
A CoV2O4 precatalyst for the oxygen evolution reaction: highlighting the importance of postmortem electrocatalyst characterization†
Samuel E.
Michaud
 a,
Michael T.
Riehs
a,
Wei-Jie
Feng
b,
Chia-Cheng
Lin
*ac and
Charles C. L.
McCrory
*ab
a,
Michael T.
Riehs
a,
Wei-Jie
Feng
b,
Chia-Cheng
Lin
*ac and
Charles C. L.
McCrory
*ab
aDepartment of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: cmccrory@umich.edu
bMacromolecular Science and Engineering Program, University of Michigan, Ann Arbor, Michigan 48109, USA
cInstitute of Mineral Resources Engineering, Department of Materials and Mineral Resources Engineering, National Taipei University of Technology, Taipei 10608, Taiwan. E-mail: johncclin@mail.ntut.edu.tw
First published on 7th December 2020
Abstract
Vanadium-doped cobalt oxide materials have emerged as a promising class of catalysts for the oxygen evolution reaction. Previous studies suggest vanadium doping in crystalline Co spinel materials tunes the electronic structure and stabilizes surface intermediates. We report a CoV2O4 material that shows good activity for the oxygen evolution reaction. However, postmortem characterization of the catalyst material shows dissolution of vanadium resulting in an amorphous CoOx material, suggesting that this vanadium-free material, and not CoV2O4, is the active catalyst. This study highlights the importance of postmortem characterization prior to mechanistic and computational analysis for this class of materials.
The sluggish kinetics of the oxygen evolution reaction (OER) hinders the development of practical water-splitting technologies, and has driven the search for efficient OER electrocatalysts comprised of earth-abundant materials that operate with high current densities at low overpotential with long-term operational stability.1–4 V-Doped Co oxide materials have recently emerged as a promising class of alkaline OER catalysts. Previous studies of V-doped Co oxide materials show increased activity for the OER at low overpotentials,5–14 and typically suggest that this increased activity is a result of changes to the catalyst's electronic structure and/or in increased stabilization of adsorbed OER intermediates.5–8
In this study, we report a CoV2O4 material that shows exceptional specific activity per BET surface area for the OER based on as-synthesized characterization data. However, materials characterization conducted after OER electrolysis shows the CoV2O4 precatalyst transforms into a V-free amorphous Co-based material during the OER. This suggests that V ions are not present in the active catalyst material, and rather, CoV2O4 serves as a template for the generation of an active amorphous CoOx species. Our study highlights the need for careful postmortem characterization of electrocatalytic materials to ensure continued catalyst integrity prior to conducting detailed mechanistic interpretations.
CoV2O4 was prepared by solid state synthesis from CoO and V2O3 metal precursors. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of CoO and V2O3 was ground and mixed, compressed into a pellet, and heated at 800 °C for 20 h under Ar. Selected characterization parameters of the as-synthesized materials are summarized in Table S2 (ESI†). The powder X-ray diffraction (PXRD) pattern of the as-synthesized CoV2O4 matches that of the CoV2O4 reference with minor contributions from V2O3 impurities as shown in Fig. 1a. Note that V2O3 is not OER active (see Fig. S1, ESI†), so we do not expect trace V2O3 impurities to affect our OER measurements. The sharp diffraction peaks suggest a highly crystalline specimen with a large domain size which might be the product of particle agglomeration due to the high temperatures and long times in the solid-state synthesis preparation. The large grain size is supported both by transmission electron microscopy (TEM) analysis which show particles > 100 nm in size (Fig. 1b) and the catalyst's small surface area of only 0.80 m2 g−1 as determined by BET gas adsorption analysis – two orders of magnitude smaller than that of Co3O4 (Table S2, ESI†). Elemental mapping experiments with TEM-energy dispersive X-ray analysis (TEM-EDX) show relatively uniform distribution of Co, V, and O in the as-synthesized material (Fig. 1b).
:1 molar ratio of CoO and V2O3 was ground and mixed, compressed into a pellet, and heated at 800 °C for 20 h under Ar. Selected characterization parameters of the as-synthesized materials are summarized in Table S2 (ESI†). The powder X-ray diffraction (PXRD) pattern of the as-synthesized CoV2O4 matches that of the CoV2O4 reference with minor contributions from V2O3 impurities as shown in Fig. 1a. Note that V2O3 is not OER active (see Fig. S1, ESI†), so we do not expect trace V2O3 impurities to affect our OER measurements. The sharp diffraction peaks suggest a highly crystalline specimen with a large domain size which might be the product of particle agglomeration due to the high temperatures and long times in the solid-state synthesis preparation. The large grain size is supported both by transmission electron microscopy (TEM) analysis which show particles > 100 nm in size (Fig. 1b) and the catalyst's small surface area of only 0.80 m2 g−1 as determined by BET gas adsorption analysis – two orders of magnitude smaller than that of Co3O4 (Table S2, ESI†). Elemental mapping experiments with TEM-energy dispersive X-ray analysis (TEM-EDX) show relatively uniform distribution of Co, V, and O in the as-synthesized material (Fig. 1b).
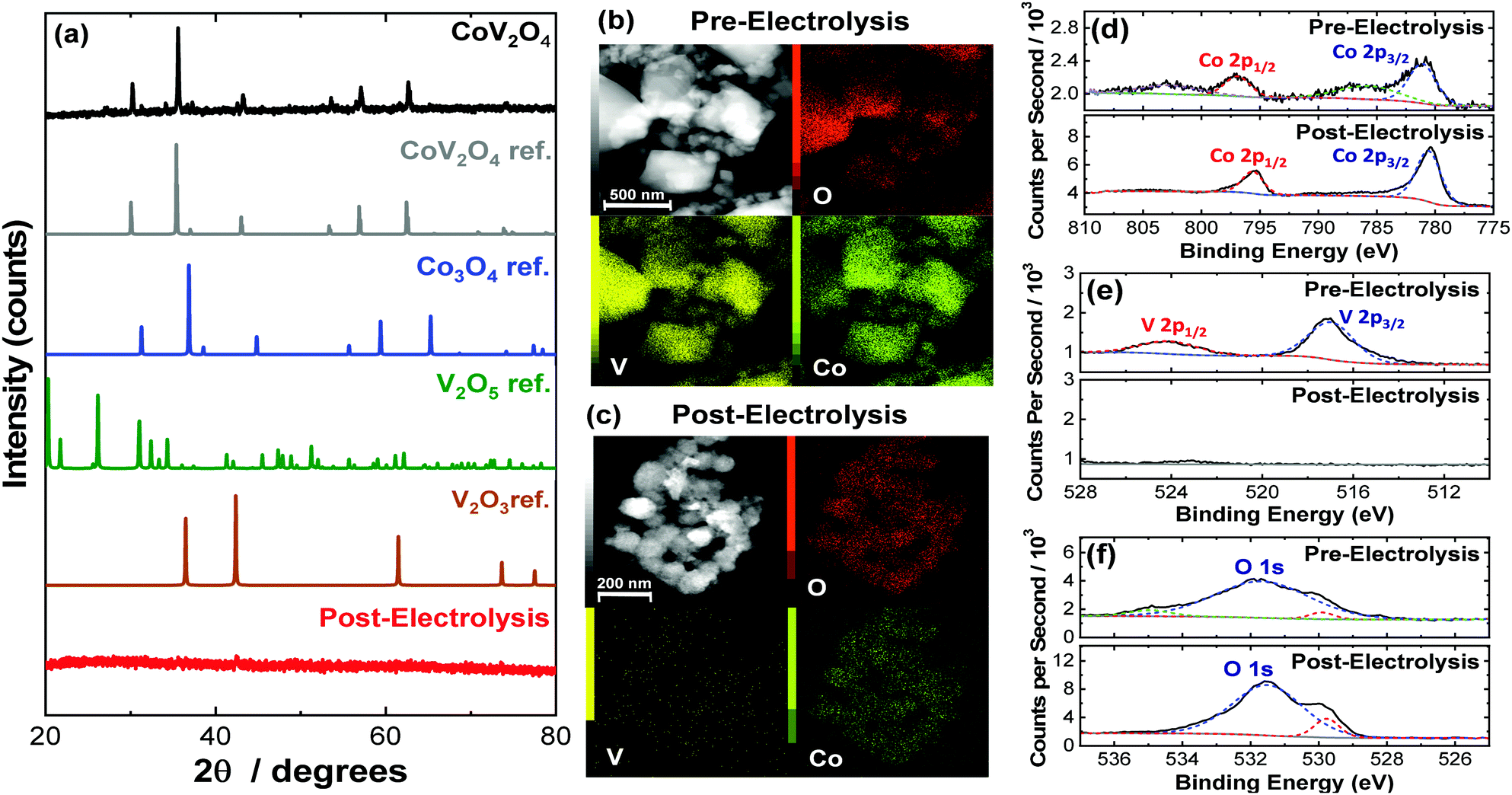 | ||
| Fig. 1 (a) PXRD patterns for as-synthesized CoV2O4 along with reference patterns for comparison. The PXRD for CoV2O4 after 28-h CCE under OER conditions is also included. (b) Representative TEM images and TEM-EDX maps of as-synthesized CoV2O4. (c) Representative TEM images and TEM-EDX maps of CoV2O4 after 28 h CCE under OER conditions. (d–f) High resolution XPS spectra of CoV2O4 both as-synthesized and after 28 h CCE in the (d) Co 2p region, (e) V 2p region, and (f) O 1s region. | ||
The as-synthesized CoV2O4 material was further characterized with X-ray photoelectron spectroscopy (XPS). XPS core scans of the as-synthesized CoV2O4 material in the Co 2p, V 2p, and O 1s regions are shown in Fig. 1d–f, and in the C 1s region in Fig. S2 (ESI†). In the Co 2p spectra of the as-synthesized material (Fig. 1d), two peaks centered at 780.8 and 796.6 eV are assigned to Co 2p3/2 and Co 2p1/2 respectively, and the two shoulder peaks at 785.3 and 802.5 eV suggest the Co is in a CoO-like (Co2+) environment.15 In the V 2p spectra (Fig. 1e), there is a peak at 516.5 eV assigned to V 2p3/2, which is resembles that of reported V2O3 samples and suggests V is in a 3+ oxidation state.16,17 There is a complicated O 1s peak at ∼531.5 eV (Fig. 1f) consistent with a mixed species of O on the surface (metal oxide/hydroxide/adsorbed water), but due to the convoluted nature, the peak cannot be used in V or Co oxidation state estimations.18 The Co/V ratio based on XPS analysis was 0.58. This XPS characterization is consistent with other synthesized CoV2O4 materials.5,19
OER activity measurements were performed using previously reported protocols.20,21 Catalyst inks were prepared by mixing the as-synthesized catalyst particles and Nafion in a water–isopropanol solution, and the resulting inks were dropcast onto polished glassy carbon electrode surfaces (0.196 cm2) resulting in films with mass loadings of 0.84 mg cm−2. The putative CoV2O4 catalyst shows promising activity for the OER, operating with increased activity per geometric area compared to the parent Co3O4 as shown in Fig. 2a, and dramatically increased specific activity per BET surface area compared to Co3O4 as shown in Fig. 2b. Activity descriptors for the putative CoV2O4 catalyst compared to other representative OER catalysts are shown in Table S3 (ESI†). The long-term performance stability of the putative CoV2O4 system was confirmed by rapid potential cycling measurements and long-term controlled-current electrolysis experiments (CCE) which showed minimal change in activity over 10000 cycles and 28 h controlled current measurements (Fig. 2c). The faradaic efficiency for the OER was measured as 90 ± 5% using an in situ O2 probe (Fig. S4, ESI†).
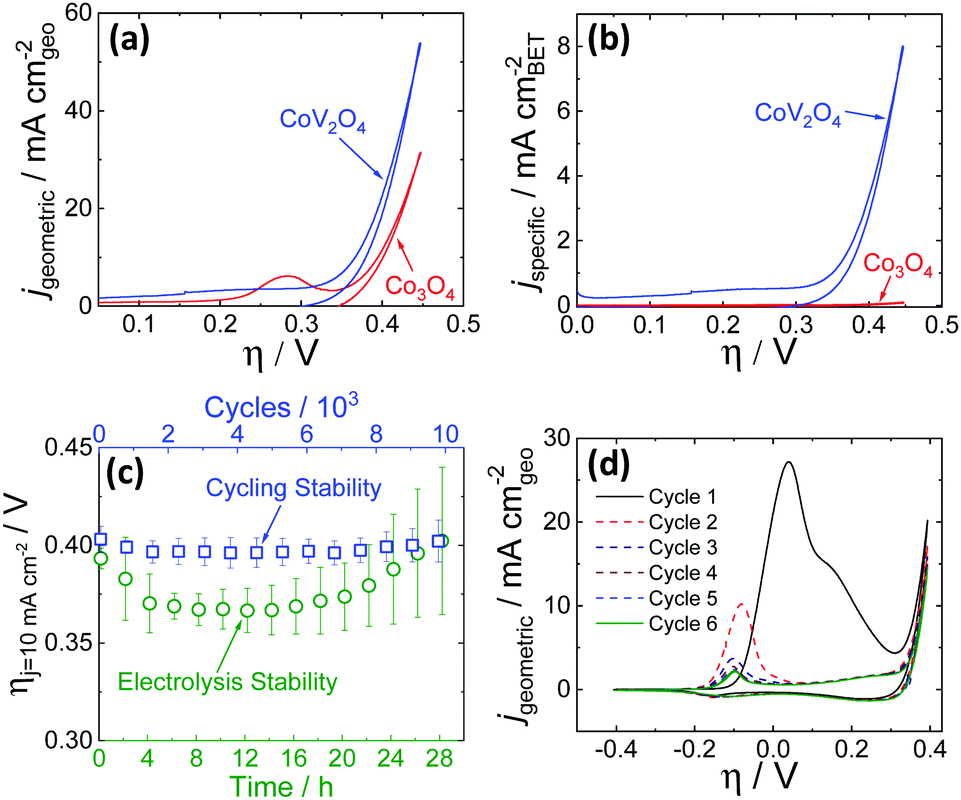 | ||
| Fig. 2 Cyclic RDEVs of the as-synthesized CoV2O4 catalyst compared to previously reported Co3O4 in O2-saturated 1 M NaOH showing (a) the current density per geometric area and (b) the specific current density per the BET surface area of the as-synthesized materials at 0.01 V s−1 scan rate and 1600 rpm rotation rate. The RDEVs shown are the 2nd cycle for each sample. The Co3O4 data is taken from ref. 20. (c) Stability studies for the putative CoV2O4 during OER. The green circles are measured overpotentials at 10 mA cm−2 geometric (ηj = 10 mA cm−2) during 28 h CCE measurements plotted vs. the polarization time on the bottom axis. The blue squares are the ηj = 10 mA cm−2 values during 10000 cycle experiments plotted vs. the number of cycles on the top x-axis, and the bottom axis also reflects the time points in the cycling measurements. The ηj = 10 mA cm−2 values were average measurements from at least three independently-prepared samples, and the error bars represent the standard deviations. (d) The first six RDEVs measured for the as-synthesized CoV2O4. There is a large pre-catalytic peak at η ≈ 0.05 V that shifts negative and decreases in intensity in subsequent scans. | ||
The activity measurements for the putative CoV2O4 catalyst suggest it shows remarkable stability and activity, particularly specific activity per BET surface area, for the OER. Sequential cycles of RDEVs at 1600 rpm of the putative CoV2O4 catalyst in 1 M NaOH are shown in Fig. 2d. In the first cycle, there is a large oxidative feature negative of the OER electrocatalytic onset, and this oxidative feature decreases with subsequent scans before approaching a steady-state peak current after six cycles. RDEVs conducted under identical conditions for V2O3 show a similar oxidative feature in the first cycle that disappears in subsequent cycles (Fig. S5, ESI†), which is consistent with previous studies that show V2O3 dissolves under oxidation when exposed to alkaline conditions.22 Our RDEV studies suggest that the large oxidative feature observed for CoV2O4 is likely due to oxidative dissolution of V, and the steady-state redox feature after six cycles is assigned to the Co2+/3+ redox couple of the resulting CoOx material.23 The stability of the catalytic performance of the putative CoV2O4 suggests that dissolution of V has a minimal effect on the OER activity.
To confirm the postulated V dissolution during our experiments, we conducted a series of postmortem characterization studies on the putative CoV2O4 catalyst. TEM-EDX conducted on a sample after the CCE measurements show a dramatic loss of V consistent with oxidative V dissolution during electrocatalytic studies. Similarly, ICP-MS measurements taken after the CCE stability measurements show almost complete disappearance of V (Table S2, ESI†). XPS measurements taken after the CCE measurements show a dramatic decrease of the V 2p peaks consistent with a loss of V from the material (Fig. 1c), and in the Co 2p region the disappearance of the shoulder peaks and the decreased FWHM of the two Co 2p peaks suggest a shift towards a Co3O4 like surrounding (Fig. 1d).24 The reduced shoulder peak of O 1s (531.5 eV), which is attributed to oxide defect sites or hydroxyl groups,25 in the post-CCE XPS suggests the loss of defect sites and surface oxyhydroxyl (M-OOH) groups while the loss of the peak at 535 eV represents the loss of H2O species. Note that the XPS measured on the post-cycling samples were analogous to the post-CCE samples (Fig. S6, ESI†).
PXRD measurements of the post-CCE catalyst material are consistent with an amorphous structure (Fig. 1), and high-resolution TEM measurements of the post-CCE material also show a broadly amorphous structure with a random dispersion of small nanocrystalline domains with d = 0.24 nm lattice spacing consistent with the (311) plane of V-free Co3O4 (Fig. S7, inset, ESI†). Selected area diffraction (SAED) measurements also show a decrease in catalyst crystallinity post-electrolysis compared to the as-synthesized material (Fig. S8, ESI†).
All electrochemical and post-CCE characterization data suggest that the active OER catalyst is not CoV2O4, but instead an amorphous, V-free CoOx material. We postulate that the CoV2O4 material undergoes a structural rearrangement under electrocatalytic conditions forming a soluble VOx phase, possibly V2O5, consistent with the Pourbaix diagram for V.26,27 Upon dissolution of VOx, the CoV2O4 lattice collapses resulting in an amorphous CoOx material that is presumed to be the active species for the OER. This process is qualitatively similar to electrochemical dealloying previously used to make porous battery materials and catalysts.28–33 We postulate that the increased observed activity for our catalyst compared to Co3O4 may be due in part to an increased surface area after V-dissolution. Unfortunately, we were not able to harvest sufficient amounts of material post-CCE for BET gas adsorption measurements. Moreover, we do not use electrochemical double-layer capacitance measurements to estimate surface area for OER materials – we believe such measurements are of questionable utility due to the well-documented fallibility of that approach applied to metal oxide catalysts.21,34–37 We did estimate average particle sizes based on the TEM images in Fig. 1, and we observe a decrease in the average (and median) particle size post-CCE that is qualitatively consistent with our postulated increase in surface area assuming no loss of material (Table S1, ESI†). However, the measured decrease in average particle size does not account for any changes in porosity or mass-loss from V dissolution that would also influence the overall surface area, and so it is reported here only as a qualitative metric.
Previous studies have reported that V-doped Co oxide materials show higher activity for the OER than their parent oxide materials.5–13 In many cases, this observed higher OER activity compared to the V-free parent materials is attributed to specific changes in the physical and/or electronic structure upon V incorporation resulting in changes to material conductivity, adsorption energy of OER intermediates, and/or the rate determining step in the catalytic mechanism. In the case of V-doped amorphous CoOx films,7,9 crystalline V-doped CoOOH8 and CoFe-based12 nanoparticles, and Co-V hydroxide nanostructures,13 postmortem characterization shows these materials retain significant concentrations of V after OER stability measurements, consistent with the assertion that the continued presence of V is an important component of the OER mechanism for these materials.7,8,12,13
However, in previous studies of crystalline V-doped Co3O4-based spinel materials, a lack of postmortem characterization introduces ambiguity as to the role of V in the catalytic mechanism. For example, in previous studies of OER by CoV2−xFexO4 and Co3−xVxO4 nanoparticles, comprehensive mechanistic and computational analysis was conducted with the assumption that V was an important component in the active catalyst species.5,6 However, in these studies, either postmortem analysis was not reported to confirm the presence of V in the material after the OER,6 or reported postmortem characterization showing the loss of V during the OER was not considered in the mechanistic analysis.5 Similarly, studies of V-doped CoP materials and Co–Mo–V catalysts both attribute increased catalytic activity to the presence of V in the materials, but did not include postmortem characterization showing the continued presence of V after the OER.10,14 This is not to say that the mechanistic arguments in these previous studies are incorrect, but rather suggests that the composition of the post-OER catalyst should be reported and discussed when considering possible catalytic mechanisms.
V-Doped Co oxide materials are an emerging class of promising electrocatalysts for the OER. In our work, we show that a putative CoV2O4 spinel catalyst shows remarkable specific activity based on as-synthesized catalyst characterization when compared to Co3O4 and other reported OER catalysts. However, postmortem characterization shows dissolution of V from the material resulting in the formation of amorphous CoOx particles, the presumed true active catalysts for the OER. Our findings introduce uncertainty into the mechanistic arguments made in previous studies of V-doped Co3O4-based spinel materials for the OER that suggest V plays an important role in catalytic mechanism without considering postmortem materials characterization to confirm the continued presence of V during the OER. Conducting compositional and structural characterization of OER materials after electrocatalytic stability studies is recommended as a minimum requirement in assigning plausible catalytic active species according to recent reviews of best practices for OER measurements.36–39 We believe our work serves as a case study highlighting the importance of postmortem characterization in determining possible catalytic species prior to in-depth mechanistic analysis.
This work was supported with funding from the University of Michigan and by a Cottrell Scholar award, a program of the Research Corporation for Science Advancement. M. T. R. acknowledges partial support by a May-Walt Summer Research Scholarship by the University of Michigan. C.-C. L. acknowledges partial support by the Ministry of Science and Technology in Taiwan (MOST 109-2113-M-027-001-MY3). XPS and TEM measurements were conducted at the Michigan Center for Materials Characterization with support from the University of Michigan College of Engineering and the National Science Foundation (DMR-0420785, DMR-0723032)
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- W.-J. Jiang, T. Tang, Y. Zhang and J.-S. Hu, Acc. Chem. Res., 2020, 53, 1111–1123 CrossRef CAS.
- R. Wei, X. Bu, W. Gao, R. A. B. Villaos, G. Macam, Z.-Q. Huang, C. Lan, F.-C. Chuang, Y. Qu and J. C. Ho, ACS Appl. Mater. Interfaces, 2019, 11, 33012–33021 CrossRef CAS.
- K. Chakrapani, G. Bendt, H. Hajiyani, T. Lunkenbein, M. T. Greiner, L. Masliuk, S. Salamon, J. Landers, R. Schlögl, H. Wende, R. Pentcheva, S. Schulz and M. Behrens, ACS Catal., 2018, 8, 1259–1267 CrossRef CAS.
- L. Liardet and X. Hu, ACS Catal., 2018, 8, 644–650 CrossRef CAS.
- Y. Cui, Y. Xue, R. Zhang, J. Zhang, X. A. Li and X. Zhu, J. Mater. Chem. A, 2019, 7, 21911–21917 RSC.
- G. Merle, I. Abrahams and J. Barralet, Mater. Today Energy, 2018, 9, 247–253 CrossRef.
- J.-F. Qin, J.-H. Lin, T.-S. Chen, D.-P. Liu, J.-Y. Xie, B.-Y. Guo, L. Wang, Y.-M. Chai and B. Dong, J. Energy Chem., 2019, 39, 182–187 CrossRef.
- J. M. Gonçalves, M. Ireno da Silva, L. Angnes and K. Araki, J. Mater. Chem. A, 2020, 8, 2171–2206 RSC.
- T. Gao, Z. Jin, M. Liao, J. Xiao, H. Yuan and D. Xiao, J. Mater. Chem. A, 2015, 3, 17763–17770 RSC.
- M. Yang, X. Fu, M. Shao, Z. Wang, L. Cao, S. Gu, M. Li, H. Cheng, Y. Li, H. Pan and Z. Lu, ChemElectroChem, 2019, 6, 2050–2055 CrossRef CAS.
- J. Bao, Z. Wang, J. Xie, L. Xu, F. Lei, M. Guan, Y. Zhao, Y. Huang and H. Li, Chem. Commun., 2019, 55, 3521–3524 RSC.
- P. W. Menezes, A. Indra, D. González-Flores, N. R. Sahraie, I. Zaharieva, M. Schwarze, P. Strasser, H. Dau and M. Driess, ACS Catal., 2015, 5, 2017–2027 CrossRef CAS.
- N. K. Nag and F. E. Massoth, J. Catal., 1990, 124, 127–132 CrossRef CAS.
- B. S. Allimi, S. P. Alpay, D. Goberman, T. Huang, J. I. Budnick, D. M. Pease and A. I. Frenkel, J. Mater. Res., 2007, 22, 2825–2831 CrossRef CAS.
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. De Gryse, J. Electron. Spectrosc. Relat. Phenom., 2004, 135, 167–175 CrossRef CAS.
- J. S. Lu, I. V. B. Maggay and W. R. Liu, Chem. Commun., 2018, 54, 3094–3097 RSC.
- C.-C. Lin and C. C. L. McCrory, ACS Catal., 2017, 7, 443–451 CrossRef CAS.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 RSC.
- F. M. Al-Kharafi and W. A. Badawy, Electrochim. Acta, 1997, 42, 579–586 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS.
- S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell, J. Mol. Catal. A: Chem., 2008, 281, 49–58 CrossRef CAS.
- J. Wei, Y. Y. Feng, Y. Liu and Y. Ding, J. Mater. Chem. A, 2015, 3, 22300–22310 RSC.
- K. Post and R. G. Robins, Electrochim. Acta, 1976, 21, 401–405 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solution, Pergamon Press, Oxford, 1966, pp. 234–245 Search PubMed.
- A. J. Forty, Nature, 1979, 282, 597–598 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450–453 CrossRef CAS.
- S. Koh and P. Strasser, J. Am. Chem. Soc., 2007, 129, 12624–12625 CrossRef CAS.
- A. Pavlišič, P. Jovanovič, V. S. Šelih, M. Šala, M. Bele, G. Dražić, I. Arčon, S. Hočevar, A. Kokalj, N. Hodnik and M. Gaberšček, ACS Catal., 2016, 6, 5530–5534 CrossRef.
- T. Fujita, H. Abe, T. Tanabe, Y. Ito, T. Tokunaga, S. Arai, Y. Yamamoto, A. Hirata and M. Chen, Adv. Funct. Mater., 2016, 26, 1609–1616 CrossRef CAS.
- X. Lu, M. Ahmadi, F. J. DiSalvo and H. D. Abruña, ACS Catal., 2020, 10, 5891–5898 CrossRef CAS.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783 RSC.
- K. Klingan, F. Ringleb, I. Zaharieva, J. Heidkamp, P. Chernev, D. Gonzalez-Flores, M. Risch, A. Fischer and H. Dau, ChemSusChem, 2014, 7, 1301–1310 CrossRef CAS.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. M. Trang and S. W. Boettcher, Chem. Mater., 2017, 29, 120–140 CrossRef CAS.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef.
- R. Frydendal, E. A. Paoli, B. P. Knudsen, B. Wickman, P. Malacrida, I. E. L. Stephens and I. Chorkendorff, ChemElectroChem, 2014, 1, 2075–2081 CrossRef CAS.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, Nat. Catal., 2018, 1, 508–515 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and details, additional characterization data for the as-synthesized and post-OER materials, cyclic voltammograms of V2O3 in alkaline conditions, measured O2 production using an in situ O2 probe, and comparative table of the OER activities of other nanoparticulate catalysts based on as-synthesized activity metrics. See DOI: 10.1039/d0cc06513g |
| This journal is © The Royal Society of Chemistry 2021 |