
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiodegradable nanocomposite of poly(ester-co-carbonate) and cellulose nanocrystals for tough tear-resistant disposable bags†
Hyeri
Kim‡
a,
Hyeonyeol
Jeon‡
 a,
Giyoung
Shin
a,
Minkyung
Lee
a,
Jonggeon
Jegal
a,
Sung Yeon
Hwang
ab,
Dongyeop X.
Oh
ab,
Jun Mo
Koo
*a,
Youngho
Eom
*c and
Jeyoung
Park
*ab
a,
Giyoung
Shin
a,
Minkyung
Lee
a,
Jonggeon
Jegal
a,
Sung Yeon
Hwang
ab,
Dongyeop X.
Oh
ab,
Jun Mo
Koo
*a,
Youngho
Eom
*c and
Jeyoung
Park
*ab
aResearch Center for Bio-based Chemistry, Korea Research Institute of Chemical Technology (KRICT), Ulsan 44429, Republic of Korea. E-mail: jmkoo@krict.re.kr; jypark@krict.re.kr
bAdvanced Materials and Chemical Engineering, University of Science and Technology (UST), Daejeon 34113, Republic of Korea
cDepartment of Polymer Engineering, Pukyong National University, Busan, 48513, Republic of Korea. E-mail: eomyh@pknu.ac.kr
First published on 2nd February 2021
Abstract
Current sustainable bioplastics need dramatic improvement to become competitive. Herein, we prepared poly(butylene succinate-co-carbonate) nanocomposites with citric acid and cellulose nanocrystals (CNCs). The dimethyl carbonate co-monomer lowers crystallinity and increases elongation; citric acid increases the strength, and the CNCs maximize mechanical performance. Accelerated biodegradation is also favorable for sustainable tough bags.
The consumption of biodegradable plastic bags has increased in response to a desire to reduce single-use non-degradable plastic waste that burdens sustainable ecosystems.1,2 However, the recent coronavirus pandemic (COVID-19) has led to the excessive use of disposable plastic commodities aimed at protecting humans against infection,3 with many countries, including Korea, losening restrictions on the use of disposable products in grocery stores, cafés, and other scenarios.4 Therefore, air pollution through incineration, soil pollution through landfill, and marine pollution through inflow and atomization, have increased as society aims to prevent the epidemic.5,6
Tough biodegradable plastic bags offer possible solutions that address both the abovementioned concerns. Meanwhile, conventional biodegradable bags are often not willingly selected by consumers because of their poor mechanical properties, high costs, and lack of necessity and justification;7 such bags are too easily torn or punctured to be stand-alone materials in the absence of environmental regulations. Inorganic fillers were used in an attempt to reinforce a poly(butylene adipate-co-terephthalate)-based (PBAT-based) film by melt-compounding;8,9 however, the effect was not significant, especially on its tear properties, while it was less eco-friendly through the inclusion of non-degradable fillers. In addition, the ductilities of rustling poly(lactic acid) (PLA) films were improved by blending with soft polymers, such as PBAT, to compensate for their inferior strengths.10,11
We previously reported the in situ synthesis of nanocomposites of poly(butylene succinate) (PBS) and cellulose nanocrystals (CNCs).12 CNCs, which can be obtained from the acidic hydrolysis of cotton and other natural resources, form whiskers with 50–3000 nm long and 3–20 nm wide, and are suitable as reinforcing agents in nanocomposites.13 While a highly dispersed nanofiller in the polymer matrix resulted in improved tensile strength and toughness,14 unfortunately, PBS is unsuitable for the fabrication of disposable bags because its linear structure along with poor intermolecular interactions prohibits high degrees of entanglement during deformation and crystallization, resulting in vulnerable tear stress along a specific axis.15,16 To highlight the biodegradability of PBS and to complement its tearing instability, blending with other aliphatic polyesters that reduces its crystallinity inevitably results in inferior mechanical properties.17,18 Another approach involves introducing carbonate groups along the main backbone, and it pays the penalty for the robustness.19,20
Herein, we boldly increased the carbonate content of poly(ester-co-carbonate) by co-polycondensation with dimethyl carbonate (DMC), dimethyl succinate (DMS), and 1,4-butanediol (BD), which resulted in a higher tensile elongation at break and tear toughness compared to PBS. In addition, the in situ incorporation of a minimal amount of citric acid (as a cross-linker) and CNCs led to an increase in tensile strength and toughness, while maintaining tear toughness. Finally, a tough biodegradable polyester film with an ultimate tensile strength of 64 MPa, an elongation at break of 690%, an ultimate tear strength of 1.4 kN cm−1, and a tear toughness of 32 J cm−1, with improved applicability to sustainable disposable bags, was prepared.
Until the early 2010s, researchers attempted to blend poly(butylene succinate-co-butylene carbonate) (PBSC) containing 10% carbonate (manufactured by the Mitsubishi Gas Chemical Company) with other biodegradable polymers, fillers, or eco-friendly additives.21,22 Alternatively, several studies investigated the characteristics of blending PBS and poly(butylene carbonate) or their multi-block copolymers linked with chain-extenders.23,24 In these cases, few studies produced films and investigated their tensile and tear properties. Based on the previously formalized PBSC structure, we synthesized PBSC with a high carbonate content, which we refer to as “PBS-1C”, in which carbonate groups are randomly distributed, using a two-step melt polycondensation procedure (Fig. 1).25 DMC, which contributes to sustainability, was used instead of diphenyl carbonate, which produces phenol as a byproduct.26 To increase the carbonate content, the initial monomer feed ratio was set to: DMS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC:BD = 0.48:0.72:1.0 ([carbonyl derivatives]/[alcohol] = 1.2). DMC (boiling point: 90 °C) is the most volatile monomer; hence, stepwise heating from 80–150 °C during esterification, and 150–200 °C in the polycondensation step, using a setup with a long condenser set to 55 °C, was required to minimize the loss of unreacted monomer.27,28 PBS was also synthesized as a standard.
:DMC:BD = 0.48:0.72:1.0 ([carbonyl derivatives]/[alcohol] = 1.2). DMC (boiling point: 90 °C) is the most volatile monomer; hence, stepwise heating from 80–150 °C during esterification, and 150–200 °C in the polycondensation step, using a setup with a long condenser set to 55 °C, was required to minimize the loss of unreacted monomer.27,28 PBS was also synthesized as a standard.
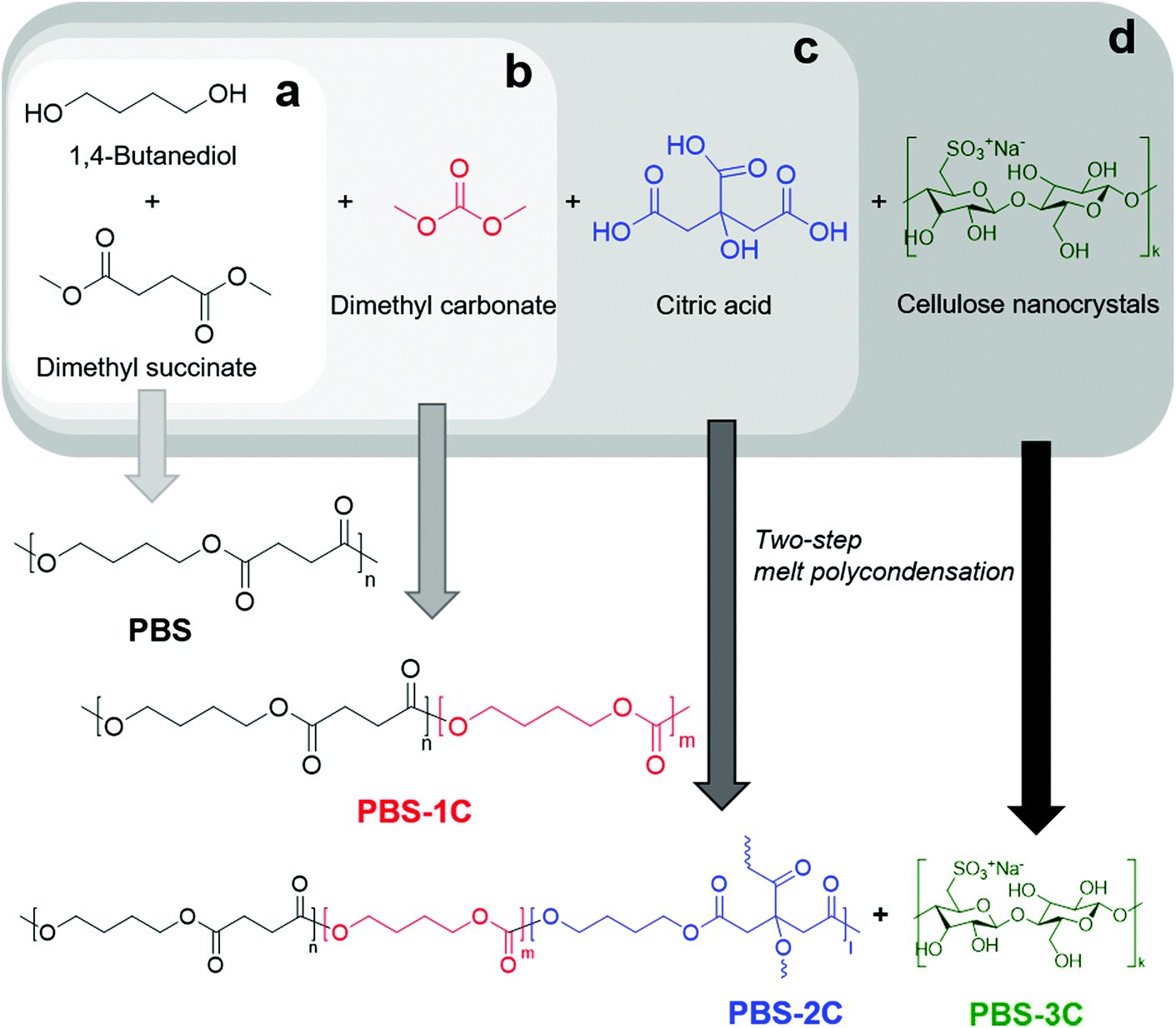 | ||
| Fig. 1 Scheme for the syntheses of: (a) PBS, (b) PBS-1C, (c) PBS-2C, and (d) PBS-3C. | ||
A large amount of DMC was fed into the reactor, as we expected it to be significantly lost during polycondensation. The incorporated carbonate content of PBS-1C was quantitatively determined by 1H NMR peak integration (Fig. S1 and S2†), and it was determined to be 24% of the total ester + carbonate groups. PBS-1C was determined by gel permeation chromatography to have a weight-average molecular weight of 158 kg per mol. This indicates that the titanium(IV)-butoxide-catalyzed transesterification of DMC and BD proceeded efficiently to produce a polymer with a sufficiently high molecular weight to enable its mechanical properties to be compared with those of the neat PBS. PBS-1C exhibited a clear single glass transition temperature (Tg) of −40 °C, which supports its random copolymer nature (Table S1†). As expected, the significant carbonate-group content greatly disassembled the crystalline features of PBS. The melting temperature of PBS-1C was more than 28 °C lower than that of PBS, while its melting enthalpy was significantly reduced to 58% of the PBS value.
To observe the effect of the carbonate structure on the macroscopic mechanical properties, PBS and PBS-1C were hot-pressed to prepare 0.5 mm thick films. The ultimate tensile strength of the PBS-1C film was measured to be 37 MPa, which is 0.9-fold lower than the 41 MPa value of PBS; meanwhile, its elongation at break was found to be 560%, which is 2.4-fold higher than that of PBS (230%) (Fig. 2a and Table S2†). The transformation from stiff PBS to soft PBS-1C was expected and is ascribable to the considerably reduced crystallinity and softened polymer chains that result from the incorporation of the carbonate groups.
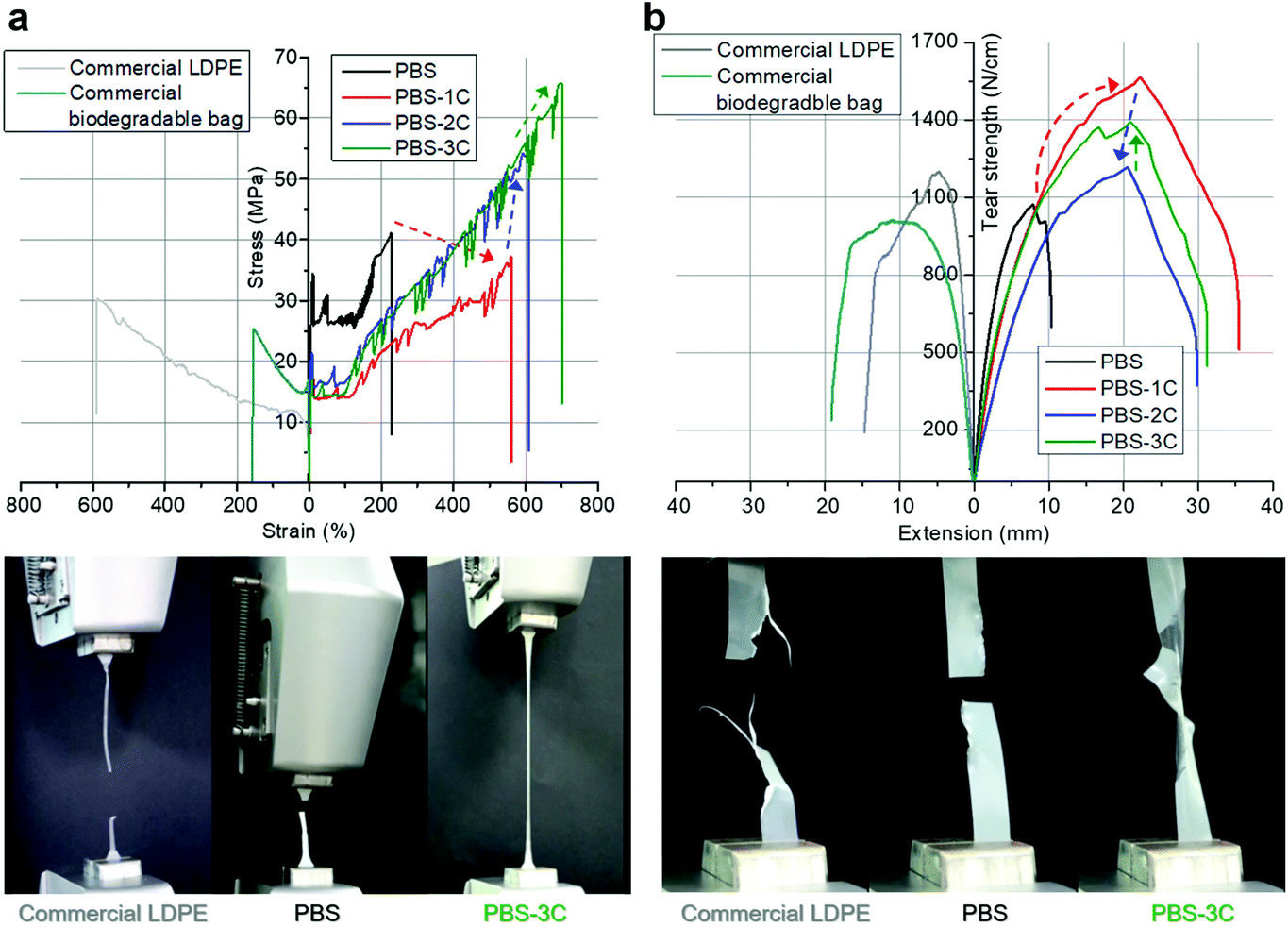 | ||
| Fig. 2 (a) Tensile stress–strain and (b) tear-strength–extension curves of prepared films and commercial reference films. Bottom: photographic images acquired during mechanical testing. | ||
While the improved mechanical stretchability of PBS-1C is applicable to disposable bags, its inferior tensile strength was a concern to us. To address this performance issue, we established two new molecular strategies involving chemical and physical tuning. First, trivalent citric acid, which forms tri-armed cross-linkages, was introduced into PBS-1C as a chemical “pinch”. To maximize the cross-linking effect while retaining melt-processability, only 0.04 mol% of citric acid per total acid- and carbonate-derived monomers was fed during polymerization. Chemically tuned poly(butylene succinate-co-butylene carbonate-co-butylene citrate) is referred to as “PBS-2C” (Fig. 1c). Interestingly, all tensile data were significantly improved with the addition of the minimal amount of citric acid (Fig. 2). The ultimate tensile strength, elongation at break, and tensile toughness values of 54 MPa, 600%, and 200 MJ m−3, are 1.5-, 1.1-, and 1.6-fold higher than those of PBS-1C, and they are ascribable to chemical tuning (Fig. 2a and Table S2†). In comparison, commercial LDPE and a biodegradable bag (PBAT/PLA blend) show strengths of only 25–31 MPa. Quite unexpectedly, the tear resistances of PBS-1C and PBS-2C were significantly higher than that of crystalline PBS; the ultimate tear strengths and extensions at break were determined to be 1.3–1.1-, and 3.5–3.0-fold higher than those of homo PBS. Tear toughness, which is a quantitative indicator of the resistance of a film to tear propagation, was improved by a factor of 4.3 for PBS-1C and 3.0 for PBS-2C over that of crystalline PBS (Fig. 2b and Table S2†).
The improved tear resistance of the carbonate-group-containing PBS derivatives (PBS-1C and 2C) is intimately related to delayed fracturing during tear testing, which can be interpreted from the perspective of stress dissipation, namely, evenly spread external stress (i.e., stress delocalization) with rapid stress dissipation. In particular, PBS, which has a linear chain structure, largely favors stress-induced crystallization upon straining.15 Therefore, crystallite evolution in the stress-concentrated region stiffens the polymer, eventually leading to fracturing. In this regard, the slow tear propagation observed for PBS-1C and 2C originates from reduced crystallizability (Table S1 and Fig. S3†). The presence of asymmetric carbonate moieties intensely disturbs regular chain folding as well as chain orientation along the stretching axis. In addition, the abilities of the highly flexible carbonate-containing chains to easily undergo conformational change result in faster energy dissipation upon stress. As a result, the addition of carbonate functional groups renders the material with high extensibility as well as strength during tearing. However, as a consequence, a lower mechanical strength was observed for PBS-1C during uniaxial tensile testing. To compensate for this loss, a minimal amount of multifunctional citric acid was added to the chains to form PBS-2C. As mentioned above, the insertion of a small amount of a chemical-junction structure (in going from 1C to 2C) remarkably improved the mechanical strength with negligible sacrifice of extension under both tensile and tearing conditions. This new approach overcomes the prejudice held against chemical cross-linking, which has a negative influence and worsens fluidity and fusibility. The crosslinking ability of citric acid results in a higher number of chain bridges, which slows down the chain-extraction process during straining and prevents premature cavitation. Such a “pinch” effect enhances mechanical performance without affecting melt processability.
In addition, aside from the chemical tuning of the polymer structure, PBS-2C was also physically tuned by incorporating CNCs as a nanocomposite reinforcement. CNCs of the spray-dried type are 136 ± 26 nm long with diameters of 19 ± 4 nm, as verified by atomic force microscopy and stable zeta sizes of 75 and 100 nm at pH 3.2 and 7.0, respectively. It is suggested that CNCs maintain good dispersibility even under the acidic conditions of polycondensation, leading to superior mechanical performance (Fig. S4†). Our previous findings revealed that the physical tuning of PBS through in situ nanocompositing with CNCs reinforced both strength and ductility. Hence, a PBS-2C/CNC nanocomposite, referred to as “PBS-3C”, was prepared using CNCs pre-dispersed in the BD monomer. The prepared PBS-3C film exhibited an ultimate tensile strength of 64 MPa, an elongation at break of 690%, and a tensile toughness of 250 MJ m−3, which are sufficient to meet the commercial requirements of tough biodegradable disposable bags (Fig. 2a and Table S2†). The ultimate tear strength also recovered to a value of 1.4 kN cm−1 to give a tear toughness of 32 J cm−1 (Fig. 2b and Table S2†). The mechanical improvement of the nanocomposite in terms of both tensile and tear performance is in line with the appreciable dispersion of the in situ incorporated CNCs. It is well known that the degree of filler dispersion plays a major role in determining the reinforcing effect.29 While an aggregated filler can act as a defect due to poor interaction with the polymer matrix, an individually well-dispersed filler can interact strongly with neighboring polymer chains through physical entanglement and interaction. As a result, highly dispersed CNCs in PBS-3C not only enhance the tensile and tear resistance owing to their stiff nature, but also impede crack propagation by disturbing chain disentanglement. These stronger and tougher features of the PBS-3C film over conventional PBS and LDPE are visualized in the tensile/tear experiments shown in Movies S1 and S2.† When observed with the naked eye, the applied force concentrated at specific spots on PBS and LDPE led to relatively clean tearing, whereas the force was evenly distributed throughout the PBS-3C specimen, leading to messy tearing, that is, the PBS-3C film was tear-stress resistant and did not tear easily. Considering that the cost of commercial LDPE is 25% of that of PBS and PBAT, the mechanical improvements in PBS-3C by more than two times may be competitive when the sustainable-plastic market leads to an increase in production volumes.
The stiff-to-tough tuning trend of the PBS series was clearly verified by observing the fracture morphologies of the tensile-tested specimens of stiff PBS and tough PBS-3C (Fig. 3a). The remarkable grain structures on the relatively smooth fractured surface of PBS reveal that cracks propagate along crystal grain boundaries during deformation; rapid and unhindered crack propagation resulted in a smooth fractured surface for pristine PBS.29,30 Meanwhile, PBS-3C exhibited an uneven and rough fractured surface accompanied by crazing phenomenon. The evolution of microfibrils through crazing promotes ductile deformation because they act as bridges that absorb external stress. As a result, crack propagation and fracturing are substantially delayed during the tensile testing of PBS-3C. The differences in ductility and the extent of crazing are further confirmed by examining the surface morphology by optical microscopy (OM; Fig. S5†). Tough PBS-3C is highly extended with a large number of elongated voids, which are the main differences observed by OM. The elongated internal voids are the result of crazing, that is, they provide another indicator of polymer robustness.31,32 The tear-tested specimens also show clear differences in fracture morphology (Fig. 3b). The neat PBS film exhibits clear-cut and distinct tear marks on its fracture surface, indicative of the rapid propagation of tearing. On the other hand, the surface of the PBS-3C film is undulated with few tear marks, consistent with delayed tearing.
 | ||
| Fig. 3 SEM images of the fractured surfaces of (a) tensile- and (b) tear-tested PBS and PBS-3C specimens. | ||
The rheological properties of the PBS series were investigated in the molten state at 140 °C in order to discuss their stress response behavior (Fig. 4). The incorporation of carbonate groups (PBS-1C) inevitably resulted in significant reductions in complex viscosity (η*) and storage modulus (G′), suggestive of weak mutual polymer-chain interference owing to enhanced chain flexibility (Fig. 4a and b). In addition, the G′ values at 0.05 rad s−1 (G′0.05) of PBS-2C and 3C are dominantly determined by the carbonate-group content because their remarkably enhanced fluidities due to their large amounts of carbonate overwhelm the influence of both chemical (citric acid) and physical (CNC) tuning (Fig. 4c). Despite the tremendous effect of the carbonate groups, the PBS series exhibits interesting rheological behavior in terms of homogeneity, which can quantitatively be determined from the slopes of their Cole–Cole plots (G′ vs. loss modulus (G′′)). Theoretically, a slope closer to two corresponds to a more homogeneous polymeric material because the slope of an ideal homogeneous polymeric melt is two.33,34 Interestingly, PBS-3C exhibited the highest slope of 1.55, which indicates that PBS-3C is the most homogeneous system. A highly homogeneous polymer nanocomposite system is unusual. Generally, the incorporation of a nanofiller (hetero-component) results in the deterioration of matrix homogeneity.35 However, we recently reported that in situ-blended CNCs improve homogeneity because the chemically modified filler acts as a plasticizer through polymer grafting.29,36 Even though PBS-3C exhibited superior homogeneity in the molten state, these viscoelastic responses are consistent with evenly spread and rapidly dissipated stress in a solid-type film.
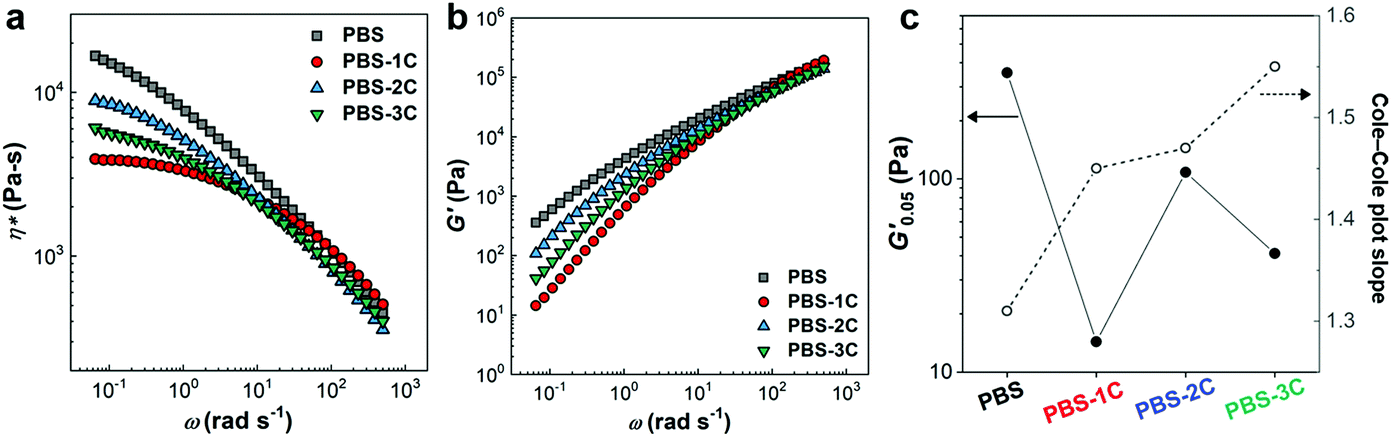 | ||
| Fig. 4 Rheological responses of the PBS series at 140 °C. (a) Complex viscosities (η*), (b) storage moduli (G′), and (c) variations in G′ at 0.05 rad s−1 (G′0.05) and Cole–Cole plot slopes. | ||
From the perspective of degradation, the lower crystallinities of the PBS derivatives achieved by incorporating carbonate groups should accelerate their decomposition because the amorphous polymer chains are more accessible to hydrolysis.37,38 Faster decomposition is advantageous for the commercialization of tough disposable bags. Under accelerated conditions (50 °C), the PBS-carbonates were rapidly and enzymatically decomposed by lipases from Pseudomonas cepacia (PS) and Thermomyces lanuginosus (TL) (Fig. S6†). Each member of the PBS-carbonate series was almost completely decomposed within 1–2 d, whereas PBS lost 10% and 55% of its weight by the actions of the PS and TL lipases, respectively, after one day.
Moreover, laboratory bio-decomposition experiments that mimic composting conditions were performed. PBS-carbonate films (80–100 μm thick) were buried in a compost and soil mixture and incubated at 50 °C. To ensure breathability, the same mass of garden soil was mixed with compost (5:5, w/w), with water content maintained at 50% (v/w) (Fig. S7†). Biodegradation was evaluated from the weight losses, film thickness, and morphological changes observed for the films. The PBS-carbonates showed significantly accelerated degradation rates compared to PBS under composting conditions due to their low crystallinities, which exceeded our expectations and are consistent with the in vitro enzymatic degradation results (Fig. 5a). Neat PBS lost 1.6% of its weight over two weeks, whereas PBS-1C and PBS-2C lost 31.2% and 26.2% of their weights, respectively. Interestingly, PBS-3C showed the highest degradation rate, with a weight loss of more than 65% and a film thickness decrease to 55% observed after 14 d (Fig. S8†). It should be noted that conventional PBS plastic generally requires an adaptation time of approximately 14 d to initiate aerobic biodegradation, with over 80% degradation observed after 45 d under composting conditions at 58 °C.39 Thus, it seems that the less-crystalline PBS-carbonates have effectively shorter initial delay times for hydrolysis and the approach of micro-organisms. Furthermore, changes in the surface morphologies of the buried films were regularly observed by OM and SEM (Fig. 5b and Fig. S9†). The PBS-carbonate series exhibited extensive damage by the naked eye, with extensive pore formation throughout the films, whereas neat PBS only exhibited surface whitening. Interestingly, more-detailed SEM observations indicate that decomposition progressed separately in each crystal-grain group of neat PBS. Meanwhile, the PBS-carbonate series decomposed with ambiguous grain boundaries. Thus, the degradation caused by microorganism activities on the PBS-carbonates led to smooth cavities compared to PBS, which indicates that the PBS-carbonates are more susceptible to depolymerizing enzymes, resulting in a significant increase in the biodegradation rate.
 | ||
| Fig. 5 (a) Bio-decomposition (weight loss) of the PBS series in composting soil. (b) Optical and SEM images of film surfaces before and after bio-decomposition (film sizes: 10 × 10 mm, scale bar: 10 μm). (c) Optical images of germinated lettuce seeds after the bio-decomposition experiments of PBS-3C and the negative control (scale bar: 1 cm). | ||
We further investigated phytotoxicity by determining the germination index (GI) of the soil after the biodegradation of each disposable film (Fig. 5c).40 To fully metabolize the carbon sources and to produce CO2, the compost mixture was aged for two months after the end of the bio-decomposition experiment. Thereafter, an aqueous soil-extract was used to determine the germination rate of Lactuca sativa L. (lettuce) seeds, with relative root lengths after 5 d of incubation reflecting the final GI. All members of the PBS series exhibited negligible ecological soil toxicities, that is, they exhibited more than 95% of the GI of the negative control (Table S3 and Fig. S10†).
Conclusions
PBS plastic, which is not suitable for film production, was reformed as an upgraded poly(butylene succinate-co-carbonate) nanocomposite for biodegradable disposable bags by incorporating significant amounts of carbonate to diminish its crystallinity, minimal amounts of citric acid as a “pinch”, and CNCs as a nanofiller. A combination of chemical and physical tuning, including lowering the crystallinity through the introduction of carbonate irregularities, chemical binding by citric acid, and physical binding by CNC, synergistically contributed to an engineered film with both high tensile and tear toughness. The additional merits of accelerated biodegradation and lack of phytotoxicity indicate that this composite is a suitable replacement material to existing non-biodegradable plastics commonly used in single-use bags.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Technology Development Program (20008628, 20010807) funded by the Ministry of Trade, Industry & Energy (MI, Korea) and the Korea Research Institute of Chemical Technology (KRICT) core project (SS2142-10). Y.E. acknowledges support from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (2020R1C1C1009340). J.J. acknowledges support from the Korea Institute for Advancement of Technology (KIAT) through the System Industrial Base Institution Support Program (P0001939). Special thanks to Jaeryeon Kang at Lotte Chemical for a fruitful discussion.Notes and references
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- Editorial, Nat. Commun., 2018, 9, 2157 CrossRef.
- J. J. Klemeš, Y. V. Fan, R. R. Tan and P. Jiang, Renewable Sustainable Energy Rev., 2020, 127, 109883 CrossRef.
- A. H. Tullo, Chem. Eng. News, 2020, 98, 24 CAS , (https://cen.acs.org/materials/polymers/Plastics-during-pandemic/98/i24).
- R. Hurley, J. Woodward and J. J. Rothwell, Nat. Geosci., 2018, 11, 251–257 CrossRef CAS.
- M. C. Rillig and A. Lehmann, Science, 2020, 368, 1430 CrossRef CAS.
- G.-Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS.
- S. Mohanty and S. K. Nayak, J. Polym. Environ., 2012, 20, 195–207 CrossRef CAS.
- Y. Someya, Y. Sugahara and M. Shibata, J. Appl. Polym. Sci., 2005, 95, 386–392 CrossRef CAS.
- K. Madhavan Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef CAS.
- N. Mallegni, T. V. Phuong, M.-B. Coltelli, P. Cinelli and A. Lazzeri, Materials, 2018, 11, 148 CrossRef.
- T. Kim, H. Jeon, J. Jegal, J. H. Kim, H. Yang, J. Park, D. X. Oh and S. Y. Hwang, RSC Adv., 2018, 8, 15389–15398 RSC.
- M. S. Reid, M. Villalobos and E. D. Cranston, Langmuir, 2017, 33, 1583–1598 CrossRef CAS.
- J. M. Koo, J. Kang, S.-H. Shin, J. Jegal, H. G. Cha, S. Choy, M. Hakkarainen, J. Park, D. X. Oh and S. Y. Hwang, Compos. Sci. Technol., 2020, 185, 107885 CrossRef CAS.
- G. Liu, L. Zheng, X. Zhang, C. Li, S. Jiang and D. Wang, Macromolecules, 2012, 45, 5487–5493 CrossRef CAS.
- H.-J. Jin, J.-K. Park, K.-H. Park, M.-N. Kim and J.-S. Yoon, J. Appl. Polym. Sci., 2000, 77, 547–555 CrossRef CAS.
- S.-M. Lai, C.-K. Huang and H.-F. Shen, J. Appl. Polym. Sci., 2005, 97, 257–264 CrossRef CAS.
- B. Palai, S. Mohanty and S. K. Nayak, Polym. Test., 2020, 83, 106130 CrossRef CAS.
- X. Cai, X. Yang, H. Zhang and G. Wang, Polymer, 2018, 134, 63–70 CrossRef CAS.
- H. Hu, R. Zhang, J. Wang, W. B. Ying and J. Zhu, ACS Sustainable Chem. Eng., 2018, 6, 7488–7498 CrossRef CAS.
- T. Ikehara, Y. Nishikawa and T. Nishi, Polymer, 2003, 44, 6657–6661 CrossRef CAS.
- M. Weng and Z. Qiu, Ind. Eng. Chem. Res., 2013, 52, 10198–10205 CrossRef CAS.
- J. Wang, L. Zheng, C. Li, W. Zhu, D. Zhang, G. Guan and Y. Xiao, Ind. Eng. Chem. Res., 2012, 51, 10785–10792 CrossRef CAS.
- J. Wang, L. Zheng, C. Li, W. Zhu, D. Zhang, Y. Xiao and G. Guan, Polym. Test., 2012, 31, 39–45 CrossRef CAS.
- H. Kim, T. Kim, S. Choi, H. Jeon, D. X. Oh, J. Park, Y. Eom, S. Y. Hwang and J. M. Koo, Green Chem., 2020, 22, 7778–7787 RSC.
- G. Fiorani, A. Perosa and M. Selva, Green Chem., 2018, 20, 288–322 RSC.
- J. H. Park, J. Y. Jeon, J. J. Lee, Y. Jang, J. K. Varghese and B. Y. Lee, Macromolecules, 2013, 46, 3301–3308 CrossRef CAS.
- W. Zhu, X. Huang, C. Li, Y. Xiao, D. Zhang and G. Guan, Polym. Int., 2011, 60, 1060–1067 CrossRef CAS.
- S.-A. Park, Y. Eom, H. Jeon, J. M. Koo, E. S. Lee, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Green Chem., 2019, 21, 5212–5221 RSC.
- Z. Lule and J. Kim, Polymers, 2019, 11, 148 CrossRef.
- L. T. Hao, Y. Eom, T. H. Tran, J. M. Koo, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Nanoscale, 2020, 12, 2393–2405 RSC.
- C. Wan, E. L. Heeley, Y. Zhou, S. Wang, C. T. Cafolla, E. M. Crabb and D. J. Hughes, Soft Matter, 2018, 14, 9175–9184 RSC.
- X.-c. Du, Y.-p. Wang, W.-b. Huang, J.-h. Yang and Y. Wang, Colloid Polym. Sci., 2015, 293, 389–400 CrossRef CAS.
- J. S. Lim, S. M. Hong, D. K. Kim and S. S. Im, J. Appl. Polym. Sci., 2008, 107, 3598–3608 CrossRef CAS.
- S. Sinha Ray, K. Okamoto and M. Okamoto, Macromolecules, 2003, 36, 2355–2367 CrossRef.
- S.-A. Park, Y. Eom, H. Jeon, J. M. Koo, T. Kim, J. Jeon, M. J. Park, S. Y. Hwang, B.-S. Kim, D. X. Oh and J. Park, ACS Macro Lett., 2020, 9, 558–564 CrossRef CAS.
- M. Gigli, A. Negroni, M. Soccio, G. Zanaroli, N. Lotti, F. Fava and A. Munari, Green Chem., 2012, 14, 2885–2893 RSC.
- K. H. Paek and S. G. Im, Green Chem., 2020, 22, 4570–4580 RSC.
- M. Kunioka, F. Ninomiya and M. Funabashi, Int. J. Mol. Sci., 2009, 10, 4267–4283 CrossRef CAS.
- P. M. S. Souza, L. R. D. Sommaggio, M. A. Marin-Morales and A. R. Morales, Chemosphere, 2020, 256, 126985 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and complete characterization data. See DOI: 10.1039/d0gc04072j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |