
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic/molecular layer deposition of Ni-terephthalate thin films†
Anish
Philip
 a,
Sami
Vasala
b,
Pieter
Glatzel
b and
Maarit
Karppinen
*a
a,
Sami
Vasala
b,
Pieter
Glatzel
b and
Maarit
Karppinen
*a
aDepartment of Chemistry and Materials Science, Aalto University, P.O. Box 16100, FI-00076 Espoo, Finland. E-mail: maarit.karppinen@aalto.fi
bESRF – The European Synchrotron, 71 Avenue des Martyrs, 38000 Grenoble, France
First published on 12th October 2021
Abstract
Atomic/molecular layer deposition (ALD/MLD) is currently strongly emerging as an intriguing route for novel metal–organic thin-film materials. This approach already covers a variety of metal and organic components, and potential applications related to e.g. sustainable energy technologies. Among the 3d metal components, nickel has remained unexplored so far. Here we report a robust and efficient ALD/MLD process for the growth of high-quality nickel terephthalate thin films. The films are deposited from Ni(thd)2 (thd: 2,2,6,6-tetramethyl-3,5-heptanedionate) and terephthalic acid (1,4-benzenedicarboxylic acid) precursors in the temperature range of 180–280 °C, with appreciably high growth rates up to 2.3 Å per cycle at 200 °C. The films are amorphous but the local structure and chemical state of the films are addressed based on XRR, FTIR and RIXS techniques.
Introduction
Hybrid metal–organic materials have attracted widespread attention as they form a promising platform for realizing unprecedented combinations of chemical and physical properties that are often difficult if not impossible to achieve with conventional materials. Atomic/molecular layer deposition (ALD/MLD) technique provides us with a scientifically elegant yet industrially feasible way to fabricate these materials in high-quality thin-film form.1–4 The method derives from the firmly established ALD (atomic layer deposition) technology for inorganic thin films, in which the atomic-level control of film thickness, composition and homogeneity stems from the self-limited gas–surface reactions of sequentially pulsed gaseous/evaporated inorganic precursors.5,6 For example, in one of the prototype ALD processes, Al2O3 thin films are grown from Al(CH3)3 and H2O precursors. In a parallel technique for purely organic thin films, i.e. MLD (molecular layer deposition), two mutually reactive organic precursors are used.7 In the combined ALD/MLD technique for the hybrid metal–organic films, the metal-entailing ALD precursor is combined with an organic MLD precursor.3,4 Moreover, the ALD/MLD approach allows any arbitrary combinations of the ALD and MLD pulses such that e.g. ultrathin (monomolecular) organic layers can be introduced between thicker inorganic layers (grown with multiple ALD cycles) into various regular (superlattice) or irregular (gradient) multilayer structures.8–12Facile ALD/MLD processes have already been developed for a rapidly expanding variety of different metal (s-block, p-block, d-block, lanthanide), and organic (allyl, aryl, pyridine, etc.) constituents.3,13–17 Transition metal based hybrids and in particular those involving the late 3d metals with partially filled d orbitals are particularly intriguing candidates for magnetically, electrically, optically or catalytically active materials. So far – among the late 3d metals – well-behaving ALD/MLD processes have been developed for Mn, Fe, Co and Cu (see Table 1),18–23 while for Ni very little efforts have been made. In previous literature, ultra-thin Ni4MP interfacial layers were grown for solar cell application from bis(dimethylamino-2-methyl-2-butoxo)nickel and 4-mercapto phenol precursors,11 but no ALD process optimization details were reported.
| Metal | Metal precursor | Organic precursor | Deposition Temp (°C) | GPC (Å per cycle), *mass gain (ng cm−2) | Ref. |
|---|---|---|---|---|---|
| Abbreviations: Metal precursor/ligand; TTIP (titanium isopropoxide), thd (2,2,6,6-tetramethyl-3,5-heptanedionate), Cp (cyclopentadienyl), CpEt (ethylcyclopentadienyl), acac (acetylacetonate), dmamb (dimethylamino-2-methyl-2-butoxo), dmap (dimethyl aminopropoxide). Organic precursor: HQ (hydroquinone), GL (glycerol), EG (ethylene glycol), 4-AP (4-aminophenol), ODA (4,4′-oxydianiline), TCNE (tetracyanoethylene), TPA (terephthalic acid), ADA (azobenzenedicarboxylic acid), MP (mercapto phenol), PPD (p-phenylenediamine), BDT (1,4-benzenedithiol). | |||||
| Titanium | TTIP | HQ | 300 | 2.5 | 35 |
| TiCl4 | GL | 210 | 2.1 | 36 | |
| GL | 130 | 2.8 | 36 | ||
| EG | 95–115 | 4.5 | 36 | ||
| EG | 135 | 1.5 | 36 | ||
| 4-AP | 120–160 | 10–11 | 37 | ||
| ODA | 160 | 0.3 | 38 | ||
| ODA | 490 | 1.1 | 38 | ||
| Vanadium | VOCl3 | EG | 90–135 | *15.2 | 39 |
| V(CO)6 | TCNE | RT | 9.8 | 40 | |
| Manganese | Mn(thd)3 | TPA | 160–280 | ≈1.0 | 18 |
| Mn(CpEt)2 | EG | 160 | 0.9 | 20 | |
| Iron | FeCl3 | TPA | 240–260 | 11 | 23 |
| ADA | 280 | 25 | 22 | ||
| Cp2Fe2(CO)4 | HQ | 180 | ≈3.7 | 41 | |
| Cobalt | Co(acac)3 | TPA | 160–280 | 1.0–1.2 | 18 |
| Co(thd)2 | TPA | 160–280 | 1.5 | 18 | |
| Nickel | Ni(dmamb)2 | 4MP | 180 | 2.26 | 11 |
| Copper | Cu(thd)2 | TPA | 180–280 | 0.7 | 19 |
| Cu(dmap)2 | HQ, TPA, ODA, PPD, BDT | 120–240 | 1.0–2.6 | 21 | |
Here our goal is to fill the gap within the late 3d transition metal based ALD/MLD processes. Besides the solar cell application,24 Ni-organic thin films could be interesting for other emerging energy applications, e.g. supercapacitors.25–27 Moreover, a facile ALD/MLD process for Ni-organics would be interesting also in the sense that it could be then combined with any of the already known ALD processes for NiO films for the fabrication of various layer-engineered NiO:organic multilayer structures. Nickel oxide as such is a wide bandgap p-type semiconductor, extensively investigated for applications such as resistive memories (memristors)28 and photovoltaics.29 For the ALD of NiO thin films, the most common nickel precursors are Ni(thd)2 (thd: 2,2,6,6-tetramethyl-3,5-heptanedionate),30,31 Ni(acac)2 (acac: acetylacetonate),32 and Ni(Cp)2 (Cp: cyclopentadienyl).33 Here we use Ni(thd)2 as the nickel precursor, as it is stable enough to allow easy handling but has shown to be reactive enough to yield NiO films even at relatively low temperatures.30,31 Of the possible organic precursors, we chose terephthalic acid (TPA; 1,4-benzenedicarboxylic acid) as it is known to be reactive enough towards the thd-based metal-bearing precursors.19,34 Another motivation for the use of TPA is the fact that it has shown to form metal–organic framework (MOF) structured materials with Ni in bulk form.26 Moreover, the combination, Ni(thd)2 + TPA, allows the straightforward comparison with the similar processes for Mn, Co and Cu.19,34 For molecular structures of the precursors, see Fig. 1.
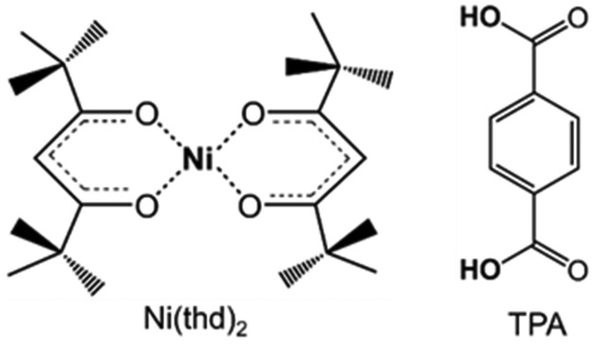 | ||
| Fig. 1 Molecular structures of the precursors employed in this study. | ||
Experimental
The Ni-TPA films were grown from the following precursors: in-house synthesized Ni(thd)2 (thd = 2,2,6,6-tetramethyl-3,5-heptanedione) and commercial TPA (benzene-1,4-dicarboxylic acid; Tokyo Chemical Industry Co. Ltd). These precursor powders were placed in open boats inside the reactor (flow-type hot-wall ALD reactor; F-120 by ASM Microchemistry Ltd) and heated for evaporation at 115 and 180 °C, respectively, based on our previous experience on these precursors in other processes.12,42 Nitrogen (>99.999%, Schmidlin UHPN 3000 N2 generator) was used as a carrier and purging gas, and the pressure inside the reactor was 3–4 mbar. The precursor pulse lengths and the deposition (substrate heating) temperature were optimized in the study. The depositions were performed on Si(100) substrates.The success of the film growth was routinely evaluated using X-ray reflectivity measurements (XRR; X'Pert MPD PRO Alfa 1, PANalytical, Cu Kα). The XRR patterns were fitted by X'Pert Reflectivity software by PANalytical for the film thickness, density and roughness, and the so-called growth-per-cycle (GPC) value was calculated by dividing the film thickness by the number of deposition cycles applied. The density (ρe) values were calculated from the critical angle (θc) as follows: ρe = (θc2π)/(λ2re), where λ is the X-ray wavelength and re is the classical electron radius. Crystallinity of the films was investigated by grazing-incidence X-ray diffraction (GIXRD; incident angle 0.5°) using the same equipment.
To address the bonding structure/local symmetry/chemical state of the films, both Fourier transform infrared (FTIR; Bruker alpha II) and resonant inelastic X-ray scattering (RIXS) experiments were carried out. The FTIR measurements were carried out in a transmission mode in the range of 400–4000 cm−1 with a resolution of 4 cm−1; each given spectrum is an average of 24 measured spectra, from which a spectrum of blank Si is subtracted.
The Ni 1s2p RIXS planes were measured at beamline ID26 of the European Synchrotron Radiation Facility in Grenoble, France. Third harmonic of the undulator source was used and the incident energy was selected and monochromatized by a pair of Si(311) crystals. The emission energy was scanned using a point-to-point Johann spectrometer; five spherically bent analyzer crystals with Si(620) reflections were used to select the emission energy, together with an avalanche photodiode detector.43 The RIXS planes were recorded by stepping through the emission energy range and scanning the incident energy at each step. Slight beam damage was found with exposure times longer than 60 s. To avoid the damage, the scan time was set to 60 s, the sample was moved between each scan and a concentration correction scan was done after recording the RIXS plane.44 In addition to the Ni-TPA sample, bulk polycrystalline NiO was measured as a reference. The Ni 1s2p RIXS planes were simulated using the crystal field multiplet code Quanty,45 together with the Cripsy user interface.46 Calculations were done for different Ni oxidation states with various local point-group symmetries (Oh, Td, D4h, D3h). Atomic Hamiltonian scaling factors, crystal field energies and peak broadening parameters were adjusted to best fit the experimental data.
Results and discussion
The Ni(thd)2 + TPA process was found to yield visually homogeneous Ni-TPA films within the entire deposition temperature range investigated. The films were also seemingly stable during storage in laboratory air, as no changes in XRR or FTIR data were observed even after elongated storage periods. Thus, the ex situ characterization of the films was straight forward and reliable.We investigated the ALD/MLD growth characteristics in detail as summarized in Fig. 2, to verify the layer-by-layer growth of the films. In the first experiments the deposition temperature was fixed to 200 °C and the changes in GPC were followed with increasing precursor pulse lengths to find the surface reaction saturation conditions. From Fig. 2a, it can be seen that the saturation is reached with the pulse/N2 purge times of 5 s/10 s for Ni(thd)2 and 10 s/20 s for TPA.
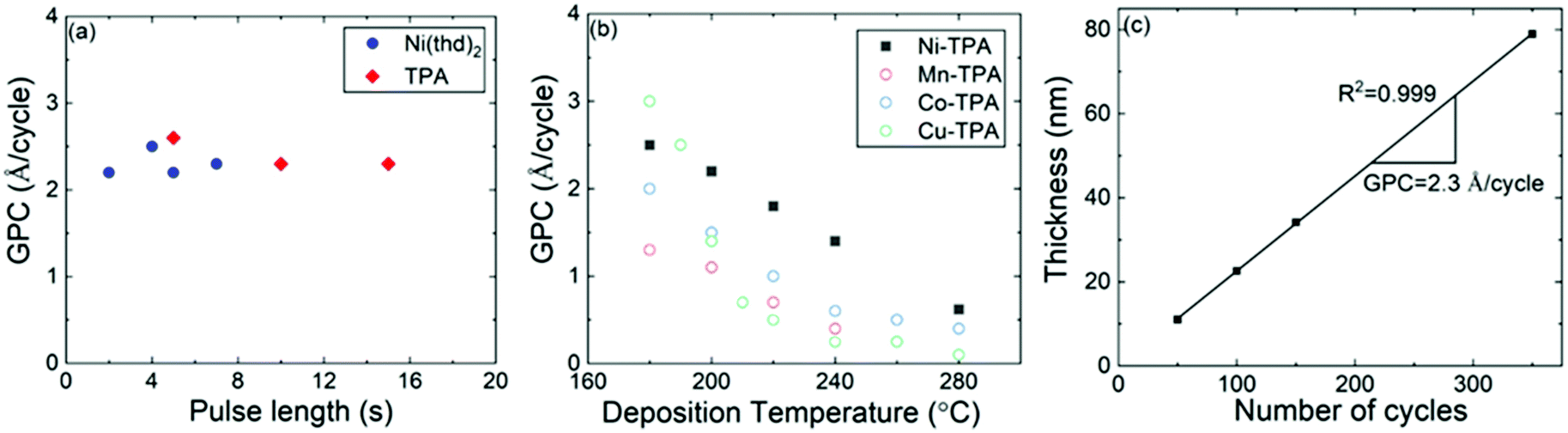 | ||
| Fig. 2 ALD/MLD growth characteristics of the Ni(thd)2 + TPA process: (a) GPC versus precursor pulse lengths, (b) GPC at different deposition temperatures; for comparison, similar data for other 3d transition metal-TPA processes are depicted from our previous works,18,19 and (c) film thickness versus number of ALD/MLD cycles; in (a) & (b) 150 ALD/MLD cycles were applied; in (a) & (c) deposition temperature was 200 °C; in (b) & (c) pulse/purge sequence was: Ni(thd)2 5 s, N2 10 s, TPA 10 s, N2 20 s. | ||
For the next experiment series, we fixed the pulse/purge times to the aforementioned values and followed the growth rate dependence on the deposition temperature in the range from 180 to 320 °C, where the lowest temperature limit was defined by the evaporation temperature of the TPA precursor.47 From Fig. 2b, the GPC value decreases with increasing deposition temperature, in a fashion seen for most of the ALD/MLD processes.48,49 This diminishing growth rate with increasing temperature is commonly attributed to (i) the tendency of the metal precursor to remain in excess in the porous organic layer more readily at low temperatures,49 and/or (ii) the tendency of the sticky low-vapor-pressure organic molecule to get incorporated as a kind of reservoir within the growing film at low temperatures;48,50 in both cases the excess precursors would act as extra reaction sites for the film growth. Despite the strong temperature dependence, the Ni(thd)2 + TPA process was found to deliver the Ni-TPA films in a highly reproducible manner at each deposition temperature. A clear indication of this is the linear control of the film thickness on the number of ALD/MLD cycles applied, see Fig. 2c for the results obtained at 200 °C. The GPC value could be accurately (R2 = 0.999) calculated at 2.3 Å per cycle from the slope of this film thickness versus number of deposition cycles curve. In Fig. 2b, we display together with the present Ni(thd)2 + TPA data our previous data for the Mn(thd)3 + TPA, Co(thd)2 + TPA and Cu(thd)2 + TPA processes, all investigated under very similar experimental conditions (including the reactor type).18,19 From this comparison it can be seen that the temperature dependency of GPC behaviours are very similar for all the four processes. However, in overall the GPC values are clearly lowest for the Mn(thd)3 + TPA process; for example, at 200 °C the GPC for the Mn-based process is 1.1 Å per cycle while it is 1.4, 1.5 and 2.3 Å per cycle, respectively, for the Cu, Co and Ni-based processes. The lower growth rate of the Mn-TPA films can be explained by the more pronounced steric hindrance caused by the bulky thd ligands left after the reaction of the metal precursor on the substrate surface:18,51,52 in the case of Mn(thd)3 two thd ligands are left while in the case of the other three precursors only one thd ligand remains.
The Ni-TPA films were found amorphous from GIXRD investigation independent of the deposition temperature used. Also, the SEM imaging revealed no macroscopic grains for these films (data not shown here). However, an interesting observation was made from the XRR data from which we estimated the film density to be ca. 1.4 g cm−3 with a very weak increasing trend upon increasing the deposition temperature from 180 to 280 °C. This density value is somewhat low compared to those (1.6–1.9 g cm−3) observed for the amorphous Mn-TPA and Co-TPA films earlier.18 Then, differently from the Mn-, Co- and Ni-based processes, the Cu(thd)2 + TPA process yielded amorphous films with densities of 2 g cm−3 or higher only at deposition temperatures higher than 200 °C, but crystalline (MOF-2 structured) Cu-TPA films with a density of ca. 1.4 g cm−3 in the lowest deposition temperature range of 180–190 °C.19 Reflecting against these previous results, we hypothesize that the present Ni-TPA films with the low densities similar to those of the crystalline Cu-TPA films could be crystalline in nanoscale. To challenge the possibility to obtain GIXRD-crystalline Ni-TPA films, we carried out additional test depositions at the “non-ideally” low deposition temperature of 170 °C (lower than the evaporation temperature of the TPA precursor); however, no sign of crystallinity was seen in the GIXRD patterns for these thin films either.
To address the bonding/local structures in the Ni-TPA films, we carried out both FTIR and RIXS experiments. In Fig. 3, FTIR spectra for a series of Ni-TPA films deposited at different temperatures are shown. In these spectra, the decreasing peak intensity with increasing deposition temperature is simply due to the lower film thicknesses of the samples deposited at the higher temperatures (as all samples were deposited using the same number of ALD/MLD cycles, and the growth rate decreased with increasing deposition temperature). Otherwise, the spectra do not show any clear differences, confirming that the local bonding structures in the films are essentially independent of the deposition temperature. Also, they are quite similar to other previously reported metal-TPA spectra.23,34,53 The fact that the spectra are essentially free of the characteristic features of hydroxy groups in the 1700–3500 cm−1 range confirms that the films do not contain any coordinated water, and also that the carboxylic acid groups of the terephthalic acid precursor have completely bonded to the Ni atoms, as expected. The characteristic C–H vibration of the benzene ring is observed at 750 cm−1, but the most indicative part of the spectra is the range from 1300 to 1600 cm−1 where the strong symmetric (νs) and asymmetric (νas) stretching bands of the carboxylate group are seen. For our Ni-TPA films these bands peak at ca. 1383 and 1571 cm−1, respectively. The separation between these peaks, i.e. Δ = νas − νs, provides us information of the bonding mode; here for Ni-TPA the Δ = 188 cm−1 value falls between 130 < Δ < 200, which is a clear indication of the bridging-type bonding,23,54 similarly to the Mn-TPA, Co-TPA and Cu-TPA cases.18,19
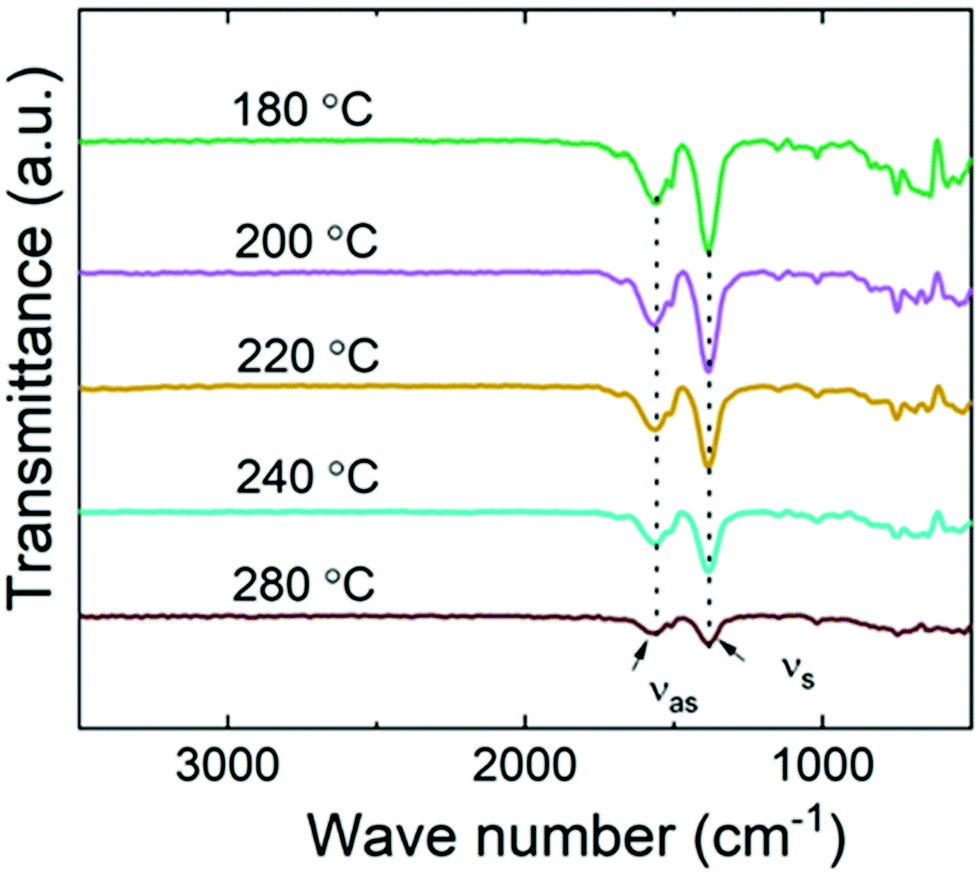 | ||
| Fig. 3 FTIR spectra for Ni-TPA films deposited at different temperatures. | ||
The measured Ni-TPA 1s2p RIXS plane is shown in Fig. 4a. In addition, Fig. 4b shows the corresponding RIXs plane for the NiO reference and Fig. 4c a simulated RIXS plane for Ni2+ in octahedral (Oh) coordination. There is a good qualitative match between the experimental and calculated spectra; calculations with tetrahedral (Td) or other non-octahedral symmetries did not result in such good matches. Fig. 4d shows a constant incident energy (CIE) cut of the Ni-TPA RIXS plane at 8.333 keV, together with the same cuts of for NiO and the calculated RIXS plane. The calculated spectrum was shifted up by 1.1 eV to match the position of the experimental spectra. The best fit between the calculated CIE cut and the experimental results was found with a crystal field splitting of 10Dq = 1.4 eV. However, a closer inspection of the measured RIXS planes revealed a tiny distortion from the ideal octahedral symmetry for both Ni-TPA and NiO (see ESI†). At room temperature, the NiO structure is known to be slightly distorted due to antiferromagnetic ordering, resulting in the Ni–O bond angles to deviate from the ideal 90° by a fraction of a degree (0.07° or less).55,56 The similarity between the Ni-TPA and NiO RIXS planes indicate a similar tiny distortion from octahedral symmetry also in case of the Ni-TPA. These results indicate that the nickel in Ni-TPA is divalent and in an almost ideal octahedral coordination. Thus, based on the RIXS and FTIR analyses, showing octahedral coordination around the Ni atoms with bridging-type bonding of the surrounding terephthalate groups, we imagine that our Ni-TPA films grow as sketched in Fig. 5.
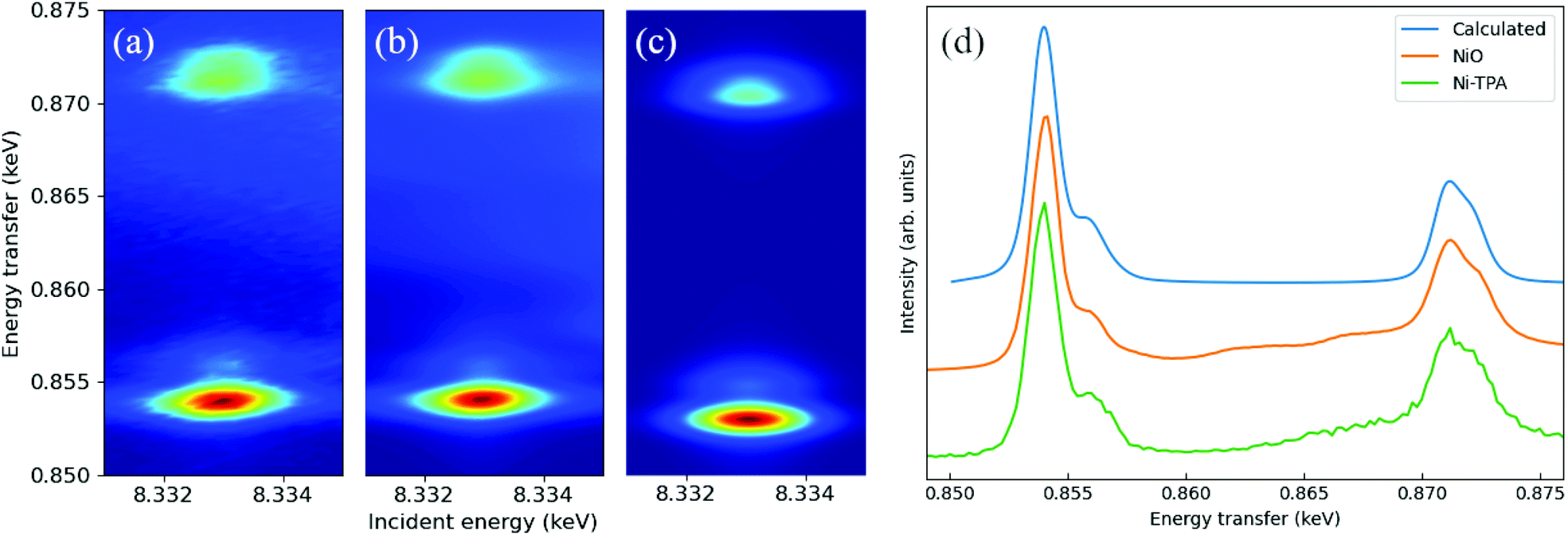 | ||
| Fig. 4 (a) Measured Ni-TPA 1s2p RIXS plane, (b) measured NiO 1s2p RIXS plane, (c) calculated 1s2p RIXS plane for Ni2+ in octahedral (Oh) symmetry, and (d) constant incident energy cuts for Ni-TPA, NiO and the calculated RIXS planes at 8.333 keV incident energy. The calculated spectrum in (d) was shifted up in energy by 1.1 eV. | ||
 | ||
| Fig. 5 Schematic illustration of the (bonding) structure of Ni-TPA films. | ||
Conclusions
In this work, we have filled the gap in ALD/MLD processes for the series of 3d transition-metal based metal–organic thin films. Our Ni-TPA films deposited from Ni(thd)2 and terephthalic acid precursors in the temperature range from 180 to 280 °C were amorphous according to GIXRD but the significantly low-density values determined with XRR suggested some local ordering indicative of a porous structure.The films grew with an appreciably high growth rate, which decreased from the value of 3.0 Å per cycle at 180 °C with increasing deposition temperature, in a fashion typical for the related Mn-TPA, Co-TPA and Cu-TPA films and also for many other ALD/MLD metal–organic thin films.
Since our Ni-TPA films were amorphous in GIXRD, we addressed the bonding/local structures using FTIR and synchrotron-based RIXS techniques, the latter experiments been so far rarely exploited for similar thin film systems. We could conclude that the terephthalate groups bond to divalent nickel ions in a bridging-type fashion to form (slightly distorted) octahedral coordination spheres around them.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the funding from Academy of Finland (Profi 3), and the use of the RawMatTERS Finland Infrastructure (RAMI) at Aalto University. The measured data of RIXS will be available for public after three years period following DOI: 10.15151/ESRF-ES-438677278.Notes and references
- Y. Zhao, L. Zhang, J. Liu, K. Adair, F. Zhao, Y. Sun, T. Wu, X. Bi, K. Amine, J. Lu and X. Sun, Chem. Soc. Rev., 2021, 50, 3889–3956 RSC.
- X. Meng, J. Mater. Chem. A, 2017, 5, 18326–18378 RSC.
- P. Sundberg and M. Karppinen, Beilstein J. Nanotechnol., 2014, 5, 1104–1136 CrossRef PubMed.
- S. M. George, B. Yoon and A. A. Dameron, Acc. Chem. Res., 2009, 42, 498–508 CrossRef CAS PubMed.
- R. W. Johnson, A. Hultqvist and S. F. Bent, Mater. Today, 2014, 17, 236–246 CrossRef CAS.
- M. Leskelä, M. Mattinen and M. Ritala, J. Vac. Sci. Technol., B, 2019, 37, 030801 CrossRef.
- T. Yoshimura, S. Tatsuura and W. Sotoyama, Appl. Phys. Lett., 1991, 59, 482–484 CrossRef CAS.
- K. H. Yoon, K. S. Han and M. M. Sung, Nanoscale Res. Lett., 2012, 7, 71 CrossRef PubMed.
- T. Tynell, A. Giri, J. Gaskins, P. E. Hopkins, P. Mele, K. Miyazaki and M. Karppinen, J. Mater. Chem. A, 2014, 2, 12150–12152 RSC.
- F. Krahl, A. Giri, J. A. Tomko, T. Tynell, P. E. Hopkins and M. Karppinen, Adv. Mater. Interfaces, 2018, 5, 1701692 CrossRef.
- L. Lee, J. Park, K. S. Han, R. S. Kumar and M. M. Sung, Appl. Surf. Sci., 2019, 476, 897–904 CrossRef CAS.
- A. Philip, J. P. Niemelä, G. C. Tewari, B. Putz, T. E. J. Edwards, M. Itoh, I. Utke and M. Karppinen, ACS Appl. Mater. Interfaces, 2020, 12, 21912–21921 CrossRef CAS PubMed.
- H. Zhu, M. H. Aboonasr Shiraz, L. Yao, K. Adair, Z. Wang, H. Tong, X. Song, T. K. Sham, M. Arjmand, X. Song and J. Liu, Chem. Commun., 2020, 56, 13221–13224 RSC.
- J. Penttinen, M. Nisula and M. Karppinen, Chem. – Eur. J., 2017, 23, 18225–18231 CrossRef CAS PubMed.
- C. MacIsaac, J. R. Schneider, R. G. Closser, T. R. Hellstern, D. S. Bergsman, J. Park, Y. Liu, R. Sinclair and S. F. Bent, Adv. Funct. Mater., 2018, 28, 1800852 CrossRef.
- K. B. Lausund, M. S. Olsen, P. A. Hansen, H. Valen and O. Nilsen, J. Mater. Chem. A, 2020, 8, 2539–2548 RSC.
- Z. Giedraityte, M. Tuomisto, M. Lastusaari and M. Karppinen, ACS Appl. Mater. Interfaces, 2018, 10, 8845–8852 CrossRef CAS PubMed.
- E. Ahvenniemi and M. Karppinen, Dalton Trans., 2016, 45, 10730–10735 RSC.
- E. Ahvenniemi and M. Karppinen, Chem. Commun., 2016, 52, 1139–1142 RSC.
- D. S. Bergsman, J. G. Baker, R. G. Closser, C. MacIsaac, M. Lillethorup, A. L. Strickler, L. Azarnouche, L. Godet and S. F. Bent, Adv. Funct. Mater., 2019, 29, 1904129 CrossRef CAS.
- D. J. Hagen, L. Mai, A. Devi, J. Sainio and M. Karppinen, Dalton Trans., 2018, 47, 15791–15800 RSC.
- A. Khayyami, A. Philip and M. Karppinen, Angew. Chem., Int. Ed., 2019, 58, 13400–13404 CrossRef CAS PubMed.
- A. Tanskanen and M. Karppinen, Sci. Rep., 2018, 8, 8976 CrossRef CAS PubMed.
- F. Ran, X. Xu, D. Pan, Y. Liu, Y. Bai and L. Shao, Nano-Micro Lett., 2020, 12, 46 CrossRef CAS PubMed.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dincǎ, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- T. Deng, X. Shi, W. Zhang, Z. Wang and W. Zheng, iScience, 2020, 23, 101220 CrossRef CAS PubMed.
- Y. Qu, C. Shi, H. Cao and Y. Wang, Mater. Lett., 2020, 280, 128526 CrossRef CAS.
- D. Ielmini, F. Nardi and C. Cagli, Nanotechnology, 2011, 22, 254022 CrossRef CAS PubMed.
- J. W. Jung, C. C. Chueh and A. K. Y. Jen, Adv. Mater., 2015, 27, 7874–7880 CrossRef CAS PubMed.
- E. Lindahl, J. Lu, M. Ottosson and J. O. Carlsson, J. Cryst. Growth, 2009, 311, 4082–4088 CrossRef CAS.
- E. Lindahl, M. Ottosson and J. O. Carlsson, Chem. Vap. Deposition, 2009, 15, 186–191 CrossRef CAS.
- M. Utriainen, M. Kröger-Laukkanen and L. Niinistö, Mater. Sci. Eng., B, 1998, B54, 98–103 CrossRef CAS.
- J. Chae, H. S. Park and S. W. Kang, Electrochem. Solid-State Lett., 2002, 5, 64–66 CrossRef.
- E. Ahvenniemi and M. Karppinen, Chem. Mater., 2016, 28, 6260–6265 CrossRef CAS.
- A. Philip, R. Ghiyasi and M. Karppinen, ChemNanoMat, 2021, 7, 253–256 CrossRef CAS.
- A. I. Abdulagatov, R. A. Hall, J. L. Sutherland, B. H. Lee, A. S. Cavanagh and S. M. George, Chem. Mater., 2012, 24, 2854–2863 CrossRef CAS.
- P. Sundberg and M. Karppinen, Eur. J. Inorg. Chem., 2014, 2014, 968–974 CrossRef CAS.
- A. Sood, P. Sundberg, J. Malm and M. Karppinen, Appl. Surf. Sci., 2011, 257, 6435–6439 CrossRef CAS.
- A. I. Abdulagatov, K. N. Ashurbekova, K. N. Ashurbekova, R. R. Amashaev, M. K. Rabadanov and I. M. Abdulagatov, Russ. J. Appl. Chem., 2018, 91, 347–359 CrossRef CAS.
- C. Y. Kao, J. W. Yoo, Y. Min and A. J. Epstein, ACS Appl. Mater. Interfaces, 2012, 4, 137–141 CrossRef CAS PubMed.
- A. Tanskanen and M. Karppinen, Dalton Trans., 2015, 44, 19194–19199 RSC.
- D. J. Hagen, T. S. Tripathi, I. Terasaki and M. Karppinen, Semicond. Sci. Technol., 2018, 33, 115015 CrossRef.
- P. Glatzel, A. Harris, P. Marion, M. Sikora, T. C. Weng, C. Guilloud, S. Lafuerza, M. Rovezzi, B. Detlefs and L. Ducotté, J. Synchrotron Radiat., 2021, 28, 362–371 CrossRef CAS PubMed.
- S. Lafuerza, M. Retegan, B. Detlefs, R. Chatterjee, V. Yachandra, J. Yano and P. Glatzel, Nanoscale, 2020, 12, 16270–16284 RSC.
- M. W. Haverkort, M. Zwierzycki and O. K. Andersen, Phys. Rev. B., 2012, 85, 165113 CrossRef.
- M. Retegan, Zenodo DOI:10.5281/zenodo.1008184.
- J. Multia, A. Khayyami, J. Heiska and M. Karppinen, J. Vac. Sci. Technol., A, 2020, 38, 052406 CrossRef CAS.
- J. Multia, J. Heiska, A. Khayyami and M. Karppinen, ACS Appl. Mater. Interfaces, 2020, 12, 41557–41566 CrossRef CAS PubMed.
- B. H. Lee, B. Yoon, A. I. Abdulagatov, R. A. Hall and S. M. George, Adv. Funct. Mater., 2013, 23, 532–546 CrossRef CAS.
- Y. Du and S. M. George, J. Phys. Chem. C, 2007, 111, 8509–8517 CrossRef CAS.
- R. L. Puurunen, J. Appl. Phys., 2005, 97, 121301 CrossRef.
- M. Ylilammi, Thin Solid Films, 1996, 279, 124–130 CrossRef CAS.
- L. Ai, C. Zhang, L. Li and J. Jiang, Appl. Catal., B, 2014, 148–149, 191–200 CrossRef CAS.
- K. B. Klepper, O. Nilsen, S. Francis and H. Fjellvåg, Dalton Trans., 2014, 43, 3492–3500 RSC.
- N. C. Tombs and H. P. Rooksby, Nature, 1950, 165, 442–443 CrossRef CAS.
- D. Rodic, V. Spasojevic, V. Kusigerski, R. Tellgren and H. Rundlof, Phys. Status Solidi B, 2000, 218, 527–536 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt02966e |
| This journal is © The Royal Society of Chemistry 2021 |