
Enhancing subcutaneous injection and target tissue accumulation of nanoparticles via co-administration with macropinocytosis inhibitory nanoparticles (MiNP)†
Trevor
Stack
,
Yugang
Liu
,
Molly
Frey
,
Sharan
Bobbala
,
Michael
Vincent
and
Evan
Scott
 *
*
Department of Biomedical Engineering, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. E-mail: evan.scott@northwestern.edu
First published on 13th April 2021
Abstract
A significant barrier to the application of nanoparticles for precision medicine is the mononuclear phagocyte system (MPS), a diverse population of phagocytic cells primarily located within the liver, spleen and lymph nodes. The majority of nanoparticles are indiscriminately cleared by the MPS via macropinocytosis before reaching their intended targets, resulting in side effects and decreased efficacy. Here, we demonstrate that the biodistribution and desired tissue accumulation of targeted nanoparticles can be significantly enhanced by co-injection with polymeric micelles containing the actin depolymerizing agent latrunculin A. These macropinocytosis inhibitory nanoparticles (MiNP) were found to selectively inhibit non-specific uptake of a second “effector” nanoparticle in vitro without impeding receptor-mediated endocytosis. In tumor bearing mice, co-injection with MiNP in a single multi-nanoparticle formulation significantly increased the accumulation of folate-receptor targeted nanoparticles within tumors. Furthermore, subcutaneous co-administration with MiNP allowed effector nanoparticles to achieve serum levels that rivaled a standard intravenous injection. This effect was only observed if the effector nanoparticles were injected within 24 h following MiNP administration, indicating a temporary avoidance of MPS cells. Co-injection with MiNP therefore allows reversible evasion of the MPS for targeted nanoparticles and presents a previously unexplored method of modulating and improving nanoparticle biodistribution following subcutaneous administration.
New conceptsPrecision nanoparticle therapeutics aim to deliver a maximum therapeutic payload to tissues of interest while minimizing off target effects. Non-specific clearance of these nanoparticles by cells of the mononuclear phagocyte system (MPS) reduces accumulation of nanoparticles in target tissues and thereby efficacy. Herein, we demonstrate the development of novel macropinocytosis inhibitory nanoparticles (MiNP) which are able to reduce non-specific MPS clearance of co-administered “effector-NPs”. MiNP are able to increase the tumor accumulation of effector-NPs in a mouse tumor model as well as increase serum accumulation of subcutaneously injected effector-NPs. These biodistribution altering effects were only seen for 24 h after MiNP administration, indicating the transient nature of this system. Combinatorial approaches are needed in order to maximize evasion of the MPS, and this adaptable, modular system could be combined with other “stealth” nanoparticle strategies to create synergistic effects. This type of transient modular platform system for MPS evasion has not been demonstrated previously. Our approach leads to a system which can be adapted to a variety of practical “effector-NPs” in order to increase their target tissue and serum accumulation, thereby enhancing existing and in-development nanoparticle therapeutics. |
Nanoparticles are versatile carriers that can improve and often specify the stability, circulation time, and biodistribution of therapeutic molecules.1 Despite these advantages, rapid clearance of nanoparticles by the mononuclear phagocyte system2(MPS) remains a significant barrier to their applications in precision medicine. The MPS consists of circulating and organ-resident phagocytic cells, which internalize nanoparticles and eventually clear them through the liver.3–6 A comprehensive survey of the literature reported that a median average of only 0.7% of administered nanoparticles successfully reach solid tumors despite the use of surface-conjugated targeting moieties like antibodies, peptides or aptamers.7 Clearance by MPS cells occurs primarily in the liver, spleen and lymph nodes through a number of endocytic pathways including clathrin-mediated and clathrin-independent pathways, macropinocytosis, and phagocytosis.8 Macropinocytosis is a process by which membranes extend and form around extracellular fluid leading to internalization of the encapsulated region, while phagocytosis is usually receptor-initiated and internalizes with or without extension of plasma membranes through the use of membrane invaginations.9 If these pathways are temporarily inhibited in MPS cells prior to or in conjunction with the introduction of therapeutic nanoparticles, the bioavailability and therapeutic efficacy of these nanoparticles would likely increase significantly.10 Developing nanoparticles with “stealth” properties to avoid this non-specific uptake remains a critical objective for nanomedicine and many different strategies such as PEGylation and CD47 “don’t eat me” peptides, have been tried with variable levels of effectiveness.11–13 Combinatorial strategies employing multiple different stealth strategies are needed to further reduce clearance by the MPS and increase nanomaterial utility in vivo.
We have previously demonstrated that diverse nanoparticle morphologies can be self-assembled from poly(ethylene glycol)-block-poly(propylene sulfide) (PEG-b-PPS) copolymers to function as customizable and non-toxic drug delivery vehicles.14,15 The nanostructure morphology and route of administration dictate the biodistribution of PEG-b-PPS nanoparticles, allowing the passive and preferential targeting of different phagocytic cell populations in vivo without the need for surface-conjugated targeting ligands.14,16–19 Of note, spherical solid core PEG-b-PPS micelles are primarily taken up by liver macrophages following intravenous (IV) administration17 and monocyte populations in draining lymph nodes and spleen following subcutaneous (SC) administration,16 both of which are key components of the MPS.20 At time points of 24 h and less, nanoparticles smaller than 100 nm in diameter primarily reach lymphoid organs directly21 as up to 48 h is typically required for trafficking of peripheral phagocytes to these locations.22 We have previously verified SC injected PEG-b-PPS micelles to reach the spleen intact after filtering through draining lymph nodes.21,23 These micelles have also been previously shown to reduce the cytotoxicity of small molecule drugs such as Celastrol.24 Here we employ macropinocytosis inhibitory nanoparticles (MiNP) to reduce specific nanoparticle uptake by the MPS and enhance their accumulation within target tissues. MiNP are comprised of PEG-b-PPS micelles containing Latrunculin A (LatA), a well-known and transient actin depolymerizing agent.25 LatA is most commonly used to temporarily inhibit macropinocytosis26 by phagocytic cells during in vitro assays to investigate mechanisms of cell endocytosis. Furthermore, LatA is hydrophobic and thus not amenable to direct administration via SC or IV routes. We have previously shown that LatA retains its inhibitory effects by disrupting the cell cytoskeleton when encapsulated in PEG-b-PPS micelles but without toxicity.27,28
We selected to investigate and optimize a co-administration strategy wherein MiNP are injected before and/or simultaneously with a model “effector” nanoparticle, which represents a nanoparticle employed for either diagnostic or therapeutic applications that will be enhanced by decreased MPS clearance. For the purposes of this proof of concept study, the employed model effector nanoparticle is a fluorescent PEG-b-PPS micelle (E-MC). The inhibitory effects of MiNP were characterized in vitro using macrophages and in vivo in a B16F10 melanoma tumor bearing mouse model. Furthermore, E-MC with surface-decorated folate (E-MC(FA)) were used to explore the ability of MiNP to enhance the accumulation of a targeted E-MC within folate receptor-expressing solid tumors (Fig. 1). We find that IV or SC injection of MiNP temporarily inhibit the non-specific MPS uptake of a subsequent chasing dose of an E-MC by increasing blood concentration and tumor accumulation compared to E-MC administered alone. Of note, this MiNP co-administration strategy significantly improved the SC injection of E-MC, achieving bioavailability of E-MC on par with IV injections.
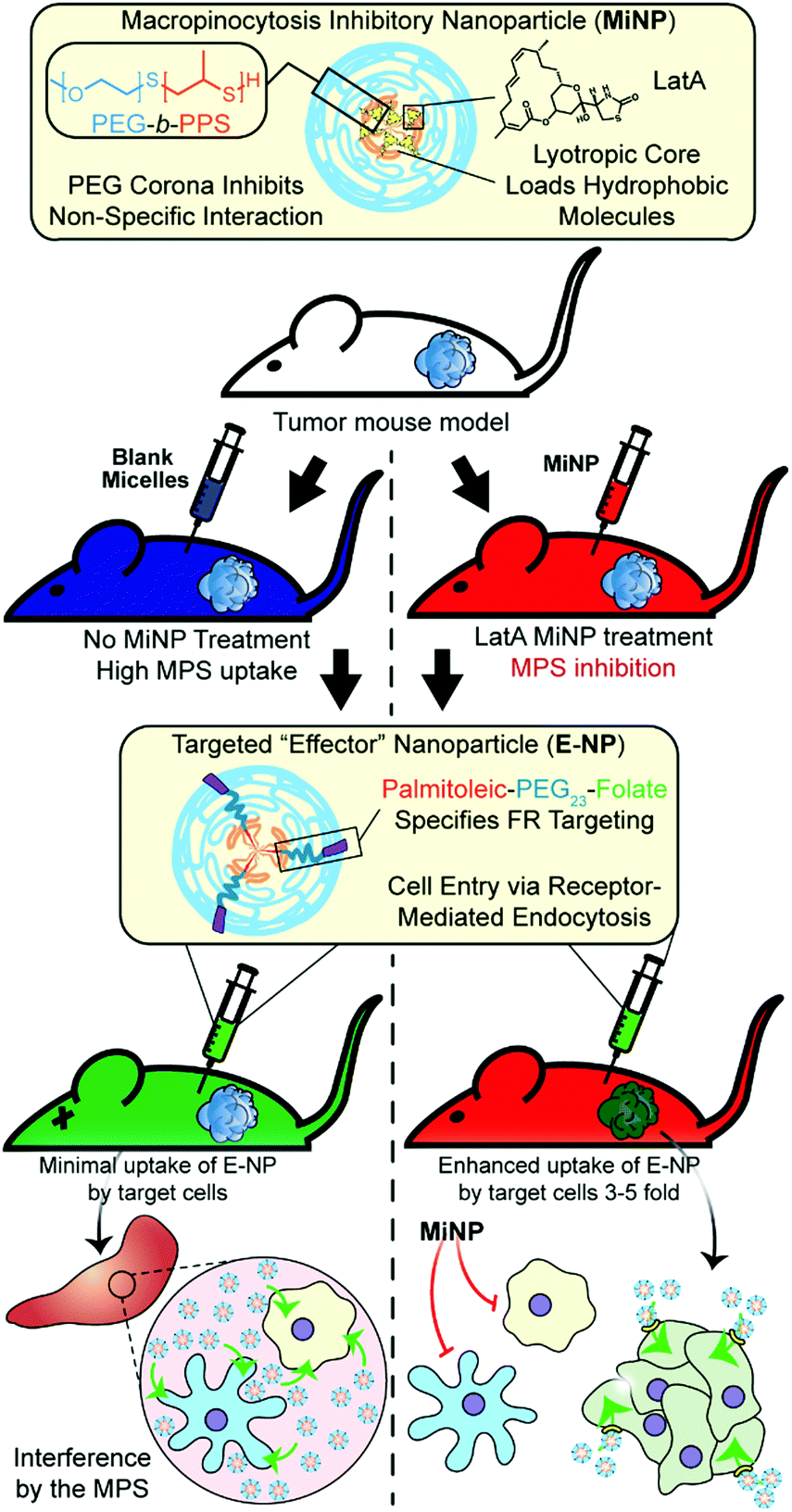 | ||
| Fig. 1 Schematic of tumor bearing mice being co-administered with latrunculin A-loaded macropinocytosis inhibitory nanoparticles (MiNP). MiNP were developed and evaluated for their effect on the accumulation of a targeted “effector” nanoparticle via subcutaneous and intravenous injection. As MiNP interferes with macropinocytosis but not receptor-mediated endocytosis, pre- and/or co-injection of MiNP with an effector nanoparticle displaying targeting ligands allows enhanced uptake by cells expressing the target receptor. As an example, MiNP are shown enhancing the targeting of receptors highly expressed within tumor microenvironments by interfering with off-target mononuclear phagocyte system (MPS) clearance. | ||
LatA-loaded ((+)MiNP) and unloaded controls ((−)MiNP) were self-assembled from PEG45-b-PPS23 copolymer using the co-solvent evaporation method.29 Dynamic light scattering was used to determine the z-average and polydispersity of the different formulations (Table S1, ESI†). Confirmation of the micelle structure was obtained using cryogenic transmission electron microscopy and small angle X-ray scattering (SAXS) studies (Fig. 2a and b). Small angle X-ray scattering curves of both (+)MiNP and (−)MiNP were successfully fitted with a micelle model indicating retention of micellar nanostructures for (+)MiNP after loading with LatA (Fig. 2b). The core radius and approximate diameter of both (−)MiNP and (+)MiNP obtained using SAXS model fits are reported in Table S2 (ESI†). LatA quantification and loading within the micelles was determined by HPLC-UV as previously reported28 and allowed for all formulations to be referenced based on their LatA content (Table S1, ESI†). These data are consistent with our previous findings that encapsulation of LatA by PEG-b-PPS micelles does not alter their physical structure or polydispersity.
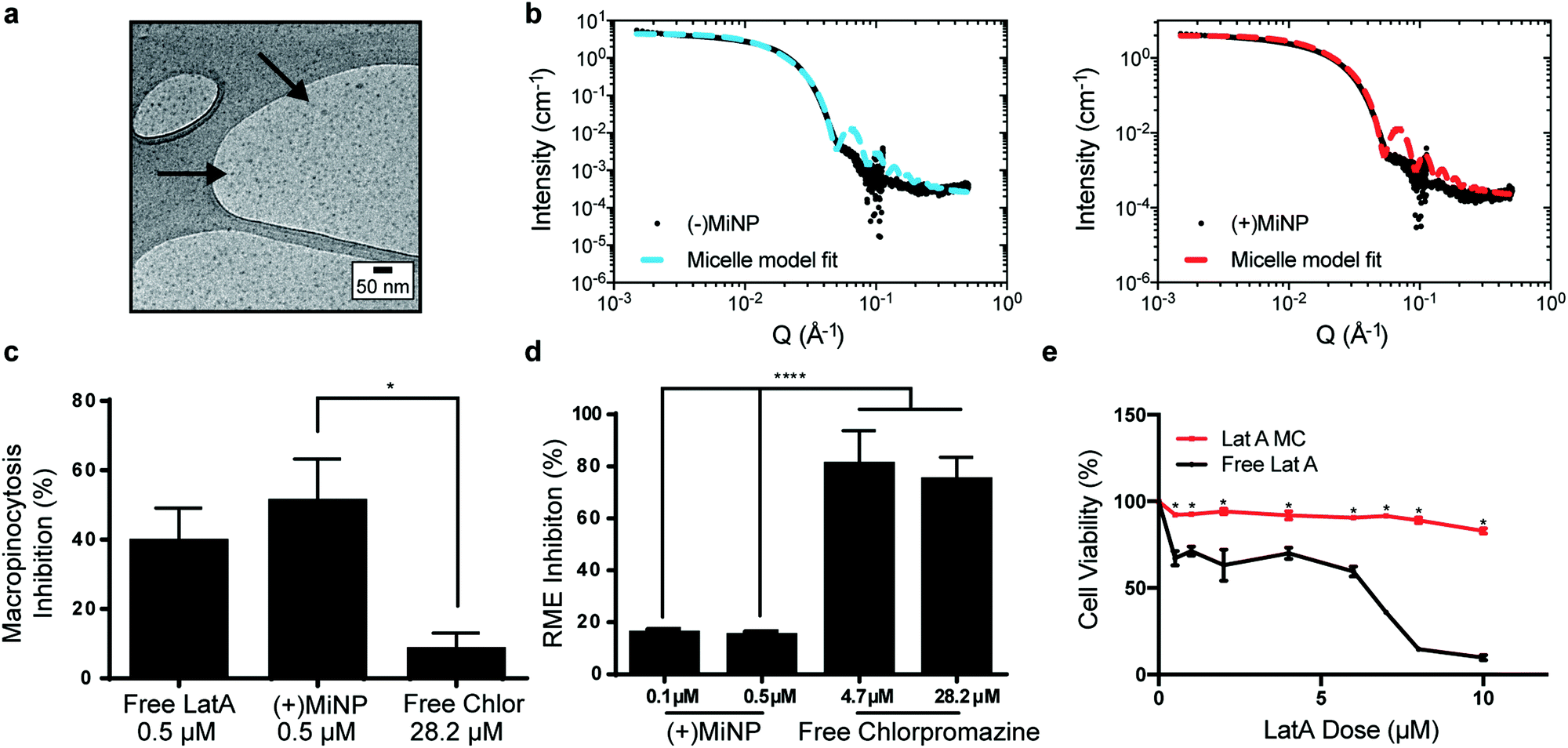 | ||
| Fig. 2 LatA retains its endocytic inhibition properties and does not change the size of PEG-b-PPS micelles when encapsulated. (a) Cryogenic transmission electron microscopy (CryoTEM) of MiNP visually confirms retention of micellar structures. (b) MiNP with ((+)MiNP) and without ((−)MiNP) loaded LatA were characterized via small angle X-ray scattering (SAXS) and fitted with a micelle model fit using SASView. (c) Free LatA and (+)MiNP significantly inhibited macropinocytosis by RAW264.7 macrophages as compared to clathrin-mediated endocytosis inhibitor chlorpromazine. Cells were treated with each inhibitor for 2 h followed by 30 min of incubation with pHrodo dextran prior to analysis by flow cytometry. Data are shown as a percentile scale of endocytosis inhibition. On this scale, 0% represents standard cell uptake with no inhibitor, while 100% represents complete inhibition with no uptake of dye. N = 3, p < 0.001. (d) In comparison, uptake of transferrin conjugated pHrodo dextran by macrophages via receptor-mediated endocytosis (RME) was significantly inhibited by chlorpromazine compared to (+)MiNP. N = 3, p < 0.001. (e) Loading within (+)MiNP significantly decreased the toxicity of LatA. Macrophages were incubated with various doses of free LatA or (+)MiNP for 4 h and assessed by flow cytometry for viability via the Zombie Aqua live/dead assay. N = 3, p < 0.05. | ||
To investigate macropinocytosis inhibition by MiNP and determine whether this inhibition still permits uptake via receptor-mediated endocytosis, (+)MiNP were compared to chlorpromazine, an inhibitor of receptor-mediated endocytosis. These distinct mechanisms of endocytosis were evaluated using dextran conjugated pHrodo dye and transferrin conjugated pHrodo dye to respectively quantify effects of (+)MiNP and chlorpromazine on macropinocytosis (dextran) and receptor-mediated endocytosis (transferrin). Free LatA, (+)MiNP, and free chlorpromazine were incubated with RAW264.7 macrophages for 2 hours and subsequently washed and then chased with dextran-pHrodo (Fig. 2c) or transferrin-pHrodo (Fig. 2d). After 30 minutes of incubation, the cells were harvested and analyzed by flow cytometry to quantify and compare uptake of dextran-pHrodo via macropinocytosis and transferrin-pHrodo by transferrin-receptors. In the case of dextran (macropinocytosis), (+)MiNP and free LatA both showed much stronger inhibition of uptake than free chlorpromazine (Fig. 2c). In the case of transferrin (receptor mediated endocytosis), free chlorpromazine at both high and low doses significantly inhibited endocytosis compared to (+)MiNP, which had minimal impact (Fig. 2d). These results demonstrate that the functional aspect of LatA is not significantly altered by encapsulation in PEG-b-PPS micelles. Importantly, MiNP did not impede uptake of transferrin via transferrin receptors, suggesting that MiNP could be employed in a multi-nanoparticle strategy to inhibit non-specific uptake of a targeted chase nanoparticle while simultaneously permitting receptor-mediated targeting of specific cell populations. As LatA has been shown to be cytotoxic at higher doses and has been used as a cytotoxic agent,30,31 we sought to evaluate the cytotoxicity of MiNP on our target cell population of macrophages. After 2 hours of exposure to various doses of (+)MiNP and Free LatA, (+)MiNP treated macrophages remained highly viable at all tested concentrations, while free LatA treated macrophages demonstrated significant toxicity at doses of 0.5 μM and above (Fig. 2e). This is consistent with our previous findings in which encapsulation of small molecule drug Celastrol reduced its cytotoxicity in vitro.24
Having characterized MiNP in vitro, we next investigated different in vivo dosing regimens to evaluate the effect of MiNP on the uptake and biodistribution of a subsequently injected blank effector nanoparticle (E-MC) without targeting moieties (Fig. 3a). Importantly, we sought to determine whether (+)MiNP could be administered SC with the E-MC in the same formulation versus administered as a separate injection prior to administration of the E-MC. For quantification of cell uptake, E-MC were labelled with DiR dye. We have previously demonstrated that lipophilic dyes are stably retained within PEG-b-PPS nanocarriers for in vivo applications and flow cytometric analysis.15,23,32 We further confirmed the stability of DiI, DiR and LatA within E-MC and (+)MiNP over the course of 7 days using membrane dialysis (Fig. S1A–C, ESI†). First, a pre-injection strategy was tested where (±)MiNP were administered twice at both 24 h and 4 h prior to injection of the chase nanoparticle. As we have previously published that the height of PEG-b-PPS micelle uptake by the MPS occurs at the 24 h timepoint,17 we suspected that this regime would extensively pre-condition and shut down the MPS to avoid non-specific clearance of the E-MC. A simplified procedure was also evaluated wherein the (±)MiNP were administered alone just once to pre-condition the mouse, which was then followed 24 h later by a co-injected dose of a (±)MiNP and E-MC multi-nanoparticle formulation (Fig. 3a). In both regimens, the same total micelle dosage of (+)MiNP or (−)MiNP and E-MC were administered. The total dosage of (−)MiNP and E-MC administered was equal to a LatA dose of 100 μL of 7 μM (+)MiNP solution (approx. 0.55 mg kg−1). This dosage was based upon previously reported intraperitoneal treatments of mice30 and our in vitro toxicity assessment in RAW264.7 macrophages (Fig. 2e). Mice were sacrificed 24 h after the chase injection, and organs were harvested for analysis by flow cytometry. Dendritic cells (DCs) and macrophages, the two key phagocytes of the MPS, were identified via antibody staining and the amount of E-MC in each cell population was quantified (Fig. S2, ESI†). In the spleen, (+)MiNP treatment with both regimes showed significantly less E-MC uptake by MPS cells than mice injected with (−)MiNP (Fig. 3b). In the liver, the multi-nanoparticle co-injection (+)MiNP/E-MC formulation had significantly less chase particle uptake than (−)MiNP/E-MC in all cell types, while the 4 h pre-injection regimen did not. These results verify that a MiNP strategy indeed inhibits uptake of a second effector nanoparticle in both the spleen and liver following SC injection. Furthermore, this indicates that in both organs, MPS phagocytes are affected by the multi-nanoparticle (+)MiNP/E-MC co-injection dosing method at a greater or equivalent level than the 4 h separate pre-injection method. The finding that the −4 h and co-injection of MiNP were equally effective, provides some indication of the time scale of the effect of the LatA-loaded nanoparticles. This suggests that the immediate effect of LatA occurs on the same time scale as the E-MC uptake, begins to wane within 4 h, and persists for at least 24 h. Additionally, the co-injection method is simpler to administer and would be preferred for any future translation of this system. As such, the co-injection multi-nanoparticle method was deemed superior and was the method of choice for future experiments. We intend to further address and optimize the MiNP injection schedule in future studies.
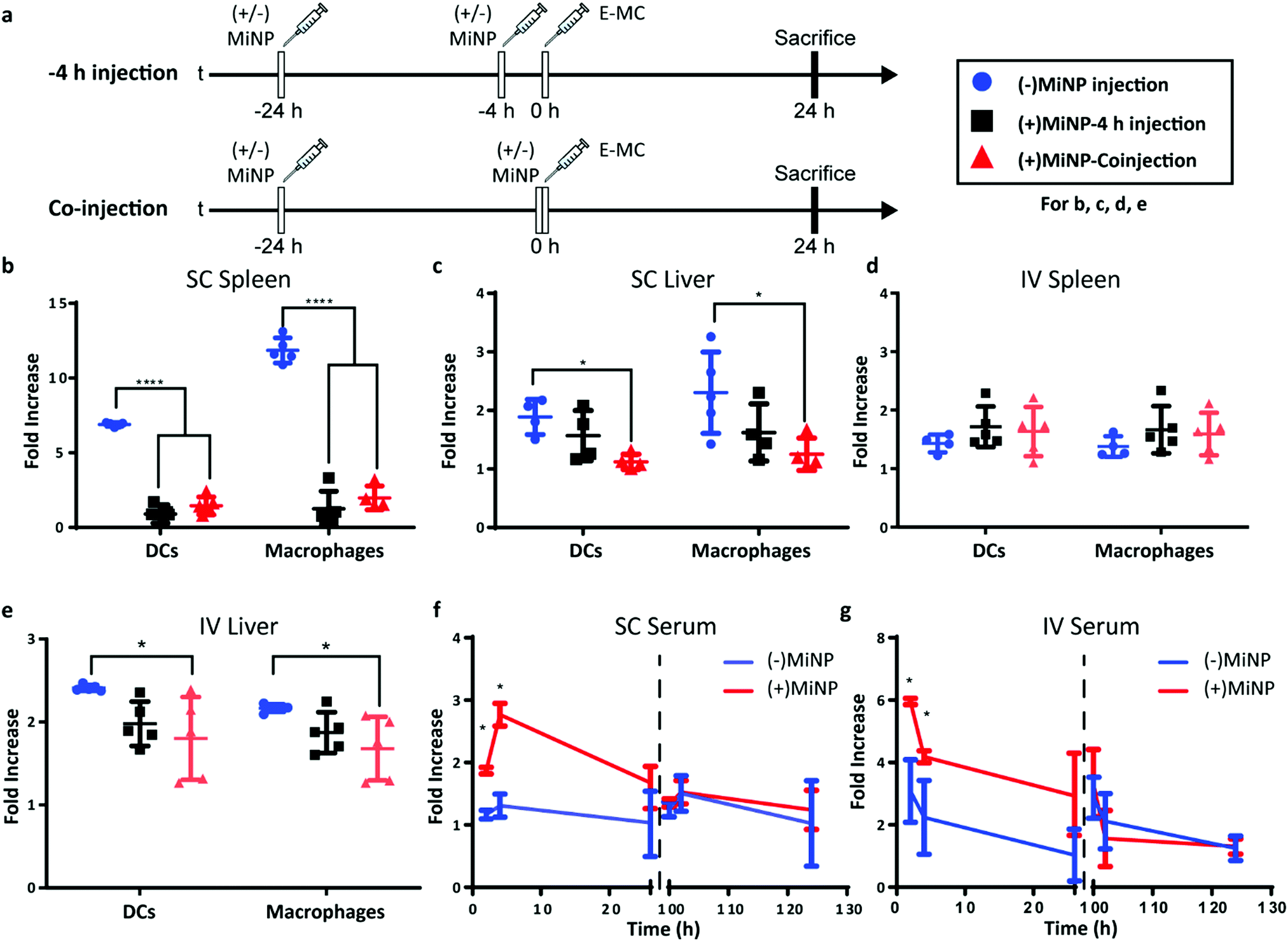 | ||
| Fig. 3 Latrunculin A loaded MC ((+)MiNP) co-injection and −4 h pre-injection lead to similar effector particle biodistributions. (a) Timeline showing the injection times for the co-injection and −4 h injection methods, which were evaluated for both subcutaneous (SC) and intravenous (IV) administration. “(±)MiNP” indicated an injection of either (+)MiNP or (−)MiNP, and effector micelle injections are indicated by “E-MC”. All mice were sacrificed at 24 h post E-MC injection. Comparisons of cell uptake in spleen and liver for the different SC (b and c) and IV (d and e) injection methods are shown. In all cases, mice were injected with 100 μL 7 μM LatA (+)MiNP or (−)MiNP and E-MC were labelled with DiR for flow cytometric quantification of cellular uptake within the spleen and liver. Data are reported as fold increase median fluorescence intensity of the E-MC over an untreated control. N = 5, p < 0.0001. To assess the transience of the MiNP effect, mice were injected SC (f) or IV (g) with (±)MiNP and E-MC according to the co-injection method, and serum levels of E-MC were evaluated by fluorescence spectroscopy. Mice were then rested for 72 hours and injected again with only E-MC to determine whether the inhibitory effect remained. N = 3 for 2 h and 4 h timepoints and N = 6 for 24 h timepoints, *p < 0.05. | ||
We next sought to compare the effects of MiNP following SC versus IV administration. A majority of nanotherapeutics are administered IV out of necessity, as nanoparticles are rapidly cleared by phagocytes during lymphatic drainage. As IV administration must be performed by healthcare professionals, enhancing SC administration to achieve IV-level biodistribution of nanoparticles would permit facile administration and more flexible dose schedules, possibly increasing patient compliance and access to treatment.
Similar to SC injections, the co-injection and −4 h injection methods were followed for IV administration and the dose remained consistent at 100 μL of 7 μM LatA (+)MiNP. IV injection had a distinctly different uptake profile than SC injections, demonstrating no difference in E-MC uptake in the spleen with either injection method (Fig. 3d). However, in the liver, treatment with (+)MiNP decreased uptake of E-MC in DC and macrophage populations when administered via the co-injection method, but not the −4 h method, indicating altered biodistribution within two MPS cell types of interest (Fig. 3e). These data further confirm the co-injection method to be equivalent or superior to the −4 h injection method.
We next evaluated serum levels of chase nanoparticles after SC and IV administration of (+)MiNP. LatA inhibits actin polymerization by binding actin at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and consuming intracellular LatA.33 Thus, its effects should decrease over time due to continuous LatA depletion without replenishment. We therefore investigated the transient effects of (+)MiNP by evaluating whether the inhibitor's effects would diminish within 100 h of the initial injection. Mice were divided into a (+)MiNP group that was administered a (+)MiNP/E-MC co-injection and a (−)MiNP control group that was administered (−)MiNP/E-MC.
:1 ratio and consuming intracellular LatA.33 Thus, its effects should decrease over time due to continuous LatA depletion without replenishment. We therefore investigated the transient effects of (+)MiNP by evaluating whether the inhibitor's effects would diminish within 100 h of the initial injection. Mice were divided into a (+)MiNP group that was administered a (+)MiNP/E-MC co-injection and a (−)MiNP control group that was administered (−)MiNP/E-MC.
Blood (100 μL) was collected from each mouse at 2 h, 4 h and 26 h post SC or IV co-injection, and serum was isolated and analysed to assess E-MC content by spectrophotometry. Following final blood collection at 26 h, mice were rested for 74 h and subsequently injected with E-MC a second time at 100 h to assess any residual effects of the original MiNP administration. Blood (100 μL) was again collected from mice at 102 h, 104 h, and 126 h relative to the initial MiNP administration to quantify E-MC content by spectrophotometry. (+)MiNP treatment increased E-MC content in serum at 2 h and 4 h post injection following both IV and SC administration. The SC injections resulted in a delay in reaching the maximum serum level of the chase, which occurred at 4 h and is indicative of the time required for the nanoparticles to drain from the SC tissue and reach systemic circulation. After the mice were rested, there was no difference in the E-MC serum content between mice administered (+)MiNP or (−)MiNP after the second chase injection (Fig. 3f and g). This indicates the mice returned to a baseline processing of E-MC by 100 h post (+)MiNP treatment for both routes of administration. Another interesting observation from this data was that the (+)MiNP SC injection group had a similar E-MC serum content at 4 h as the (−)MiNP IV injection group, suggesting that (+)MiNP administered subcutaneously are able to achieve a similar amount of E-MC serum concentration as the typically used IV injection ((−)MiNP treatment). Upon observing this, we measured the total area under the curve over the course of the first 26 h and found that SC (+)MiNP treatment had a value of 53.5 h, an increase over the IV (−)MiNP value of 41.2 h (Table S3, ESI†). This further confirms the enhanced serum levels of E-MC in response to SC injection of (+)MiNP. This effect in conjunction with a therapeutic payload would allow access to a host of different dosing strategies for existing nanotherapeutics as well as easier administration.
Having shown in vitro that (+)MiNP can transiently inhibit macropinocytosis while still allowing receptor-mediated endocytosis, we next sought to investigate whether (+)MiNP could enhance the uptake of chase nanoparticles targeting a specific cell receptor in vivo. The well-established B16F10 melanoma mouse model was chosen to compare the targeting of intratumoral folate receptors following IV and. SC routes of administration. B16F10 mouse melanoma cells have increased expression of folate receptors and folate decorated nanoparticles have been used by other groups to successfully target these cells.34 We therefore synthesized a [folate]–[PEG linker]–[palmitoleic acid lipid anchor] (FA–PEG–PA) amphiphilic construct for stable incorporation into self-assembled PEG-b-PPS micelles (Fig. 1). Briefly, folate was attached to a PEG1k-amine spacer that was then linked to a palmitoleic acid tail using EDC (1-ethyl-3-[3-dimethylaminopropyl]-carbodiimide hydrochloride) chemistry. The resulting FA–PEG–PA construct was incorporated into micelles by shaking overnight in phosphate buffered saline, allowing the palmitoleic acid anchor to partition into the hydrophobic PPS core of the micelle. We have previously demonstrated that such lipid anchored constructs could be stably retained within self-assembled PEG-b-PPS nanoparticles for controlled surface display of targeting moieties, such as peptides.35 The formation of folate displaying micelles (E-MC(FA)) at controllable molar ratios of PEG-b-PPS polymer to FA–PEG–PA construct was confirmed using UV-Vis spectroscopy (Fig. S3, ESI†). Additionally, the stability of the incorporation of the FA–PEG–PAlmitoleic acid construct and DiR dye was confirmed out to 7 days after initial formulation (Fig. S1D and E, ESI†) An initial in vitro assessment by flow cytometry confirmed a significantly higher uptake of E-MC(FA) compared to E-MC following incubation with B16F10 melanoma cells for 1 h (Fig. S4, ESI†).
To investigate the enhancement in targeted delivery of MiNP co-administration, we compared the effect of our strategy on the uptake of folic-acid targeted E-MC(FA) vs. non-targeted E-MC. Mice were first inoculated with B16F10 melanoma cells, which were allowed to grow for approximately two weeks. Following an initial (+)MiNP or (−)MiNP injection at the −24 h timepoint, E-MC(FA) and E-MC were then coinjected with (+)MiNP or (−)MiNP using either SC or IV routes of administration (Fig. 4a). E-MC(FA) and E-MC uptake was assessed in the liver, lymph nodes, spleen and tumor using flow cytometry. DiR dye was used to identify both E-MC and E-MC(FA). IV injection of (+)MiNP increased E-MC uptake in tumor and decreased uptake in lymph node CD45-cells (non-immune) as well as lymph node macrophages (Fig. 4b–d). SC injection of (+)MiNP increased E-MC content in serum at 24 h while decreasing E-MC in liver and splenic DCs and macrophages (Fig. S5, ESI† and Fig. 4e–g). No differences were found in any of the tumor cell subsets using SC administration. These data indicate that IV injection of MiNPs could facilitate increased tumor targeting of an effector therapeutic, but that SC would not.
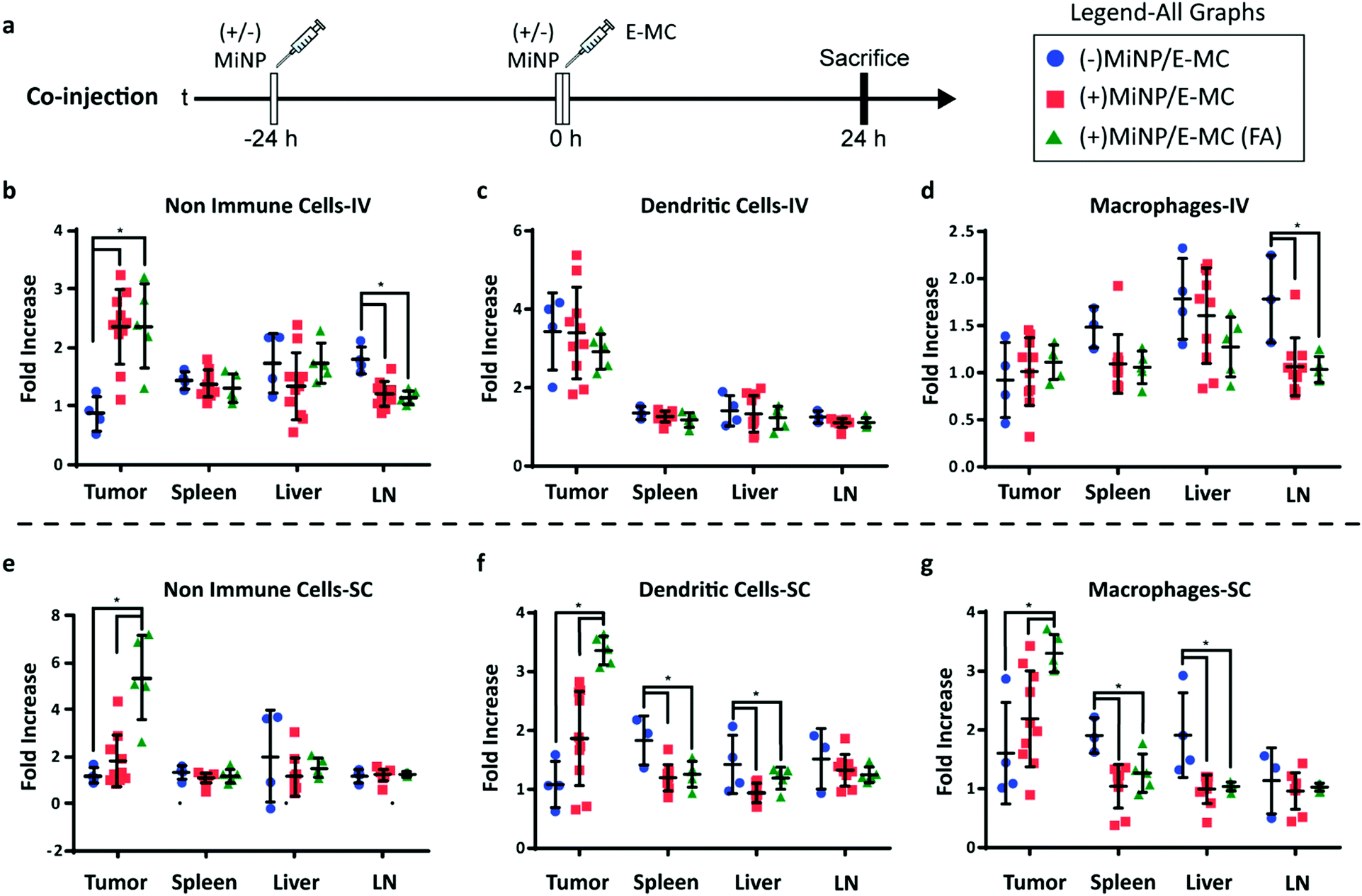 | ||
| Fig. 4 (+)MiNP treatment increases the accumulation of folate-targeted E-MC (E-MC(FA)) in B16F10 tumors following SC injection. (a) Timeline of injection protocol assessing the tumor-targeting co-injection method. (±)MiNP indicates an injection of either (+)MiNP or (−)MiNP. Mice were sacrificed 24 h after the co-injection for analysis by flow cytometry. Results are shown for IV (b–d) and SC (e–g) injections of 3 co-injection modalities: (−)MiNP treatment/E-MC, (+)MiNP treatment/E-MC, and (+)MiNP/E-MC(FA). Fluorescent E-MC and E-MC(FA) uptake by 3 different cell subsets were quantified: non-immune cells (b and e), dendritic cells (c and f), and macrophages (d and g) for 4 different organs. Data are reported as fold increase median fluorescence intensity of E-MC or E-MC(FA) over a PBS baseline control. N = 4–10, p < 0.05. Significance was determined within each organ by separate unpaired student's t-tests. | ||
The administering of folate receptor targeted E-MC(FA) instead of E-MC had no significant effect when administered IV, but when administered SC, there was significantly increased uptake of E-MC(FA) in all tumor cell subsets (Fig. 4b–g). This increased tumor accumulation for targeted nanoparticles following only one of the routes of administration was unexpected, but may be explained by differences in E-MC clearance in MPS organs. Only the SC injection of (+)MiNP resulted in significant decreases in macrophage and dendritic cell uptake of E-MC in the spleen and liver, which would account for the additional E-MC(FA) available for accumulate in tumors. Our in vitro data demonstrated that MiNP inhibits macropinocytosis but does not strongly impact receptor mediated endocytosis (Fig. 2c, d and Fig. S6, ESI†), which we hypothesized would enhance the uptake of a receptor-targeted chase nanoparticle in vivo. Thus our co-administration of MiNP with E-MC possessing an additional folate receptor targeting element validated these in vitro results by significantly enhancing the accumulation of E-MC(FA) in B16F10 tumors up to 8-fold (Fig. 4e–g). This significant increase in uptake for E-MC(FA) versus E-MC was only observed within the solid tumors and in no other organs.
Conclusions
Here, we demonstrate the nanoparticle biodistribution-altering effects of MiNP that encapsulate a small molecule inhibitor of macropinocytosis, LatA. We have characterized and evaluated these nanoparticles both in vitro and in vivo as key mediators in a co-administration strategy to increase the targeting efficacy of a second “effector” nanoparticle, E-MC. Clearance by the MPS remains a critical issue for many drug delivery applications beyond nanoparticles, suggesting a potentially broad range of applications for MiNP. For example, MPS organs are major sites of off-target accumulation for monoclonal antibodies36 and decreasing this effect may allow enhanced efficacy with lower dosages and fewer side effects during cancer therapy. Strategies for blocking the clearance of therapeutic antibodies have long been under investigation,37 yet inhibiting non-specific uptake via macropinocytosis remains underexplored. Recently, the depletion of subcapsular sinus macrophages via liposomes loaded with clodronate and other agents was employed to investigate the role of these cells during nanovaccination.38 Results showed that removal of these cells prior to immunization enhanced delivery of the nanovaccine to lymph node follicles for improved humoral responses. Our work supports such strategies while additionally demonstrating that inhibition of MPS cells can be performed in a reversible and nontoxic manner without killing phagocytes, many of which play critical downstream roles in the generation of an immune response. Furthermore, SC administration of MiNP increased the serum concentration of E-MC to levels similar to IV administration, potentially opening up new routes of administration and dosing regimens previously unavailable to many nanotherapeutics and controlled delivery strategies.In a tumor model, we found that MiNP increase target tissue and cell accumulation through reduction of uptake by phagocytic cells such as macrophages and dendritic cells in the liver and spleen, which accounts for >90% of MPS cells. Our results validate LatA loaded PEG-b-PPS MiNP as a promising platform to improve the performance of other, paired effector nanoparticle therapeutic and diagnostic platforms. These proof-of-concept results justify the exploration of alternative MiNP formulations encapsulating inhibitors in addition to or in combination with LatA, as well as the investigation of MiNP as part of functional strategies employing effector nanoparticles loaded with diagnostic and/or therapeutic agents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for support from the Center for Computation & Theory of Soft Materials, the BioCryo facility of Northwestern University's NUANCE Center, the Integrated Molecular Structure Education and Research Center, Structural Biology Facility, NU Atomic, the Nanoscale Characterization Experimental Center, the Louis A. Simpson & Kimberly K. Querrey Center for Regenerative Nanomedicine, the Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core, and the Biological Imaging Facility at Northwestern University. SAXS experiments were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by Northwestern University, E. I. DuPont de Nemours & Co., and The Dow Chemical Company. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Funding: this work was supported by the National Institutes of Health Director's New Innovator Award (grant no. 1DP2HL132390-01), and the National Science Foundation (CBET-1806007 and CAREER Award no. 1453576).References
- R. F. Pagels and R. K. Prud’homme, J. Controlled Release, 2015, 219, 519–535 CrossRef CAS PubMed.
- N. Desai, AAPS J., 2012, 14, 282–295 CrossRef CAS PubMed.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- R. van Furth, Z. A. Cohn, J. G. Hirsch, J. H. Humphrey, W. G. Spector and H. L. Langevoort, Bull. W. H. O., 1972, 46, 845–852 CAS.
- N. Bertrand and J.-C. Leroux, J. Controlled Release, 2012, 161, 152–163 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- D. Dutta and J. G. Donaldson, Cell. Logist., 2012, 2, 203–208 CrossRef PubMed.
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2009, 78, 857–902 CrossRef CAS PubMed.
- S. Xu, B. Z. Olenyuk, C. T. Okamoto and S. F. Hamm-Alvarez, Adv. Drug Delivery Rev., 2013, 65, 121–138 CrossRef CAS PubMed.
- N. G. Sosale, K. R. Spinler, C. Alvey and D. E. Discher, Curr. Opin. Immunol., 2015, 35, 107–112 CrossRef CAS PubMed.
- N. G. Sosale, T. Rouhiparkouhi, A. M. Bradshaw, R. Dimova, R. Lipowsky and D. E. Discher, Blood, 2015, 125, 542–552 CrossRef CAS PubMed.
- K. P. García, K. Zarschler, L. Barbaro, J. A. Barreto, W. O’Malley, L. Spiccia, H. Stephan and B. Graham, Small, 2014, 10, 2516–2529 CrossRef.
- S. Allen, O. Osorio, Y.-G. Liu and E. Scott, J. Controlled Release, 2017, 262, 91–103 CrossRef CAS.
- S. D. Allen, Y.-G. Liu, S. Bobbala, L. Cai, P. I. Hecker, R. Temel and E. A. Scott, Nano Res., 2018, 11, 5689–5703 CrossRef CAS.
- D. J. Dowling, E. A. Scott, A. Scheid, I. Bergelson, S. Joshi, C. Pietrasanta, S. Brightman, G. Sanchez-Schmitz, S. D. Van Haren, J. Ninković, D. Kats, C. Guiducci, A. de Titta, D. K. Bonner, S. Hirosue, M. A. Swartz, J. A. Hubbell and O. Levy, J. Allergy Clin. Immunol., 2017, 140, 1339–1350 CrossRef CAS PubMed.
- S. Yi, S. D. Allen, Y.-G. Liu, B. Z. Ouyang, X. Li, P. Augsornworawat, E. B. Thorp and E. A. Scott, ACS Nano, 2016, 10, 11290–11303 CrossRef CAS PubMed.
- E. A. Scott, A. Stano, M. Gillard, A. C. Maio-Liu, M. A. Swartz and J. A. Hubbell, Biomaterials, 2012, 33, 6211–6219 CrossRef CAS PubMed.
- S. Bobbala, S. D. Allen and E. A. Scott, Nanoscale, 2018, 10, 5078–5088 RSC.
- A. M. Bannunah, D. Vllasaliu, J. Lord and S. Stolnik, Mol. Pharmaceutics, 2014, 11, 4363–4373 CrossRef CAS PubMed.
- S. T. Reddy, A. J. van der Vlies, E. Simeoni, V. Angeli, G. J. Randolph, C. P. O’Neil, L. K. Lee, M. A. Swartz and J. A. Hubbell, Nat. Biotechnol., 2007, 25, 1159–1164 CrossRef CAS PubMed.
- V. Manolova, A. Flace, M. Bauer, K. Schwarz, P. Saudan and M. F. Bachmann, Eur. J. Immunol., 2008, 38, 1404–1413 CrossRef CAS PubMed.
- N. B. Karabin, S. Allen, H.-K. Kwon, S. Bobbala, E. Firlar, T. Shokuhfar, K. R. Shull and E. A. Scott, Nat. Commun., 2018, 9, 624 CrossRef.
- S. D. Allen, Y.-G. Liu, T. Kim, S. Bobbala, S. Yi, X. Zhang, J. Choi and E. A. Scott, Biomater. Sci., 2019, 7, 657–668 RSC.
- M. C. Louzao, I. R. Ares and E. Cagide, FEBS J., 2008, 275, 6067–6074 CrossRef CAS PubMed.
- W. M. Morton, K. R. Ayscough and P. J. McLaughlin, Nat. Cell Biol., 2000, 2, 376–378 CrossRef CAS PubMed.
- I. Spector, N. R. Shochet, D. Blasberger and Y. Kashman, Cell Motil. Cytoskeleton, 1989, 13, 127–144 CrossRef CAS PubMed.
- T. Stack, A. Vahabikashi, M. Johnson and E. Scott, J. Biomed. Mater. Res., Part A, 2018, 106, 1771–1779 CrossRef CAS PubMed.
- S. Cerritelli, C. P. O’Neil, D. Velluto, A. Fontana, M. Adrian, J. Dubochet and J. A. Hubbell, Langmuir, 2009, 25, 11328–11335 CrossRef CAS PubMed.
- H. Konishi, S. Kikuchi, T. Ochiai, H. Ikoma, T. Kubota, D. Ichikawa, H. Fujiwara, K. Okamoto, C. Sakakura, T. Sonoyama, Y. Kokuba, H. Sasaki, T. Matsui and E. Otsuji, Anticancer Res., 2009, 29, 2091–2097 CAS.
- A. Doller, A. Badawi, T. Schmid, T. Brauß, T. Pleli, D. M. zu Heringdorf, A. Piiper, J. Pfeilschifter and W. Eberhardt, Exp. Cell Res., 2015, 330, 66–80 CrossRef CAS PubMed.
- S. D. Allen, S. Bobbala, N. B. Karabin, M. Modak and E. A. Scott, ACS Appl. Mater. Interfaces, 2018, 10, 33857–33866 CrossRef CAS PubMed.
- D. R. Overby, E. H. Zhou, R. Vargas-Pinto, R. M. Pedrigi, R. Fuchshofer, S. T. Braakman, R. Gupta, K. M. Perkumas, J. M. Sherwood, A. Vahabikashi, Q. Dang, J. H. Kim, C. R. Ethier, W. D. Stamer, J. J. Fredberg and M. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2014, 201410602, DOI:10.1073/pnas.1410602111.
- L. Cheng, H. Ma, M. Shao, Q. Fan, H. Lv, J. Peng, T. Hao, D. Li, C. Zhao and X. Zong, Mol. Med. Rep., 2017, 16, 1101–1108 CrossRef CAS PubMed.
- S. Yi, X. Zhang, M. H. Sangji, Y. Liu, S. D. Allen, B. Xiao, S. Bobbala, C. L. Braverman, L. Cai, P. I. Hecker, M. DeBerge, E. B. Thorp, R. E. Temel, S. I. Stupp and E. A. Scott, Adv. Funct. Mater., 2019, 29, 1904399 CrossRef CAS.
- C. Halma, M. R. Daha and L. A. van Es, Clin. Exp. Immunol., 1992, 89, 1–7 CrossRef CAS PubMed.
- S. B. Clarkson, R. P. Kimberly, J. E. Valinsky, M. D. Witmer, J. B. Bussel, R. L. Nachman and J. C. Unkeless, J. Exp. Med., 1986, 164, 474–489 CrossRef CAS PubMed.
- Y.-N. Zhang, W. Poon, E. Sefton and W. C. W. Chan, ACS Nano, 2020, 14, 9478–9490 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00679c |
| This journal is © The Royal Society of Chemistry 2021 |