
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein–protein interaction based substrate control in the E. coli octanoic acid transferase, LipB†
Thomas G.
Bartholow
a,
Terra
Sztain
a,
Megan A.
Young
a,
Tony D.
Davis
 a,
Ruben
Abagyan
b and
Michael D.
Burkart
*a
a,
Ruben
Abagyan
b and
Michael D.
Burkart
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, CA 92093-0358, USA. E-mail: mburkart@ucsd.edu
bSchool of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA 92093, USA
First published on 28th July 2021
Abstract
Lipoic acid is an essential cofactor produced in all organisms by diverting octanoic acid derived as an intermediate of type II fatty acid biosynthesis. In bacteria, octanoic acid is transferred from the acyl carrier protein (ACP) to the lipoylated target protein by the octanoyltransferase LipB. LipB has a well-documented substrate selectivity, indicating a mechanism of octanoic acid recognition. The present study reveals the precise protein–protein interactions (PPIs) responsible for this selectivity in Escherichia coli through a combination of solution-state protein NMR titration with high-resolution docking of the experimentally examined substrates. We examine the structural changes of substrate-bound ACP and determine the precise geometry of the LipB interface. Thermodynamic effects from varying substrates were observed by NMR, and steric occlusion of docked models indicates how LipB interprets proper substrate identity via allosteric binding. This study provides a model for elucidating how substrate identity is transferred through the ACP structure to regulate activity in octanoyl transferases.
Introduction
De novo lipoic acid biosynthesis occurs in all organisms as a branch point from type II fatty acid biosynthesis (FAB), and lipoic acid is the only known essential product of human mitochondrial FAB.1–5 Octanoic acid is transferred from within the FAB onto a lipoylated target protein, whereupon thiol moieties are subsequently added through the activity of iron–sulfur cluster enzymes.1,6–8 Control of octanoate transfer from the FAB must be maintained, as the role and structure of lipoic acid is reliant on the proper chain length and oxidative state of the fatty acid from which it is derived. In E. coli, the fidelity to select a single fatty acid from within the 30–35 potential acyl substrates attached to ACP poses a selectivity mechanism9–11 that remains elusive.12,13FAB is an iterative, multi-enzyme pathway in which each reaction step is catalyzed upon a fatty acid precursor that is covalently attached to the acyl carrier protein (AcpP in E. coli).14 AcpP is a small, 77 amino acid protein with a four α-helical bundle structure.15,16 The acyl substrates are carried on a 4′-phosphopantetheine cofactor attached to serine 36 of the AcpP, which carries each fatty acyl intermediate attached as a thioester. In solution, AcpP sequesters acyl cargo within a hydrophobic pocket between its α-helices, only presenting the hydrolyzable thioester once it favorably interacts with a partner protein through protein–protein interactions (PPIs).17 In the case of LipB, the sourcing of octanoyl-ACP must occur after enoyl reduction but before the substrate can re-enter the elongation cycle for another iteration (Fig. 1A). Here we have pursued an understanding for how the LipB accomplishes this highly selective interaction, where LipB rapidly intercepts C8-AcpP with high fidelity.
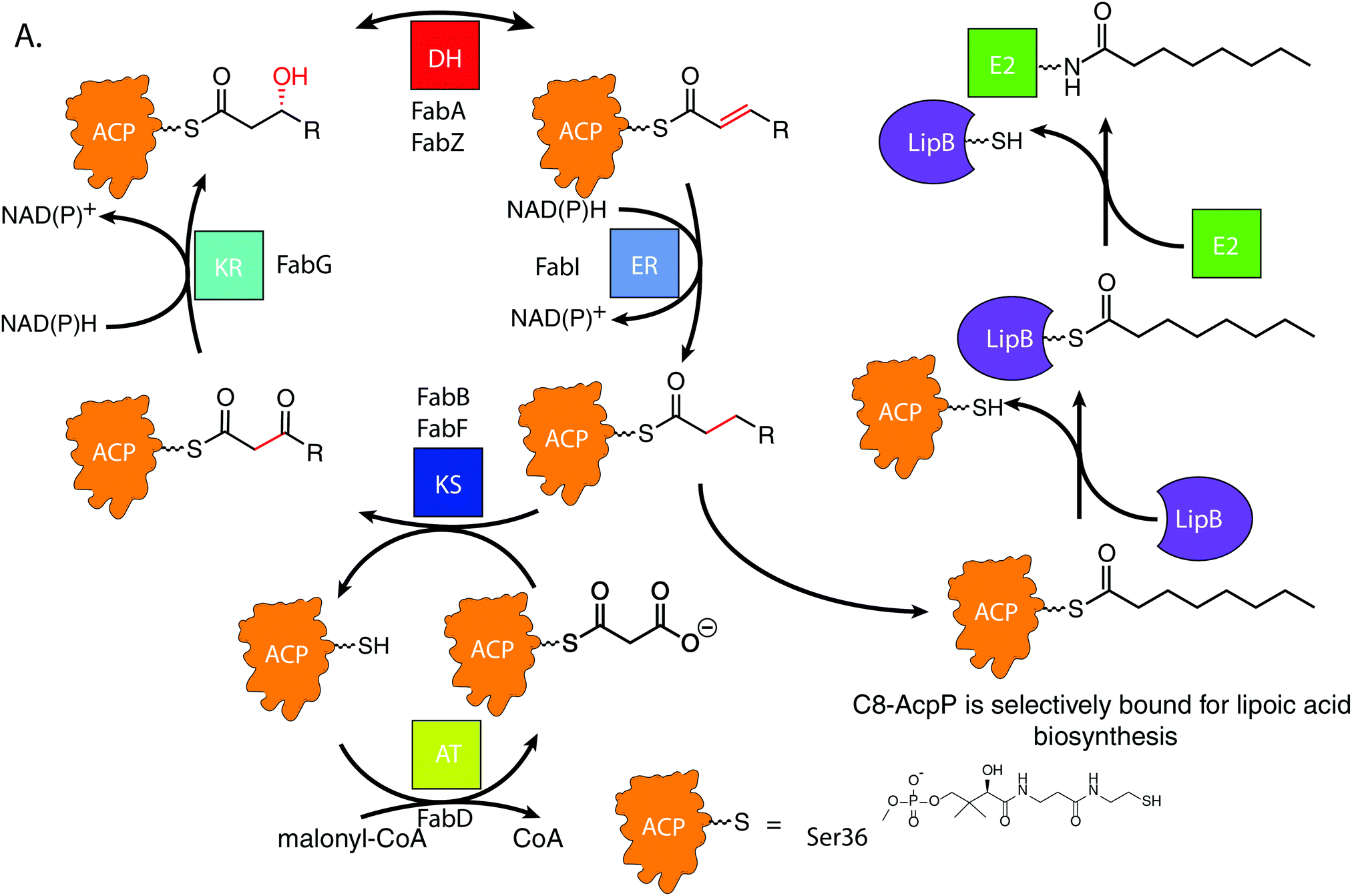 | ||
| Fig. 1 Fatty acid biosynthetic cycle of E. coli showing LipB transferase activity. KS: ketosynthase, KR: ketoreductase, DH: dehydratase, ER: enoylreductase, TE: thioesterase, AT: malonyl acyltransferase. The central steps occur, with ACPs carrying all substrates between partners. LipB must selectively transfer a saturated eight carbon ACP. The acyl chain is transferred onto LipB to be carried to E2 domains and subsequent lipoic acid biosynthetic enzymes. | ||
LipB transfers octanoyl groups scavenged from AcpP to an active site cysteine 169′ (residues of LipB will hereafter be labeled as prime), with the LipB's hydrophobic pocket sheltering the lipid tail before transferring it to E2 or other lipoyl domains. This creates an octanoyl-modified enzyme, freeing the LipB to scavenge more octanoic acid substrates.18 LipB is required to source and attach octanoic acid from AcpP, as neither free octanoic acid nor octanoyl-CoA are substrates, requiring LipB to carefully select substrates attached to the AcpP or risk inactivating downstream enzymes.19
Recent evidence has suggested that the AcpP·LipB interaction can exert allosteric control over the substrates it interacts with prior to catalysis through control of the “chain flipping” event.20 Chain flipping is the term applied to the exit of the acyl chain from the carrier protein pocket.21 This creates a “control” step which must occur prior to any catalysis. Prior studies have reported decanoic acid crystallized within the LipB active site, suggesting that it is possible for the C10-acyl chain to fit into the LipB pocket.22 Further, we recently demonstrated that the acyl chain of dodecanoyl-AcpP does not chain flip when attached to the E. coli AcpP, but mutation of the LipB interface residue R145′ can induce loss of chain length selectivity for chain flipping,20 indicating both a substrate selectivity by wild-type LipB and the importance of the proper protein–protein interface for this selectivity. To resolve this PPI-based control mechanism at atomic detail, we chose to perform NMR studies of AcpP with C6, C8, and C10 acyl chains titrated with LipB to observe interaction changes based on chain length. These chain lengths represent the known LipB substrate and two most similar chain lengths seen in the cell. This data was then used to guide high-resolution in silico docking to identify the surface features responsible for the experimentally identified binding differences. We have shown that the implementation of NMR titration experiments to guide docking algorithms can accurately and reproducibly deduce PPI poses in ACP-dependent pathways.23
It has been suggested that unique features imparted by identity of the acyl chain can likely serve as a source for binding discrimination by enzymes.24–26 Furthermore, control of reactivity based on substrate has been seen by crosslinking27 and NMR,20 but developing a structural model requires atomic level detail. Here we identify the structural features of LipB that allow interaction with C8-AcpP while inducing structural hindrance to C6- and C10-AcpP binding, further elucidating the mechanism and role of PPIs in carrier protein-dependent enzymes.
Results
NMR titration to examine residue-by-residue interaction of acyl-AcpP with LipB
It has been established that solution-state 15N–1H HSQC NMR spectroscopy can function in appreciating the transient dynamic interactions between AcpP and partner proteins.28,29 Given the known specificity of LipB, we sought to elucidate how substrate specificity is conferred by PPI with AcpP carrying different acyl cargo. In order to detect small functional differences in the interactions of LipB, AcpPs of different chain lengths were prepared as C6-, C8-, and C10-linked 4′-pantetheinamide probes30 (Fig. S1–S3 and Tables S1–S3, ESI†). These AcpP species were titrated with increasing concentrations, from zero to beyond saturation: 2 molar equivalents of LipB in the case of C6-AcpP, 1.5 equivalents of LipB in the case of C8-AcpP, and 2 molar equivalents of LipB in the case of C10-AcpP. The effect of increasing LipB concentration on each AcpP species was examined.The first observation made when examining perturbations against one another was the difference in chemical shift perturbation (CSP) magnitude between the chain lengths (Fig. 2 and Fig. S4, ESI†). It was immediately clear that the degree of perturbation was greatest in C8-AcpP, despite all experiments being titrated to saturation. Further, the CSPs of C8-AcpP revealed unique interactions. Whereas most titrations have been noted to have little effect on helix I of the AcpP, LipB effected strong CSPs throughout the early residues and lasting through to residue 18. After this, there was a drop-off in CSPs through the end of loop 1 to residue 30. Small perturbations rose above background at residues 34 and 35, and CSPs continued consistently through helix 2. Next, there were consistent perturbations through loop 2, helix 3, and helix 4. The most unique region of CSP was the very strong migrations occurring in helix I (Fig. 2A). These perturbations appeared consistent in these regions among C6-, C8- and C10-AcpP, with varying magnitudes.
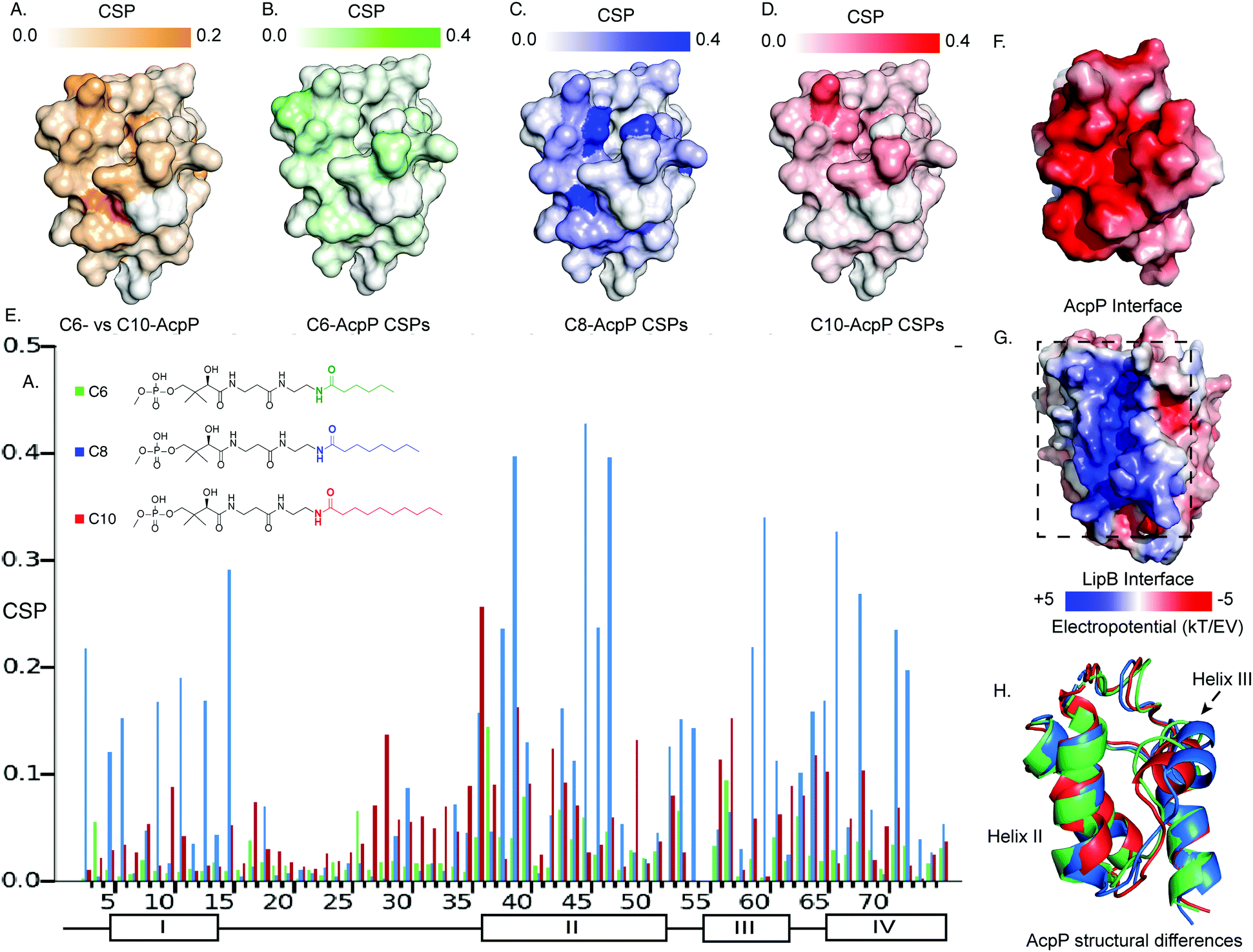 | ||
| Fig. 2 Titration NMR to probe the substrate selectivity of LipB. (A) Chemical shift differences between the C6- and C10-AcpP without LipB. CSPs are colored based on the magnitude of the CSP. Highlighting those residues which should be expected to be most different between the two proteins in solution. (B) CSPs of C6-AcpP titrated with LipB. CSPs are colored based on the magnitude of perturbation. (C) CSPs of the C8-AcpP titrated with LipB. (D) CSPs of C10-AcpP titrated with LipB. (E) Chemical shift perturbations (CSPs) of C6-, C8-, and C10-AcpP titrated with LipB. Individual spectra and bar charts are shown in Fig. S1–S3 (ESI†). (F) Electrostatic surface of AcpP, showing the electronegative surface of AcpP. Values were calculated using the APBS electrostatics plugin in PyMOL. (G) Electrostatic surface of LipB, highlighting the positive surface for the AcpP interaction. (H) The structures of C6 (Green), C8 (Blue), and C10 (Red) AcpP derived from molecular dynamics. The most significant differences are seen in the orientation of helix III, with C8 AcpP having the most space between helix III and helix II. | ||
Binding thermodynamics and kinetics demonstrate specificity experimentally
Though CSPs are not a quantitative measure of binding, C8-AcpP binding LipB displayed considerably stronger perturbations than C6- or C10-AcpP. In order to deduce quantitative binding parameters of AcpP tethering the three chain lengths, we examined the NMR titration data by applying TITAN line shape analysis.31 Here, C8-AcpP exhibited a 47.2 ± 5.1 μM Kd and a low off rate of 633 ± 98 s−1. The C6-AcpP bound less strongly, with a 189.9 ± 15.21 μM Kd and off rate of 4237 ± 2544 s−1. C10-AcpP bound slightly better than the C6-AcpP with a 134.8 ± 34.0 M Kd and off rate of 1521 ± 225 s−1 (Fig. S5–S7, ESI†). This difference in binding constant was supported by the comparative magnitude of the respective CSP data. TITAN analysis quantitatively supported control that LipB maintains over interactions with AcpPs tethering different acyl chains. The difference in binding strength suggested differences in binding surfaces which we next explored by high-resolution docking.Docking analysis to identify chain length specific interactions
LipB was first modeled by homology modeling to the 2QHS Thermus thermophilus lipoyltransferase with ICM Homology32,33 (Fig. S8, ESI†). The Thermus thermophilus structure was chosen because it shared the highest sequence identity of available crystal structures. AcpP structures were derived from molecular dynamics (MD) simulations and represented the highest population state seen in the simulation20 (Fig. S9, ESI†).To perform in silico docking experiments, the ICM Fast Fourier Transform protocol was used to generate high quality docking poses and scores for the AcpP·LipB interface and used to sample AcpP conformations across the entire LipB protein surface. The resulting docking poses were organized based on RMSD from the previously published model of the C8-AcpP·LipB docked complex20 (Fig. S10, ESI†). The methodology used was the same as we previously developed to model six ACP-partner protein interfaces from E. coli FAB and benchmark them with established crystal crosslinked structures.23
C8-AcpP adopted a low energy structure at 4.03 Å RMSD, with an energy of −50.5 kcal mol−1 (Fig. 3A) based on the ICM energy function. There was a second low energy docked conformation at 15.8 Å RMSD, which was ruled out as inactive with serine 36 17 Å from the LipB active site and the AcpP rotated away from the LipB pocket (Fig. 3B and C). The C6-AcpP had significantly higher energy poses (Fig. 3A) at low RMSD, with the only similarly stable state at 20 Å RMSD. Like C6-AcpP, C10-AcpP had most stable poses at 18.4 Å and 24.5 Å RMSD, with no similarly stable state near the C8-AcpPs.
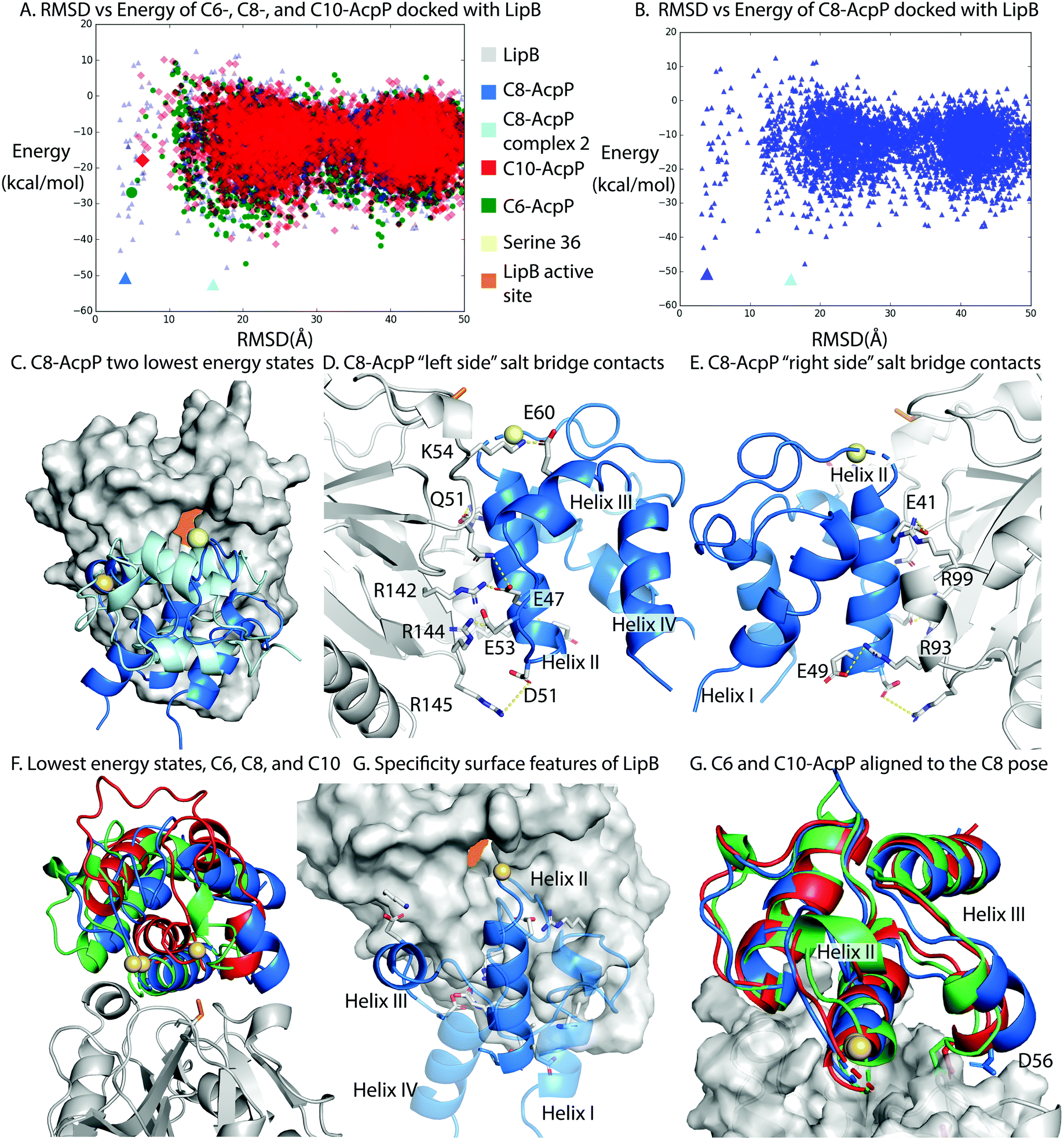 | ||
| Fig. 3 Structural analysis of the AcpP·LipB surface. (A) RMSD vs. energy plot of the tested AcpPs. RMSD was determined against the previously published Cluspro model.20 Individual models examined in other panels are highlighted by larger colored icons. (B) RMSD vs energy plot of the C8-AcpP with LipB. RMSD was determined against the previously published model. The two lowest energy states are examined in panel (C). (C) Comparison of the two lowest energy states of the C8-AcpP docked to LipB. The ∼4 Å RMSD pose (Lime green) is positioned with Ser36 in position for substrate delivery. The ∼16 Å pose (Light blue) can be seen binding with the substrate positioned far from the active site. (D) Salt bridge contacts formed between C8-AcpP and LipB. The “left side” is presented here, representing the majority of the contacts. Individual contacts and distances are reported in ESI,† Table S1. (E) Salt bridge contacts formed between C8-AcpP and LipB on the “right side.” A table of all contacts formed is reported in ESI,† Table S1. (F) “Active” energy states of the C6-, C8-, and C10-AcpP LipB docked poses. The most stable structure which was similar in RMSD to the low energy state in panel (A) was chosen. (G) Architecture of the AcpP·LipB interaction. The interacting helices are shown with the precise orientation necessary to form a stable interaction. Helices I&IV are made transparent to better appreciate the surface of interaction of the other two helices. (H) C6- and C10-AcpP are aligned to the docked orientation of C8-AcpP. This demonstrates the steric hindrance which makes C8 able to properly position into the active site, while the C6 and C10 must form the less stable poses seen in panel (F). | ||
The high RMSD states of C8-AcpP·LipB interaction are clearly not oriented for substrate delivery (Fig. S9, ESI†), with the 4′-phosphopantetheine positioned away from the active site. Therefore, lower RMSD states were examined to explore how the difference in AcpP structure translated to different energetics for binding. C8-AcpP bound tightly onto the LipB surface, with six arginine or lysine residues available for salt bridges with discrete AcpP residues. The most stable state of C8-AcpP had each of these residues coordinating closely, within 5 Å. The C8-AcpP·LipB interactions began at helix II, with E41 nearby R99′, E47 with R142′, and E49 with R93′. D51 and R145′ remained 4.5 Å apart, but side chain rotation could bring the residues within range of a salt bridge. E53 with R144′ and E60 with K54′ finished the total salt bridges.
The C6-AcpP model displayed a significantly poorer binding surface with LipB (Fig. 3A and F), matching the results of the thermodynamic and CSP data. Specifically, E41, E48, and D51 lost the proper orientation of interaction (Fig. S3, ESI†). This was due to the structural effects of chain length shortening upon AcpP, with the structure most perturbed on helix II, loop II, and the orientation of helix III. There were two residues within range for a salt bridge in the C6-AcpP·LipB complex: E47 with the pair R142′ and R144′, and E57 with K54′. It is interesting to not how distinctly the binding surface of LipB could be effected by the small structural changes between C6 and C8-AcpP. The C10-AcpP model similarly bound more poorly to the LipB surface, displaying an ability to only bind four residues at the LipB surface. E41 and E47 appeared to be out of any vicinity to interact with R99′ or R142′, but E48 and D51 were within interacting distance of R93′. E53 lies within 3.6 Å of R142′, and E60 is 3.8 Å from K54′ (Fig. S4, ESI†).
The disparities in possible interactions were further highlighted by aligning the C6- and C10-AcpP structures onto the C8 docked pose to reveal their structural differences. The most immediate difference between them was the orientation of helix III. In the C8-AcpP, the helix was oriented outwards, creating space around the bottom of AcpP helix III for the residues 50′–55′ of LipB (Fig. 3F). For example, residue D56 lay nearby Q51′ on LipB in the C8-AcpP, but the more closed structure of C6- and C10-AcpP placed D56 in direct steric clash with LipB. This reflected a structural filtering mechanism, where the surface appeared to be arranged to interact with the structural features of C8-AcpP. Other orientations of helix III disallowed proper binding for chain flipping from helix II. These regions of selectivity overlayed with the differences in structure seen in the NMR data.‡ Though the C6- and C10-AcpPs could likely relax their structure to better bind the LipB surface, this initial instability and necessary relaxation could slow the association, explaining the poor binding of non-substrate AcpPs. The effects of this selectivity were evident in the respective CSPs, thermodynamics, and conformational landscape of the docking calculations.
Comparing CSPs to the docked model
To combine both data sets, the NMR titration of C8-AcpP was compared to the docked model (Fig. S11, ESI†). Beginning at helix I there were perturbations occurring through most of the helix, though they were most prominent at the end of the helix. These likely correspond to the interactions across the “right” side of the channel which binds helix II. There were several residues at the base of the helix which would likely experience movement while the ACP adopts a bound state, but the residues at the end were close enough to be in direct contact. This direct contact would lead to a higher degree of perturbation. The CSPs almost fully dropped off until helix II, matching the pose where there was no surface of the LipB in proximity. Helix II showed large perturbations throughout until its end, this agreed well with the model, where most of helix II is deep into the LipB channel. Especially significant were the uncharged residues such as T39, V43, and A45 which usually saw small CSPs but were buried against hydrophobic regions of LipB. The perturbations ended at the bottom of the helix, to be seen again at E53, which bound R144′. On helix III further down the ACP D56 had little perturbation, though it was usually participating in interactions. However, this matches the model, where the acidic side chain is either binding a backbone or associating with Q68. However, at the end of helix III strong perturbations continued with a salt bridge at E60. Helix IV saw perturbations which were likely linked to the movement of the helix upon chain flipping. Seen especially in V65, A64, Q66, and Y71, the perturbed residues at the top of helix IV may also have been due to movement of the dynamic helix III upon binding, especially when all the residues on the loop before and after helix III saw some small perturbation.The C6 and C10-AcpP perturbations were very different in their distribution. There were a limited number of electrostatic CSPs at D35, E49, D56, and E57 in the C10-AcpP titration. There were more perturbations of hydrophobic residues at I10, V17, S27, P28, T39, L42, V43, T63, T64, and V65. These C10-AcpP perturbations were consistent with C6-AcpP, though C6-AcpP's CSPs had significantly smaller magnitudes. The largest effects were seen on helix II and III, seeming to indicate interactions at the interface. The only region which saw interactions greater than C8 was the loop preceding helix II and the very top of helix II. The difference in hydrogen bonding matched the docked model for C10, where the helix III angle better aligned D56 and E57 to form interactions. This contrasts with C8-AcpP where D56 was not aligned for a strong interaction. This lower number of salt bridges and poorer interaction suggested a model where there are sets of interactions which must be sufficiently strong to induce chain flipping. Chain flipping is a large dynamic event, and we propose that the deeply buried interface and multiple salt bridges are essential to drive this, with sufficient interactions shifting the residues such as Y71 and I54 to close the acyl pocket and initiate flipping. The stronger surface interactions also explain the higher degree of measured CSPs. The weak interactions by C6 and C10-AcpP lowered the degree of structural perturbation upon binding and decreased the time spent in the “bound” state in solution, resulting in smaller shifts on the magnet. Surprisingly, the C8-pantethenamide used in this study saw larger chemical shifts than the C8-pantetheine probe used in a previous study.20 We hypothesize that this was due to a disruption at the interface of the mutant LipB C169A utilized in that study. With the intricate binding mechanism identified, a mutation nearby the interaction site could significantly affect the interface.
Discussion
A significant kinetic advantage for any protein interacting with ACP, currently tallied at 27 known enzymes and regulatory proteins in E. coli,9–11 is the ability to discern acyl identity without the requirement of chain flipping. For LipB, the ability to differentiate acyl chains based on the initial PPIs significantly increases the efficiency of this selection process and provides thermodynamic control to maintain the fidelity of lipoic acid biosynthesis. We have recently demonstrated how discrete salt bridge interactions at the protein interface can allow differentiation between C8- and C12-AcpP for chain flipping with LipB.20 We have now determined the comparative binding constants and CSPs of LipB with acyl-AcpPs of both shorter and longer chain lengths with that of the natural C8 substrate, indicating a clear ability of LipB to select for interaction with C8-AcpP. This is accomplished by possessing a surface that can complement the specific shape of octanoyl-sequestered (C8-) AcpP, while simultaneously deterring interactions with C6- and C10-AcpP. This selectivity is primarily reliant on helix III perturbations in response to the sequestered acyl chain lengths, previously identified in numerous experimental and theoretical studies. Structurally, this model leverages unique conformational features of AcpP induced by different chain lengths, an incredibly useful evolutionary feature when selectivity for a single chain length is required.Understanding that the ACP·LipB reaction requires chain flipping controlled by PPIs unlocks the potential to modify these essential and sensitive interactions through inhibition or engineering. The high sequence identity of LipB shared between bacteria implies that the observations made here in E. coli will likely extend to other species. Furthermore, targeting the protein interface of AcpP·LipB in a pathogen could avoid potential side effects from activity against the human mitochondrial LipB (Fig. S8, ESI†). These data also suggest an important factor to consider when engineering FAB and related acetate pathway proteins. Where poor interface complementarity could lead to a loss of activity, understanding and optimizing these interactions may prove essential. While these transient PPIs can be challenging to observe, we have developed an approach that merges new data with prior observations and formed a model that explains both the specificity and efficiency for lipoic acid biosynthesis.
Author contributions
T. B. performed NMR experiments and Docking experiments. T. S. provided MD structures for docking. M. A. Y. assisted protein preparation and analysis. T. D. D. provided probes for chemoenzymatic loading. R. A. provided support and guidance. M. D. B. supervised the study and analysis. All authors drafted the manuscript.Funding sources
This work was funded by NSF IOS-1516156 and NIH R01GM095970. T. G. B. was supported in part by the NIH CBI T32GM112584. T. S. was supported by NSF GRFP DGE-1650112. T. D. D. was supported by NIH K12GM068524 and K99GM129454.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank S. Opella, X. Huang, and the UCSD Biomolecular NMR facility for support and guidance.Notes and references
- R. M. Cicchillo and S. J. Booker, Mechanistic Investigations of Lipoic Acid Biosynthesis in Escherichia Coli:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Both Sulfur Atoms in Lipoic Acid Are Contributed by the Same Lipoyl Synthase Polypeptide, J. Am. Chem. Soc., 2005, 127(9), 2860–2861, DOI:10.1021/ja042428u.
Both Sulfur Atoms in Lipoic Acid Are Contributed by the Same Lipoyl Synthase Polypeptide, J. Am. Chem. Soc., 2005, 127(9), 2860–2861, DOI:10.1021/ja042428u. - S. J. Booker, Unraveling the Pathway of Lipoic Acid Biosynthesis, Chem. Biol., 2004, 11(1), 10–12, DOI:10.1016/j.chembiol.2004.01.002.
- M. S. Schonauer, A. J. Kastaniotis, V. A. S. Kursu, J. K. Hiltunen and C. L. Dieckmann, Lipoic Acid Synthesis and Attachment in Yeast Mitochondria, J. Biol. Chem., 2009, 284(35), 23234–23242, DOI:10.1074/jbc.M109.015594.
- F. Habarou, Y. Hamel, T. B. Haack, R. G. Feichtinger, E. Lebigot, I. Marquardt, K. Busiah, C. Laroche, M. Madrange, C. Grisel, C. Pontoizeau, M. Eisermann, A. Boutron, D. Chrétien, B. Chadefaux-Vekemans, R. Barouki, C. Bole-Feysot, P. Nitschke, N. Goudin, N. Boddaert, I. Nemazanyy, A. Delahodde, S. Kölker, R. J. Rodenburg, G. C. Korenke, T. Meitinger, T. M. Strom, H. Prokisch, A. Rotig, C. Ottolenghi, J. A. Mayr and P. de Lonlay, Biallelic Mutations in LIPT2 Cause a Mitochondrial Lipoylation Defect Associated with Severe Neonatal Encephalopathy, Am. J. Hum. Genet., 2017, 101(2), 283–290, DOI:10.1016/j.ajhg.2017.07.001.
- E. Bernardinelli, R. Costa, G. Scantamburlo, J. To, R. Morabito, C. Nofziger, C. Doerrier, G. Krumschnabel, M. Paulmichl and S. Dossena, Mis-Targeting of the Mitochondrial Protein LIPT2 Leads to Apoptotic Cell Death, PLoS One, 2017, 12(6), e0179591, DOI:10.1371/journal.pone.0179591.
- C. O. Rock, Opening a New Path to Lipoic Acid, J. Bacteriol., 2009, 191(22), 6782–6784, DOI:10.1128/JB.01151-09.
- J.-P. Carreau, [32] Biosynthesis of Lipoic Acid via Unsaturated Fatty Acids, Methods in Enzymology; Vitamins and Coenzymes Part D, Academic Press, 1979, vol. 62, pp. 152–158 DOI:10.1016/0076-6879(79)62212-7.
- A. Solmonson and R. J. DeBerardinis, Lipoic Acid Metabolism and Mitochondrial Redox Regulation, J. Biol. Chem., 2018, 293(20), 7522–7530, DOI:10.1074/jbc.TM117.000259.
- D. Gully, D. Moinier, L. Loiseau and E. Bouveret, New Partners of Acyl Carrier Protein Detected in Escherichia Coli by Tandem Affinity Purification, FEBS Lett., 2003, 548(1–3), 90–96, DOI:10.1016/s0014-5793(03)00746-4.
- N. R. D. Lay and J. E. Cronan, In Vivo Functional Analyses of the Type II Acyl Carrier Proteins of Fatty Acid Biosynthesis, J. Biol. Chem., 2007, 282(28), 20319–20328, DOI:10.1074/jbc.M703789200.
- D. Gully and E. Bouveret, A Protein Network for Phospholipid Synthesis Uncovered by a Variant of the Tandem Affinity Purification Method in Escherichia Coli, Proteomics, 2006, 6(1), 282–293, DOI:10.1002/pmic.200500115.
- J. E. Cronan and J. Thomas, Bacterial Fatty Acid Synthesis and Its Relationships with Polyketide Synthetic Pathways, Methods Enzymol., 2009, 459, 395–433, DOI:10.1016/S0076-6879(09)04617-5.
- K. Magnuson, S. Jackowski, C. O. Rock and J. E. Cronan, Regulation of Fatty Acid Biosynthesis in Escherichia Coli, Microbiol. Mol. Biol. Rev., 1993, 57(3), 522–542 CAS.
- A. Shokri and G. Larsson, Characterisation of the Escherichia Coli Membrane Structure and Function during Fedbatch Cultivation, Microb. Cell Fact., 2004, 3, 9, DOI:10.1186/1475-2859-3-9.
- J. Crosby and M. P. Crump, The Structural Role of the Carrier Protein – Active Controller or Passive Carrier, Nat. Prod. Rep., 2012, 29(10), 1111–1137, 10.1039/C2NP20062G.
- D. M. Byers and H. Gong, Acyl Carrier Protein: Structure–Function Relationships in a Conserved Multifunctional Protein Family, Biochem. Cell Biol., 2007, 85(6), 649–662, DOI:10.1139/O07-109.
- J. Beld, H. Cang and M. D. Burkart, Visualizing the Chain-Flipping Mechanism in Fatty-Acid Biosynthesis, Angew. Chem., Int. Ed., 2014, 53(52), 14456–14461, DOI:10.1002/anie.201408576.
- T. W. Morris, K. E. Reed and J. E. Cronan, Lipoic Acid Metabolism in Escherichia Coli: The LplA and LipB Genes Define Redundant Pathways for Ligation of Lipoyl Groups to Apoprotein, J. Bacteriol., 1995, 177(1), 1–10, DOI:10.1128/jb.177.1.1-10.1995.
- X. Zhao, J. R. Miller, Y. Jiang, M. A. Marletta and J. E. Cronan, Assembly of the Covalent Linkage between Lipoic Acid and Its Cognate Enzymes, Chem. Biol., 2003, 10(12), 1293–1302, DOI:10.1016/j.chembiol.2003.11.016.
- T. Sztain, T. G. Bartholow, D. J. Lee, L. Casalino, A. Mitchell, M. A. Young, J. Wang, J. A. McCammon and M. D. Burkart, Decoding Allosteric Regulation by the Acyl Carrier Protein, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(16), e2025597118, DOI:10.1073/pnas.2025597118.
- J. E. Cronan, The Chain-Flipping Mechanism of ACP (Acyl Carrier Protein)-Dependent Enzymes Appears Universal, Biochem. J., 2014, 460(2), 157–163, DOI:10.1042/BJ20140239.
- Q. Ma, X. Zhao, A. N. Eddine, A. Geerlof, X. Li, J. E. Cronan, S. H. E. Kaufmann and M. Wilmanns, The Mycobacterium Tuberculosis LipB Enzyme Functions as a Cysteine/Lysine Dyad Acyltransferase, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(23), 8662–8667, DOI:10.1073/pnas.0510436103.
- T. G. Bartholow, T. Sztain, A. Patel, D. J. Lee, M. A. Young, R. Abagyan and M. D. Burkart, Elucidation of Transient Protein-Protein Interactions within Carrier Protein-Dependent Biosynthesis, Commun. Biol., 2021, 4(1), 1–10, DOI:10.1038/s42003-021-01838-3.
- E. Płoskoń, C. J. Arthur, A. L. P. Kanari, P. Wattana-amorn, C. Williams, J. Crosby, T. J. Simpson, C. L. Willis and M. P. Crump, Recognition of Intermediate Functionality by Acyl Carrier Protein over a Complete Cycle of Fatty Acid Biosynthesis, Chem. Biol., 2010, 17(7), 776–785, DOI:10.1016/j.chembiol.2010.05.024.
- G. A. Zornetzer, J. Tanem, B. G. Fox and J. L. Markley, The Length of the Bound Fatty Acid Influences the Dynamics of the Acyl Carrier Protein and the Stability of the Thioester Bond, Biochemistry, 2010, 49(3), 470–477, DOI:10.1021/bi9014659.
- A. Roujeinikova, W. J. Simon, J. Gilroy, D. W. Rice, J. B. Rafferty and A. R. Slabas, Structural Studies of Fatty Acyl-(Acyl Carrier Protein) Thioesters Reveal a Hydrophobic Binding Cavity That Can Expand to Fit Longer Substrates, J. Mol. Biol., 2007, 365(1), 135–145, DOI:10.1016/j.jmb.2006.09.049.
- K. Finzel, C. Nguyen, D. R. Jackson, A. Gupta, S.-C. Tsai and M. D. Burkart, Probing the Substrate Specificity and Protein-Protein Interactions of the E. Coli Fatty Acid Dehydratase, FabA, Chem. Biol., 2015, 22(11), 1453–1460, DOI:10.1016/j.chembiol.2015.09.009.
- C. Nguyen, R. W. Haushalter, D. J. Lee, P. R. L. Markwick, J. Bruegger, G. Caldara-Festin, K. Finzel, D. R. Jackson, F. Ishikawa, B. O’Dowd, J. A. McCammon, S. J. Opella, S.-C. Tsai and M. D. Burkart, Trapping the Dynamic Acyl Carrier Protein in Fatty Acid Biosynthesis, Nature, 2014, 505(7483), 427–431, DOI:10.1038/nature12810.
- J. C. Milligan, D. J. Lee, D. R. Jackson, A. J. Schaub, J. Beld, J. F. Barajas, J. J. Hale, R. Luo, M. D. Burkart and S.-C. Tsai, Molecular Basis for Interactions between an Acyl Carrier Protein and a Ketosynthase, Nat. Chem. Biol., 2019, 15(7), 669–671, DOI:10.1038/s41589-019-0301-y.
- A. S. Worthington and M. D. Burkart, One-Pot Chemo-Enzymatic Synthesis of Reporter-Modified Proteins, Org. Biomol. Chem., 2006, 4(1), 44–46, 10.1039/b512735a.
- C. A. Waudby, A. Ramos, L. D. Cabrita and J. Christodoulou, Two-Dimensional NMR Lineshape Analysis, Sci. Rep., 2016, 6, 24826, DOI:10.1038/srep24826.
- M. A. C. Neves, M. Totrov and R. Abagyan, Docking and Scoring with ICM: The Benchmarking Results and Strategies for Improvement, J. Comput. -Aided Mol. Des., 2012, 26(6), 675–686, DOI:10.1007/s10822-012-9547-0.
- ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation, Abagyan, Journal of Computational Chemistry, Wiley Online Library, 1994, https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.540150503, accessed 2020-08-31.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00125f |
| ‡ The NMR data have been made available on the Biological Magnetic Resonance data Bank (BMRB) under the ascension codes: 50755, 50756, and 50757. Additional information on the methods, figures, and tables can be found in the ESI.† |
| This journal is © The Royal Society of Chemistry 2021 |