
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microfluidic organ-on-a-chip model of the outer blood–retinal barrier with clinically relevant read-outs for tissue permeability and vascular structure†
Yusuf B.
Arık
 *ab,
Wesley
Buijsman
ab,
Joshua
Loessberg-Zahl
b,
Carlos
Cuartas-Vélez
c,
Colin
Veenstra
c,
Sander
Logtenberg
ac,
Anne M.
Grobbink
a,
Piet
Bergveld
b,
Giuliana
Gagliardi
d,
Anneke I.
den Hollander
de,
Nienke
Bosschaart
c,
Albert
van den Berg
b,
Robert
Passier
af and
Andries D.
van der Meer
a
*ab,
Wesley
Buijsman
ab,
Joshua
Loessberg-Zahl
b,
Carlos
Cuartas-Vélez
c,
Colin
Veenstra
c,
Sander
Logtenberg
ac,
Anne M.
Grobbink
a,
Piet
Bergveld
b,
Giuliana
Gagliardi
d,
Anneke I.
den Hollander
de,
Nienke
Bosschaart
c,
Albert
van den Berg
b,
Robert
Passier
af and
Andries D.
van der Meer
a
aApplied Stem Cell Technologies, Technical Medical Centre, University of Twente, PO Box 217, 7500 AE, Enschede, The Netherlands. E-mail: y.b.arik@utwente.nl
bBIOS Lab on a Chip group, Technical Medical Centre, MESA+ Institute for Nanotechnology, University of Twente, Enschede, The Netherlands
cBiomedical Photonic Imaging Group, Technical Medical Centre, University of Twente, Enschede, The Netherlands
dDepartment of Ophthalmology, Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Centre, Nijmegen, The Netherlands
eDepartment of Human Genetics, Donders institute for Brain, Cognition and Behaviour, Radboud University of Medical Centre, Nijmegen, The Netherlands
fDepartment of Anatomy and Embryology, Leiden University Medical Centre, Leiden, The Netherlands
First published on 15th December 2020
Abstract
The outer blood–retinal barrier (oBRB) tightly controls the transport processes between the neural tissue of the retina and the underlying blood vessel network. The barrier is formed by the retinal pigment epithelium (RPE), its basal membrane and the underlying choroidal capillary bed. Realistic three-dimensional cell culture based models of the oBRB are needed to study mechanisms and potential treatments of visual disorders such as age-related macular degeneration that result from dysfunction of the barrier tissue. Ideally, such models should also include clinically relevant read-outs to enable translation of experimental findings in the context of pathophysiology. Here, we report a microfluidic organ-on-a-chip model of the oBRB that contains a monolayer of human immortalized RPE and a microvessel of human endothelial cells, separated by a semi-permeable membrane. Confluent monolayers of both cell types were confirmed by fluorescence microscopy. The three-dimensional vascular structures within the chip were imaged by optical coherence tomography: a medical imaging technique, which is routinely applied in ophthalmology. Differences in diameters and vessel density could be readily detected. Upon inducing oxidative stress by treating with hydrogen peroxide (H2O2), a dose dependent increase in barrier permeability was observed by using a dynamic assay for fluorescence tracing, analogous to the clinically used fluorescence angiography. This organ-on-a-chip of the oBRB will allow future studies of complex disease mechanisms and treatments for visual disorders using clinically relevant endpoints in vitro.
Introduction
Vision loss and blindness are estimated to affect approximately 314 million people globally.1 Visual impairment dramatically affects quality of life for patients, and causes major direct and indirect costs related to healthcare. In order to study disease mechanisms and to develop new treatment strategies, experimental models that realistically mimic tissues in the human eye are essential. The outer blood–retinal barrier (oBRB) is one of the key ocular structures for which the development of new model systems is needed, as it is involved in the pathophysiology of various visual disorders, of which age-related macular degeneration (AMD) is the one with the highest prevalence. AMD is a progressive chronic disease that affects vision in nearly 9% of the worldwide population. This number is expected to increase even further as the global population ages in the coming decades.2AMD is a result of dysfunction of the key tissues in the oBRB: the retinal pigment epithelium (RPE), the underlying collagenous membrane, known as ‘Bruch's membrane’ and the adjacent choroidal capillary bed. There are two types of AMD: the “dry” and “wet” forms. Dry AMD is a chronic disease that can progress into severe vision loss. It is characterized by accumulation of insoluble, extracellular aggregates of proteins and lipids in the retina, called “drusen”. As the disease progresses to a late stage called “geographic atrophy”, there is a considerable loss of RPE cells as well as overlying photoreceptors, which rely on the RPE for nourishment and waste disposal.3 In contrast, “wet” AMD only corresponds to 15% of the cases of AMD but is responsible for the majority of cases of AMD-related vision loss.4 This form is characterized by choroidal neovascularization (CNV), in which new blood vessels arise and breach the normal tissue barriers of the outer retina from the underlying choroid. These new vessels also leak fluid below or within the retina, which can cause sudden loss of central vision.
Environmental and genetic factors are involved in the pathogenesis of AMD. Non-genetic risk factors include cigarette smoking, older age and obesity.5 Furthermore, genetic studies have identified associations of several important biological pathways with AMD pathology: the complement system, extracellular matrix remodeling, lipid metabolism, and angiogenesis signaling pathways.6,7
Oxidative stress due to accumulation of reactive oxygen species (ROS) is a key factor in the pathophysiology of AMD.8,9 Patient retinas have increased local accumulation of lipofuscin which generates ROS,10 they have mitochondrial DNA damage due to ROS,11 and there is a marked increase in glycation end-products and peroxidized lipids.12 As this oxidative stress due to ROS production is toxic to the cells, this might lead to increased permeability of the choroidal blood vessels (e.g. leakage), which is a hallmark of wet AMD.
From a clinical perspective, it is important to closely monitor the disease progression of AMD in a patient, for example to detect when the disease progresses from dry AMD, for which there are no treatments, to wet AMD, which can be treated with intraocular injections of anti-angiogenic drugs. The clinical assessment is performed using medical imaging modalities, particularly optical coherence tomography (OCT) and fluorescence-based angiography (FA).13,14 OCT is used to reconstruct high-resolution two-dimensional (2D), or three-dimensional (3D)-images that visualize (abnormalities in) the individual retinal cell layers and blood vessels of the choroid. It is based on the measurement of ‘light echoes’ as a function of tissue depth, through the interference of a reference beam with the light that has been backscattered from the retina.15 FA relies on the perfusion, clearance and leakage of intravenously injected hydrophilic fluorescent tracers.13 As normally these dyes do not cross the blood–retinal barrier, any defect that compromises the barrier integrity will result in abnormalities on FA.16
To better understand the pathophysiology of AMD, experimental models are required in which the morphological changes of the tissues can easily be observed, and the experimental conditions can be readily manipulated. Rodent models are disadvantageous due to the lack of a macula and interspecies anatomical differences. Non-human primates are more realistic models, but the time required for the disease progression, the high costs and the ethical issues make primate models less suitable for fundamental biomedical research.17
Due to the recent developments in human pluripotent stem cell technology, in vitro models of the oBRB are becoming increasingly sophisticated. However, most of the these models still rely on simple monolayers of cells (typically monocultures of human pluripotent stem cell-derived RPE) on plastic surfaces or membranes, and therefore these models do not fully recapitulate the 3D and tissue-level physiology.18
In recent years, organ-on-a-chip devices have proven to be promising disease models.19–26 Organs-on-chips are microfluidic cell culture devices with engineered microchannels that are continuously perfused and inhabited by living cells to form tissues that exhibit organ-level physiology. Depending on the research question, complexity of these systems can be adjusted by systematically including cell types and physico-chemical parameters of the tissue microenvironment in a well-controlled manner. The simplest systems consist of a single perfused microfluidic chamber and one type of cultured cell, whereas more complex devices have two or more microchannels that are separated by porous membranes lined by two or more cell types, which then simulate the interface between different cell types.22 Mimicry of tissue complexity can be further increased by the inclusion of 3D structures, for example by including organoids or hydrogels based on extracellular matrix proteins.27–29
Recently, multiple organ-on-a-chip models of the outer layers of the human retina have been reported.30 One of the models relies on 2D co-cultures of RPE and endothelial monolayers in microfluidic chips.31 One reported model also includes co-cultured retinal organoids in addition to a monolayer of RPE.32 The other two models consist of co-cultures of RPE and self-developed 3D capillary endothelial networks.33,34 Due to their 3D nature, these latter models allow studies of on-chip neovascularization in response to hypoxia and growth factor stimulation, thereby clearly demonstrating the potential of applying organ-on-a-chip models of the oBRB in studies of AMD. However, because these models rely on endothelial cell self-organization into microvascular networks, researcher control over 3D vessel geometries is limited. More control over vessel geometry would strongly improve the reproducibility of these models.
In terms of read-outs, some of the current organ-on-a-chip models do include tracking of fluorescent tracers in order to assess the cell coverage by comparison of cell-containing channels with empty devices.35 In contrast, none of the current organ-on-chip models of the oBRB use OCT as an additional read-out. Previously, OCT has been used in organs-on-chips as a functional tool to monitor and characterize cell specific functions, demonstrating the feasibility of using this imaging modality as a read-out.36 The inclusion of clinically relevant read-outs will be especially important for validation of devices and to facilitate the transfer of knowledge between researchers and ophthalmologists.
In the present study, we report an organ-on-a-chip model of the oBRB, based on co-cultures of RPE and human umbilical vein endothelial cells (HUVECs) in a microfluidic chip that contains a microchannel and an open-top culture chamber separated by a polyester membrane. In the microchannel, we created a microvessel with a well-defined geometry within a collagen I hydrogel using a subtractive method of micropatterning.37 We used fluorescent tracers to track permeability of the microvessels and the RPE. Moreover, we implemented OCT as an innovative and clinically relevant read-out for our organ-on-chip model and used it to confirm sizes and structural changes of the engineered microvessels. Using this model, in contrast to existing models of AMD, we investigated one of the early hallmarks of wet AMD: the increased permeability of blood vessels due to ROS exposure.
Materials and methods
Chip fabrication
Poly(methyl methacrylate) (PMMA) (Altuglass, France) master molds of the channel structures were designed with SolidWorks and fabricated with a computer numerical control (CNC) milling machine (Datron Neo, Datron AG, Germany). PDMS base and curing agent were mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 wt ratio (Sylgard 184 Silicone elastomer kit, Dow corning, USA). After degassing the mixture, it was poured onto the fabricated positive PMMA molds and cured for at least 3 hours at 60 °C. Following curing, PDMS was removed from each one of the molds as slabs. PDMS slabs with microchannel imprints (1 × 1 mm) were cut from each side to generate side inlets for the insertion of needles for microvessel patterning. After that, using biopsy punches (Robbins Instruments, USA) 2 inlets and 1 culture chamber (1.2 mm and 3 mm in diameter respectively) were punched into the PDMS slab which corresponds to the middle part of the final assembled device (Fig. 1A-iii). In addition, 3 reservoirs were punched (5 mm in diameter) into another PDMS slab for the top compartment of the assembled device (Fig. 1A-iv). After that, all three slabs were aligned and cut into device-sized pieces. Before assembly of the parts, dust was removed using Scotch tape (3M, USA). Leakage-free bonding of the parts with a membrane (Fig. 1A-ii) in between them was achieved by using a PDMS/toluene mortar (5:3 wt ratio) (toluene from Merck, Germany) as reported previously.38,39 This mixture was spin-coated onto a glass coverslip (1500 rpm, 60 s, 1000 rpm s−1, Spin150, Polos, The Netherlands) and transferred to the device parts using an ink roller (Fig. 1A-i and iii). After that, a ∼36 mm2 piece of polyester membrane with a pore size of 8 μm (GVS Life Sciences, USA) was aligned and sandwiched between the center of the bottom (Fig. 1A-i) and middle (Fig. 1A-iii) compartments. This was followed by overnight baking the parts at 60 °C. Final assembly of the device was done by exposing the surfaces of top compartment (Fig. 1A-iv) and the pre-assembled device (Fig. 1A-i–iii) to air plasma (50 W) for 40 seconds (Cute, Femto Science, South Korea). After plasma treatment, activated surfaces were pressed together.
:1 wt ratio (Sylgard 184 Silicone elastomer kit, Dow corning, USA). After degassing the mixture, it was poured onto the fabricated positive PMMA molds and cured for at least 3 hours at 60 °C. Following curing, PDMS was removed from each one of the molds as slabs. PDMS slabs with microchannel imprints (1 × 1 mm) were cut from each side to generate side inlets for the insertion of needles for microvessel patterning. After that, using biopsy punches (Robbins Instruments, USA) 2 inlets and 1 culture chamber (1.2 mm and 3 mm in diameter respectively) were punched into the PDMS slab which corresponds to the middle part of the final assembled device (Fig. 1A-iii). In addition, 3 reservoirs were punched (5 mm in diameter) into another PDMS slab for the top compartment of the assembled device (Fig. 1A-iv). After that, all three slabs were aligned and cut into device-sized pieces. Before assembly of the parts, dust was removed using Scotch tape (3M, USA). Leakage-free bonding of the parts with a membrane (Fig. 1A-ii) in between them was achieved by using a PDMS/toluene mortar (5:3 wt ratio) (toluene from Merck, Germany) as reported previously.38,39 This mixture was spin-coated onto a glass coverslip (1500 rpm, 60 s, 1000 rpm s−1, Spin150, Polos, The Netherlands) and transferred to the device parts using an ink roller (Fig. 1A-i and iii). After that, a ∼36 mm2 piece of polyester membrane with a pore size of 8 μm (GVS Life Sciences, USA) was aligned and sandwiched between the center of the bottom (Fig. 1A-i) and middle (Fig. 1A-iii) compartments. This was followed by overnight baking the parts at 60 °C. Final assembly of the device was done by exposing the surfaces of top compartment (Fig. 1A-iv) and the pre-assembled device (Fig. 1A-i–iii) to air plasma (50 W) for 40 seconds (Cute, Femto Science, South Korea). After plasma treatment, activated surfaces were pressed together.
 | ||
| Fig. 1 PDMS-based organ-on-a-chip device allows 3D co-cultures of monolayers of human RPE cells with human endothelial cells lining a microvessel. (A) The device contains (i) a bottom compartment with a defined (1 mm × 1 mm) microchannel, (ii) a polyester membrane with 8 μm pore size (∼10 μm thickness), (iii) a middle compartment which contains an open-top culture chamber (3 mm in diameter) and 2 inlets (1.2 mm in diameter) and (iv) a top compartment that consists of three reservoirs for media storage (5 mm in diameter). (B) Schematic representation of patterning collagen I hydrogel using the subtractive method of patterning. This process starts with the hydrogel being pipetted into the channels from side inlets. This was followed by the insertion of two blunt needles (outer diameter of 0.9 mm) each inserted from side inlets and a syringe needle (outer diameter of 0.5 mm) inserted through the needles along the channel. After gelation of the collagen inside the microchannel, needles were removed to reveal the microvessel structure. For illustrative purposes, reservoir layer was removed. (C) Immunolabelling of the cells revealed a continuous distribution of each cell type. (i) 3D construction of confocal microscopy image of the co-culture area stained for nuclei and actin filaments indicates a monolayer alongside the channel. (ii) Top surface of the microvessel shows an equal distribution of HUVECs along the microchannel. (iii) Cross-sectional microscopy image of the co-culture area as indicated by DAPI (nuclei) and actin filaments staining reveals the circular structure of the microvessel occupied by HUVECs, as well as ARPE-19 cells that are located above the microvessel. These cells were positive for their respective cell–cell adhesion markers; (iv) VE-cadherin expression for HUVEC, (v) ZO-1 expression for ARPE-19. Scale bars: 50 μm. | ||
Surface functionalization of PDMS
Internal surfaces of PDMS devices (microchannels and culture chambers) were functionalized using (3-aminopropyl)triethoxysilane (APTES, Sigma Aldrich, Germany) and glutaraldehyde (Sigma Aldrich, Germany). Following the aforementioned assembly of the final device with plasma activation of surfaces, 3% (v/v) APTES mixed in ultrapure H2O (ELGA, UK) was added into the channel and reservoir, and incubated at room temperature (RT) for 5 min. Following APTES coating, the chips were rinsed thoroughly with 100% ethanol and incubated for 5 min in 100% ethanol to eliminate the remaining APTES. After air-drying the devices, 10% glutaraldehyde was added to the channel and the culture chamber, and the devices were incubated for 5 min at RT. This was followed by thorough rinsing with distilled H2O and drying for 2 hours at 60 °C prior to collagen patterning.Hydrogel patterning
Cell culture and seeding in chips
Fibroblasts (Lonza), HUVECs (Lonza) and ARPE-19 (ATCC) cells were cultured with fibroblast growth media (FGM-3: basal medium with supplement mix from PromoCell Gmbh, Germany and 50 U ml−1 P/S (ThermoFisher, USA), ECGM-2 and DMEM/F12 (with GlutaMAX, ThermoFisher, USA) supplemented with 10% fetal bovine serum (FBS; ThermoFisher, USA) and 50 U ml−1 P/S), respectively. HUVECs were cultured in T75 culture flasks coated with 0.1 mg ml−1 collagen I (rat tail collagen I, ThermoFisher, USA) in PBS (ThermoFisher, USA), whereas ARPE-19 cells and fibroblasts were cultured in non-coated T75 flasks. The cells were incubated at 37 °C in humidified air with 5% CO2. Flasks with confluent monolayers were either used for experiments or subcultured. HUVECs and ARPE-19 were kept in culture up to passage number 6 and 30, respectively. Prior to cell seeding, the culture chamber was rinsed with PBS and coated with 0.1 mg ml−1 collagen I for 30 min at 37 °C. After that, the chamber was washed with cell medium to remove non-bound collagen. This was followed by obtaining HUVECs from a confluent flask using 0.05% trypsin–EDTA (ThermoFisher). Suspended in fresh ECGM-2 media at 4.5 × 106 cells per ml, cell suspension was pipetted into the channel via the inlet. After optically confirming the cell suspension in the channels, the devices were placed upside down for gravity-driven seeding of the top of the lumens and incubated at 37 °C and 5% CO2 for 60 min. After that, devices were taken out of the incubator, and non-bound cells were removed by flushing the channels with fresh cell media. Next, to seed the bottom of the lumens and the culture chamber, HUVECs and ARPE-19 were freshly obtained from confluent flasks using 0.05% trypsin–EDTA. After that, suspended in ECGM-2 at a concentration of 5 × 105 cells per ml for ARPE-19 and 4.5 × 106 cells per ml for HUVECs, cells were pipetted into the culture chamber and microvessel, respectively. After 60 min of static incubation at 37 °C and 5% CO2, non-attached cells were washed away with fresh media, and reservoirs were filled with ECGM-2 medium to prevent drying. For media refreshing, a filtered pipette tip full of media was placed on the inlet, whereas an empty one placed in the outlet. Furthermore, the culture chamber was filled with media and sealed with Scotch tape. The devices were then placed on a custom-built rocking platform inside a 37 °C and 5% CO2 humidified incubator. The rocking platform tilted back and forth at an angle of 60° at an interval of 45 seconds to facilitate a gravity driven medium flow in channels. Media was replaced with fresh media daily.Cell staining
On-chip co-cultures were stained for actin filaments and nuclei for confirmation of cell monolayers on microvessel walls and inside the culture chamber. For that, devices were first washed with PBS and fixed with 4% formaldehyde (in PBS, ThermoFisher, USA) for 15 min at RT. The fixative was washed away with PBS and the cells were incubated in permeabilization buffer (PB), which contains 0.1% Triton X-100 (Sigma Aldrich, Germany) and 10 mg ml−1 bovine serum albumin (Sigma Aldrich, Germany) in PBS for 60 min at RT. Afterwards, the cells were incubated for 2 hours at RT with 12.5 μg ml−1 4′,6-diamino-2-phenylindole (DAPI, ThermoFisher, USA) and 4 drops per ml ActinGreen (binds to actin filaments, ThermoFisher, USA) in PB. Following incubation, the cells were rinsed three times with PBS and then washed three times with PBS for 10 min at RT.For assessment of cell morphology under the exposure of H2O2, HUVECs on culture well plates were fixed for 10 min at RT with 4% formaldehyde, followed by permeabilization in 0.1% Triton X-100 at RT for 15 min. Afterwards, cells were stained by incubating with 1.25 μg ml−1 DAPI and 3.75 U ml−1 Alexa Fluor 633 phalloidin in PBS for 1 hour at RT. After each step cells were washed 3 times with PBS.
As a confirmation of expression of specific adhesion markers, each cell type was stained on a glass cover slip coated with collagen I. Cells were seeded at a density of 5 × 104 cells per cm2 and kept in culture conditions for a day to grow to a monolayer. This was followed by washing with PBS and fixing with 4% formaldehyde for 10 min at RT. The fixative was washed away with PBS and the cells were incubated in PB for 15 min at RT. Afterwards, HUVECs and ARPE-19 were incubated with goat anti-human VE-cadherin IgG (1 μg ml−1 in PB, R&D Systems) and mouse anti-human ZO-1 IgG (5 μg ml−1 in PB, BD Transduction Laboratories) for 2 hours at room temperature, respectively. Following incubation, the cells were rinsed three times with PBS and then washed three times with PBS for 10 min at RT. After that, the cells were incubated for 1 hour at RT with donkey anti-goat IgG Alexa Fluor 546 (2.5 μg ml−1, ThermoFisher) and chicken anti-rabbit IgG Alexa Fluor 488 (2.5 μg ml−1, ThermoFisher) in PB, respectively. After staining, cells were washed again with PBS. Afterwards, the coverslips were transferred to microscope slides for imaging.
The cells were imaged with phase contrast, fluorescence microscopy using the EVOS FL Cell Imaging System (Life Technologies; RFP filter (ex 531/40 em 593/40) for VE-cadherin, GFP filter (ex 470/22 em 510/42) for ZO-1, Cy5 filter (ex 628/40 em 692/40) for phalloidin and DAPI filter (ex 357/44 em 447/60) for DAPI) and Nikon Confocal A1 Microscope (Nikon, Japan). Z-stacks, 3D reconstruction and Z-projections were all analyzed and reconstructed with Fiji software.40
Tracking permeability in co-cultures on-chip
The permeability of cell layers in the organ-on-a-chip co-cultures was visualized by a method analogous to the clinically used FA. Devices were cultured for 72 hours prior to experimentation. Afterwards, devices were first treated with ECBM-2 (with 2% FBS and 1% P/S) overnight. The following day, HUVECs were either left untreated or were exposed to 800 μM and 10 mM H2O2 dissolved in ECBM-2 (with 2% FBS and 1% P/S) for 1 day. Culture medium in devices was refreshed twice daily using the rocking platform with freshly made H2O2 solutions and ECBM-2. After 1 day of exposure, first, each device was mounted on EVOS FL Cell Imaging System (Life Technologies) with a 4× air objective. Then the bottom half of a 200 μl pipette tip, acting as a reservoir, was fitted on the inlet. After that, a syringe pump was connected to the device via Tygon tubing (1 mm inner diameter, Saint-Gobain Performance Plastics, France) which was placed on the outlet. After that, the reservoir was filled with 100 μl of 60 μg ml−1 40 kDa FITC-dextran diluted in ECBM-2. This was followed by withdrawal of the dextran along the microchannel 25 μl min−1 using a syringe pump (PHD ULTRA, Harvard apparatus, USA). After ∼4 min of FITC-dextran perfusion, the reservoir was emptied and replaced with 100 μl ECBM-2 with which the channels were perfused (25 μl min−1). During the whole procedure, sequential images were taken with 10 s intervals for 9 min. Using these images, the barrier formed by HUVECs was evaluated. First, intensities of a 20 by 20 pixel area of the gel (Fig. 2B-top between white and red lines) and microvessel (Fig. 2B-top between red lines) were corrected for background intensity by subtracting the average intensity of a 20 by 20 pixel background area (Fig. 2B-top outside of white lines). Second, intensity of the gel was normalized to intensity of the microvessel. Using the fluorescence profile of normalized gel over time, the slope of the increase in intensity during the perfusion phase was calculated by linear fitting (MS Excel). This normalized slope was used as a measure for permeability. | ||
| Fig. 2 Assessment of barrier integrity by ‘fluorescein angiography’ in devices. (A) Device schematics illustrating the location used in acquisition. (B) Illustration of the fluorescein angiography procedure (top) and sequential top-view images taken during the experimentation (bottom). Each column represents different stages of the procedure: before the dye perfusion (‘Background’), during the perfusion of the dye (‘Perfusion’), during the media perfusion (‘Removal’). As the dye was perfused through the microvessel (red dashed lines), diffusion occurs into the gel depending on the integrity of the barrier. Untreated (UT) is a positive control with HUVECs and ARPE-19 cultured in the microvessel and the culture chamber respectively. The ‘800 μM’ and ‘10 mM’ rows represent the treatment concentrations of HUVECs with H2O2 for 5 days. Contrast in raw images were enhanced for illustrative purposes using a false-color lookup table (bottom). Scale bar: 200 μm (C) a typical fluorescence intensity profile during FA is summarized using ‘untreated’ condition as an example, with different phases labeled in their respective colors, matched to the phases in panel B. The slope in the ‘Perfusion’ phase (‘α’) is used as a measure of permeability. (D) Normalized to the intensity in the microvessel, slope of the increasing gel intensity in the ‘Perfusion’ phase is summarized as a bar graph. Compared to untreated cells (UT), a significant increase (UT vs. condition, Student's t-test, p < 0.05) in permeability (denoted by an asterisk) was detected in longer exposures of higher H2O2 concentrations. Error bars represent standard error of the mean, all experiments were performed at least 6 times. | ||
Optical coherence tomography measurements
OCT images were acquired with a home-built visible light system (Fig. S2†) previously described.41,42 Briefly, the setup was based on an open-air Michelson interferometer, with a supercontinuum light source (SuperK Extreme EXB-6, NKT Photonics, Denmark). A beam splitter (BS028, Thorlabs, USA) guided 10% and 90% of the light towards the sample and reference arm respectively. Achromatic lenses (AC127-025-A, Thorlabs, USA) with a focal length of 25 mm focused the light on the sample and on a piezo-driven reference mirror. Those lenses determine the lateral resolution of the system, in this case 2.5 μm in air. After interaction of light with the sample and the reference mirror, backscattered light was guided through a single mode fiber (S405-XP, Thorlabs, USA) into a custom-built spectrometer. The spectrometer dispersed light onto a line-scan camera (Sprint spL4096-140km, Basler, Germany), achieving a spectral resolution δλ = 0.1 nm over a range of 440–600 nm (Fig. S2A†). The spectrum acquired by the camera can be related to depth after processing, producing depth-intensity profiles known as A-scans. The system parameters described above led to a theoretical axial resolution of 1.4 μm in air and an imaging depth of ∼±0.6 mm. Chips were mounted on a motorized stage (T-LS13M, Zaber, CA), controlled by a custom-built LabView algorithm in order to allow lateral scanning. Cross-sectional images (B-scans) were obtained by concatenating 350 A-scans spaced at 3 μm, leading to a lateral scan range of 1.05 mm. At every lateral position, 100 spectra were acquired with an exposure time of 800 μs. The piezo-driven reference mirror allowed full extension of the imaging depth to ∼1.2 mm, by removal of the DC-component and mirror-image in each A-scan. This was achieved by applying a procedure similar to our previous zero-delay acquisition.42,43 In short, the movement of the piezo induces a frequency modulation as a function of time for each A-scan. By means of a band-pass filter in the frequency domain, the DC component and mirror image can be removed. Note that unlike our method for zero-delay acquisition, here the position of the reference mirror was kept constant, and the depth-axis was calculated from the spectrum. Finally, in order to mitigate the loss in resolution caused by dispersion of the PDMS layers in the chip, a flat layer of PDMS with the same thickness of the chip was introduced in the optical path of the reference beam.Microvessel quality was evaluated with a custom-built Matlab44 script. First, OCT images were recorded at 4 different cross-sections alongside the microchannels containing various sized microvessels (n = 32 for each size). Second, images were filtered using a Gaussian convolution to reduce speckle and then binarized. Third, a rough region of interest (ROI) was drawn on the microvessel. The extremes of the microvessel were detected and their respective locations marked as coordinates, physical coordinates were calculated assuming a refractive index of 1.4 as for PDMS. Using these locations, a circular shape was drawn around the central point of the four marks with theoretical diameters of 500, 300 or 120 μm. Fourth, the contents of the ROI were analyzed by comparing the features in the image to the desired shape of the microvessel. Finally, we defined a quality factor Qvessel, where 1 indicates perfectly circularly microvessels. The quality of the microvessel (Qvessel) was calculated as ratios between areas inside and outside the circular ROIs, as follows,
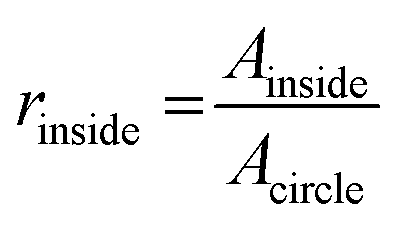

where Ainside and Aoutside are the areas of features inside and outside the theoretical circle respectively, as well as Acircle which is the area of the circle itself. A minimum threshold of 0.5 was set for selecting proper microvessels.

Diameters of the branching vessels within patterned fibrin hydrogel were calculated by fitting circles into each observed cavity and measuring their diameter using a custom-built Matlab script.44
Results and discussion
Co-culture of retinal pigment epithelium and vascular endothelium in the microfluidic chip
In the present study we aim to mimic the organization of the outer lining of the retina in an organ-on-a-chip model. The chips were designed to have two compartments separated by a porous membrane (Fig. 1A), resembling the layered structure of the RPE and the underlying choroid in the human retina. After the patterning of microvessels, HUVECs and ARPE-19 were introduced in the device. After 72 hours of culture, confocal microscopy inspection of the co-cultures stained for actin cytoskeleton and nuclei revealed a homogeneous distribution of the cells, which suggests that there were continuous cell layers (Fig. 1C). To confirm each of these specific cell types, ARPE-19 and HUVECs were fluorescently stained for the specific cell–cell adhesion molecules ZO-1 and VE-cadherin, respectively (Fig. 1C). Subcellular localizations were as expected, with localization of the respective proteins at cellular junctions.We chose to set up our model with ARPE-19 and HUVEC, because they have both been used extensively in modelling the oBRB in vitro.45–47 The purpose of our current study was to establish relevant read-outs, and we therefore chose well-characterized cells as a point of reference. Still, it is clear that ARPE-19 has only limited relevance when modelling the RPE, as it lacks pigmentation and other hallmarks of RPE in vivo. Similarly, HUVECs are a popular source of primary human endothelial cells, but its relevance for modelling the choroid is limited to cell type. Modelling the choroid is challenging in any case, because even primary human choroidal endothelium loses choroidal endothelial characteristics, e.g. vascular markers and fenestrations, when cells are isolated and expanded in culture.48,49 Undoubtedly, stem cell-derived choroidal endothelial and RPE cells are what is ultimately needed to create an organ-on-a-chip model that mimics the human outer-blood retinal barrier as closely as possible. However, the goal of our current study is to establish well-characterized read-outs that are relevant for clinical translation. Such read-outs will also be essential when studying organ-on-chip models of the oBRB that integrate stem cell-derived tissue.
Fluorescein angiography on 3D microvessels in the microfluidic chip
In order to explore the relevant conditions of oxidative stress to cause endothelial dysfunction in our cultured cells, we first treated HUVECs in regular culture systems to different concentrations of H2O2 (Fig. S1†). Here, our logic for using H2O2 as a disease stimulus was motivated by the fact that RPE cells generate hydrogen peroxide during photoreceptor outer segment digestion.50 Disrupted metabolic activities by aging may cause accumulation of free radicals in the native microenvironment that eventually damages nearby tissues such as the choroid. It has been reported that hydrogen peroxide is neutralized by HUVECs (1 nmol H2O2 per 103 cells per hour),51 however continuous exposure causes the damage to accumulate. Here, we studied the long-term effects of peroxide exposure on cells.In ophthalmology, FA is a powerful imaging modality commonly used to assess circulation in the eye (e.g. fluid leakage from the choroidal blood vessels) as well as to find vessel defects that are not detectable otherwise.52 Therefore, many clinical trials of AMD rely on FA to evaluate the potency of treatments. FA involves intravenous injection of a fluorescent tracer like fluorescein, which then flows through the circulation and is visible by fluorescence imaging within the choroidal microvessels within seconds. After a mid-stage, in which all retinal vessels light up for a few minutes, the dye is gradually eliminated from the system. If a retinal vascular defect or a defect in the RPE is present, the dye fills up the intercellular space from the lesion and is retained even after most of the dye has been cleared from the retinal vasculature and choroid. This remaining hyperfluorescence in the late phase of the angiogram is a clinical indication of barrier tissue damage.53
We combined FA with organ-on-a-chip technology to examine the effect of the previously established concentrations of H2O2 (see ESI† Methods) on intercellular leakage of co-cultures in our model (Fig. 2). To minimize the possible damaging effect of flow on the cell barrier, we opted for shear rates (∼34 s−1) much lower than those found in arteries (∼300–1000 s−1).54 Since the volume of the channels of our chip was much lower than well plate cultures, we made use of a rocking platform in order to maintain cell viability for multiple days in our devices. This platform was able to rotate from side to side allowing cyclic patterns of hydrostatic pressure from the attached reservoirs, and this in turn made it possible for cells to access a larger volume of medium and hydrogen peroxide. In our experiments, we exposed the co-cultures on-chip for 1, 2 and 5 days to concentrations of 800 μM and 10 mM H2O2, prior to performing the ‘on-chip FA’. The on-chip FA relied on introducing a fluorescently labelled dextran into the patterned microvessel, which could be detected by fluorescence microscopy. After perfusing the system with the dye for a few minutes (Fig. 2B, ‘Perfusion’), the model was perfused with control medium to clear the dye from the system and subsequently inspected for signs of hyperfluorescence, the late stage (Fig. 2B, ‘Removal’).
Using fluorescence intensity data from the ‘Perfusion’ phase of the on-chip FA, we calculated the permeability of the endothelial monolayers. For that, first we corrected the images for background intensity, to eliminate the effect of ambient light. Afterwards, fluorescence intensities of the gel were normalized to microvessel intensities. Using these normalized intensities, we calculated the slope of the gradual increase in fluorescence (Fig. 2C). Normalizing intensities minimized the inconsistencies of the fluorescence intensity of dye perfused through the microvessel, and provided an objective method to compare data from multiple chips and multiple days.
Based on FA analysis of untreated samples, our on-chip cultures maintained their barrier over the course of the treatment (Fig. 2D). Moreover, 800 μM treatment did not cause significant damage to the barrier in shorter exposures (1 day). However, significant damage occurred upon 2 and 5 days of exposure. As a positive control, we used a very high concentration of H2O2 10 mM, which caused significant damage to the cellular barrier after 1, 2 and 5 days of exposure as well. Here, our FA setup was only used for semi-quantitative determination of permeability, as the diffusion time for labeled dextran to travel from the microvessel-gel border to the channel wall (∼8 min) was longer than the time of the ‘Perfusion’ phase (∼4 min). As a result, the gradients within the gel were still significant at the end of this phase. Therefore, any quantitative estimation of the permeability from the rate of change of this average intensity would be significantly skewed. The technical impossibility of full quantification prevents a direct comparison of permeability measurements in our system with other in vitro culture systems.
In addition to performing this semi-quantitative analysis of the fluorescence data, our method based on microscopic imaging also allows a qualitative inspection of the microvessels for local defects. Normally, the dye leaks from the microvessel in a uniform pattern, but occasionally, we found patterns of enhanced dye accumulation at specific sites of the microvessel (Fig. S3†).
In order to reveal the cell morphology upon peroxide exposure, we fluorescently stained HUVECs inside the microvessel for nuclei and actin filaments. In line with our observations in the FA analysis, we observed a dose-dependent effect on the cell morphology and monolayer integrity (Fig. S4†). At 800 μM H2O2, cells acquired an elongated morphology with strong F-actin stress fibers and damaged monolayers with scattered intercellular holes. At 10 mM of H2O2, cells displayed a shrunken morphology, and intercellular spaces became so large that the monolayer disappeared almost completely. Interestingly, the observed patterns of monolayer damage (particularly in the condition of 800 μM H2O2) were not as severe as what was observed in our experiments with HUVECs in culture plates, in which we already observed strongly reduced cell numbers upon treatment with 800 μM H2O2 (Fig. S1C†). This is presumably due to the low internal volume of the chips, which causes reduced cellular exposure to H2O2 and its short-lived radical oxygen species compared to culture plates with high absolute amounts of H2O2. Alternatively, endothelial cells in chips could be better protected from damaging stimuli because their culture conditions better recapitulate the native in vivo conditions of these cells.
In our experiments with co-cultures, even with high concentrations of H2O2, no considerable damage to the layer of RPE was observed as there was no dye accumulation in the culture chamber. A possible reason for this might be that the effective concentrations of H2O2 to which the RPE cells are exposed is strongly reduced because they were shielded from the H2O2 in the patterned microvessel by both the cultured endothelium and the collagen hydrogel. Another reason may be the specific RPE cell line we utilized in this study; ARPE-19 has been reported to be highly resistant to oxidative stress.55 Even though our current disease stimulus does not have a damaging effect on RPE, we confirmed that our FA analysis is suitable to evaluate this type of damage. For this, we performed experiments in devices with monocultures of endothelial cells only. The absence of the ARPE-19 layer in the culture chamber enabled us to simulate the severe late stages of AMD where both endothelial and epithelial layers are damaged.3 In these experiments, we found leakage of dye towards the culture chamber upon treatment with H2O2 (Fig. S5†).
Our results demonstrate that an FA can be carried out on the organ-on-chip co-cultures, and that this method allows both semi-quantitative evaluation of the endothelial barrier by analyzing the slope of the fluorescence increase in the ‘Perfusion’ phase, as well as qualitative assessment of lesions and defects by analyzing local accumulation of the fluorescent dye in the ‘Removal’ phase.
In the clinic, two dyes are typically used in fluorescence-based angiography: fluorescein and indocyanine. Fluorescein has a low molecular weight (0.3 kDa) and therefore readily leaks out of the fenestrated choroidal capillaries, leading to a diffuse ‘choroidal flush’ early in the angiogram that reveals few details about the choroidal vasculature. It is therefore mostly used to find defects in the retinal pigment epithelium and the retinal vasculature. In contrast, indocyanine binds strongly to plasma proteins and is therefore retained in the lumens of the fenestrated choroidal microvessels. Patterns in the indocyanine angiogram like delayed filling of vessels or focal hyperfluorescence can therefore be used to diagnose defects in the choroidal vasculature. For example, hyperpermeability of choroidal vessels is often observed in diseases like central serous retinopathy and AMD.56–60 The dye that we used in our studies was a fluorescein-labeled 40 kDa dextran, which diffuses over the endothelial barrier at a rate much lower than fluorescein, but which is not retained fully in the vessels like high molecular weight plasma proteins. These properties allow us to reliably track the diffusion of the dye into the collagen gel over time by time-lapse microscopy, which enables sensitive semi-quantitative analysis of vascular permeability. Fluorescein would diffuse too rapidly to perform a reliable quantification, while a very high-molecular weight dye (e.g. fluorescently labelled albumin, 66 kDa) would instead only allow qualitative identification of extreme vascular defects. Since our assay gives information on the state of the ‘choroidal’ vessel in the organ-on-chip, the observed increase in permeability (Fig. 2D) upon treatment with hydrogen peroxide can be considered to be analogous to choroidal hyperpermeability observed in clinical indocyanine angiography.
Optical coherence tomography on 3D microvessels in the microfluidic chip
OCT is a method in ophthalmology to assess the structure of the tissue layers of the retina. Because it is a non-invasive readout, and allows for in vivo imaging, it is a routine tool for diagnosis and follow-up after AMD treatment. OCT is based on a pattern that is formed by backscattered light from the sample. Through the interference of this pattern with the reference beam (Fig. S2†), an optical map of the structures of the retina can be drawn.In this study, we explored whether our organ-on-chip model of the outer tissue of the retina is compatible with OCT, and whether structural changes in the on-chip hydrogel structures could be visualized. To characterize the sensitivity of using OCT as a read-out, we first used it to measure patterned microvessels of defined, but different sizes: 500, 300 and 120 μm. Horizontal and vertical axes lengths showed a low variance and average lengths were approximately the same size as the intended diameters (Fig. 3B). In addition, we evaluated the quality of these microvessels, Qvessel (see Materials and methods), using OCT data. Consistent with our optical observations, analysis of vessel quality revealed a higher number of microvessels in good quality (Qvessel > 0.5) for larger sized (500 μm) patterned microvessels (high quality: 15/32), as compared to smaller ones: 5/48 and 1/32 for 300, 120 μm microvessels, respectively (Fig. 3C-i). Any microvessel below Qvessel of 0.5 was deemed improper as these were partially destroyed and non-perfusable (Fig. 3C-ii and iii).
 | ||
| Fig. 3 Imaging matrix structure and microvessels in the organ-on-chip system using OCT. (A) Schematic overview of the organ-on-a-chip device with inset showing the cross-section view of the channel. (B) Measured diameters of collagen I hydrogel patterned microvessels with different needle dimensions. Horizontal (Hor, x-axis in (A)) and vertical (Vert, y-axis in (A)) are measured separately. Each dot represents a different cross-section along the channel. Line showing mean with error bars denoting standard error of mean. Theoretical distances were labelled as dotted lines at 500, 300, and 120 μm. (C-i) Distribution of quality assessment of microvessels with different diameters. Values below 0.5 were considered as improper. (C-ii and iii) OCT images of two different vessels (500 μm) with qualities at opposite extremities were shown with their respective quality factors. (D) OCT cross section of a microvessel containing collagen I patterned hydrogel without cells (D-i) and fibrin, HUVECs and fibroblasts with white arrows showing cavities representing branches within the hydrogel (D-ii). Blue arrows represent OCT imaging artefacts. Scale bars: 100 μm. (D-iii) Distribution of vessel sizes within the fibrin hydrogel based on image analysis of multiple OCT cross-sections. | ||
We then set out to study whether OCT can not only be used for analysis of pre-patterned microvessels, but for self-developed microvessels of physiological sizes as well. For this, we compared our regular organ-on-a-chip with those in which the collagen I hydrogel was replaced with a fibroblast-containing fibrin hydrogel. It is known from literature that fibrin hydrogels can strongly induce cultured endothelial cells to form network branches in vitro.61–64 After 7 days of culturing, the matrix (Fig. 3D) in our organs-on-chips was imaged by OCT. Typical cross-sectional scans of the patterned microvessel in a collagen I matrix revealed a lumen with the same cross-section as the needle used for patterning, with clearly delineated borders (Fig. 3D-i). In contrast, scans of the fibrin gel (Fig. 3D-ii) revealed an enlarged lumen and a damaged matrix structure with cavities (Fig. 3D-ii, white arrows), which coincided with endothelial growth into the fibrin hydrogel (Fig. S2C†). Here it is worth noting that the resolution was considerably lower in the upper part of the damaged matrix due to the thick PDMS layer below the microchannel. By analyzing the OCT scans, we determined the size distribution of the observed cavities. The majority of cavities consisted of smaller vessels (∼20 μm, Fig. 3D-iii), sizes which are consistent with choroidal capillaries in vivo.65 Note that because of the 3D acquisition scheme described in Materials and methods, artefacts were prone to appear in OCT images (Fig. 3D), due to specular reflections and incomplete removal of DC component in the signal which were consistent with recent relevant studies.66,67
Our data demonstrate that OCT can be used to visualize the physical structure of the matrix, as well as formation of new microvessels in our organ-on-chip device. The resolution of the imaging technique is high enough to not only detect large structural defects (Fig. 3C-iii), but also microvessels and cavities of physiological sizes (Fig. 3D-iii). Given the importance of OCT as a clinical tool, this read-out should in the future enable the comparison and correlation of data from our in vitro assay with clinical observations. In the clinic, OCT provides information about structural defects in the RPE in the form of drusen formation and fluid accumulation, as well as neovascularization from the choroid.68 Currently, the lack of resolution in the area of RPE culture in our device prevents us from screening for sub-retinal fluid accumulation or drusen formation. However, the technique does allow us to observe changes in capillary density and neovascularization, which are key steps in the pathophysiology of AMD preceding RPE dysfunction and photoreceptor degradation.69 Future studies will focus on improving the imaging depth of the set-up.
Conclusions
Microfluidic organ-on-a-chip systems have great potential to investigate basic mechanisms of disease pathology and organ-level physiology due to their flexibility of incorporating various cell types and physiologically relevant biochemical readouts.22 In addition, they can be used to test the effect of disease triggers and drugs. Here, we report an organ-on-a-chip model of the oBRB, and we use it to track pathophysiological processes relevant for AMD.Our model consists of a microvessel that is defined by patterning a collagen I hydrogel to recapitulate the in vivo choroidal microenvironment. Using H2O2 to mimic oxidative stress, one of the well-known disease factors in AMD pathophysiology, we explored the effects on co-cultures in our organ-on-a-chip devices. Using a readout that is analogous to the clinically used FA, we showed that on-chip co-cultures were affected by the H2O2 stimulus. In addition, we used OCT to image the hydrogel matrix of our organ-on-chip model, as well as 3D microvascular structures formed by cells within the hydrogel. Together, our results demonstrate the added value of integrating relevant read-outs in complex in vitro models like organs-on-chips when studying the oBRB.
There are various challenges in investigating AMD pathophysiology in vivo due to AMD being a multifactorial disease.5 The model, including read-outs, presented in this study demonstrates the potential added value of using organs-on-chips for future studies of disease mechanisms and treatment development for AMD.
As a next step, co-cultures of patient stem cell-derived endothelial cells and RPE cells can be integrated into the chip to generate a more representative model of AMD. Moreover, the integration of clinically relevant read-outs in our model will strongly facilitate future side-by-side comparison of in vitro findings with patient data. Together, this would make our organ-on-a-chip model of the outer tissues of the retina a powerful functional test for evaluating potential treatments for specific patients or patient sub-groups in the context of precision medicine.70
Author contributions
Y. B. A. and A. D. M. designed the experiments and critically analyzed data. Y. B. A. and W. B. performed chip design and developed the microvessel patterning protocol. J. L.-Z. and Y. B. A. developed and performed the fibrin gel patterning for OCT measurements. A. G. and Y. B. A. performed and analyzed endothelial seeding and morphology experiments. C. C.-V., C. V., S. L., N. B. developed OCT setup, performed OCT analysis. Y. B. A. and A. D. M. wrote the manuscript. P. B., A. I. H., G. G., R. P., and A. B. advised on key experimental design criteria, critically advised on data analysis and reviewed the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge Duco Schriemer and Karin Roelofs for kindly providing ARPE-19 cells. The authors acknowledge the funding received from Stichting Toegepast Wetenschappelijk Instituut voor Neuromodulatie (TWIN) under the project ‘Inflammation and Edema in an Organ-on-a-Chip Model of Wet Age-Related Macular Degeneration’, from Top sector ‘Life Sciences and Health’ Health – Holland under the ‘PLURIMACULA’ grant (grant no. LSHM19001) of Dr. Andries van der Meer, from the Dutch Science Foundation (NWO) under the Gravitation Grant ‘NOCI’ Program (grant no. 024.003.001) of Prof. Christine Mummery and ‘VESCEL’ Program (grant no. 669768) of Prof. Albert van den Berg.References
- F. K. Alswailmi, Pak. J. Med. Sci., 2018, 34, 751 Search PubMed.
- W. L. Wong, X. Su, X. Li, C. M. G. Cheung, R. Klein, C.-Y. Cheng and T. Y. Wong, Lancet Glob Health, 2014, 2, e106–e116 CrossRef.
- J. Ambati and B. J. Fowler, Neuron, 2012, 75, 26–39 CrossRef CAS PubMed.
- C. L. Hagedorn and R. A. Adelman, in Ocular Angiogenesis, Springer, 2006, pp. 3–22 Search PubMed.
- L. S. Lim, P. Mitchell, J. M. Seddon, F. G. Holz and T. Y. Wong, Lancet, 2012, 379, 1728–1738 CrossRef.
- R. Ratnapriya and E. Y. Chew, Clin. Genet., 2013, 84, 160–166 CrossRef CAS PubMed.
- L. G. Fritsche, W. Igl, J. N. C. Bailey, F. Grassmann, S. Sengupta, J. L. Bragg-Gresham, K. P. Burdon, S. J. Hebbring, C. Wen and M. Gorski, Nat. Genet., 2016, 48, 134–143 CrossRef CAS PubMed.
- Z. Ablonczy, M. Dahrouj, P. H. Tang, Y. Liu, K. Sambamurti, A. D. Marmorstein and C. E. Crosson, Invest. Ophthalmol. Visual Sci., 2011, 52, 8614–8620 CrossRef CAS PubMed.
- S. G. Jarrett and M. E. Boulton, Mol. Aspects Med., 2012, 33, 399–417 CrossRef CAS PubMed.
- M. Boulton, M. Rózanowska, B. Rózanowski and T. Wess, Photochem. Photobiol. Sci., 2004, 3, 759–764 CrossRef CAS PubMed.
- S. G. Jarrett, A. S. Lewin and M. E. Boulton, Ophthalmic Res., 2010, 44, 179–190 CrossRef CAS PubMed.
- J. V. Glenn and A. W. Stitt, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 1109–1116 CrossRef CAS PubMed.
- H. R. Novotny and D. L. Alvis, Circulation, 1961, 24, 82–86 CrossRef CAS PubMed.
- M. Castillo, G. Mowatt, N. Lois, A. Elders, C. Fraser, W. Amoaku, J. Burr, A. Lotery, C. Ramsay and A. Azuara-Blanco, Eye, 2014, 28, 1399 CrossRef CAS PubMed.
- M. Adhi and J. S. Duker, Curr. Opin. Ophthalmol., 2013, 24, 213 CrossRef PubMed.
- L. Arias and J. Monés, in AMD book, 2010, ch 7, pp. 61–80 Search PubMed.
- C. Zeiss, Vet. Pathol., 2010, 47, 396–413 CrossRef CAS PubMed.
- A. Maminishkis, S. Chen, S. Jalickee, T. Banzon, G. Shi, F. E. Wang, T. Ehalt, J. A. Hammer and S. S. Miller, Invest. Ophthalmol. Visual Sci., 2006, 47, 3612–3624 CrossRef PubMed.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- A. D. van der Meer and A. van den Berg, Integr. Biol., 2012, 4, 461–470 CrossRef CAS PubMed.
- G. Du, Q. Fang and J. M. den Toonder, Anal. Chim. Acta, 2016, 903, 36–50 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- P. Loskill, T. Sezhian, K. M. Tharp, F. T. Lee-Montiel, S. Jeeawoody, W. M. Reese, P.-J. H. Zushin, A. Stahl and K. E. Healy, Lab Chip, 2017, 17, 1645–1654 RSC.
- M. S. Hutson, P. G. Alexander, V. Allwardt, D. M. Aronoff, K. L. Bruner-Tran, D. E. Cliffel, J. M. Davidson, A. Gough, D. A. Markov and L. J. McCawley, Appl. In Vitro Toxicol., 2016, 2, 97–102 CrossRef.
- L. Bergers, T. Waaijman, T. De Gruijl, A. Van De Stolpe, R. Dekker and S. Gibbs, Tissue Eng., Part A, 2015, 21, S337–S338 Search PubMed.
- U. Marx, H. Walles, S. Hoffmann, G. Lindner, R. Horland, F. Sonntag, U. Klotzbach, D. Sakharov, A. Tonevitsky and R. Lauster, ATLA, Altern. Lab. Anim., 2012, 40, 235 CrossRef CAS PubMed.
- M. L. Moya, Y.-H. Hsu, A. P. Lee, C. C. Hughes and S. C. George, Tissue Eng., Part C, 2013, 19, 730–737 CrossRef CAS PubMed.
- A. Herland, A. D. van der Meer, E. A. FitzGerald, T.-E. Park, J. J. Sleeboom and D. E. Ingber, PLoS One, 2016, 11, e0150360 CrossRef PubMed.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–754 CrossRef CAS PubMed.
- J. C. Haderspeck, J. Chuchuy, S. Kustermann, S. Liebau and P. Loskill, Expert Opin. Drug Discovery, 2019, 14, 47–57 CrossRef CAS PubMed.
- L.-J. Chen, S. Ito, H. Kai, K. Nagamine, N. Nagai, M. Nishizawa, T. Abe and H. Kaji, Sci. Rep., 2017, 7, 3538 CrossRef PubMed.
- K. Achberger, C. Probst, J. Haderspeck, S. Bolz, J. Rogal, J. Chuchuy, M. Nikolova, V. Cora, L. Antkowiak and W. Haq, eLife, 2019, 8, e46188 CrossRef PubMed.
- M. Chung, S. Lee, B. J. Lee, K. Son, N. L. Jeon and J. H. Kim, Advanced Healthcare Materials, 2017 Search PubMed.
- J. Paek, S. E. Park, Q. Lu, K. T. Park, M. Cho and J. M. Oh, et al. , ACS Nano, 2019, 13(7), 7627–7643 CrossRef CAS PubMed.
- J. Yeste, M. García-Ramírez, X. Illa, A. Guimerà, C. Hernández, R. Simó and R. Villa, Lab Chip, 2018, 18, 95–105 RSC.
- Z. Liu, S. Mackay, D. M. Gordon, J. D. Anderson, D. W. Haithcock, C. J. Garson, G. J. Tearney, G. M. Solomon, K. Pant and B. Prabhakarpandian, Biomed. Opt. Express, 2019, 10, 5414–5430 CrossRef CAS PubMed.
- J. Pauty, R. Usuba, I. G. Cheng, L. Hespel, H. Takahashi, K. Kato, M. Kobayashi, H. Nakajima, E. Lee and F. Yger, EBioMedicine, 2018, 27, 225–236 CrossRef PubMed.
- L. Griep, F. Wolbers, B. De Wagenaar, P. M. ter Braak, B. Weksler, I. A. Romero, P. Couraud, I. Vermes, A. D. van der Meer and A. van den Berg, Biomed. Microdevices, 2013, 15, 145–150 CrossRef CAS PubMed.
- M. W. van der Helm, M. Odijk, J. P. Frimat, A. D. van der Meer, J. C. Eijkel, A. van den Berg and L. I. Segerink, J. Visualized Exp., 2017, 127, e56334 Search PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld and B. Schmid, Nat. Methods, 2012, 9, 676 CrossRef CAS PubMed.
- C. Veenstra, S. Kruitwagen, D. Groener, W. Petersen, W. Steenbergen and N. Bosschaart, Sci. Rep., 2019, 9, 1–8 CAS.
- C. Veenstra, W. Petersen, I. M. Vellekoop, W. Steenbergen and N. Bosschaart, Biomed. Opt. Express, 2018, 9, 3581–3589 CrossRef CAS PubMed.
- N. Bosschaart, M. C. Aalders, T. G. van Leeuwen and D. J. Faber, Biomed. Opt. Express, 2012, 3, 2263–2272 CrossRef PubMed.
- MATLAB, 9.7.0.1247435 (R2019b), The MathWorks Inc., Natick, Massachusetts, 2019 Search PubMed.
- R. Hamilton, A. Foss and L. Leach, J. Anat., 2007, 211, 707–716 CrossRef CAS PubMed.
- E. Mannermaa, M. Reinisalo, V.-P. Ranta, K.-S. Vellonen, H. Kokki, A. Saarikko, K. Kaarniranta and A. Urtti, Eur. J. Pharm. Sci., 2010, 40, 289–296 CrossRef CAS PubMed.
- H. Nevala, T. Ylikomi and H. Tähti, Hum. Exp. Toxicol., 2008, 27, 741–749 CrossRef CAS PubMed.
- C. Spencer, S. Abend, K. J. McHugh and M. Saint-Geniez, J. Cell. Mol. Med., 2017, 21, 2542–2552 CrossRef CAS PubMed.
- A. F. Moleiro, G. Conceição, A. F. Leite-Moreira and A. Rocha-Sousa, J. Ophthalmol., 2017, 2017, 3034953 CAS.
- S. W. Ballinger, B. Van Houten, C. A. Conklin, G.-F. Jin and B. F. Godley, Exp. Eye Res., 1999, 68, 765–772 CrossRef CAS PubMed.
- N. Goncharov, M. Terpilowski, A. Nadeev, I. Kudryavtsev, M. Serebriakova, V. Zinchenko and P. Avdonin, Biochem. Suppl. Ser. A Membr. Cell Biol., 2018, 12, 180–188 Search PubMed.
- R. N. Johnson, A. D. Fu, H. R. McDonald, J. M. Jumper, E. Ai, E. T. Cunningham and B. J. Lujan, in Retina, Elsevier Inc., 5th edn, 2012 Search PubMed.
- J. F. Arevalo, Retinal angiography and optical coherence tomography, Springer, 2008 Search PubMed.
- T. G. Papaioannou and C. Stefanadis, Hellenic J. Cardiol., 2005, 46, 9–15 Search PubMed.
- T. Kurz, M. Karlsson, U. T. Brunk and S. E. Nilsson, Autophagy, 2009, 5, 494–501 CrossRef CAS PubMed.
- J. H. Kim, Y. S. Chang, T. G. Lee and C. G. Kim, Invest. Ophthalmol. Visual Sci., 2015, 56, 1909–1915 CrossRef CAS PubMed.
- T. Iida, S. Kishi, N. Hagimura and K. Shimizu, Retina, 1999, 19, 508–512 CrossRef CAS PubMed.
- S. E. Chung, S. W. Kang, J. H. Lee and Y. T. Kim, Ophthalmology, 2011, 118, 840–845 CrossRef PubMed.
- P. Jirarattanasopa, S. Ooto, I. Nakata, A. Tsujikawa, K. Yamashiro, A. Oishi and N. Yoshimura, Invest. Ophthalmol. Visual Sci., 2012, 53, 3663–3672 CrossRef CAS PubMed.
- M. Miyake, A. Tsujikawa, K. Yamashiro, S. Ooto, A. Oishi, H. Tamura, I. Nakata, F. Matsuda and N. Yoshimura, Invest. Ophthalmol. Visual Sci., 2014, 55, 3223–3230 CrossRef PubMed.
- M. B. Chen, J. A. Whisler, J. Fröse, C. Yu, Y. Shin and R. D. Kamm, Nat. Protoc., 2017, 12, 865 CrossRef CAS PubMed.
- J. A. Whisler, M. B. Chen and R. D. Kamm, Tissue Eng., Part C, 2012, 20, 543–552 CrossRef PubMed.
- A. Cochrane, H. J. Albers, R. Passier, C. L. Mummery, A. van den Berg, V. V. Orlova and A. D. van der Meer, Adv. Drug Delivery Rev., 2019, 140, 68–77 CrossRef CAS PubMed.
- S. Kim, H. Lee, M. Chung and N. L. Jeon, Lab Chip, 2013, 13, 1489–1500 RSC.
- S. Roh and J. Weiter, Retinal and Choroidal Circulation, 2009, (https://www.semanticscholar.org/paper/Retinal-and-Choroidal-Circulation-Roh-Weiter/fe640af3a95bd22565b48a1a71a1a6d3502ad45c#citing-papers) Search PubMed.
- L. Kuehlewein, M. Bansal, T. L. Lenis, N. A. Iafe, S. R. Sadda, M. A. Bonini Filho, E. Talisa, N. K. Waheed, J. S. Duker and D. Sarraf, Am. J. Ophthalmol., 2015, 160, 739–748.e732 CrossRef PubMed.
- L. Roisman, Q. Zhang, R. K. Wang, G. Gregori, A. Zhang, C.-L. Chen, M. K. Durbin, L. An, P. F. Stetson and G. Robbins, Ophthalmology, 2016, 123, 1309–1319 CrossRef PubMed.
- A. Garcia-Layana, G. Ciuffo, J. Zarranz-Ventura and A. Alvarez-Vidal, Optical Coherence Tomography in Age-related Macular Degeneration, 2017, https://amdbook.org/content/optical-coherence-tomography-age-related-macular-degeneration Search PubMed.
- L. H. L. Koh, R. Agrawal, N. Khandelwal, L. Sai Charan and J. Chhablani, Acta Ophthalmol., 2017, 95, e597–e601 CrossRef PubMed.
- A. Van Den Berg, C. L. Mummery, R. Passier and A. D. Van der Meer, Lab Chip, 2019, 19, 198–205 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00639d |
| This journal is © The Royal Society of Chemistry 2021 |