSC; (e) K. Manabe and S. Kobayashi, Chem. – Eur. J., 2002, 8, 4094 CrossRef CAS; (f) C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS; (g) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS; (h) R. N. Butler and A. G. Coyne, Chem. Rev., 2009, 110, 6303 Search PubMed; (i) R. N. Butler and A. G. Coyne, J. Org. Chem., 2015, 80, 1809 CrossRef CAS; (j) R. N. Butler and A. G. Coyne, Org. Biomol. Chem., 2016, 14, 9945 RSC; (k) F. Zhou and C.-J. Li, Chem. Sci., 2019, 10, 34 RSC; (l) T. Kitanosono and S. Kobayashi, Chem. – Eur. J., 2020, 26, 9408 CrossRef CAS.
- (a) C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2014, 53, 5969 CrossRef CAS; (b) V. Mallardo, R. Rizzi, F. C. Sassone, R. Mansueto, F. M. Perna, A. Salomone and V. Capriati, Chem. Commun., 2014, 50, 8655 RSC; (c) F. C. Sassone, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Commun., 2015, 51, 9459 RSC; (d) L. Cicco, J. M. Rodríguez-Álvarez, F. M. Perna, J. García-Álvarez and V. Capriati, Green Chem., 2017, 19, 3069 RSC; (e) L. Cicco, S. Sblendorio, R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Sci., 2016, 7, 1192 RSC; (f) C. Vidal, J. García-Álvarez, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Angew. Chem., Int. Ed., 2016, 55, 16145 CrossRef CAS; (g) G. Dilauro, M. Dell'Aera, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2017, 56, 10200 CrossRef CAS; (h) A. Sánchez-Condado, G. A. Carriedo, A. Presa Soto, M. J. Rodríguez-Álvarez, J. García-Álvarez and E. Hevia, ChemSusChem, 2019, 12, 3134 CrossRef; (i) L. Cicco, A. Fombona-Pascual, A. Sánchez-Condado, G. A. Carriedo, F. M. Perna, V. Capriati, A. Presa Soto and J. García-Álvarez, ChemSusChem, 2020, 13, 4967 CrossRef CAS; (j) S. Ghinato, G. Dilauro, F. M. Perna, V. Capriati, M. Blangetti and C. Prandi, Chem. Commun., 2019, 55, 7741 RSC; (k) A. F. Quivelli, G. D'Addato, P. Vitale, J. García-Álvarez, F. M. Perna and V. Capriati, Tetrahedron, 2020 DOI:10.1016/j.tet.2020.131898.
- N. Ríos-Lombardía, L. Cicco, K. Yamamoto, J. A. Hernández-Fernández, F. Morís, V. Capriati, J. García-Álvarez and J. González-Sabín, Chem. Commun., 2020, 15, 15165 RSC.
- (a) R. A. Sheldon, Chem. – Eur. J., 2016, 22, 12984 CrossRef CAS; (b) L. Cicco, N. Ríos-Lombardía, M. J. Rodríguez-Álvarez, F. Morís, F. M. Perna, V. Capriati, J. García-Álvarez and J. González-Sabín, Green Chem., 2018, 20, 3468 RSC; (c) V. Gotor-Fernández and C. E. Paul, J. Biotechnol., 2019, 293, 24 CrossRef; (d) P. Vitale, F. Lavolpe, F. Valerio, M. Di Biase, F. M. Perna, E. Messina, G. Agrimi, I. Pisano and V. Capriati, React. Chem. Eng., 2020, 5, 859 RSC.
- (a) R. Martínez, L. Berbegal, G. Guillena and D. J. Ramón, Green Chem., 2016, 18, 1724 RSC; (b) D. Brenna, E. Massolo, A. Puglisi, S. Rossi, G. Celentano, M. Benaglia and V. Capriati, Beilstein J. Org. Chem., 2016, 12, 2620 CrossRef CAS; (c) A. Torregrosa-Chinillach, A. Sánchez-Laó, E. Santagostino and R. Chinchilla, Molecules, 2019, 24, 4058 CrossRef CAS; (d) E. Massolo, S. Palmieri, M. Benaglia, V. Capriati and F. M. Perna, Green Chem., 2016, 18, 792 RSC.
- L. Millia, V. Dall'Asta, C. Ferrara, V. Berbenni, E. Quartarone, F. M. Perna, V. Capriati and P. Mustarelli, Solid State Ionics, 2018, 323, 44 CrossRef CAS.
- F. Milano, L. Giotta, M. R. Guascito, A. Agostiano, S. Sblendorio, L. Valli, F. M. Perna, L. Cicco, M. Trotta and V. Capriati, ACS Sustainable Chem. Eng., 2017, 5, 7768 CrossRef CAS.
- (a) C. L. Boldrini, N. Manfredi, F. M. Perna, V. Trifiletti, V. Capriati and A. Abbotto, Energy Technol., 2017, 5, 345 CrossRef CAS; (b) C. L. Boldrini, N. Manfredi, F. M. Perna, V. Capriati and A. Abbotto, Chem. – Eur. J., 2018, 24, 17656 CrossRef CAS; (c) C. L. Boldrini, N. Manfredi, F. M. Perna, V. Capriati and A. Abbotto, ChemElectroChem, 2020, 7, 1707 CrossRef CAS.
- (a) J. García-Álvarez, E. Hevia and V. Capriati, Eur. J. Org. Chem., 2015, 6779 CrossRef; (b) J. García-Álvarez, E. Hevia and V. Capriati, Chem. – Eur. J., 2018, 24, 14854 CrossRef; (c) E. Hevia, Chimia, 2020, 74, 681 CrossRef CAS; (d) D. Elorriaga, M. J. Rodríguez-Álvarez, N. Ríos-Lombardía, F. Morís, A. Presa Soto, J. González-Sabín, E. Hevia and J. García-Álvarez, Chem. Commun., 2020, 56, 8932 RSC.
- (a) B. H. Lipshutz and S. Ghorai, Aldrichimica Acta, 2008, 41(3), 59 CAS; (b) B. H. Lipshutz and S. Ghorai, Aldrichimica Acta, 2012, 45(1), 3 CAS; (c) G. Cravotto, E. Borretto, M. Oliverio, A. Procopio and A. Penoni, Catal. Commun., 2015, 63, 2 CrossRef CAS; (d) A. Chatterjee and T. R. Ward, Catal. Lett., 2016, 146, 820 CrossRef CAS; (e) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249 CrossRef CAS; (f) B. H. Lipshutz, S. Ghorai and M. Cortes-Clerget, Chem. – Eur. J., 2018, 24, 6672 CrossRef CAS; (g) S. Serrano-Luginbühl, K. Ruiz-Mirazo, R. Ostaszewski, F. Gallou and P. Walde, Nat. Rev. Chem., 2018, 2, 306–327 CrossRef; (h) M. Sassi, S. Mattiello and L. Beverina, Eur. J. Org. Chem., 2020, 3942 CrossRef CAS.
- (a) A. Suzuki, Pure Appl. Chem., 1985, 57, 1749 CAS; (b) R. F. Heck, Org. React., 2004, 27, 345 Search PubMed; (c) K. Sonogashira, Comprehensive Organic Synthesis, Pergamon Press, New York, 1991, vol. 3, p. 521 Search PubMed; (d) C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef.
- (a) A. J. Cocuzza, D. R. Chidester, S. Culp, L. Fitzgerald and P. Gilligan, Bioorg. Med. Chem. Lett., 1999, 9, 1063 CrossRef CAS; (b) A. Yokoyama, H. Suzuki, Y. Kubota, K. Ohuchi, H. Higashimura and T. Yokozawa, J. Am. Chem. Soc., 2007, 129, 7236 CrossRef CAS.
- (a) C. Reichardt and T. Welton, in Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, 4th edn, 2010 CrossRef; (b) J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164 RSC.
- R. A. Sheldon, Green Chem., 2017, 19, 18 RSC.
- S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601 RSC.
- S. E. Hooshmand, R. Afshari, D. J. Ramón and R. S. Varma, Green Chem., 2020, 22, 3668 RSC.
- M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133, 19505 CrossRef CAS.
- G. Imperato, S. Höger, D. Lenoir and B. König, Green Chem., 2006, 8, 1051 RSC.
- X. Marset, A. Khoshnood, L. Sotorríos, E. Gómez-Bengoa, D. A. Alonso and D. J. Ramón, ChemCatChem, 2017, 9, 1269 CrossRef CAS.
- D. J. Ramón, G. Guillena, X. Marset, B. Saavedra, N. González-Gallardo, A. Beaton, M. M. León and R. Luna, Front. Chem., 2019, 7, 700 CrossRef.
- B. Saavedra, N. González-Gallardo, A. Meli and D. J. Ramón, Adv. Synth. Catal., 2019, 361, 3868 CrossRef CAS.
- G. Dilauro, S. M. García, D. Tagarelli, P. Vitale, F. M. Perna and V. Capriati, ChemSusChem, 2018, 11, 3495 CrossRef CAS.
- V. Pelliccioli, G. Dilauro, S. Grecchi, S. Arnaboldi, C. Graiff, F. M. Perna, P. Vitale, E. Licandro, A. Aliprandi, S. Cauteruccio and V. Capriati, Eur. J. Org. Chem., 2020, 6981 CrossRef CAS.
- I. Hoffmann, B. Blumenröder, S. O. Thumann, S. Dommer and J. Schatz, Green Chem., 2015, 17, 3844 RSC.
- L. Liu, M. Liu, J. Yang, B. Zhu and X. Wang, Curr. Org. Chem., 2018, 22, 2023 CrossRef CAS.
- Y. Zhang, Z. Wang, Z. Tang, Z. Luo, H. Wu, T. Liu, Y. Zhu and Z. Zeng, Eur. J. Org. Chem., 2020, 1620 CrossRef CAS.
- K. C. Nicolaou and W.-M. Dai, Angew. Chem., Int. Ed. Engl., 1991, 30, 1387 CrossRef.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS.
- F. Ilgen and B. König, Green Chem., 2009, 11, 848 RSC.
- Y. Li, L. Sun and T. Zhu, J. Mol. Liq., 2019, 296, 111958 CrossRef CAS.
- P. Thiyagamurthy and F. R. N. Khan, ChemistrySelect, 2020, 5, 2610 CrossRef CAS.
- F. Messa, G. Dilauro, F. M. Perna, P. Vitale, V. Capriati and A. Salomone, ChemCatChem, 2020, 12, 1979 CrossRef CAS.
- S. Bhattacharya and S. Sengupta, Tetrahedron Lett., 2004, 45, 8733 CrossRef CAS.
- B. Soberats, L. Martínez, M. Vega, C. Rotger and A. Costa, Adv. Synth. Catal., 2009, 351, 1727 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS.
- M. Nasrollahzadeh, S. M. Sajadi, M. Maham and A. Ehsani, RSC Adv., 2015, 5, 2562 RSC.
- M. Esmaeilpour, A. Sardarian and J. Javidi, Catal. Sci. Technol., 2016, 6, 4005 RSC.
- Y. Dong, Y. Chen, J. Jv, Y. Li, W. Li and Y. Dong, RSC Adv., 2019, 9, 21671 RSC.
- (a) Tetrahedron Organic Chemistry Series, Vol 23, Organo- lithiums: Selectivity for Synthesis, ed. J. Clayden, Pergamon, Amsterdam, 2002 Search PubMed; (b) The Chemistry of Organolithium Com- pounds, ed. Z. Rappoport and I. Marek, Wiley-VCH, 2004 Search PubMed; (c) V. Capriati, S. Florio, R. Luisi, F. M. Perna and A. Spina, J. Org. Chem., 2008, 73, 9552–9564 CrossRef CAS; (d) V. Capriati, S. Florio, F. M. Perna and A. Salomone, Chem. – Eur. J., 2010, 16, 9778 CrossRef CAS; (e) Lithium Compounds in Organic Synthesis – From Fundamentals to Applications, ed. R. Luisi and V. Capriati, Wiley-VCH, 2014 Search PubMed; (f) V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204 RSC; (g) U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194 CrossRef CAS.
- For a review on lithium compounds in cross-coupling reactions, see: M. Shimizu, in Lithium Compounds in Organic Synthesis – From Fundamentals to Applications, ed. R. Luisi and V. Capriati, Wiley-VCH, 2014, ch. 16, p. 463 Search PubMed.
- S.-I. Murahashi, M. Yamamura, K.-i. Yanagisawa, N. Mita and K. Kondo, J. Org. Chem., 1979, 44, 2408 CrossRef CAS.
- Selected publications: (a) M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667 CrossRef CAS; (b) V. Hornillos, M. Giannerini, C. Vila, M. Fañanás-Mastral and B. L. Feringa, Org. Lett., 2013, 15, 5114 CrossRef CAS; (c) M. Giannerini, V. Hornillos, C. Vila, M. Fañanás-Mastral and B. L. Feringa, Angew. Chem., Int. Ed., 2013, 52, 13329 CrossRef CAS; (d) C. Vila, M. Giannerini, V. Hornillos, M. Fañanás-Mastral and B. L. Feringa, Chem. Sci., 2014, 5, 1361 RSC; (e) V. Hornillos, M. Giannerini, C. Vila, M. Fañanás-Mastral and B. L. Feringa, Chem. Sci., 2015, 6, 1394 RSC; (f) D. Heijnen, J. P. Gualtierotti, V. Hornillos and B. L. Feringa, Chem. – Eur. J., 2016, 22, 3991 CrossRef CAS; (g) D. Heijnen, F. Tosi, C. Vila, M. C. A. Stuart, P. H. Elsinga, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 3354 CrossRef CAS.
- E. B. Pinxterhuis, M. Giannerini, V. Hornillos and B. L. Feringa, Nat. Commun., 2016, 7, 11698 CrossRef.
- G. Dilauro, A. F. Quivelli, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2019, 58, 1799 CrossRef CAS.
- (a) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS; (b) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CrossRef CAS.
- F. Christoffel and T. R. Ward, Catal. Lett., 2018, 148, 489 CrossRef CAS.
- K. V. Kutonova, M. E. Trusova, A. V. Stankevich, P. S. Postnikov and V. D. Filimonov, Beilstein J. Org. Chem., 2015, 11, 358 CrossRef CAS.
- H.-S. Lee, S.-H. Pai, W.-T. Liao, X.-J. Yang and F.-Y. Tsai, RSC Adv., 2017, 7, 34293 RSC.
- F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 2382 CrossRef.
- (a) H.-J. Cristau, P. P. Cellier, J.-F. Spindler and M. Taillefer, Chem. – Eur. J., 2004, 10, 5607 CrossRef CAS; (b) H.-J. Cristau, A. Ouali, J.-F. Spindler and M. Taillefer, Chem. – Eur. J., 2005, 11, 2483 CrossRef CAS; (c) A. Shafir and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 8742 CrossRef CAS; (d) D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450 CrossRef CAS; (e) J. W. Tye, Z. Weng, A. M. Johns, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 9971 CrossRef CAS; (f) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS; (g) C. Sambiagio, S. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525 RSC.
- (a) X. Li, D. Yang, Y. Jiang and H. Fu, Green Chem., 2010, 12, 1097 RSC; (b) L. Huang, R. Hu, X. Zhu and Y. Wan, Tetrahedron, 2013, 69, 8974 CrossRef CAS; (c) G. Chakraborti, S. Paladhi, T. Mandal and J. Dash, J. Org. Chem., 2018, 83, 7347 CrossRef CAS.
- J. Jiao, X. R. Zhang, N.-H. Chang, J. Wang, J.-F. Wei, X.-Y. Shi and Z.-G. Chen, J. Org. Chem., 2011, 76, 1180 CrossRef CAS.
- F. Ferlin, V. Trombettoni, L. Luciani, S. Fusi, O. Piermatti and S. Santoro, Green Chem., 2018, 20, 1634 RSC.
- A. Shaabani and R. J. Afshari, Colloids Interface Sci., 2018, 510, 384 CrossRef CAS.
- A. F. Quivelli, P. Vitale, F. M. Perna and V. Capriati, Front. Chem., 2019, 7, 723 CrossRef CAS.
- S. Merenyi, REACH: Regulation (EC) No 1907/2006: Consolidated version (June 2012) with an introduction and future prospects regarding the area of Chemicals legislation, 2012 Search PubMed.
- (a) A. D. Curzons, D. C. Constable and V. L. Cunningham, Clean Prod. Processes, 1999, 1, 82 Search PubMed; (b) C. Jiménez-González, A. D. Curzons and D. C. Constable, Clean Technol. Environ. Policy, 2005, 7, 42 CrossRef.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517 CrossRef CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31 RSC.
- (a) C. Lu, J. Cao, N. Wang and E. Su, Med. Chem. Commun., 2016, 7, 955 RSC; (b) Z. Li and P. I. Lee, Int. J. Pharm., 2016, 505, 283 CrossRef CAS; (c) S. N. Pedro, M. G. Freire, C. S. R. Freire and A. J. D. Silvestre, Expert Opin. Drug Delivery, 2019, 16, 497 CrossRef; (d) I. M. Aroso, J. C. Silva, F. Mano, A. S. D. Ferreira, M. Dionisio, I. Sanogueira, S. Barreiros, R. L. Reis, A. Paiva and A. R. C. Duarte, Eur. J. Pharm. Biopharm., 2016, 98, 57 CrossRef CAS; (e) C. W. Park, Y. J. Kim, Y. S. Rhee, T. O. Oh, J. M. Ha, N. Y. Choi, S. C. Chi and E. S. Park, Arch. Pharmacal Res., 2012, 35, 1935 CrossRef CAS; (f) A. P. Abbott, E. I. Ahmed, K. Prasad, I. B. Qader and K. S. Ryder, Fluid Phase Equilib., 2017, 448, 2 CrossRef CAS; (g) P. Berton, D. K. R. Bona, D. Yancey, S. A. A. Rizvi, M. Gray, G. Gurau, J. L. Shamshina, J. F. Rasco and R. D. Rogers, ACS Med. Chem. Lett., 2017, 8, 498 CrossRef CAS; (h) A. R. C. Duarte, A. S. D. Ferreira, S. Barreiros, E. Cabrita, R. L. Reis and A. A. Paiva, Eur. J. Pharm. Biopharm., 2017, 114, 296 CrossRef CAS; (i) S. Nazzal, I. I. Smalyukh, O. D. Lavrentovich and M. A. Khan, Int. J. Pharm., 2002, 235, 247 CrossRef CAS.
- G. Hosie and H. Bird, J. Rheumatol. Inflammation, 1999, 14, 21 Search PubMed.
- (a) R. N. Brogden, R. C. Heel, G. E. Pakes, T. M. Speight and G. S. Avery, Drugs, 1980, 19, 84 CrossRef CAS; (b) J. N. Parker and P. M. Parker, in Diflunisal: A Medical Dictionary, Bibliography, and Annotated Research Guide to Internet References, ICON Group International, Inc, 2004 Search PubMed.
- A. Palasi and D. Ciez, Eur. J. Med. Chem., 2015, 97, 582 CrossRef.
- L. Cicco, A. Salomone, P. Vitale, N. Ríos-Lombardía, J. González-Sabín, J. García-Álvarez, F. M. Perna and V. Capriati, ChemSusChem, 2020, 13, 3583 CrossRef CAS.
- D. A. Handley, C. H. Senanayake, W. Dutczak, J. L. Benovic, T. Walle, R. B. Penn, H. S. Wilkinson, G. J. Tanoury, R. G. G. Andersson, F. Johansson and J. Morley, Pulm. Pharmacol. Ther., 2002, 15, 135 CrossRef CAS.
- (a) F. Campos, M. Pilar and A. Guerrero, Tetrahedron: Asymmetry, 2000, 11, 2705 CrossRef CAS; (b) L. Huang, J. Liu, W. Shan, B. Liu, A. Shi and X. Li, Chirality, 2010, 22, 206 CAS.
- E. F. Nemeth, M. E. Steffey, L. G. Hammerland, B. C. P. Hung, B. C. Van Wagenagen, E. G. DelMar and M. F. Balandrin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4040 CrossRef CAS.
- L. Marx, N. Ríos-Lombardía, J. F. Farnberger, W. Kroutil, A. I. Benítez-Mateos, F. López-Gallego, F. Morís, J. González-Sabín and P. Berglund, Adv. Synth. Catal., 2018, 360, 2157 CrossRef CAS.
- O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Chem. Commun., 2010, 46, 1541 RSC.
- (a) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS; (b) B. Bang-Andersen, T. Ruhland, M. Jørgensen, G. Smith, K. Frederiksen, K. G. Jensen, H. Zhong, S. M. Nielsen, S. Hogg, A. Mørk and T. B. Stensbøl, J. Med. Chem., 2011, 54, 3206 CrossRef CAS.
- C. B. Jacobsen, M. Meldal and F. Diness, Chem. – Eur. J., 2017, 23, 846 CrossRef.
- S. A. Zisopouloua, A. E. Pafilia, P. Gkizisb, T. Andreoub, T. V. Koftisb, A. Lithadiotib, E. Neokosmidisb and J. K. Gallos, Synthesis, 2020, 2662 Search PubMed.
- (a) K. Sujatha, P. T. Perumal, D. Muralidharan and M. Rajendran, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2009, 48, 267 Search PubMed; (b) P. K. Dubey and T. V. Kumar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2006, 45, 2128 Search PubMed; (c) A. Kumar, D. Kumar, M. Akram and H. Kaur, Int. J. Pharm. Biol. Arch., 2011, 2, 744 Search PubMed.
- M. Imran, A. Bakht, A. Khan, T. Alam, El H. Anouar, M. B. Alshammari, N. Ajmal, A. Vimal, A. Kumar and Y. Riadi, Molecules, 2020, 25, 1118 CrossRef CAS.
- (a) D. Nauduri and G. B. Reddy, Chem. Pharm. Bull., 1998, 46, 1254 CrossRef CAS; (b) P. Mondal, S. Jana, A. Balaji, R. Ramakrishna and L. K. Kanthal, J. Young Pharm., 2012, 4, 38 CrossRef CAS; (c) I. Filali, J. Bouajila, M. Znati, F. Bousejra-El Garah and H. B. Jannet, J. Enzyme Inhib. Med. Chem., 2015, 30, 371 CrossRef CAS; (d) M. Shaharyar, M. A. Ali, M. A. Bakht and V. Murugan, J. Enzyme Med. Chem., 2008, 23, 432 CrossRef CAS; (e) L. Shi, R. Hu, Y. Wei, Y. Liang, Z. Yang and S. Ke, Eur. J. Med. Chem., 2012, 47, 549 CrossRef.
- M. A. Bakht, M. J. Ansari, Y. Riadi, N. Ajmal, M. J. Ahsan and M. S. Yar, J. Mol. Liq., 2016, 224, 1249 CrossRef CAS.
- (a) W. P. Smith, L. S. Sollis, D. P. Howes, C. P. Cherry, D. I. Starkey and N. K. Cobley, J. Med. Chem., 1998, 41, 787 CrossRef; (b) K. Mazaahir, S. Shilpi, R. K. Khalilur and S. T. Sharanjit, Bioorg. Med. Chem. Lett., 2005, 15, 4295 CrossRef; (c) M. E. A. Zaki, E. M. Morsy and M. Abdul, Heterocycl. Commun., 2004, 10, 97 CAS; (d) M. E. A. Zaki, H. A. Saliman, O. A. Hickal and A. E. Rashad, Z. Naturforsch., C: Biosci., 2006, 61, 1 CAS.
- M. R. Tipale, L. D. Khillare, A. R. Deshmukh and M. R. Bhosle, J. Heterocycl. Chem., 2018, 55, 716 CrossRef CAS.
- V. K. Singh and A. Parle, Int. J. Pharma Sci. Res., 2019, 10, 1540 CrossRef CAS.
- Salahuddin, M. Shaharyar and A. Mazumder, Arabian J. Chem., 2017, 10, S157 CrossRef CAS.
- (a) H. O. Gulcan, A. Mavideniz, M. F. Sahin and I. E. Orhan, Curr. Med. Chem., 2019, 26, 3260 CrossRef CAS; (b) L. Piemontese, G. Vitucci, M. Catto, A. Laghezza, F. M. Perna, M. Rullo, F. Loiodice, V. Capriati and M. Solfrizzo, Molecules, 2018, 23, 2182 CrossRef.
- L. Piemontese, R. Sergio, F. Rinaldo, L. Brunetti, F. M. Perna, M. A. Santos and V. Capriati, Molecules, 2020, 25, 574 CrossRef CAS.
- (a) L. Piemontese, D. Tomás, A. Hiremathad, V. Capriati, E. Candeias, S. M. Cardoso, S. Chaves and M. A. Santos, J. Enzyme Inhib. Med. Chem., 2018, 33, 1212 CrossRef CAS; (b) S. Chaves, A. Hiremathad, D. Tomás, R. S. Keri, L. Piemontese and M. A. Santos, New J. Chem., 2018, 42, 16503 RSC.
- B. C. C. Cantello, M. A. Cawthorne, G. P. Cottam, P. T. Duff, D. Haigh, R. M. Hindley, C. A. Lister, S. A. Smith and P. L. Thurlby, J. Med. Chem., 1994, 37, 3977 CrossRef CAS.
- D. V. Jawale, U. R. Pratap and R. A. Mane, Bioorg. Med. Chem. Lett., 2012, 22, 924 CrossRef CAS.
- (a) C. Ma, Y. Li, S. Niu, H. Zhang, X. Liu and Y. Che, J. Nat. Prod., 2011, 74, 32 CrossRef CAS; (b) C.-S. Li, C.-Y. An, X.-M. Li, S.-S. Gao, C.-M. Cui, H.-F. Sun and B.-G. Wang, J. Nat. Prod., 2011, 74, 1331 CrossRef CAS.
- S. K. Ghosh and R. Nagarajan, RSC Adv., 2016, 6, 27378 RSC.
- (a) C. Wattanapiromsakul, P. I. Forster and P. G. Waterman, Phytochemistry, 2003, 64, 609 CrossRef CAS; (b) Y. Rao, H. Liu, L. Gao, H. Yu, J.-H. Tan, T.-M. Ou, S.-L. Huang, L.-Q. Gu, J.-M. Ye and Z.-S. Huang, Bioorg. Med. Chem., 2015, 23, 4719 CrossRef CAS.
- R. Filip, T. A. Shaw, A. Nishida and J. P. Pezacki, MedChemComm, 2019, 10, 985 RSC.
- M. Viji and R. Nagarajan, J. Chem. Sci., 2014, 126, 1075 CrossRef CAS.
- K. Uehata, N. Kimura, K. Hasegawa, S. Arai, M. Nishida, T. Hosoe, K.-I. Kawai and A. Nishida, J. Nat. Prod., 2013, 76, 2034 CrossRef CAS.
- (a) Z. Maugeri and P. Dominguez de Maria, ChemCatChem, 2014, 6, 1535 CrossRef CAS; (b) Y. Dai, B. Huan, H. S. Zhang and Y. C. He, Appl. Biochem. Biotechnol., 2016, 181, 1347 CrossRef.
- L. Huang, J. P. Bittner, P. Domìnguez de Marìa, S. Jakobtorweihen and S. Kara, ChemBioChem, 2020, 21, 811 CrossRef CAS.
- Z. Maugeri and P. Domínguez de María, J. Mol. Catal. B: Enzym., 2014, 107, 120 CrossRef CAS.
- J. Tan and Y. Dou, Appl. Microbiol. Biotechnol., 2020, 104, 1481 CrossRef CAS.
- M. L. Toledo, M. M. Pereira, M. G. Freire, J. P. A. Silva, J. A. P. Coutinho and A. P. M. Tavares, ACS Sustainable Chem. Eng., 2019, 7(13), 11806 CrossRef CAS.
- (a) F. Peng, Y. Zhao, F. Z. Li, M. H. Zong and W. Y. Lou, Bioresour. Bioprocess., 2018, 5, 5 CrossRef; (b) J. T. Gorke, F. Srienc and R. J. Kazlauskas, Chem. Commun., 2008, 1235 RSC.
- A. K. Schweiger, N. Ríos-Lombardía, C. K. Winkler, S. Schmidt, F. Morís, W. Kroutil, J. González-Sabín and R. Kourist, ACS Sustainable Chem. Eng., 2019, 7, 16364 CrossRef CAS.
- C. R. Müller, I. Lavandera, V. Gotor-Fernández and P. Domínguez de María, ChemCatChem, 2015, 7, 2654 CrossRef.
- P. Xu, G.-W. Zheng, M.-H. Zong, N. Li and W.-Y. Lou, Bioresour. Bioprocess., 2017, 4, 34 CrossRef.
- (a) Ö. Erol and F. Hollmann, Adv. Bot. Res., 2020 DOI:10.1016/bs.abr.2020.09.004 , Academic Press; (b) P. Vitale, F. M. Perna, G. Agrimi, I. Pisano, F. Mirizzi, R. V. Capobianco and V. Capriati, Catalysts, 2018, 8, 55 CrossRef.
- M. C. Gutirrez, M. L. Ferrer and F. del Monte, Angew. Chem., Int. Ed., 2010, 49, 2158 CrossRef.
- (a) M. C. Bubalo, M. Mazur, K. Radosevic and I. R. Redovnikovic, Process Biochem., 2015, 50, 1788 CrossRef; (b) P. Vitale, V. M. Abbinante, F. M. Perna, A. Salomone, C. Cardellicchio and V. Capriati, Adv. Synth. Catal., 2017, 359, 1049 CrossRef CAS.
- L. Zheng, S. Zhang and X. Tian, J. Microbiol. Biotechnol., 2016, 26, 80 CrossRef.
- H. Monhemi, M. R. Housaindokht, A. A. Moosavi-Movahedi and M. R. Bozorgmehr, Phys. Chem. Chem. Phys., 2014, 16, 14882 RSC.
- (a) A. Zaks and A. M. Klibanov, J. Biol. Chem., 1988, 263, 8017 CrossRef CAS; (b) N. M. Micaelo and C. M. Soares, FEBS J., 2007, 274, 2424 CrossRef CAS; (c) S. B. Lee and K.-J. Kim, J. Ferment. Bioeng., 1995, 79, 473 CrossRef CAS; (d) R. Wedberg, J. Abildskov and G. N. H. Peters, J. Phys. Chem. B, 2012, 116, 2575 CrossRef CAS.
- S. Mao, L. Yu, S. Ji, X. Liu and F. Lu, J. Chem. Technol. Biotechnol., 2016, 91, 1099 CrossRef CAS.
- S. Mao, X. Wang, Z. Zhang, S. Wang, K. Li, F. Lu and H. Qin, Biochem. Eng. J., 2020, 164, 107781 CrossRef CAS.
- D. Wang, P. Zheng, P. Chen and D. Wu, ACS Sustainable Chem. Eng., 2020, 8, 14905 CrossRef CAS.
- F. Zhang, C.-T. Zhu, Q.-M. Peng, F.-Q. Wang, S. Sheng, Q.-Y. Wu and J. Wang, J. Chem. Technol. Biotechnol., 2020, 95, 384 CrossRef CAS.
- N. Xia, L. Xiong, S. Bi, F. Qian and P. Wang, Bioprocess Biosyst. Eng., 2020, 43, 1987 CrossRef CAS.
- Y. He, Q. Huang and P. Wang, J. Chem. Technol. Biotechnol., 2020, 95, 1980 CrossRef CAS.
- P. Xu, Y. Xu, X.-F. Li, B.-Y. Zhao, M.-H. Zong and W.-Y. Lou, ACS Sustainable Chem. Eng., 2015, 3, 718 CrossRef CAS.
- T. X. Yang, L. Q. Zhao, J. Wang, G. L. Song, H. M. Liu, H. Cheng and Z. Yang, ACS Sustainable Chem. Eng., 2017, 5, 5713 CrossRef CAS.
- (a) T. Murakami, K. Murakami, T. Ogiya and K. Shibuya, JP 2013112674, 2013; (b) O. Tadaaki, M. Takaeshi, M. Katsutoshi, Y. Koichi and K. Taichi, U.S. Patent, 20130225814A, 2013 Search PubMed.
- Z. Zhu, S. Bi, N. Ye and P. Wang, Molecules, 2020, 25, 1855 CrossRef CAS.
- S. Zou, D. Hua, Z. Jiang, X. Han, Y. Xue and Y. Zheng, Bioresour. Technol., 2021, 320, 124392 CrossRef CAS.
- (a) H. Pfruender, M. Amidjojo, U. Kragl and D. Weuster-Botz, Angew. Chem., Int. Ed., 2004, 43, 4529 CrossRef; (b) D. Leis, B. Lauß, R. Macher-Ambrosch, A. Pfennig, B. Nidetzky and R. Kratzer, J. Biotechnol., 2017, 257, 110 CrossRef CAS.
- (a) C. R. Müller, I. Meiners and P. Domínguez de María, RSC Adv., 2014, 4, 46097 RSC; (b) C. R. Müller, A. Rosen and P. Domínguez de María, Sustain. Chem. Process., 2015, 3, 12 CrossRef.
- (a) N. Ríos-Lombardía, M. J. Rodríguez-Álvarez, F. Morís, R. Kourist, N. Comino, F. López-Gallego, J. González-Sabín and J. García-Álvarez, Front. Chem., 2020, 8, 139 CrossRef; (b) E. Liardo, R. González-Fernández, N. Ríos-Lombardía, F. Morís, J. García-Álvarez, V. Cadierno, P. Crochet, F. Rebolledo and J. González-Sabín, ChemCatChem, 2018, 10, 4676 CrossRef CAS; (c) N. Ríos-Lombardía, J. García-Álvarez and J. González-Sabín, Catalysts, 2018, 8, 75 CrossRef; (d) M. J. Rodríguez-Álvarez, N. Ríos-Lombardía, S. Schumacher, D. Pérez-Iglesias, F. Morís, V. Cadierno, J. García-Álvarez and J. González-Sabín, ACS Catal., 2017, 7, 7753 CrossRef; (e) N. Ríos-Lombardía, C. Vidal, E. Liardo, F. Morís, J. García-Álvarez and J. González-Sabín, Angew. Chem., Int. Ed., 2016, 55, 8691 CrossRef; (f) N. Ríos-Lombardía, C. Vidal, M. Cocina, F. Morís, J. García-Álvarez and J. González-Sabín, Chem. Commun., 2015, 54, 10937 RSC.
- N. Guajardo and P. Domínguez de María, ChemCatChem, 2019, 11, 3128 CrossRef CAS.
- K. S. Egorova and V. P. Ananikov, Biophys. Rev., 2018, 10, 881 CrossRef CAS.
- R. Kourist and J. González-Sabín, ChemCatChem, 2020, 12, 1903 CrossRef CAS.
- D. E. Crawford, L. A. Wright, S. L. James and A. P. Abbott, Chem. Commun., 2016, 52, 4215 RSC.
- P. Zamani and A. R. Khosropour, Green Chem., 2016, 18, 6450 RSC.
- G. Oliveira, J. P. Wojeicchowski, F. Oliveira Farias, L. Igarashi-Mafra, R. de Pelegrini Soares and M. R. Mafra, J. Mol. Liq., 2020, 318, 114266 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |