
Recent advances in the development of covalent inhibitors
Hyunsoo
Kim†
a,
Yoon Soo
Hwang†
 a,
Mingi
Kim
a and
Seung Bum
Park
*abc
a,
Mingi
Kim
a and
Seung Bum
Park
*abc
aCRI Center for Chemical Proteomics, Department of Chemistry, Seoul National University, Seoul 08826, Korea. E-mail: sbpark@snu.ac.kr
bDepartment of Biophysics and Chemical Biology, Seoul National University, Seoul 08826, Korea
cSPARK Biopharma, Inc., Seoul 08791, Korea
First published on 4th May 2021
Abstract
The use of covalent inhibitors in the field of drug discovery has attracted considerable attention in the 2000s. As a result, more than 50 covalent drugs are currently on the market, and numerous covalent drug candidates are now under development. Therefore, interest in covalent drugs is expected to continue in the future. The purpose of this focused review is to provide an understanding of the development of covalent inhibitors by describing their inherent characteristics, possibilities, and limitations based on their mechanistic differences from noncovalent inhibitors. We also introduce the latest covalent warheads that can be applied to the development of potential covalent inhibitors.
1. Introduction
Drugs that form covalent interactions with a target, such as penicillin and omeprazole (a proton pump inhibitor), are continuously developed and widely used.1 However, with the increase in the interest in covalent drugs,1,2 highlighted in the early 2000s, the paradigm of approaches for their development has changed. A survey of drugs approved from the mid-1900s to early 2000s revealed that there were few cases in which drugs were developed by intentionally introducing apparent covalent moieties, such as Michael acceptors; in most cases, the introduction of a covalent moiety during the developmental stage has not been deliberate; rather, the moiety is covalently bonded to the target by metabolism. In contrast, since the early 2000s, the development of most covalent drugs, which have pharmacological advantages like prolonged duration of action and enhanced potency over noncovalent drugs, included the intentional introduction of a covalent bonding moiety that can interact with the target.1,2 Among them, the acrylamide moiety has been the most commonly used.3Although covalent drugs have been employed since a long time, the paradigm is currently shifting from an era of accidental discovery to a phase of rational design and development.4 This shift has been facilitated by the recent advancements in basic science and technology for drug discovery, including proteomics, which enables the improvement of covalent drug-target selectivity by identifying their binding sites. The value of the rational design approach for covalent drugs is expected to increase due to the continued increase in demand for covalent small molecules in the fields of medicinal chemistry and chemical biology, the advantages of covalent drugs, and their clinical effectiveness.
Here, we will review the most recent and relevant studies investigating the development of covalent binding small-molecules. Specifically, this review will focus on rationally designed covalent inhibitors (targeted covalent inhibitors, TCIs) with a covalent moiety (warhead), rather than a structure with electrophilic moieties generated via metabolic reactions. Based on the types of electrophilic functional groups on the covalent inhibitor, we will provide actual inhibitors for each type along with their co-crystal structure to demonstrate their mode of actions. Throughout the manuscript, the term covalent inhibitor will include both reversible and irreversible inhibitors, unless otherwise specified.
In the first part of this focused review, we present the mechanisms, characteristics, and limitations of covalent inhibitors. Using the mechanistic properties of covalent inhibitors that differentiate them from noncovalent inhibitors, we will illustrate the differences between actual efficacy and expected activity which is inferred from pharmacokinetic parameters such as area under curve (AUC) and clearance via independent consideration of pharmacokinetics (PK) and pharmacodynamics (PD). In addition, we will introduce target occupancy (TO), a factor that predicts the activity of covalent inhibitors, and then describe the limitations of covalent inhibitors. In the second part, we will present the covalent warheads used and reported to date. This review will highlight the recent studies conducted after the comprehensive review by Gehringer et al.5 We would like to introduce previously reported covalent moieties that have not been incorporated into approved drugs and present the research stage and potential use of each structure. Some of the specific warheads include sulfur(VI) motifs, heteroaromatic motifs, strain-released motifs, and terminal alkynes, which are all novel proposals. Our focused review intends to characterize the flow of development of covalent inhibitors by introducing state-of-the-art studies related to covalent inhibitors.
2. Mechanism and characteristics of covalent inhibitors
2.1. Mechanism and evaluation
Distinct from a noncovalent inhibitor, a covalent inhibitor operates in two steps to inhibit its target protein. The general mechanism underlying the action of a covalent inhibitor is shown in Fig. 1. First, the covalent inhibitor binds to its target protein by forming specific noncovalent interactions between the protein and the compound, which is similar to the binding mechanism of noncovalent inhibitors. This step is an equilibrium process represented by the inhibition constant (Ki), which indicates the binding affinity of the ligand. Once ligands interact with their target proteins, direct bond formation occurs between the covalent inhibitor and its target protein. During the second step, the electrophilic warhead of the covalent inhibitor is appropriately oriented and positioned proximal to the nucleophilic residues on the target protein. This step is a non-equilibrium process that is used to classify covalent inhibitors. If the rate constant of the reverse reaction (k−2) in which the inhibitor–protein complex dissociates is equal to or approaching zero, the inhibitor is called an irreversible covalent inhibitor exemplified by penicillin (Fig. 1a). In this extreme case, nearly perfect inhibition would be observed if the compound interacts with the target protein for enough time. Conversely, a reversible covalent inhibitor such as bortezomib (a proteasome inhibitor) is associated with a more practical k−2 value, showing an intermediate behavior between noncovalent inhibitors and irreversible covalent inhibitors (Fig. 1b).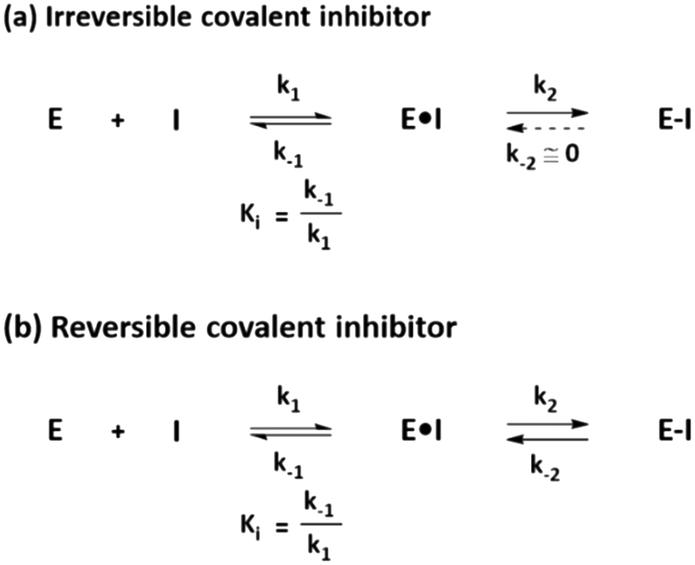 | ||
| Fig. 1 General mechanism underlying the action of (a) irreversible covalent inhibitors and (b) reversible covalent inhibitors. | ||
Target proteins bound by covalent inhibitors can lose their original functions and exhibit the desired inhibitory activity. For reversible covalent inhibitors, the reverse reaction should occur to recover the original functions of the target protein. Otherwise, de novo synthesis of the protein should occur after the covalent inhibitor is cleared. In this case, covalent inhibition is directly associated with the re-synthesis rate of the target protein. Mechanistically, the residence time of the covalent inhibitor should be longer than the turnover of the target protein to exhibit significant covalent inhibition.1
Normally, the inhibitory potency is determined by measuring the Ki or the half maximal inhibitory concentration (IC50), which is also applied to covalent inhibitors. However, these methods are not appropriate for measuring the potency of covalent inhibitors because, in the case of these inhibitors, the Ki and IC50 value are time-dependent6 and can result in a misleading evaluation during the incubation time. To rank the potency of covalent inhibitors, kinetics of the second step should be considered. k2/Ki is an accurate metric for irreversible covalent inhibitors, whereas 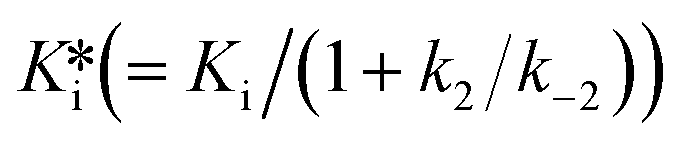 is an accurate metric for reversible covalent inhibitors.7
is an accurate metric for reversible covalent inhibitors.7
2.2. PK/PD
During metabolism and clearance from the body, drugs reach and bind their targets according to their binding kinetics and affinity, leading to the desired PD response for therapeutic efficacy of the drug. Usually, noncovalent inhibitors bind to their targets and form a rapid equilibrium, making PK an important influencing factor of PD. Specifically, the increase in AUC and decrease in clearance would allow the drug concentration to reach the threshold for therapeutic effects to be manifested. For a favorable PD profile, PK parameters and binding properties of a noncovalent inhibitor should be optimized together.Conversely, covalent inhibitors show distinct features based on the non-equilibrium step of covalent bond formation. This characteristic also affects the PK/PD profile for covalent inhibitors, and covalent inhibitors exhibit a distinctive PK/PD profile. With this nonequilibrium process, covalent inhibitors maintain their efficacy while unreacted compounds are metabolised and cleared from the body; this characteristic improves the actual efficacy of covalent inhibitors when compared to noncovalent inhibitors possessing the same PK parameters (Fig. 2). In this respect, the PD of covalent inhibitors is decoupled from their PK and covalent inhibitors exhibit a prolonged duration of action. For example, omeprazole shows a short half-life about 1 h but its efficacy last for more than 12 h.8 Thus, researchers do not have to consider optimising processes to improve the PK parameters to induce prolonged exposure. Owing to their prolonged duration of action, covalent inhibitors often exhibit good potency.1,2,9,10 Furthermore, covalent inhibitors can be administered at lower doses and frequencies, reducing the off-target events and toxicity.2,10,11
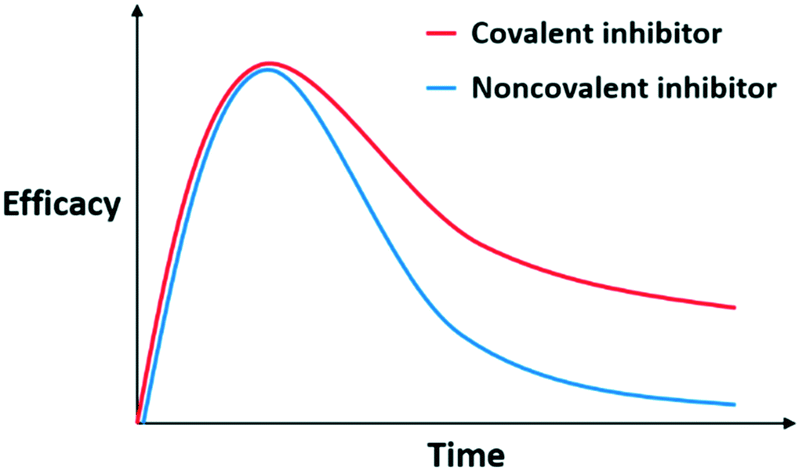 | ||
| Fig. 2 Hypothetical pharmacokinetic (PK)/pharmacodynamic (PD) profiles of noncovalent and covalent inhibitors with the same PK parameters. | ||
Recently, a PK/PD model correlating drug–target binding kinetics to predict in vivo efficacy of covalent inhibitors was generated by Tonge and Fisher et al.12–14 In 2015, they developed the PK/PD model to simulate slow-binding inhibitors that behave like reversible covalent inhibitors of the UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC).12 By measuring the PK profiles and binding kinetic parameters using in vitro assays, the authors successfully predicted the in vivo efficacy of LpxC inhibitor, PF5081090. Later, they modified the PK/PD model to simulate an irreversible covalent inhibitor for Bruton's tyrosine kinase (BTK).14 In this study, they considered not only the PK profiles and binding kinetics of the drug, but also the turnover of the target, to quantitatively correlate the engagement of BTK by the irreversible covalent inhibitor CC-292. Based on this modelling, Tonge and Fisher et al. successfully predicted the in vivo efficacy of the BTK inhibitor CC-292. One of the advantages of using PK/PD models for covalent inhibitors is the ability to directly and accurately quantify the TO, which represents the degree to which a drug occupies a given target. This approach mainly provides a prediction of the PD that matches the actual PD. Thus, optimal dosage of covalent inhibitors can be predicted, and the success rate of drug development can be improved.
TO is an important concept that is directly associated with the pharmacological effects of covalent inhibitors. Each target requires a different level of TO to elicit the desired pharmacological effect, called target vulnerability.15 A high TO is required to elicit the desired pharmacological effect in low vulnerability targets, although a low TO is enough to elicit the desired pharmacological effect in high vulnerability targets. For low vulnerability targets, a covalent inhibitor can be an effective strategy to occupy the target binding site for a prolonged period.
2.3. Limitation and selectivity issues
The use of covalent inhibitors can represent an appropriate strategy for overcoming the drug-resistance mutations in the gene encoding the target protein. This has been demonstrated for the epidermal growth factor receptor (EGFR) protein.16,17 If the binding sites of EGFR (T790M and/or L858R) are mutated, noncovalent inhibitors such as gefitinib lose their activity, while covalent inhibitors such as osimertinib remain potent and inhibit cell proliferation by forming covalent bonds with Cys797. However, a mutation that occurs at the targeted amino acid residue would be fatal for the covalent inhibitor. For EGFR, a C797S mutation has a severe negative impact on the capacity of osimertinib of binding its target protein.18Ideally, covalent inhibitors would bind only to their desired target proteins and form drug–protein complexes. However, electrophilic warheads of a covalent inhibitor can react with nucleophilic residues on the other proteins, especially within the same family or cellular metabolites. For instance, covalent inhibitors that target a cysteine residue on a protein can conjugate with glutathione (GSH), thus perturbing cellular redox environments.9,10,19 This type of off-target effect is associated with hepatotoxicity due to liver protein binding and immunogenicity caused by drug–protein complexation, leading to idiosyncratic toxicity or haptenisation.1,2,9,10,20 Compared with irreversible covalent inhibitors, reversible covalent inhibitors are theorized to have less toxicity issues, which has attracted special attention in the drug development process.21 This is attributed to the dissociative ability of reversible covalent inhibitors, while the labelling of off-target proteins by irreversible covalent inhibitors is permanent.
The selectivity of covalent inhibitors has therefore been considered an important issue. Covalent inhibitors operate through two steps, and the selectivity of the first step is determined by Ki. The affinity of the compound to the desired target protein can be increased through optimization. Conversely, the selectivity of the second step is determined by both the target residue of the protein and the warhead of the covalent inhibitor. When deciding the target residue, it is critical to know whether the target residue is catalytic or noncatalytic. If the covalent inhibitor targets catalytic residues, such as cysteine or serine, it cannot distinguish between proteins within the same family. Thus, targeting poorly conserved noncatalytic amino acids can be an attractive strategy for improving the selectivity within the same target protein family.22,23 After choosing an appropriate target residue, an appropriate warhead should be selected for the target residue to minimize concerns regarding off-target issues. Normally, the reactivity of the warhead depends on its electronic character and steric factor. Based on the properties of each warhead, delicate tuning of reactivity and selectivity improvement are feasible in the process of drug development.5
3. Promising warheads for covalent inhibitors
An electrophilic functional group on the covalent inhibitor, also known as a “warhead”, forms a covalent bond with a nucleophilic residue such as cysteine, lysine, or tyrosine on targeted proteins.5 In this part, we review the recently developed α,β-unsaturated carbonyl moieties and the recently reported promising electrophilic warheads such as sulfur(VI) motifs, heteroaromatic motifs, strain-released motifs, and terminal alkynes producing either reversible or irreversible inhibition.3.1. α,β-Unsaturated carbonyl
Various electrophilic warheads exhibiting various reactivities have been introduced for covalent inhibitors. The most commonly used electrophilic building blocks in a covalent inhibitor targeting cysteine are Michael acceptors, such as acrylamides and other α,β-unsaturated carbonyl compounds (Fig. 3a). The olefin moiety of the acrylamide warhead reacts irreversibly with the thiol group from the cysteine via Michael addition and forms conjugated adducts (Fig. 3b). Neratinib (2017), dacomitinib (2018), and zanubrutinib (2019) are notable examples of recent FDA-approved drugs bearing acrylamide for irreversible covalent reaction with cysteine, and are capable of inhibiting human protein kinases (Fig. 3c).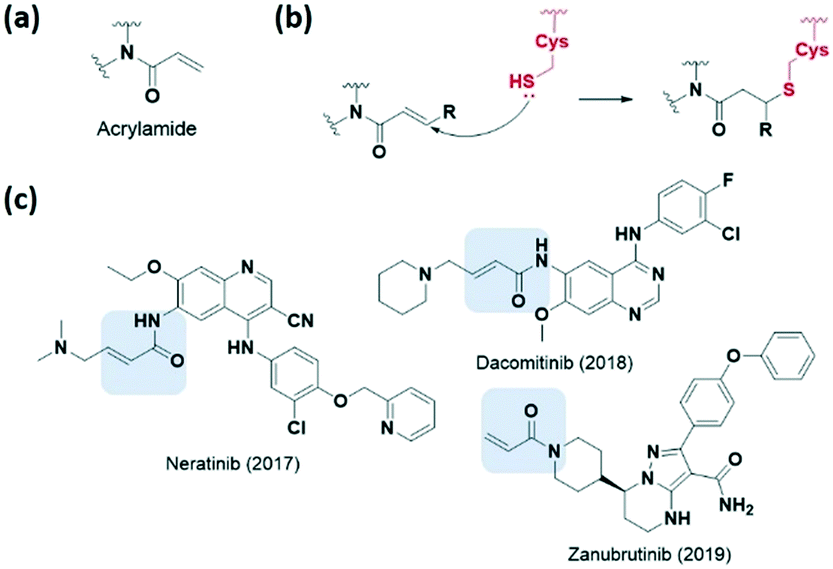 | ||
| Fig. 3 α,β-Unsaturated carbonyl moiety acrylamide as cysteine-targeting covalent warheads. (a) Chemical structure of acrylamide. (b) Mechanism of acrylamide forming a covalent adduct with cysteine. (c) Chemical structure and year of approval of acrylamide-based FDA-approved drugs neratinib, dacomitinib, zanubrutinib. | ||
To avoid the off-target effects of acrylamide covalent inhibitors, numerous cases have been reported where acrylamide has been replaced with other reactive groups to improve reactivity and safety profiles. The introduction of a strong electron-withdrawing cyano group at the α-position was shown to induce a tight reversible covalent bond between the inhibitor and the cysteine of the targeted protein (Fig. 4a).23,24 Increased acidity of the α-position by the cyano group favoured both the thiol addition and thiol adduct elimination (Fig. 4b). Taunton et al. were the first to present reversible p90 ribosomal S6 kinase 2 (RSK2) inhibitors involving the cyano-substituted acrylamide motif, which exhibited long-lasting inhibitory effect and high potency as well as selectivity.23 They further developed BTK inhibitors containing end-capped α-cyanoacrylamide by systematic structural modification of the α-cyanoacrylamide warhead, which exhibited long target residence times (Fig. 4c and d).25 Also, in 2019, Park et al. applied a cyanoacrylamide warhead to the peroxisome proliferator-activated receptor γ (PPARγ) phosphorylation inhibitor to induce a reversible covalent bond with Cys285 in helix H3 of the PPARγ ligand-binding domain and successfully demonstrated the reversible covalent inhibition of the cyanoacrylamide moiety through mass spectrometry and X-ray crystallography studies (Fig. 4c and d).26,27
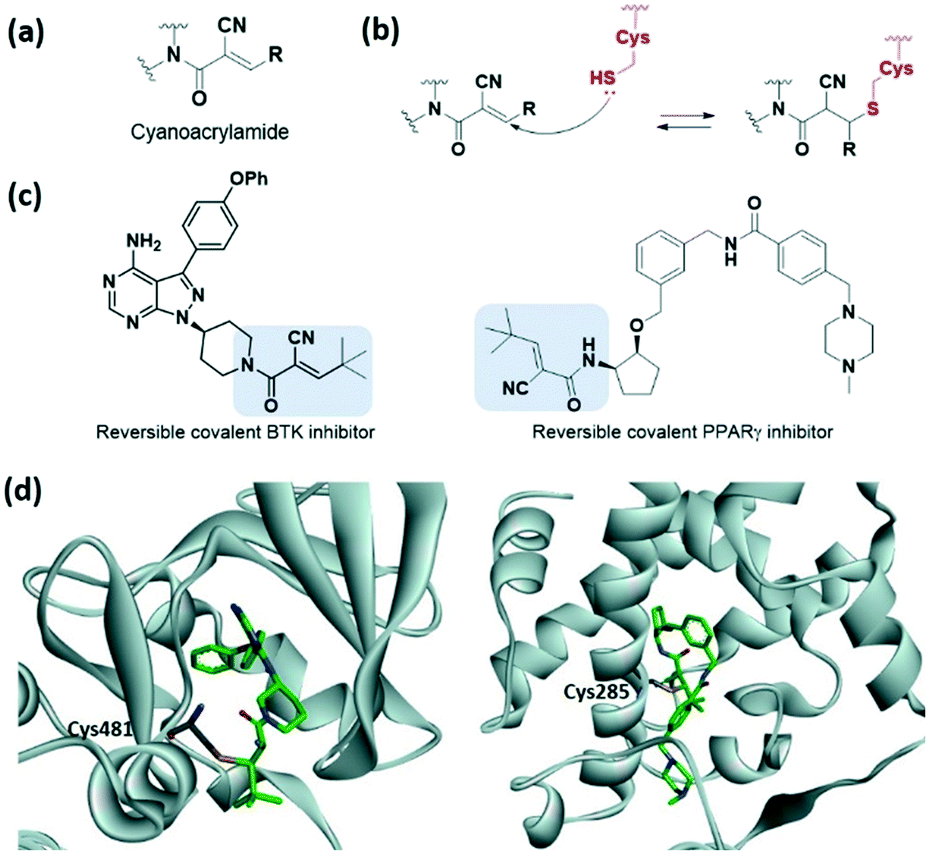 | ||
| Fig. 4 α,β-Unsaturated carbonyl moiety cyanoacrylamide as cysteine-targeting covalent warheads. (a) Chemical structure of cyanoacrylamide. (b) Mechanism of cyanoacrylamide forming a reversible covalent adduct with cysteine. (c) Chemical structure of a cyanoacrylamide-based reversible covalent BTK inhibitor and PPARγ. (d) X-ray crystal structure of cyanoacrylamide-based reversible covalent inhibitor bound to Cys481 in BTK (PDB code: 4YHF) and bound to Cys285 in PPARγ (PDB code: 6IJR). | ||
The allenamide group, which exhibits weaker electron-deficient characters, has been reported as a reactive bioisostere for replacing the acrylamide group (Fig. 5a and b). In 2018, Zhao et al. presented a protocol for the synthesis and optimisation of the allenamide covalent inhibitor, which efficiently inhibited the enzymatic activity of T790M/L858R double mutant and wild-type EGFR kinases at a low nM range (Fig. 5c).28 In addition, they obtained a reaction rate constant of 302.5 × 10−3 min−1 by measuring the conjugated adduct formed by the allenamide functional group and GSH. The formation of the conjugated adduct between the allenamide functional group and GSH was about 7- to 28-fold higher than that of the marketed acrylamide-containing covalent drugs. However, allenamide-containing compounds showed poor pharmacokinetic properties and low stability in plasma and in vivo.
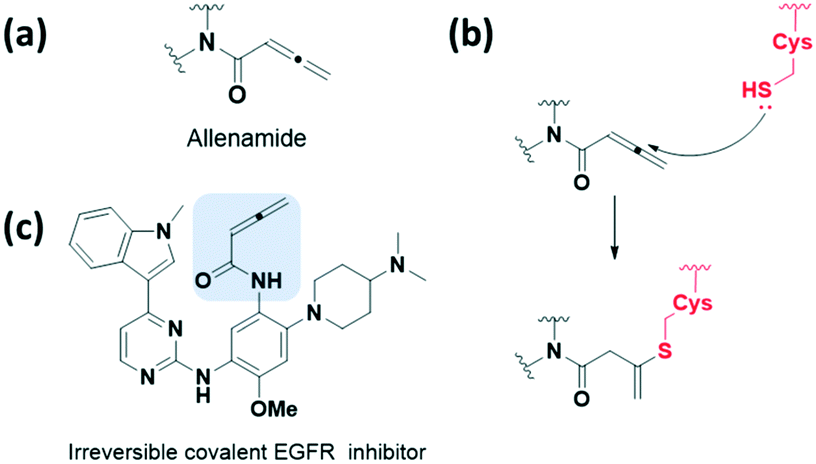 | ||
| Fig. 5 α,β-Unsaturated carbonyl moiety allenamide as cysteine-targeting covalent warheads. (a) Chemical structure of allenamide. (b) Mechanism of allenamide forming a covalent adduct with cysteine. (c) Chemical structure of allenamide-based irreversible covalent EGFR inhibitor. | ||
3.2. Sulfur(VI) warheads
We also detail the less common but recently employed electrophilic covalent reactive groups that could serve as alternative warheads to the Michael acceptor system in covalent drug design. Recently, inverse drug discovery (IDD) was found to be a promising strategy for developing covalent inhibitors. In 2018, Kelly et al. proposed and developed the concept of IDD based on latent warheads.29 IDD utilizes a small collection of diverse organic compounds that harbour a weak but activatable electrophile. This electrophile reacts with the human proteome in cell lysate and ultimately leads to the identification of the targeted protein(s). In other words, these compounds find their target proteins by primarily using a series of covalent inhibitors rather than discovering inhibitors of the targeted protein.In 2016 and 2018, Kelly et al. applied the IDD strategy with aryl fluorosulfates which had been underexplored sulfur(VI) halides (Fig. 6a).29,30 The aryl fluorosulfates showed increased kinetic stability, improved selectivity, and minimized off-target effects compared to sulfonyl fluorides in sulfur(VI) fluoride exchange (SuFEx) reaction. The latency of the aryl fluorosulfates binding to target proteins was largely affected by reversible binding of the warhead, appropriate orientation of residues, and transition-state stabilization of the reaction. Furthermore, X-ray crystal structure data revealed that these aryl fluorosulfates chemoselectively react with lysine as well as tyrosine side chains (Fig. 6b). Thus, they can be used to target largely non-overlapping groups of human proteins such as neutrophil elastase31 and HIV-1 reverse transcriptase.32 In 2020, Kelly et al. further demonstrated that sulfuramidimidoyl fluorides, a class of weak electrophiles, undergo SuFEx chemistry by applying the IDD approach (Fig. 6a).33 Binding of a sulfuramidimidoyl fluoride to the targeted protein can allow the nucleophilic attack of a specific amino acid side chain, leading to conjugate formation (Fig. 6b). After incubation of small molecules bearing a sulfuramidimidoyl fluoride electrophile with human cell lysate, the resulting protein-small molecule conjugates can be identified by affinity chromatography-mass spectrometry. The resulting compound involving the sulfuramidimidoyl fluoride electrophile, which covalently binds to and irreversibly inhibits the activity of poly(ADP-ribose)polymerase 1 (PARP1), an important anticancer target, was explored using the IDD approach (Fig. 6c).
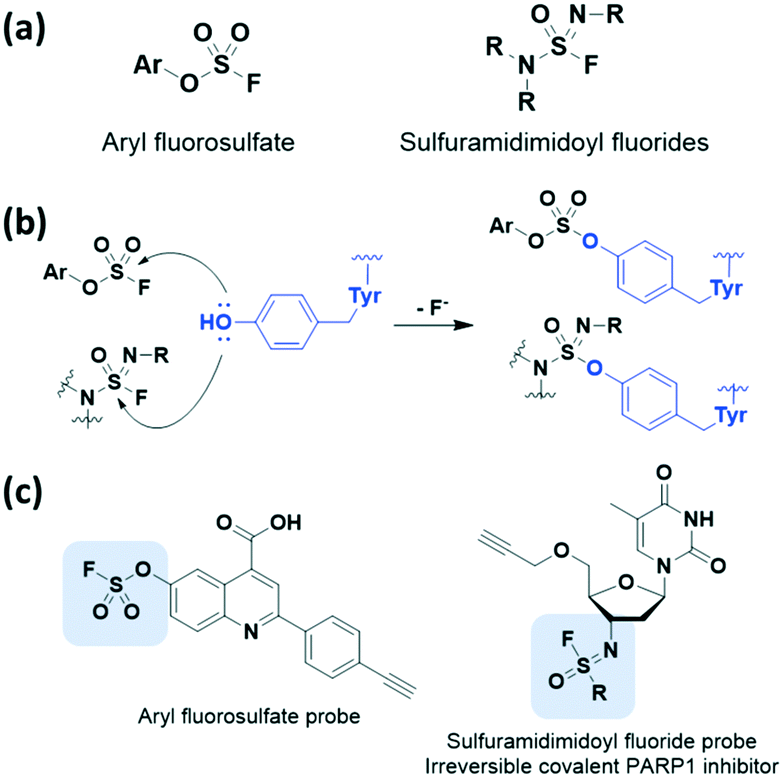 | ||
| Fig. 6 Sulfur(VI) warheads used in IDD approaches. (a) Chemical structure of aryl fluorosulfate and sulfuramidimidoyl fluorides. (b) Mechanism of aryl fluorosulfate and sulfuramidimidoyl fluorides forming covalent adduct with tyrosine. (c) Chemical structure of an aryl fluorosulfate probe and sulfuramidimidoyl fluoride probe which is an irreversible covalent PARP1 inhibitor discovered by the IDD approach. | ||
3.3. Heteroaromatic warheads
Heteroaromatic electrophiles have received relatively little attention throughout the design of covalent inhibitors; however, the interest in exploring novel aromatic moieties as a noble warhead capable of cysteine capture continues to grow. These heteroaromatic electrophiles react with thiols of cysteine residues via nucleophilic aromatic substitution (SNAr) reactions or nucleophilic addition. The potential of heteroaromatic motifs in developing TCI was revealed by screening a heterocyclic electrophilic fragment library of small heterocycles by Keserű et al. in 2018.34 This approach validated cysteine reactivity as well as aqueous stability, and characterized this motif as a UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) inhibitor.Heteroaromatic sulfones and sulfoxides have newly emerged as thiol-reactive electrophiles, which undergo SNAr reactions with thiols. In 2020, Martin et al. evaluated a library of heteroaromatic sulfides with different oxidation states, heteroatom substitutions, and a series of electron-donating and electron-withdrawing substituents.35 They demonstrated that the live-cell application of heteroaromatic sulfones, particularly methylsulfonylbenzothiazole, led to cysteine engagement and direct enrichment without the need for further click chemistry conjugation. In 2020, using cell-based screening, Bollong et al. identified a series of 2-sulfonylpyridines as a thiol-reactive electrophilic group allowing SNAr-based cysteine modification (Fig. 7a and b).36 The electrophilicity of this series was tuned by chemical structural optimisation of the 2-sulfonylpyridine scaffold, through the introduction of electron-withdrawing groups at the C3 position. By discovering a selective covalent modifier of adenosine deaminase (ADA), they demonstrated the potential of the 2-sulfonylpyridine reactive group as a covalent inhibitor (Fig. 7c).
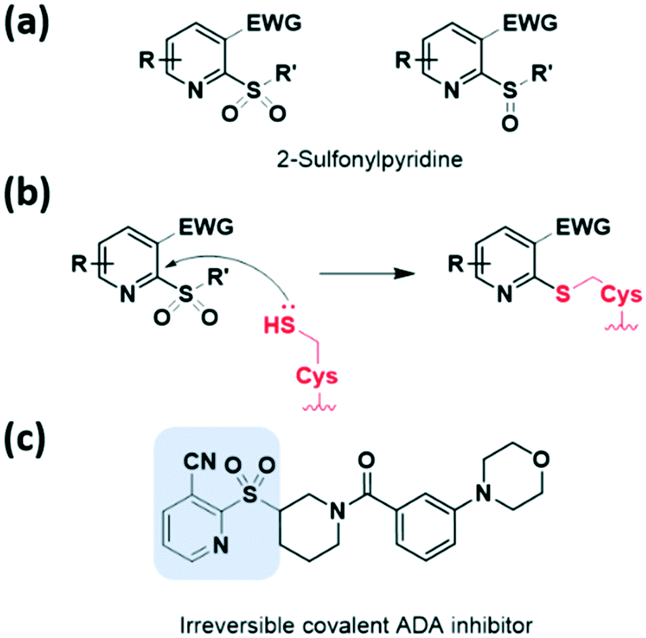 | ||
| Fig. 7 Heteroaromatic sulfones reported as thiol-reactive electrophiles. (a) Chemical structure of 2-sulfonylpyridine. (b) Mechanism of 2-sulfonylpyridine forming a covalent adduct with cysteine. (c) Chemical structure of 2-sulfonylpyridine-based irreversible covalent ADA modifier. | ||
Recently, McAulay et al. identified alkynyl benzoxazines and dihydroquinazolines as new reactive and selective heterocyclic moieties capable of cysteine capture (Fig. 8a and b).37 The conjugation of the Michael acceptor with the aromatic ring was extended with the introduction of benzoxazine motif. They demonstrated the application of these new moieties in Janus kinase 3 (JAK3) kinase drug scaffolds and also their improved in vitro PK profiles relative to those of acrylamide-based inhibitors (Fig. 8c). Improvements in clearance at human liver microsomes and both human and rathepatocytes with a ∼9-fold difference and also in human ether-a-go-go related gene (hERG) IC50 were observed. This novel motif showed comparable reactivity toward GSH range to osimertinib. Moreover, the cysteine of receptor tyrosine kinase c-KIT could be successfully targeted by these heterocycle moieties, while targeting by acrylamide had previously failed, as verified using X-ray crystal structure determination (Fig. 8d).
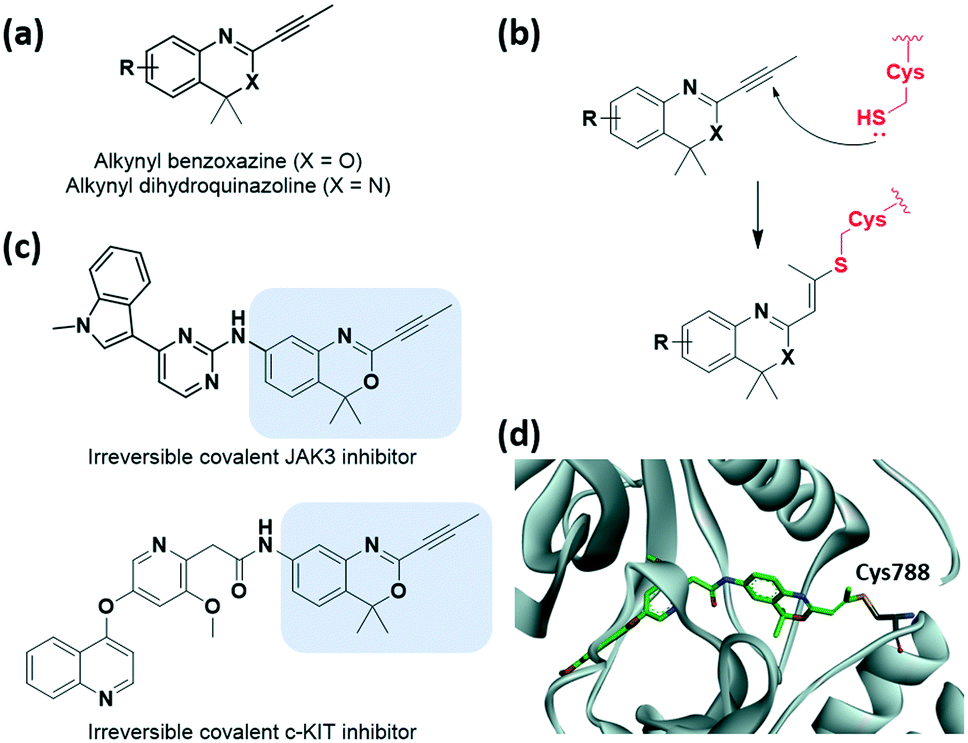 | ||
| Fig. 8 Alkynyl benzoxazine and dihydroquinazoline reported as cysteine-targeting warheads. (a) Chemical structure of alkynyl benzoxazine and dihydroquinazoline. (b) Mechanism of alkynyl benzoxazine and dihydroquinazoline forming covalent adduct with cysteine. (c) Chemical structure of alkynyl benzoxazine-based JAK3 and c-KIT covalent inhibitor (d) X-ray crystal structure of c-KIT covalent inhibitor bound to Cys788 in c-KIT (PDB code: 6XVB). | ||
3.4. Strain-released motifs
Among the strain-released motifs, bicyclo[1.1.0]butane (BCB) derivatives were explored as potential strained electrophiles (Fig. 9a). BCB is a highly strained ring that undergoes nucleophilic ring-opening. In 2017, Baran et al. proposed BCB sulfone as a cysteine-directed warhead for chemoselective covalent labelling of the target peptide.38 Moreover, in 2020, Ojida et al. used the BCB amide as a new class of strained electrophilic warheads.39 The strain-driven nucleophilic addition to BCB amides allowed selective covalent targeting of cysteine residues in live cells (Fig. 9b). Both covalent bond formation with cysteine thiols and tuning of the thiol reactivity with BCB were controlled by attaching electron-withdrawing groups. Furthermore, the BCB amide was applied to BTK inhibitor, and exhibited higher selectivity toward BTK in human cells than acrylamide-based probes (Fig. 9c).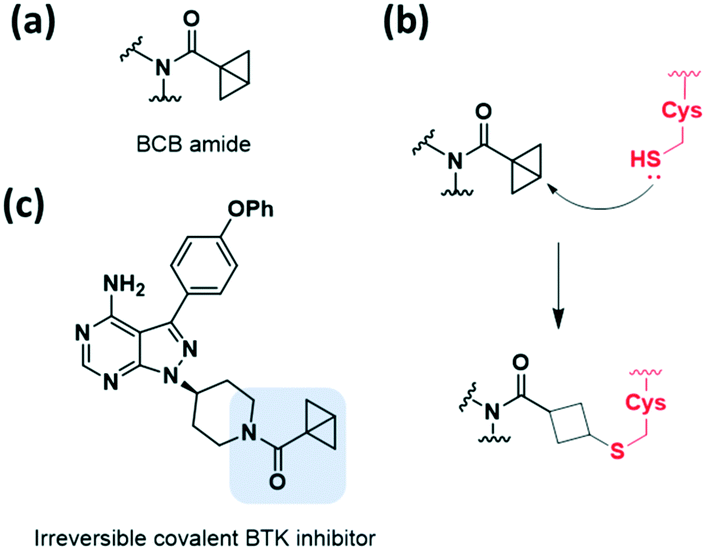 | ||
| Fig. 9 Strain-released motif of BCB amide reported as a warhead of cysteine-targeting covalent inhibitor. (a) Chemical structure of the BCB amide. (b) Mechanism of the BCB amide forming a covalent adduct with cysteine. (c) Chemical structure of the BCB amide-based irreversible covalent BTK inhibitor. | ||
Aziridine is one of the most notable strained electrophiles that undergoes nucleophilic ring-opening (Fig. 10a). This moiety has been studied and applied as mechanism-based inhibitors of glycosidase and cysteine protease (Fig. 10b).40,41 Recently, aziridine demonstrated potential as a warhead for other classes of target proteins. In 2020, Cierpicki et al. developed the first-in-class irreversible covalent inhibitors of nuclear receptor-binding SET domain protein 1 (NSD1) histone methyltransferase with aziridine moiety (Fig. 10c).42 In this study, they revealed that the conformation of the autoinhibitory loop of NSD1 was changed upon binding of the covalent inhibitor, as confirmed by crystal structure analysis. With this knowledge, covalent warheads were introduced to target Cys2062 of the autoinhibitory loop which is buried in the hydrophobic site. Among the warheads evaluated, the aziridine moiety could successfully label Cys2062, which was not possible using the acrylamide moiety. Due to its relatively small size and hydrophobic character, aziridine was suitable for targeting the buried binding site.
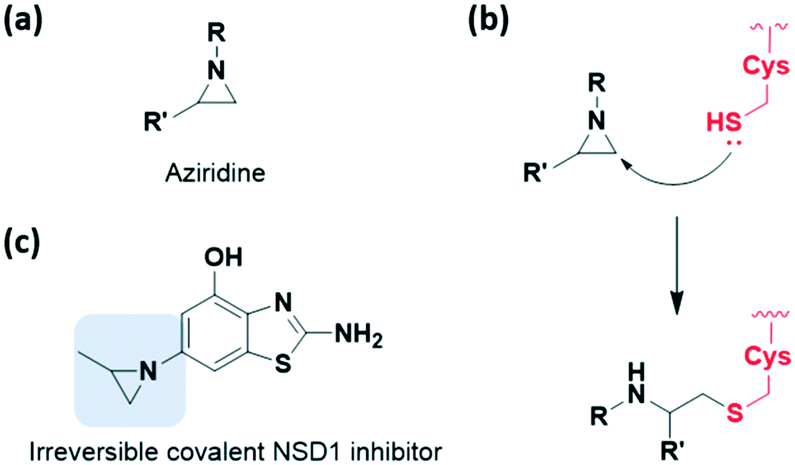 | ||
| Fig. 10 Strain-released motif aziridine reported as a warhead of cysteine-targeting covalent inhibitor. (a) Chemical structure of aziridine. (b) Mechanism of aziridine forming covalent adduct with cysteine. (c) Chemical structure of aziridine-based irreversible covalent NSD1 inhibitor. | ||
3.5. Terminal alkynes
Although terminal alkynes are generally used as a bioorthogonal click handle, they have been recently classified as a warhead of cysteine-targeting covalent inhibitors (Fig. 11a). In 2013, Ovaa et al. showed that a C-terminal propargyl moiety of ubiquitin reacts in an activity-based manner with the catalytic cysteine residue of deubiquitinating enzymes (DUBs), forming an irreversible thioether bond via an in situ thiol–alkyne addition (Fig. 11b).43 Moreover, in 2019, the alkyne moiety was introduced into small-molecule covalent inhibitors of cathepsin K (CatK) as a latent electrophile (Fig. 11c).44 While the alkyne-based covalent inhibitors are non-reactive against thiols, indicating proximity-driven reactivity, they formed an irreversible covalent bond with Cys25 in CatK, which was confirmed using X-ray crystal structure analysis (Fig. 11d). The reaction between the internal carbon of the alkyne moiety and cysteine forms a Markovnikov-type product. Inhibition of CatK-mediated bone resorption was also observed in human osteoclasts.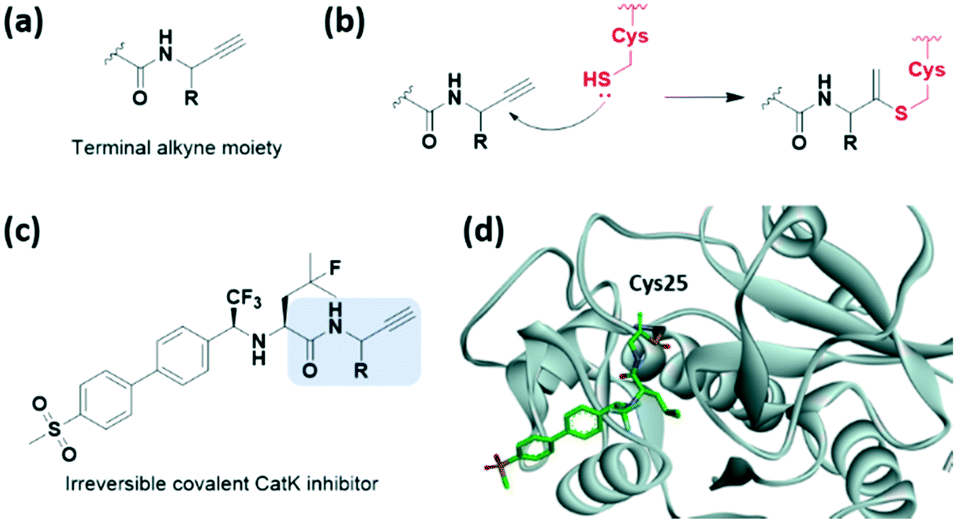 | ||
| Fig. 11 Terminal alkyne moieties reported as cysteine-targeting warheads. (a) Chemical structure of the terminal alkyne moiety. (b) Mechanism of the terminal alkyne moiety forming a covalent adduct with cysteine. (c) Chemical structure of the terminal alkyne moiety-based CatK covalent inhibitor. (d) X-ray crystal structure of the CatK covalent inhibitor bound to Cys25 in CatK (PDB code: 6QBS). | ||
4. Conclusion
Substantial evidence has indicated that covalent drugs exhibit an ideal advantage by inhibiting protein target reactivation until the regeneration of the protein, compared to noncovalent drugs which only transiently bind target proteins before dissociating. This behaviour has been thoroughly investigated, validated, and utilised for application in the clinical field. In addition, with the development of covalent inhibitors, chemical approaches for protein targets that were previously considered to be undruggable have been proposed. Additionally, the development of reactive groups that selectively attach amino acids such as lysine, tyrosine, and aspartate as well as cysteine is an interesting topic for future research. The rational development of covalent inhibitors will continue to be of great interest to researchers and is a field of sufficient value for future investigations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Creative Research Initiative Grant (2014R1A3A2030423), the Bio & Medical Technology Development Program (2012M3A9C4048780), and the international cooperation program (2016K2A9A2A10005504) through the National Research Foundation of Korea (NRF) funded by the Korean Government (Ministry of Science & ICT). H. K., Y. S. H., and M. K. are grateful for the fellowship by BK21 Plus Program.Notes and references
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 Search PubMed.
- R. A. Bauer, Drug Discovery Today, 2015, 20, 1061–1073 Search PubMed.
- E. D. Vita, Future Med. Chem., 2021, 13, 193–210 Search PubMed.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 Search PubMed.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 Search PubMed.
- B.-F. Krippendorff, R. Neuhaus, P. Lienau, A. Reichel and W. Huisinga, J. Biomol. Screening, 2009, 14, 913–923 Search PubMed.
- S. De Cesco, J. Kurian, C. Dufresne, A. K. Mittermaier and N. Moitessier, Eur. J. Med. Chem., 2017, 138, 96–114 Search PubMed.
- J. M. Shin and N. Kim, J. Neurogastroenterol. Motil., 2013, 19, 25–35 Search PubMed.
- T. Barf and A. Kaptein, J. Med. Chem., 2012, 55, 6243–6262 Search PubMed.
- T. A. Baillie, Angew. Chem., Int. Ed., 2016, 55, 13408–13421 Search PubMed.
- A. J. T. Smith, X. Zhang, A. G. Leach and K. N. Houk, J. Med. Chem., 2009, 52, 225–233 Search PubMed.
- G. K. Walkup, Z. You, P. L. Ross, E. K. H. Allen, F. Daryaee, M. R. Hale, J. O'Donnell, D. E. Ehmann, V. J. A. Schuck, E. T. Buurman, A. L. Choy, L. Hajec, K. Murphy-Benenato, V. Marone, S. A. Patey, L. A. Grosser, M. Johnstone, S. G. Walker, P. J. Tonge and S. L. Fisher, Nat. Chem. Biol., 2015, 11, 416–423 Search PubMed.
- F. Daryaee, A. Chang, J. Schiebel, Y. Lu, Z. Zhang, K. Kapilashrami, S. G. Walker, C. Kisker, C. A. Sotriffer, S. L. Fisher and P. J. Tonge, Chem. Sci., 2016, 7, 5945–5954 Search PubMed.
- F. Daryaee, Z. Zhang, K. R. Gogarty, Y. Li, J. Merino, S. L. Fisher and P. J. Tonge, Chem. Sci., 2017, 8, 3434–3443 Search PubMed.
- F. Daryaee and P. J. Tonge, Curr. Opin. Chem. Biol., 2019, 50, 120–127 Search PubMed.
- E. L. Kwak, R. Sordella, D. W. Bell, N. Godin-Heymann, R. A. Okimoto, B. W. Brannigan, P. L. Harris, D. R. Driscoll, P. Fidias, T. J. Lynch, S. K. Rabindran, J. P. McGinnis, A. Wissner, S. V. Sharma, K. J. Isselbacher, J. Settleman and D. A. Haber, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7665–7670 Search PubMed.
- C.-H. Yun, K. E. Mengwasser, A. V. Toms, M. S. Woo, H. Greulich, K.-K. Wong, M. Meyerson and M. J. Eck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2070–2075 Search PubMed.
- T. Grabe, J. Lategahn and D. Rauh, ACS Med. Chem. Lett., 2018, 9, 779–782 Search PubMed.
- Y. Shibata and M. Chiba, Drug Metab. Dispos., 2015, 43, 375–384 Search PubMed.
- J. Uetrecht, Chem. Res. Toxicol., 2008, 21, 84–92 Search PubMed.
- A. Bandyopadhyay and J. Gao, Curr. Opin. Chem. Biol., 2016, 34, 110–116 Search PubMed.
- K. K. Hallenbeck, D. M. Turner, A. R. Renslo and M. R. Arkin, Curr. Top. Med. Chem., 2017, 17, 4–15 Search PubMed.
- I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frödin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471–476 Search PubMed.
- R. M. Miller, V. O. Paavilainen, S. Krishnan, I. M. Serafimova and J. Taunton, J. Am. Chem. Soc., 2013, 135, 5298–5301 Search PubMed.
- J. M. Bradshaw, J. M. McFarland, V. O. Paavilainen, A. Bisconte, D. Tam, V. T. Phan, S. Romanov, D. Finkle, J. Shu, V. Patel, T. Ton, X. Li, D. G. Loughhead, P. A. Nunn, D. E. Karr, M. E. Gerritsen, J. O. Funk, T. D. Owens, E. Verner, K. A. Brameld, R. J. Hill, D. M. Goldstein and J. Taunton, Nat. Chem. Biol., 2015, 11, 525–531 Search PubMed.
- H. Kim, A. Jo, S.-S. Choi, H. Nam, W. G. Byun, H. Bae, J. H. Choi and S. B. Park, Asian J. Org. Chem., 2019, 8, 1698–1706 Search PubMed.
- J. Y. Jang, H. Kim, H.-J. Kim, S. W. Suh, S. B. Park and B. W. Han, Sci. Rep., 2019, 9, 11168 Search PubMed.
- D. Chen, D. Guo, Z. Yan and Y. Zhao, MedChemComm, 2018, 9, 244–253 Search PubMed.
- D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 Search PubMed.
- W. Chen, J. Dong, L. Plate, D. E. Mortenson, G. J. Brighty, S. Li, Y. Liu, A. Galmozzi, P. S. Lee, J. J. Hulce, B. F. Cravatt, E. Saez, E. T. Powers, I. A. Wilson, K. B. Sharpless and J. W. Kelly, J. Am. Chem. Soc., 2016, 138, 7353–7364 Search PubMed.
- Q. Zheng, J. L. Woehl, S. Kitamura, D. Santos-Martins, C. J. Smedley, G. Li, S. Forli, J. E. Moses, D. W. Wolan and K. B. Sharpless, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18808–18814 Search PubMed.
- J. A. Ippolito, H. Niu, N. Bertoletti, Z. J. Carter, S. Jin, K. A. Spasov, J. A. Cisneros, M. Valhondo, K. J. Cutrona, K. S. Anderson and W. L. Jorgensen, ACS Med. Chem. Lett., 2021, 12, 249–255 Search PubMed.
- G. J. Brighty, R. C. Botham, S. Li, L. Nelson, D. E. Mortenson, G. Li, C. Morisseau, H. Wang, B. D. Hammock, K. B. Sharpless and J. W. Kelly, Nat. Chem., 2020, 12, 906–913 Search PubMed.
- A. Keeley, P. Ábrányi-Balogh, M. Hrast, T. Imre, J. Ilaš, S. Gobec and G. M. Keserű, Arch. Pharm., 2018, 351, 1800184 Search PubMed.
- H. F. Motiwala, Y. H. Kuo, B. L. Stinger, B. A. Palfey and B. R. Martin, J. Am. Chem. Soc., 2020, 142, 1801–1810 Search PubMed.
- C. Zambaldo, E. V. Vinogradova, X. Qi, J. Iaconelli, R. M. Suciu, M. Koh, K. Senkane, S. R. Chadwick, B. B. Sanchez, J. S. Chen, A. K. Chatterjee, P. Liu, P. G. Schultz, B. F. Cravatt and M. J. Bollong, J. Am. Chem. Soc., 2020, 142, 8972–8979 Search PubMed.
- K. McAulay, E. A. Hoyt, M. Thomas, M. Schimpl, M. S. Bodnarchuk, H. J. Lewis, D. Barratt, D. Bhavsar, D. M. Robinson, M. J. Deery, D. J. Ogg, G. J. L. Bernardes, R. A. Ward, M. J. Waring and J. G. Kettle, J. Am. Chem. Soc., 2020, 142, 10358–10372 Search PubMed.
- J. M. Lopchuk, K. Fjelbye, Y. Kawamata, L. R. Malins, C.-M. Pan, R. Gianatassio, J. Wang, L. Prieto, J. Bradow, T. A. Brandt, M. R. Collins, J. Elleraas, J. Ewanicki, W. Farrell, O. O. Fadeyi, G. M. Gallego, J. J. Mousseau, R. Oliver, N. W. Sach, J. K. Smith, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 3209–3226 Search PubMed.
- K. Tokunaga, M. Sato, K. Kuwata, C. Miura, H. Fuchida, N. Matsunaga, S. Koyanagi, S. Ohdo, N. Shindo and A. Ojida, J. Am. Chem. Soc., 2020, 142, 18522–18531 Search PubMed.
- W. W. Kallemeijn, M. D. Witte, T. Wennekes and J. M. F. G. Aerts, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. Horton, Academic Press, 2014, vol. 71, pp. 297–338 Search PubMed.
- R. Vicik, H. Helten, T. Schirmeister and B. Engels, ChemMedChem, 2006, 1, 1021–1028 Search PubMed.
- H. Huang, C. A. Howard, S. Zari, H. J. Cho, S. Shukla, H. Li, J. Ndoj, P. González-Alonso, C. Nikolaidis, J. Abbott, D. S. Rogawski, M. A. Potopnyk, K. Kempinska, H. Miao, T. Purohit, A. Henderson, A. Mapp, M. L. Sulis, A. Ferrando, J. Grembecka and T. Cierpicki, Nat. Chem. Biol., 2020, 16, 1403–1410 Search PubMed.
- R. Ekkebus, S. I. van Kasteren, Y. Kulathu, A. Scholten, I. Berlin, P. P. Geurink, A. de Jong, S. Goerdayal, J. Neefjes, A. J. Heck, D. Komander and H. Ovaa, J. Am. Chem. Soc., 2013, 135, 2867–2870 Search PubMed.
- E. Mons, I. D. C. Jansen, J. Loboda, B. R. van Doodewaerd, J. Hermans, M. Verdoes, C. A. A. van Boeckel, P. A. van Veelen, B. Turk, D. Turk and H. Ovaa, J. Am. Chem. Soc., 2019, 141, 3507–3514 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |