
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrigin, toxicity and characteristics of two amyloid oligomer polymorphs
Chamani
Niyangoda
a,
Jeremy
Barton
a,
Nabila
Bushra
a,
Kanchana
Karunarathne
a,
Graham
Strauss
b,
Fadia
Fakhre
a,
Piyush
Koria
b and
Martin
Muschol
 *a
*a
aDept. of Physics, University of South Florida, Tampa, FL 33620, USA. E-mail: mmuschol@usf.edu
bDept. of Chemical and Biomedical Engineering, University of South Florida, Tampa, FL 33620, USA
First published on 25th August 2021
Abstract
There is compelling evidence that small oligomeric aggregates, emerging during the assembly of amyloid fibrils and plaques, are important molecular pathogens in many amyloid diseases. While significant progress has been made in revealing the mechanisms underlying fibril growth, understanding how amyloid oligomers fit into the fibril assembly process, and how they contribute to the pathogenesis of amyloid diseases, has remained elusive. Commonly, amyloid oligomers are considered to be metastable, early-stage precursors to fibril formation that are either on- or off-pathway from fibril growth. In addition, amyloid oligomers have been reported to colocalize with late-stage fibrils and plaques. Whether these early and late-stage oligomer species are identical or distinct, and whether both are relevant to pathogenesis remains unclear. Here we report on the formation of two distinct oligomer species of lysozyme, formed either during the early or late-stages of in vitro fibril growth. We further observe that the pH change from in vitro growth conditions to cell media used for toxicity studies induced distinct mesoscopic precipitates, two of which resemble either diffuse or neuritic plaques seen in Alzheimer's histology. Our biophysical characterization indicates that both oligomer species share morphological and tinctorial features considered characteristic for amyloid oligomers. At the same time, their sizes, morphologies, their immunostaining, detailed tinctorial profiles and, most prominently, their biological activity are clearly distinct from each other. Probing the conditions promoting the formation of these two distinct oligomer species suggests distinct roles of charge interactions, hydrophobicity and monomer flexibility in directing oligomer assembly.
1. Introduction
Amyloidoses refer to a group of human disorders all characterized by the deposition of protein fibril plaques appearing in different, disease-specific tissues and organs.1–4 While the proteins that form these amyloid plaques are unrelated and disease-specific, the resulting fibrils have a shared cross-beta sheet architecture.3,5 This structural commonality has been linked to the underlying propensity of polypeptide chains to form intermolecular hydrogen bonds across their backbone.5,6 However, the role of amyloid fibrils as the underlying cause of disease symptoms is much less clear. In several non-neuropathic amyloidoses fibril deposits have been implicated as the primary cause of disease pathology.7 Conversely, there is compelling evidence that small oligomers, emerging during the assembly of amyloid fibrils and plaques, are potent molecular pathogens particularly in neurodegenerative diseases.8–13 Establishing causality between amyloid formation and disease, however, has been complicated by recent findings that both fibrils and oligomers can exist in multiple polymorphic forms with distinct pathogenic potentials.14–18 In addition, there is evidence that different types of amyloid aggregates can interact with and alter the accumulation of one another.19–23 For Alzheimer's disease, there are strong indications that Aβ aggregates interact with tau, α-syn and TDP-43, each of which is associated with distinct neurodegenerative diseases of its own, thereby adding to the complexity of amyloid species and their relation to pathogenesis.20,24Detailed models of the molecular steps and mechanisms regulating fibril nucleation and growth, together with analytical solutions to the resulting kinetics of fibril assembly, have significantly enhanced our mechanistic understanding of amyloid fibril self-assembly.25,26 In contrast, the conditions and mechanisms inducing or modulating the formation of metastable amyloid oligomers, in vitro or in vivo, remain poorly understood. In addition, there is evidence for the presence of multiple, distinct oligomer polymorphs formed in vitro and in vivo.27–31 This raises the questions of how oligomer species come about, what distinguishes them from one another, which conditions promote the formation of one over the other, and how they fit into the overall fibril assembly process. Commonly, amyloid oligomers are considered to be metastable, early-stage precursors that are either on- or off-pathway from fibril growth.19,27,32,33 At the same time, late-stage fibrils and plaques have been implicated as reservoirs for amyloid oligomer formation.34 Whether those two modes of oligomer formation are identical or distinct, and whether both are relevant to pathogenesis is unclear. Understanding the mechanisms underlying the formation of various oligomer polymorphs, and how they relate to each other, would provide important insights into their contributions to pathogenesis in different cell types and tissues, and open up new strategies for therapeutic interventions. Similarly, elucidating the relation between oligomer to fibril formation directly affects the approaches taken for targeting oligomers vs. fibrils as pathogenic species in amyloid disease.35
Here we describe the formation of two distinct amyloid oligomer species formed in vitro by the amyloid protein lysozyme. Intriguingly, these two distinct amyloid species form under nearly identical growth conditions, but at different time points of the fibril assembly process. While one oligomer species assembles during the initial lag phase of fibril formation, the second population emerges at the late-stages of fibril growth. For the sole purpose of distinguishing these two oligomer species, we will refer to them as early-stage versus late-stage oligomers (ESOs vs. LSOs). We had previously characterized ESOs as off-pathway competitors and inhibitors of fibril formation.19,23 We therefore focused on characterizing the sizes, morphologies and composition of LSOs, and determined their tinctorial and immunological signatures relative to those of fibrils and ESOs. Using a live/dead fluorescence assay we established that ESOs, LSOs and fibrils all display distinct levels of cell toxicity. We further observed that transfer from growth solutions to cell media induced prominent changes in morphology for each aggregate species, with fibrils and ESOs showing striking similarities to neuritic fibrillar vs. diffuse plaque associated with Alzheimer's disease. Tracking the relationship of fibrils to LSOs indicated that the latter, just like their ESO counterparts, emerge as off-pathway byproduct of fibril growth conditions. Our findings provide some mechanistic insights into how distinct oligomer species can emerge as result of compromises between intermolecular charge repulsion, hydrophobicity and structural flexibility.
2. Results
We have previously reported that in vitro amyloid assembly of lysozyme under acidic conditions proceeds along two distinct assembly pathways. One pathway is characterized by lag-free oligomer formation, but only emerges upon crossing a (protein and salt-dependent) threshold concentration. Due to its similarity with micelle formation by lipids, we refer to this threshold as ‘critical oligomer concentration’ or COC. The rate at which these metastable globular early-stage oligomers (ESOs) and curvilinear fibrils (CFs) form increases as a power law of the protein concentration above the threshold. The subsequent fibril nucleation event induces the biphasic ThT kinetics, which is characteristic for this pathway (Fig. 1A).19,36,37 Below the aforementioned threshold, the nucleated growth of amyloid filaments and fibrils (we collectively refer to as rigid fibril, or RFs) displays the ubiquitous sigmoidal ThT kinetics. During the extended lag phase of this pathway, no discernible populations of oligomeric precursors are observed. However, the cell toxicity experiments detailed below revealed that the fibril samples generated during sigmoidal fibril growth contained a second class of “late-stage” oligomers (LSOs). Here we isolated each of these oligomeric and fibrillar species for quantitative cell toxicity assays, and for determining their respective morphological, tinctorial, and immunological characteristics. We have previously characterized multiple features of the fibrils and ESO/CFs generated during sigmoidal and biphasic growth, respectively. We therefore focused on elucidating the origin of the LSOs formed during sigmoidal fibril growth, and on characterizing the new LSOs relative to RFs and ESOs.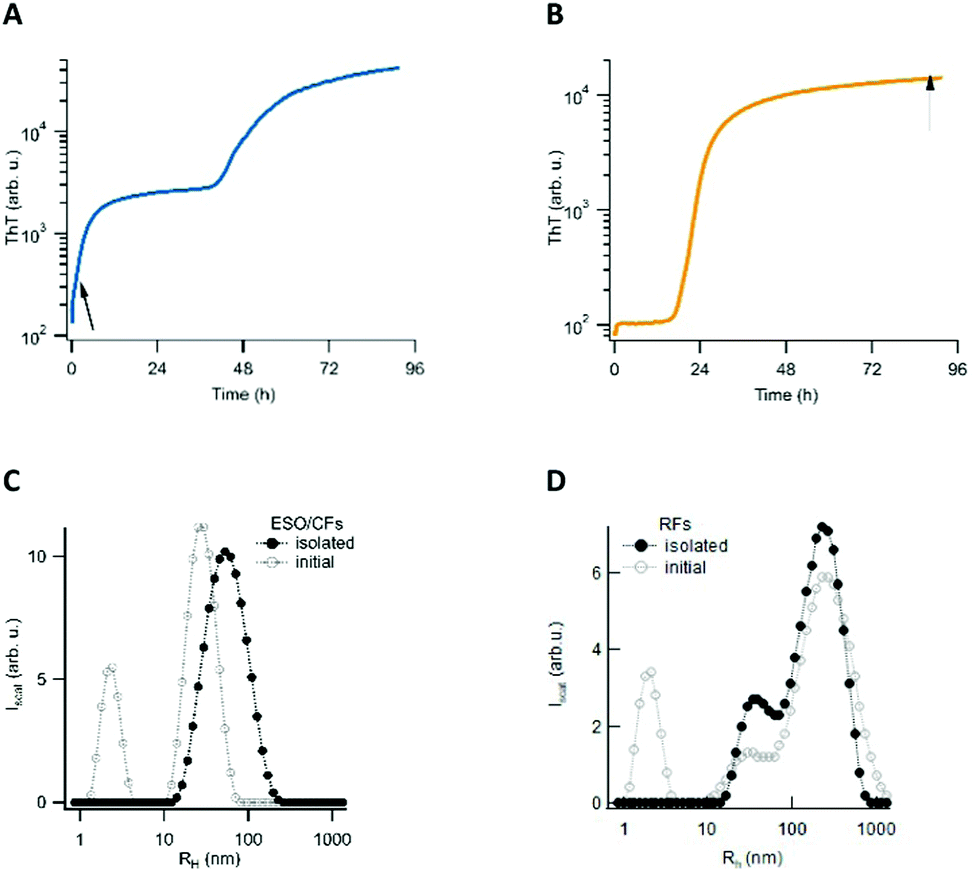 | ||
| Fig. 1 Biphasic vs. sigmoidal ThT kinetics and distinct particle size distributions indicate early-stage oligomer (ESO) vs. rigid fibril (RF) dominated amyloid growth. (A) Biphasic ThT kinetics associated with formation of ESO/CFs during initial phase of amyloid assembly (1.4 mM hewL at 250 mM NaCl). (B) Typical sigmoidal ThT kinetics indicating RF dominated amyloid formation (1.4 mM hewL at 50 mM NaCl). The small, initial step of ThT in B results from surface binding of denatured hewL upon heating. Both solutions were incubated at pH 2, 52 °C. The arrows indicate typical time points for the collection of either ESO/CFs or RFs for toxicity measurements (C) DLS particle size distribution (PSD) of early-stage oligomers (ESO) and curvilinear fibrils (CFs), after incubation (open circles), and after their isolation from monomers (filled circles). (D) Typical tri-modal PSD of hewL RFs grown under sigmoidal conditions, after incubation (open circles), and after their isolation from monomers (filled circles). | ||
2.1 Growth and isolation of oligomeric and fibrillar amyloids
Aggregates were generated under similar in vitro growth conditions, and without addition of any denaturants. In both cases, 1.4 mM of hewL was dissolved in water adjusted to pH 2 with HCl, and was incubated at T = 52 °C. Fibril- vs. oligomer-dominated growth, as indicated by their respective ThT kinetics or particle-size distribution profiles (Fig. 1), was induced by using either 50 mM or 250 mM NaCl in the solutions.19,36–38 The progress of the aggregation reaction was monitored using dynamic light scattering. The reaction was stopped at time points appropriate to obtain suspensions of rigid fibrils (RFs) and late-stage oligomers (LSOs), typically after 6–8 days at 50 mM NaCl. Early-stage oligomers (ESOs), in turn, were harvested within 2–8 hours of incubation at 250 mM NaCl. We have previously shown that ESOs have a strong tendency to self-assemble into large curvilinear fibrils (CFs) which become the dominant aggregate species present in solution.38,39 Transmission electron microscope images of the ESO/CFs grown under bimodal conditions show the dominant CF population (Fig. 2A). TEM images of RFs formed during sigmoidal growth, in turn, display the characteristic rigid fibril shapes (Fig. 2B). The cell toxicity studies described below indicated the presence of an additional “late-stage” oligomer (LSO) species formed during the prolonged sigmoidal RF growth. Fig. 2C shows AFM images of LSO following their isolation from the simultaneously present RFs.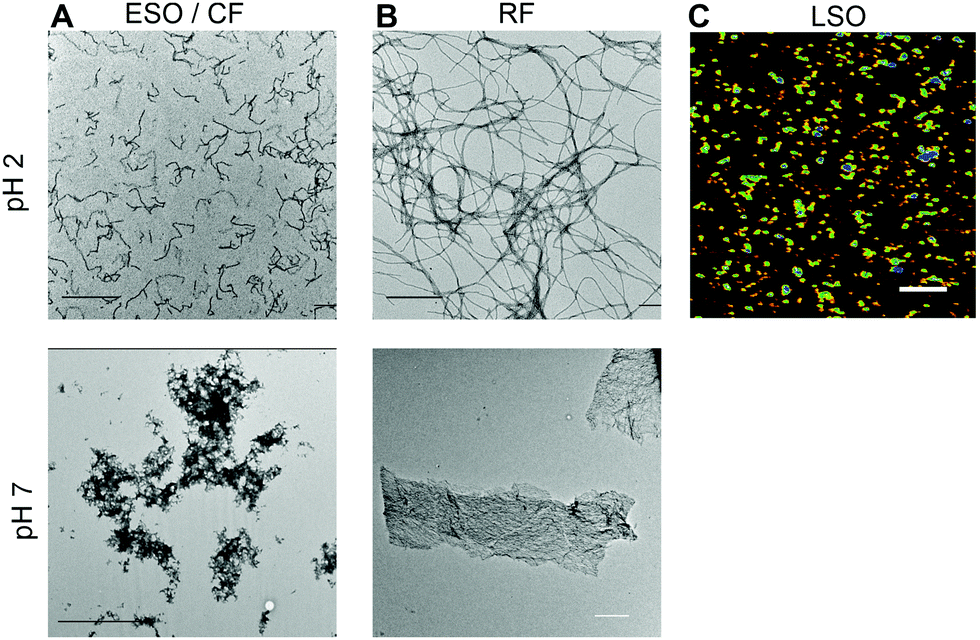 | ||
| Fig. 2 Solution transfer of aggregates creates precipitates with distinct morphologies. (A) TEM images of separated early-stage oligomers (ESOs) and their polymerized curvilinear fibrils (CFs) formed at pH 2 (top), and following transfer to pH 7 (bottom). (B) TEM images of rigid fibrils (RFs) formed under sigmoidal growth conditions at pH 2 (top) and after transfer to pH 7 (bottom). (C) AFM image of late-stage oligomers, isolated from RFs after incubation at pH 2. Precipitates formed at pH 7 were too large for imaging with AFM or TEM (scale bars: 500 nm). | ||
For quantification of their cell toxicity, we isolated oligomeric and fibrillar aggregates from their monomeric background, and each other, using a combination of centrifugation and molecular weight cut-off filters. Fibrils were centrifuged overnight at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and the pellet re-suspended. This process was repeated three times in order to obtain a pure RF suspension. Successful removal of residual monomers and oligomers was confirmed by the absence of UV-absorption from the supernatant after the third spin, and by the loss of the monomer peak in DLS (Fig. 1D). The same approach was used to isolate ESO/CFs. Isolation of LSOs started with the fibril supernatant obtained after the first centrifugation step. Residual fibrils were removed by passing the supernatant through a 50 nm syringe filter. Monomers were removed by passing this filtered supernatant through a 50 kDa molecular weight cut off filter. This latter filtration step was repeated twice, with oligomers collected on top of the filter being re-suspended in solution. In all cases, we saw no significant aggregate dissociation into monomers over the course of our experiments. This implies that either the rates of dissociation are slow or monomer equilibria are very small. The effects of solution transfer discussed next only further shifted this balance towards the aggregated states.
000g and the pellet re-suspended. This process was repeated three times in order to obtain a pure RF suspension. Successful removal of residual monomers and oligomers was confirmed by the absence of UV-absorption from the supernatant after the third spin, and by the loss of the monomer peak in DLS (Fig. 1D). The same approach was used to isolate ESO/CFs. Isolation of LSOs started with the fibril supernatant obtained after the first centrifugation step. Residual fibrils were removed by passing the supernatant through a 50 nm syringe filter. Monomers were removed by passing this filtered supernatant through a 50 kDa molecular weight cut off filter. This latter filtration step was repeated twice, with oligomers collected on top of the filter being re-suspended in solution. In all cases, we saw no significant aggregate dissociation into monomers over the course of our experiments. This implies that either the rates of dissociation are slow or monomer equilibria are very small. The effects of solution transfer discussed next only further shifted this balance towards the aggregated states.
2.2 Formation of ordered vs. diffuse amyloid plaques upon solution transfer
At pH 2, aggregates typically remained in suspension and displayed the morphologies of individual rigid or curvilinear fibrils (Fig. 2, top row). After transfer to pH 7.4 cell medium, solutions consistently turned turbid indicating that all aggregate populations started to precipitate. The rate of precipitate formation and the resulting precipitate structures, however, were markedly different. RFs and ESO/CFs precipitated rapidly, while LSO precipitation evolved over several hours. RFs bundled into highly organized fibril sheets (Fig. 2B, bottom row). These bundles displayed bright ThS fluorescence and prominent birefringence in optical microscopy, without the need for Congo Red staining (Fig. 3, top row). Both features are considered diagnostic of amyloid plaques in vivo. ESOs and CFs, in contrast, precipitated into open-structured diffuse clusters (Fig. 2A, bottom row). These clusters exhibited no birefringence, indicating the lack of long-range alignment, retained their fractal-like morphology and showed only weak ThS affinity (Fig. 3, center row). Precipitates of LSOs formed compact aggregates too heavy for TEM imaging and, similarly to ESOs, displayed weak ThS fluorescence (Fig. 3, bottom row). The absence of optical activity again indicated the absence of long-range organization. These different amyloid precipitates showed noticeable differences in their affinity for cell membranes during the subsequent cell toxicity experiments. While fibril plaques strongly adhered to cell surfaces, both types of oligomeric precipitates could be mostly washed out following incubation.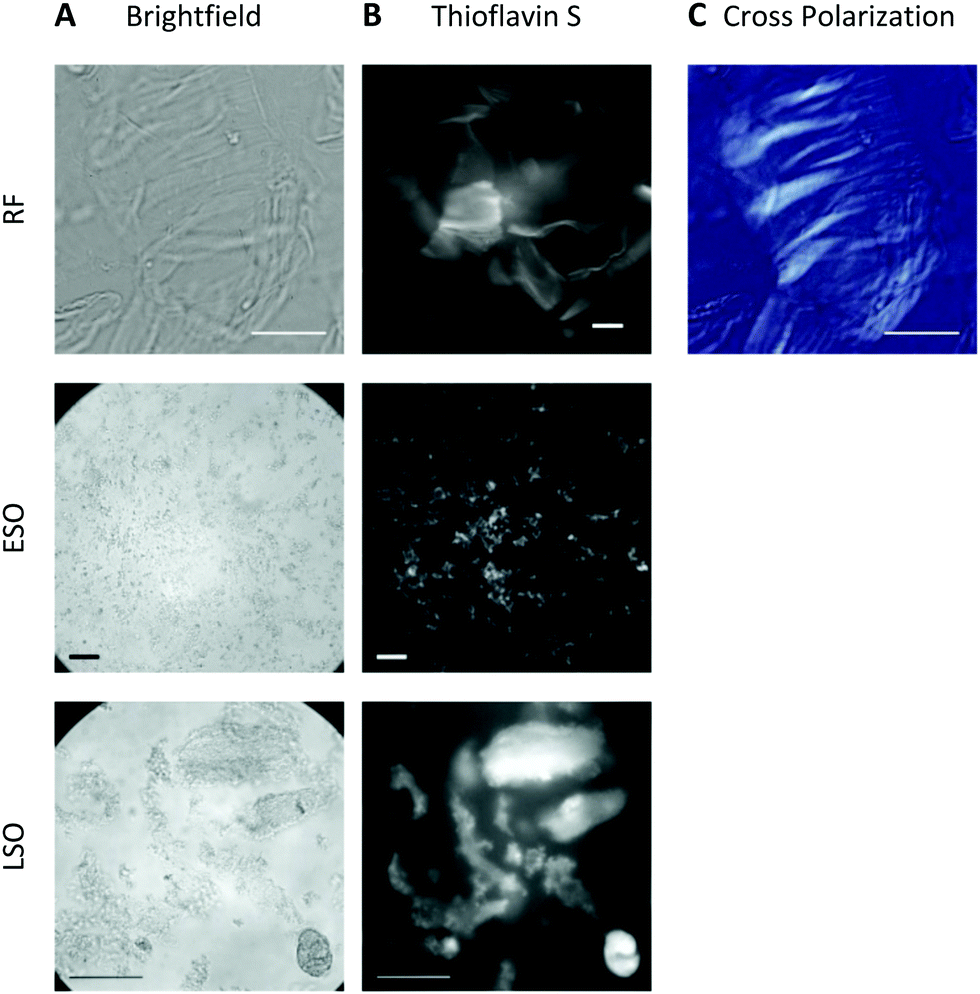 | ||
| Fig. 3 Optical microscopy of amyloid precipitates formed at pH 7. (A) Optical brightfield images (left column) of RFs (top), early-stage (middle) and late-stage oligomers (bottom). (B) Thioflavin S fluorescence of aggregates in A (center column; intensities not calibrated). (C) Cross-polarized image of RF bundles. Scale bars: 100 μm. | ||
The morphologies of the various hewL amyloid precipitates formed at pH 7.4 resemble basic features of neuritic and dense core vs. diffuse plaques commonly observed in Alzheimer's disease and Down syndrome pathology.40–44 Both of these Alzheimer-associated plaques are composed of amyloid-β, with diffuse plaques often considered innocuous byproducts of Aβ accumulation. Dense and neuritic plaques show intense ThS staining and optical birefringence while diffuse plaques only stain weakly, if at all, with ThS, and lack the long-range alignment required for optical birefringence.17,42,43,45,46 To our knowledge, though, the specific origin of diffuse plaques and their relation to fibrillar plaques has remained unresolved. The above observation with fibrillar vs. oligomeric precipitates suggests that the corresponding Alzheimer plaques might originate from these two distinct amyloid aggregate species and, therefore, provide insights into the progression of amyloid aggregation in vivo. In light of the toxicity data presented next, it is worth noting that diffuse plaques are not associated with tissue damage in AD or Down syndrome patients.
2.3 Cell viability and altered cell morphology
Cell viability measurements were performed with adenocarcinomic human alveolar basal epithelial (A549) cells using an imaging live–dead fluorescence assay. Upon growing cells to confluence, the cells were exposed to either isolated fibrillar or oligomeric aggregates. Our preliminary experiments revealed the presence of highly toxic, non-fibrillar species present in late-stage fibril solutions. We isolated the corresponding LSOs from their fibril background and included them as separate oligomer species in subsequent toxicity studies. Cells were exposed to amyloid aggregates ranging in concentrations from 1 μM up to 100 μM, with the most prominent increase in toxicity between 5 μM and 25 μM. Together with various controls, Fig. 4A shows the relative toxicity of the two oligomeric vs. fibrillar amyloid species at 5 μM vs. 25 μM concentration. We found the toxicity of hewL RFs to be quite modest. Since RF plaques did not readily wash out and were intensely stained by the ethidium homodimer and Hoechst 33342 dyes we could not evaluate the viability of A549 cells directly located underneath RF plaques.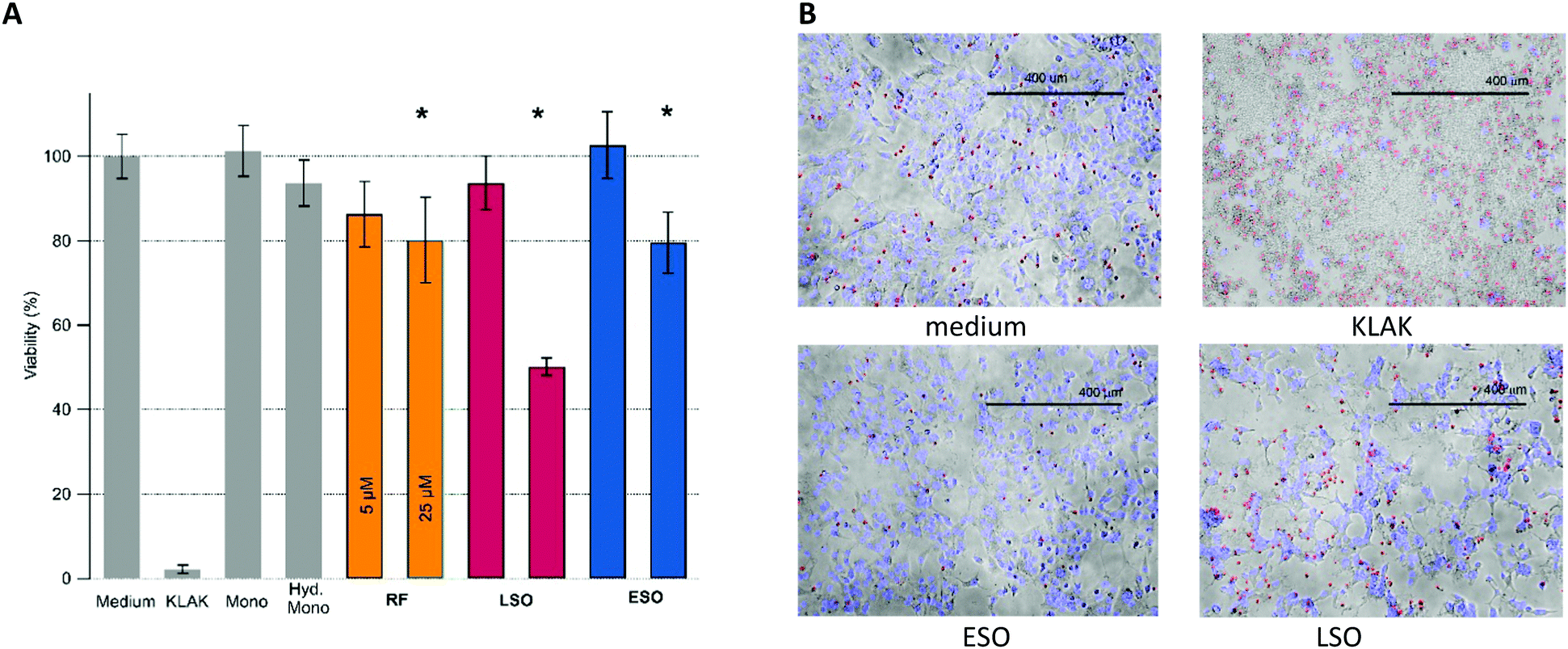 | ||
| Fig. 4 Amyloid induced cell toxicity and altered cell morphology and connectivity. (A) Viability of A549 cells exposed to controls (cell medium, apoptotic toxin KLAK (50 μM), native and hydrolyzed hewL monomers (25 μM) as well as RFs, LSOs, and ESOs at 5 and 25 μM, each. Error bars indicate one standard deviation (N = 5). Stars indicate p-values <0.05. (B) Superposition of brightfield and fluorescence images of cells exposed to controls, ESOs or LSOs. Images for RFs were comparable to those of ESOs. LSOs, in contrast, altered both cell morphologies and disrupted cell layer connectivity, leading to islands of crowded cells with large voids in between. Scale bar: 400 μm. | ||
Most striking, though, was the wide disparity in the relative toxicity of the two oligomeric species. While ESO/CF toxicity was comparable to the modest toxicity of RFs, LSOs were highly toxic. The imaging assay also revealed dramatic changes to the overall connectivity and organization of the confluent cell layer induced by LSOs. These were absent in cells exposed to either ESOs or RFs. Specifically, LSOs caused the formation of prominent voids in the confluent cell layer and a concurrent compaction of the remaining cells into misshapen clusters (Fig. 4B). The disruption of the overall connectivity by LSOs hints at their potential at disrupting epithelial cell layers in systemic amyloidoses and endothelial cell layers in vascular AD. It also indicates that multiple amyloid oligomer species with distinct biological activity can form under conditions supportive of amyloid fibril formation.
2.4 Origin of late-stage oligomers
One obvious question is what causes the emergence of LSOs under growth conditions very similar to those used to generate ESOs, the latter of which we had characterized in our prior work.36,37,39 In contrast to ESOs formed above the COC, neither ThT fluorescence nor dynamic light scattering measurements provided any indication for LSO formation during the lag phase of fibril growth under sigmoidal growth conditions (i.e. below the threshold for ESO formation). Toxic oligomers of IAPP and Aβ have been shown to be ThT negative but to form during the lag phase of fibril growth.32,47 We therefore explored whether hewL LSOs could have been formed throughout the assembly process but were invisible to ThT, or whether they were indeed late-stage products. In the latter case we wanted to determine whether LSOs emerged as direct result of fibril formation or were off-pathway from it. To probe the correlation between LSO and fibril formation, we utilized our observation that incubating solutions at decreasing surface-to-volume ratios accelerated fibril nucleation and growth, most likely due to preferential fibril nucleation at the air–water interface.We monitored the time course of LSO formation during fibril growth conditions identical to those used for cell toxicity experiments. To probe the correlation between RF and ESO formation, we incubated two identical sets of seven 1.4 mM hewL solutions at 50 mM NaCl. One set of solutions was incubated in individual 1.5 ml centrifuge tubes while the second set was distributed across 42 wells of a 96 well plate, filled with 250 μl of solution each. Each day, the incubation of one centrifuge tube and six 250 μl wells was stopped, with the latter combined into one 1.5 ml solution. LSO populations from these identical solution volumes were isolated following the same separation protocol used for cell toxicity measurements, and their concentrations measured using UV-absorbance. For the 1.5 ml solutions, we had observed that fibril nucleation and growth required at least 4 days of incubation. Incubation of the 250 μl wells in a fluorescence plate reader, in turn, allowed us to monitor the time course for fibril growth with ThT and relate it directly to the emergence of late-stage oligomers. Fig. 5A shows the time course for the build-up of LSOs in the single 1.5 ml vs. six 250 μl incubation volumes. The ThT signal monitoring RF fibril formation in the 96 well plates is shown as well. In both incubation volumes the onset of toxic oligomer formation is delayed by several days, with LSO concentrations increasing rapidly thereafter. LSOs formed at 1.5 ml solution volume do not become detectable until about 4 days of incubation, which is comparable to the onset of noticeable fibril formation under those conditions. In the 96 well plates, fibril growth rapidly accelerated already after 2 days. This, instead of accelerating, delayed and depressed LSO formation.
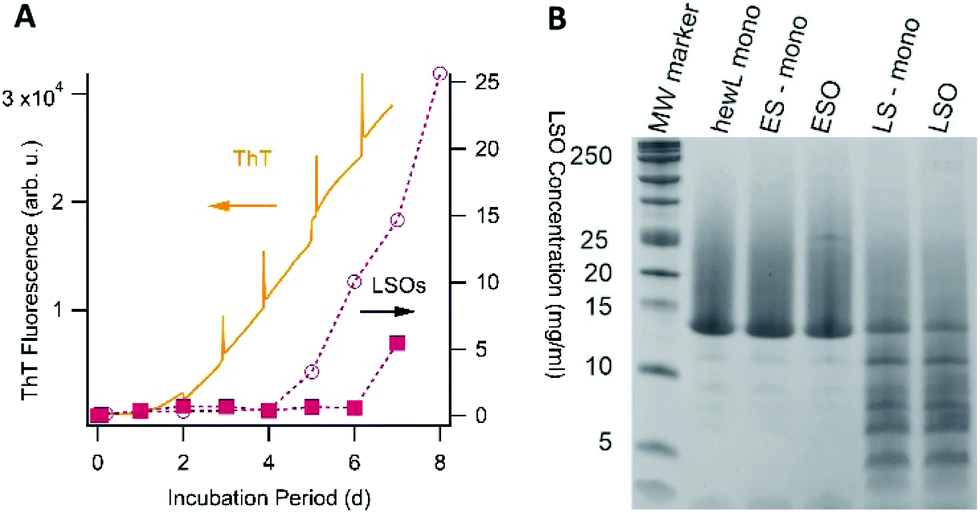 | ||
| Fig. 5 LSOs are off-pathway and are composed of full-length, partially hydrolyzed monomers. (A) Two sets of seven lysozyme solutions were incubated under identical fibril/LSO growth conditions, but at different volumes (1.5 ml or 250 μl), resulting in different lag phases for fibril formation. Each day one 1.5 ml solution and six 250 μl wells were removed, the solution from the 250 μl wells combined, and their oligomer content measured. LSO formation in the 1.5 ml volume (open circles) commenced after 4 day, comparable with the lag period required for fibril formation under those conditions. Fibril formation in the plate reader (monitored via ThT, orange trace) started after just two days, with LSOs (filled squares) only emerging at day 7. (B) Reducing SDS-PAGE gel of freshly dissolved lysozyme monomers (hewL-mono), isolated early stage monomers (ES-mono) and oligomers (ESO), and isolated late-stage monomers (LS-mono) and oligomers (LSO). MW markers (in kDa) are shown as reference. The identical intensity of LS-mono and LSO bands indicates that LSOs are not composed of a specific fragment but contain the nicked but full-length LS-monos. | ||
In addition, we confirmed that LSOs neither emerged as de-polymerization products of fibrils, nor did fibrils catalyze LSO formation from monomers. To do so, we seeded isolated RFs either in buffer alone (de-polymerization) or in buffer with added hewL monomers (catalytic oligomer formation) and incubated them under RF growth conditions for a time period of two days. This period was short enough to avoid “spontaneous” LSO formation from monomers. However, neither approach resulted in any detectable LSO formation (data not shown). Combining the above results, we conclude that toxic oligomers are neither fibril precursors, nor fibril de-polymerization products. In addition, fibrils do not catalyze toxic oligomer formation from monomers. These experiments imply that LSO represent late-stage, off-pathway amyloid oligomers emerging under fibril growth conditions, and competing for their shared pool of monomers to grow from. Hence, just as we had established previously for ESOs, LSO formation is off-pathway from fibril growth. However, they are distinct in their biological activity from off pathway ESOs formed under biphasic growth conditions.
The above data raise the question what causes the formation of this distinct oligomer species, and how to explain their delayed onset? One possibility is that LSOs form from either partially or fully hydrolyzed hewL monomers.37,48 HewL hydrolysis progresses slowly but steadily at the acidic pH and 52 °C incubation temperature used in our experiments. If so, LSOs might form because the monomers, while still tethered by their disulfide bonds, become partially nicked and acquire enhanced structural flexibility.48 Alternatively, LSOs might form from a fully hydrolyzed hewL fragment. To test these two alternatives, we determined the composition of LSOs and ESOs vs. the hydrolyzed monomers present at the same time point with SDS-PAGE under reducing conditions. Using reducing conditions identifies all hydrolyzed fragments, whether still tethered by disulfide bonds or not. Consistent with our prior observation, Fig. 5B indicates that ESOs form from the mostly intact monomers present during the early phases of incubation.37 In contrast, hewL monomers present during LSO formation are significantly hydrolyzed. Nevertheless, LSOs display the same distribution of hydrolysis fragments as the monomers present in solution. If a fully cleaved monomeric fragment were the building block for LSOs, this fragment would become enriched in LSOs. This implies that LSOs are formed from full-length but partially nicked monomers.
2.5 LSO characterization
Given the dramatic difference in the toxicity of ESOs vs. LSOs, we explored some of the basic biophysical, morphological, tinctorial, and immunological characteristics of this oligomer species in order to contrast it with the previously identified ESOs.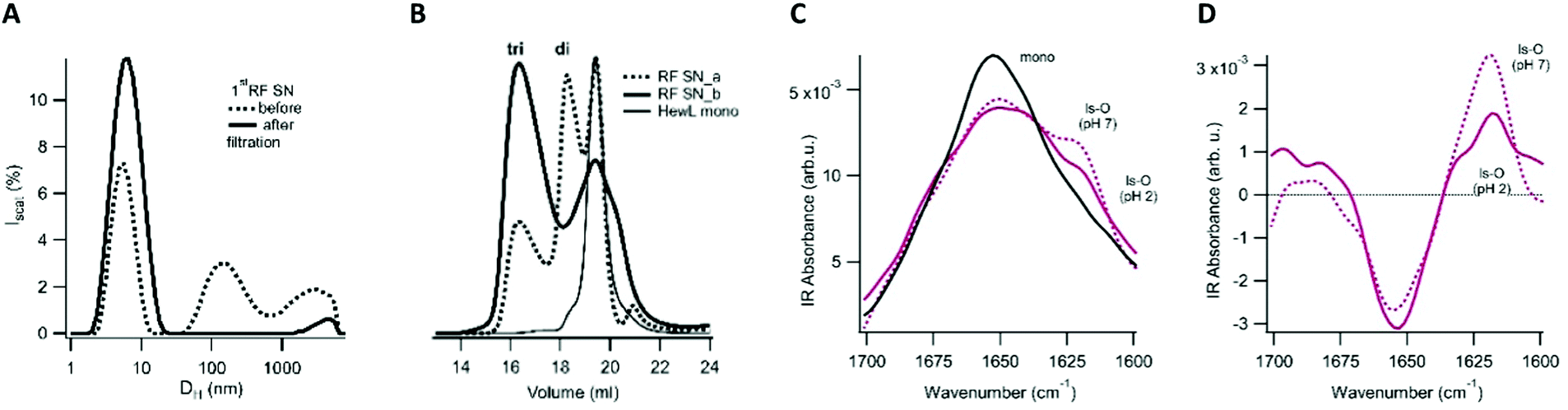 | ||
| Fig. 6 Size and structural features of LSOs. Characterization of 1st supernatant (SN) from centrifugation of RFs grown under sigmoidal growth conditions. (A) DLS particle size distribution before (dotted line) and after (solid line) filtration through a 50 nm syringe filter to remove residual fibrils. The hydrodynamic diameter DH of the first peak suggests it contains a mixture of hewL monomers and small oligomers (B) FPLC of 1st supernatant (SN) from hewL fibrils grown for either (a) 6 or (b) 8 days. Dependent on the incubation period, one or two larger aggregates peaks emerge. Both SNs show a monomer peak and some hydrolyzed fragments. Aggregate peaks are consistent with hewL dimers (di) and trimers (tri). (C) Amide-I band of the FTIR spectra for LSO grown at pH 2 and after transfer to pH 7, compared to hewL monomers. (D) Difference spectra of LSOs following subtraction of monomer reference spectra. A positive/negative value indicates an increase/decrease over the corresponding value over the monomer reference spectrum. | ||
Staining with the oligomer-selective antibody A11 antibody is a common assay to confirm the presence of amyloid oligomers.57,58 For the dot-blots, the amount of hewL RFs deposited had to be titrated to account for the much stronger absorption and subsequent retention of RFs on the nitrocellulose membrane than for all other protein samples (see Methods for details). A11 did show noticeable cross-reactivity with hewL RFs, which we presume is due to the same non-specific stickiness the fibrils displayed for the nitrocellulose membrane, and for cells in the toxicity assay. Nevertheless, the A11 antibody preferentially recognized LSOs and reacted only very weakly with ESOs (Fig. 7). This lines up with the difference in cellular toxicity and supports an underlying structural difference between these two oligomer species.
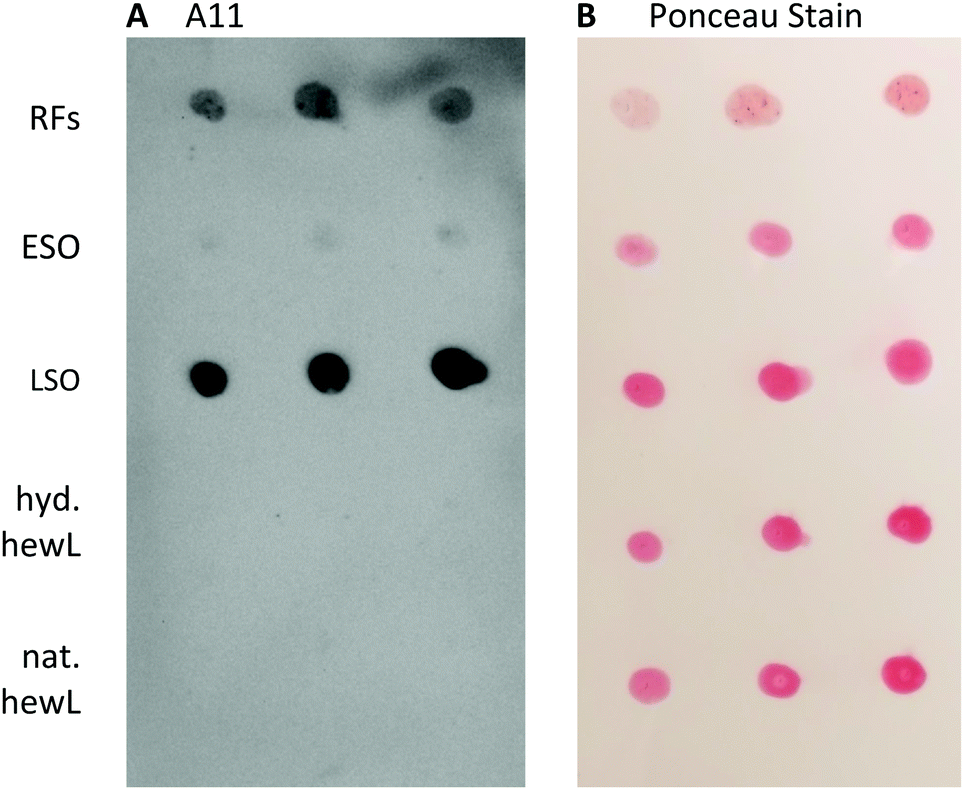 | ||
| Fig. 7 Immuno-staining of early vs. late stage amyloid oligomers. Dot blots of different amyloid aggregates and controls at three different concentrations. (A) Reactivity of A11 with RFs (top row) and late-stage oligomers (middle row). (B) Ponceau staining of all samples blotted onto membrane. Concentrations for the various samples that yielded equivalent protein retention were (from left to right) RFs: 2.0, 1.0 and 0.5 μg; all others: 12, 8 and 4 μg. | ||
Tinctorial profiles
Recently, differences in the emission of multiple fibril-specific dyes have been used to identify different fibril polymorphs isolated from patients with familiar vs. sporadic Alzheimer's.59 Here we extended this approach by using a combination of fibril-selective (ThT, FSB and curcumin) and oligomer-sensitive (bis-ANS, crystal violet) fluorescent dyes to determine the fluorescence profiles of ESOs and LSOs relative to those of hewL RFs. Bis-ANS has been recognized as oligomer-selective by multiple investigators.60–62 Our lab recently identified crystal violet as another oligomer-selective dye.63 As expected, the fibril-selective amyloid dyes all showed significantly higher fluorescence enhancements upon binding to RFs than to ESOs or LSOs at the same concentration (Fig. 8). While quite different in amplitude to RF responses, fibril dye fluorescence was enhanced by both LSOs and ESOs. This suggests that fibrils and oligomers do share some structural characteristics, most likely the formation of intermolecular hydrogen bonds. The modest but noticeable ThT response evoked by LSOs, combined with the absence of a ThT increase in the lag phase of sigmoidal fibril growth, further supports our contention that LSOs are absent during the lag phase of sigmoidal fibril growth. In contrast to the fibril-selective dyes, fluorescence responses of bis-ANS and crystal violet to ESOs and LSOs were similar to those evoked by RFs. The significant hydrophobicity of RFs might underlie their inherent tendency to form sheet-like plaques upon their decrease in charge repulsion with increasing pH. For all dyes, though, ESOs and LSOs show clearly distinguishable fluorescence enhancements. Several of the dyes also display modest but noticeable changes in their peak emission wavelength or spectral shape in the presence of the three aggregate species (see inserts). The enhanced response to LSOs vs. ESOs by the environmentally sensitive dye bis-ANS and crystal violet points to an underlying difference in the hydrophobicity of these two oligomeric species that likely correlates with their noticeable differences in cell toxicity.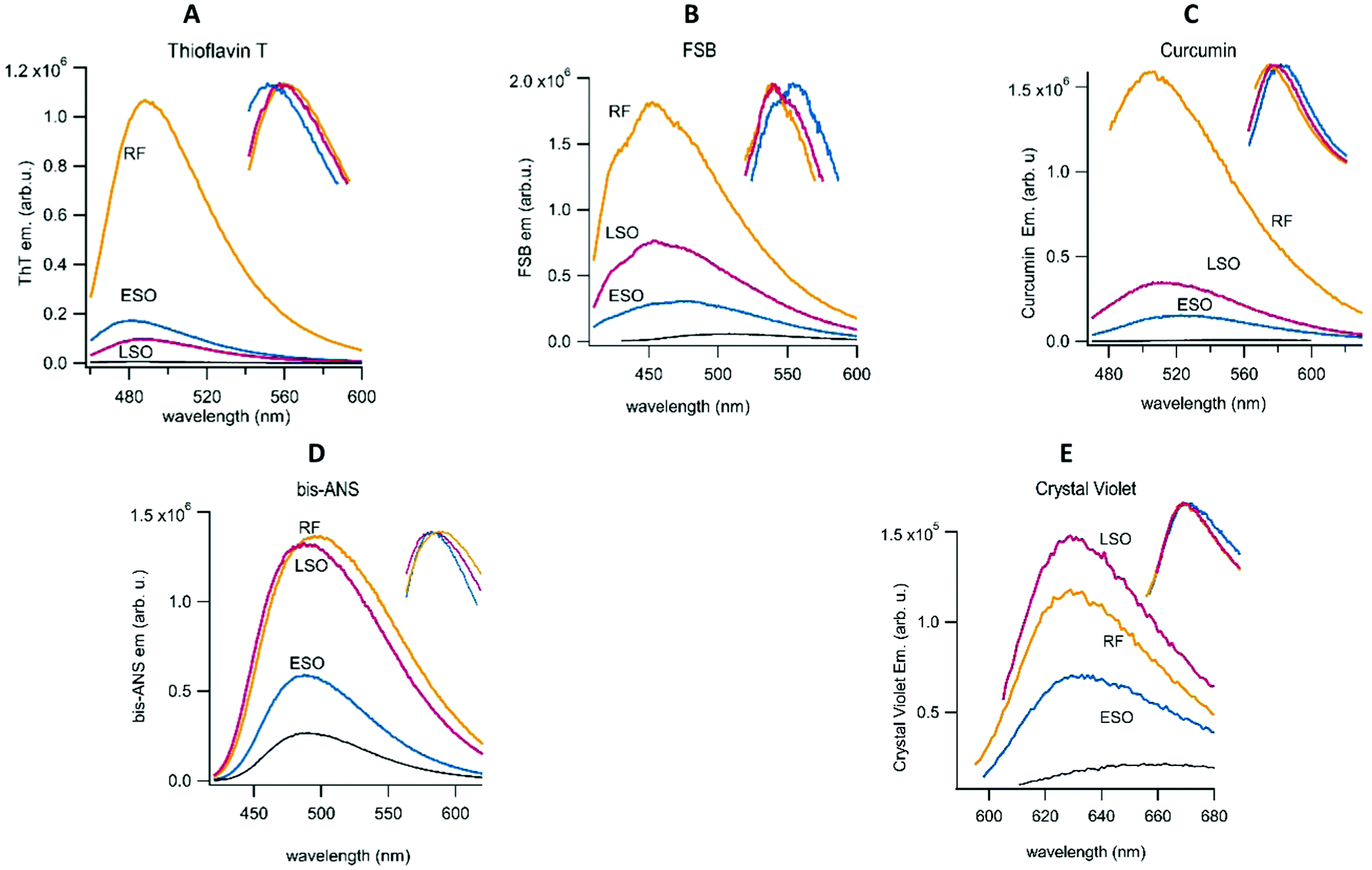 | ||
| Fig. 8 Responses of fibril- vs. oligomer-sensitive dyes to RFs, ESOs and LSOs. Fluorescence emission spectra of (A) the amyloid indicator dyes thioflavin T (B) the fluorescent Congo Red derivative FSB and (C) the amyloid binding dye curcumin (D) the environment-sensitive dye bis-ANS and (E) the oligomer dye crystal violet. Dye and aggregate concentrations were typically 15 μM and 1 mg ml−1, respectively. Monomer reference spectra are shown in black. Spectral shifts to the emission peaks are highlighted in the insert to each spectrum. | ||
3. Discussion and conclusion
Our experiments indicate that slight changes in the conditions supportive of amyloid fibril formation by lysozyme can generate two distinct conformers of amyloid oligomers we have labelled early-stage (ESOs) and late-stage oligomers (LSOs). ESOs emerge as “precursor” to fibrils during the lag phase of fibril formation. We have previously shown that the process of ESO formation in lysozyme is identical to that observed with amyloid-β, that they are off-pathway from fibril formation, and that their emergence requires the crossing of a threshold concentration we called the “critical oligomer concentration” or COC.19,36 We further confirmed that ESOs and curvilinear fibrils (CFs) of both Aβ and lysozyme actively inhibit secondary nucleation of amyloid fibrils by binding to the lateral surfaces of fibril seeds.23 In contrast to ESOs, LSOs require extended incubation at concentrations below the COC of ESOs. For some other amyloid proteins, such as IAPP, toxic oligomers emerged without ThT response during the lag phase of fibril growth. LSOs formed by hewL, instead, did not form during the lag phase of fibril growth.47 While overlapping with the time course for fibril formation, our time-resolved measurements of RF vs. LSO formation indicate that LSOs, just as their ESO counterparts, are forming off-pathway during fibril growth. Reducing SDS-PAGE indicates that ESOs and LSOs are both composed of full-length lysozyme instead of some hydrolysis-induced peptide fragment. However, while ESOs assemble from essentially intact monomers, LSOs are formed from severely nicked monomers. This matches with prior results documenting hydrolysis effects on lysozyme fibril formation upon extended incubation at pH 2.48Isolating each of the three different amyloid aggregate species (ESOs, LSOs and RFs) from monomers, and from each other, allowed us to assess their relative cell toxicity. Interestingly, while ESOs and RFs were only mildly noxious, LSOs proved to be highly toxic to cells. There are two prior reports of lysozyme oligomers formed after weeks of incubation under acidic growth conditions. These oligomers were shown to be highly toxic to pheochromocytoma (PC12) cells64 and to induce toxicity and tau hyperphosphorylation in cortical neuronal slices at levels comparable to Aβ42 oligomers.65 We suspect that these oligomers were similar to the LSOs generated here. However, the growth temperature and incubation periods were sufficiently different to prevent a direct comparison. The authors also did not indicate whether pH transfer affected the structure of their aggregates.
The interpretation of aggregate toxicity is complicated by the inherent changes in aggregate structures upon transfer from pH 2 growth conditions to cell medium, but also provide some intriguing parallels to AD plaques. The precipitates of hewL RFs and ESOs closely resemble the morphological, optical and tinctorial characteristics of neuritic vs. diffuse plaques seen in post-mortem AD histology. The fact that hewL ESOs emerge as precursors to RFs also matches with the observation that diffuse plaques are considered benign precursors of late-stage neuritic plaques.43 At the same time, the rapid assembly of RFs and ESO/CFs into larger structures might underlie the low levels of resulting cytotoxicity. Imaging the morphology of the cell layer revealed that LSOs not only were considerably more toxic than either RFs or ESOs, it also indicated that the former significantly altered cell morphology and cell layer connectivity. This effect suggests that amyloid toxicity measurements should not only account for single-cell responses but for the changes in tissue structures often seen in pathology. It is worth mentioning that the precipitation of LSOs proceeded relatively slowly, suggesting that significant populations of small oligomers remained in solution during cell exposure. While we suspect that the underlying structural differences between ESOs and LSOs are the main reason for their distinct cell toxicity, we can't exclude that the precipitation and differences in solubility upon transfer to cell medium contribute to the observed differences in cell toxicity. It is interesting to note, though, that amyloid formation in cells is likely to involve different cellular compartments with distinct environments. Hence those differences in morphology under different growth conditions might well be physiologically relevant.
Generating two oligomer polymorphs with distinct biological activity allowed us to investigate to what extend they share, or differ in, various characteristics associated with amyloid oligomers, and to explore the mechanism(s) promoting their formation. The molecular weights of both oligomer species fall in the range of just a few monomers, with ESOs estimated to contain eight monomers,39 while FLPC of LSOs suggested that they are as small as two to three monomers. Among the characteristics distinguishing these oligomers are their relative propensities towards further polymerization. While ESOs readily polymerize into curvilinear fibrils,39 also often referred to as protofibrils, LSOs did not form any discernible polymeric assemblies under their acidic growth conditions. Similar to our prior observations with ESOs, the infrared spectra of LSOs do contain amyloid-like intermolecular hydrogen bonds, as indicated by their peaks in the amyloid region (1610–1630 cm−1) of the amide-I bands.50 Equally important, a weak peak around 1680–90 cm−1, considered indicative of anti-parallel β-sheets, is present as well.51,52 In contrast to populations of ordered vs. disordered oligomers observed with α-synuclein, and of disordered oligomers of Aβ40,66,67 both LSOs and ESOs appear to be structured.
The fluorescence responses of fibril-selective vs. oligomer-sensitive dyes support several conclusions. Both ESOs and LSOs induce relatively muted fluorescence enhancements of fibril-selective dyes. At the same time, the fluorescence of the oligomer-specific dyes bis-ANS and crystal violet show robust enhancements in the presence of ESOs and LSOs. This pattern is characteristic of amyloid oligomers, in general, which are structurally distinct from amyloid fibrils. Equally important, LSOs consistently evoke more prominent fluorescence responses from both crystal violet and bis-ANS than ESOs. The enhanced response to bis-ANS and the smaller size of LSOs vs. ESOs matches with suggestions that increased hydrophobicity and reduced size of amyloid oligomers correlates with increases in cell toxicity.68,69 The difference in LSO vs. ESO structure and size as underlying cause for their distinct cellular toxicity is further buttressed by the preferential response of the anti-oligomer antibody A11 to LSOs.
Our in vitro experiments indicate that two distinct amyloid oligomer species can emerge under nearly identical solution conditions. This, in turn, raises the question what mechanisms regulate the formation of these distinct oligomer species? In the case of ESOs, we had previously shown that (a) lysozyme needed to be partially unfolded, and (b) ESO formation was limited by a protein/salt-dependent threshold related to the inherent charge repulsion among the monomers.36 In contrast, LSOs require significant partial hydrolysis of lysozyme monomers. We hypothesize that it is this increased flexibility and concurrent exposure of hydrophobic residues of the partially hydrolyzed monomers that are essential requirements for the assembly into LSOs. Interestingly, the point mutants of lysozyme implicated in human lysozyme amyloidosis, while structurally identical to the native state, also display increased structural flexibility and reduced thermal stability.70 The partial hydrolysis of hewL monomers under our growth conditions seems to provide an alternative way to promote such structural flexibility. This would imply that the flexibility of the amyloid monomer plays an important role in the type of amyloid oligomers that will assembly from them. Our data suggested that LSO are formed exclusively from hydrolyzed monomers and not as depolymerization product of fibrils. However, we can't exclude that prolonged incubation of RFs or changes in solution conditions, as described for β-microglobulin, could result in the release of monomers capable of forming “plaque-associated” LSOs.71 It is intriguing that folded proteins implicated in amyloidoses are all extracellular, and therefore more prone to degradation and enhanced structural flexibility during their life cycle.
Overall, the ability to generate two distinctly different and homogeneous oligomer populations without the use of denaturants or drugs can provide insights into the in vivo conditions promoting the formation of either relatively innocuous or highly toxic oligomer species. This could open up new approaches in targeting cellular pathways underlying pathogenic oligomer formation.
4. Materials and methods
4.1 Chemicals and cell lines
Two times recrystallized, dialyzed and lyophilized hen egg white lysozyme (hewL) was purchased from Worthington Biochemicals (Lakewood NJ). Ultrapure grade thioflavin T (ThT) was obtained from Anaspec (Freemont, CA). Bis-ANS was obtained from Tocris Bioscience (Minneapolis, MN) and FSB from Calbiochem (San Diego, CA). Other chemicals were from Fisher Scientific (Pittsburgh, PA) and were reagent grade or better. All solutions were prepared using 18 MΩ water from a reverse osmosis unit (Barnstead E-pure, Dubuque, IA). A549 cells for toxicity were obtained from American Type Culture Collection (Manassas, VA).4.2 Growth and separation of amyloid aggregates
The amyloid aggregate species generated from hewL included rigid fibrils (RFs), early-stage oligomers (ESOs) and their associated curvilinear fibrils (CFs), as well as late-stage oligomers (LSOs) were all formed at acidic pH. Solution pH was established using either 25 mM KH2PO4 buffer or by adjusting the pH of the water/protein/salt solutions to pH 2 by adding small amounts of hydrochloric acid. hewL was at a fixed concentration of 1.4 mM in the pH 2, 50 mM NaCl solution (for formation of RFs and LSOs) or 250 mM NaCl (for formation of ESO/CFs). Solutions were filtered through 220 nm PVDF syringe filters (Fisherbrand, Fisher Scientific) prior to incubation at 52 °C. Protein samples with 50 mM NaCl were incubated for 6–8 days for RF formation while 250 mM NaCl hewL solutions were incubated for 2–8 hours and ESO/CF formation monitored with dynamic light-scattering (DLS). For quantification of their toxicity, individual aggregate species were isolated from monomers via centrifugation and/or use of molecular weight cut-off (MWCO) filters.000 g and 15 °C. The RF supernatant (SN) and RF pellets were collected. Aggregate pellets were re-suspended in their respective solutions and spun again for 22–24 hours. This process was repeated until the monomer concentration in the supernatant was less than 1% of the aggregate concentration in the pellet. This typically required 3 cycles.
000 g for 20 min. The filtrate after the first spin was collected for subsequent isolation of hydrolyzed monomers. The residue on top of the filter was washed adding 300 μl of 50 mM NaCl pH 2 solution. After three washes, the filter was flipped, put into a new centrifuge tube and spun at 1000 g for 5 minutes to collect isolated toxic oligomers.
Isolation of hydrolyzed monomers. The filtrate after the 1st spin of the RF supernatant through the 50 kDa MWCO filter was used to isolate hydrolyzed monomers. This filtrate was brought to pH 7 in a two-step process: it was first diluted in DMEM no serum media in 1
:1 proportion (500 μl DMEM and 500 μl of the filtrate) and spun for 22–24 h at 15000 g. The resultant supernatant was collected and filtered through 30 kD MWCO filter for 20 min at 15000 g. The residue on the top of the filter was collected and the pH adjustment completed by adding 300 μl DMEM to the top of the filter. To remove toxic oligomers induced by the pH transfer, this suspension was filtered again through a 50 kDa MWCO filter at 15000 g for 20 min, and the filtrate was collected.
4.3 Cell viability assay
The cytotoxicity of the various hewL aggregates was investigated using adenocarcinomic human alveolar basal epithelial cells (A549). Cell viability was assessed using a live/dead fluorescence assay. A549 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (Gibco), 1% antibiotic–antimycotic (100×) and 2 g l−1 NaHCO3. Cells were maintained in a 5% CO2 humidified atmosphere at 37 °C and seeded at a density of 1.5 × 104 cells per well in 96 well plates. After 24 hours, the cell medium was replaced with DMEM without FBS and the plate incubated for another 24 hours. HewL aggregates were added to the cells and incubated for 48–72 hours. Dilutions of all aggregates were chosen so that the same dilutions of the growth medium (50 or 250 mM NaCl at pH 2) with the same concentrations of monomeric hewL did not affect cell viability. Cell medium was used as negative control and the apoptotic peptide toxin KLAK73 at 50–100 μM was used as positive toxicity control. After washing the cells twice in PBS, any dead cells were stained with 4 μM of the membrane impermeable nuclear stain ethidium homodimer-1 (Life Technologies) for 1 h at 37 °C. Subsequently, live cells were stained with the cell permeant dye Hoechst 33342 (NucBlue, Life Technologies) for 30 minutes. Five non-overlapping regions were imaged from each well using an EVOS inverted fluorescence microscope at 10× magnification. NucBlue and ethidium homodimer fluorescence were imaged using a DAPI and RFP filter cube, respectively. Both fluorescence images were overlayed with brightfield images of the same region. Live/dead cell counting was performed using NIH ImageJ. Fluorescence and bright-field images in Fig. 4 were adjusted for contrast and intensity for display purposes.4.4 FPLC and SDS PAGE analysis
For fast liquid protein chromatograph (FPLC) 1–2 mg of protein was dissolved in 500 μl of DI water with 50 mM NaCl, adjusted to pH 2 with HCl, as running buffer. Samples were injected into a Superdex 75 10/300 GL column (GE Healthcare) mounted on an ATKA PURE (GE Healthcare) FPLC. The column was typically rinsed with 2 column volumes of DI water, and then washed with running buffer for one column volume before injection of the sample. For separation, UV absorption was monitored at 280 nm during column filtration and samples were fractionated into test tubes.For reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE), samples of fresh monomers, early-stage oligomers ESOs) and late-stage oligomers (LSOs) were prepared and isolated as described above. All samples were analyzed using 16.5% gradient Tris-tricine gels (Criterion, Bio-Rad) with an SDS running buffer without glycine. An aliquot of each sample (2 mg ml−1) was mixed with Tricine sample buffer (Bio-Rad) in the presence of 2% (v/v) 2-mercaptoethanol (Fisher Scientific) in 1:1 ratio and boiled for 5 minutes at 90 °C. 30 μl of each sample was loaded onto the gel and run for about 1.5 hours at 105 V. The gel was fixed in a solution of 40% methanol and 10% acetic acid for 30 minutes, stained for 2 hours using Coomassie blue (G-250 stain, Bio-Rad) for 1 hour, and was washed overnight with DI water.
4.5 Antibody dot blots
HewL monomer, and isolated RFs, ESO/CFs, LSOs and hydrolyzed monomer (2 μl each) were spotted on nitrocellulose membranes (Thermofisher Pierce). HewL RFs absorbed onto the nitrocellulose membrane strongly while all other aggregates became diluted during washing steps. Using Ponceau staining, we adjusted the amount of protein deposited to obtain comparable total protein retained by the membrane. Membranes were blocked with 7% milk in 1× Tris-buffered saline (TBS) at room temperature for 1 h. After three washing steps in 1× TBST (5 min each), membranes were incubated overnight at 4 °C with 1:1000 dilution of the amyloid oligomer antibody A11 (Novus Biologicals). Membranes were washed in 1× TBST for 3 times (5 min each) and were incubated in 1:1000 dilution of Ig goat anti-rabbit antibody (Southern biotech) for 1 h at room temperature. After 1 h, membranes were washed in 1× TBST for 3 times and once in 1× TBS. Then, ECL western blotting substrate (Thermo Fisher Pierce) was added for detection. Sample photoluminescence was detected using a Chemidoc imager (Bio-Rad).
4.6 Tinctorial profiles
The fluorescence emission spectra of RFs, ESOs and LSOs relative to monomers were obtained using the fibril-selective dyes Thioflavin T, FSB and curcumin,59 as well as the oligomer-selective dyes bis-ANS and Crystal Violet.62,63 Concentrations of dyes used were typically 15 μM and aggregate concentrations were adjusted to 1 mg ml−1. Dye emissions for aggregates were excited at 445 nm for ThT, 377 (ESO) or 395 nm for FSB, 449 nm for curcumin, 395 nm for bis-ANS, and 583 (ESO) or 593 nm for CV, respectively, and emission spectra collected using a spectrofluorometer (Fluoromax-4, Horiba Instruments, USA) using excitation/emission slit width of 5 mm each.Author contributions
C. N., J. B., N. B., K. K., F. F. and G. S. all participated in the investigation and formal analysis of the data. They also reviewed and edited the manuscript. P. K. provided input to the methodology and resources. M. M. conceptualized the project, secured funding, provided resources and wrote the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge support and helpful suggestions from Drs. Laura Blair and Shannon Hill with the immunostaining assays. We are also grateful for access to the Chemidoc Imager by Dr Kristina Schmidt's and the FPLC instrumentation of Dr Jianjun Pan. This research was funded, in part, by NIH grant 2R15GM097723-02.References
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188–1203 CrossRef CAS.
- J. N. Buxbaum, Curr. Opin. Rheumatol., 2004, 16, 67–75 CrossRef CAS PubMed.
- C. M. Dobson, Trends Biochem. Sci., 1999, 24, 329–332 CrossRef CAS PubMed.
- M. Fändrich and C. M. Dobson, EMBO J., 2002, 21, 5682–5690 CrossRef PubMed.
- M. B. Pepys, Annu. Rev. Med., 2006, 57, 223–241 CrossRef CAS PubMed.
- E. N. Cline, M. A. Bicca, K. L. Viola and W. L. Klein, J. Alzheimer's Dis., 2018, 64, S567–S610 CAS.
- U. Sengupta, A. N. Nilson and R. Kayed, EBioMedicine, 2016, 6, 42–49 CrossRef.
- R. Kayed and C. A. Lasagna-Reeves, J. Alzheimer's Dis., 2013, 33, S67–S78 Search PubMed.
- E. K. Pickett, R. M. Koffie, S. Wegmann, C. M. Henstridge, A. G. Herrmann, M. Colom-Cadena, A. Lleo, K. R. Kay, M. Vaught, R. Soberman, D. M. Walsh, B. T. Hyman and T. L. Spires-Jones, J. Alzheimer's Dis., 2016, 53, 787–800 CAS.
- F. Hefti, W. F. Goure, J. Jerecic, K. S. Iverson, P. A. Walicke and G. A. Krafft, Trends Pharmacol. Sci., 2013, 34, 261–266 CrossRef CAS.
- R. M. Koffie, M. Meyer-Luehmann, T. Hashimoto, K. W. Adams, M. L. Mielke, M. Garcia-Alloza, K. D. Micheva, S. J. Smith, M. L. Kim, V. M. Lee, B. T. Hyman and T. L. Spires-Jones, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4012–4017 CrossRef CAS PubMed.
- R. Tycko, Neuron, 2015, 86, 632–645 CrossRef CAS PubMed.
- W. Qiang, K. Kelley and R. Tycko, J. Am. Chem. Soc., 2013, 135, 6860–6871 CrossRef CAS PubMed.
- W. Close, M. Neumann, A. Schmidt, M. Hora, K. Annamalai, M. Schmidt, B. Reif, V. Schmidt, N. Grigorieff and M. Fändrich, Nat. Commun., 2018, 9, 699 CrossRef.
- P. Liu, M. N. Reed, L. A. Kotilinek, M. K. O. Grant, C. L. Forster, W. Qiang, S. L. Shapiro, J. H. Reichl, A. C. A. Chiang, J. L. Jankowsky, C. M. Wilmot, J. P. Cleary, K. R. Zahs and K. H. Ashe, Cell Rep., 2015, 11, 1760–1771 CrossRef CAS PubMed.
- R. Kayed, I. Canto, L. Breydo, S. Rasool, T. Lukacsovich, J. Wu, R. Albay, A. Pensalfini, S. Yeung, E. Head, J. L. Marsh and C. Glabe, Mol. Neurodegener., 2010, 5, 57 CrossRef CAS PubMed.
- F. Hasecke, T. Miti, C. Perez, J. Barton, D. Schölzel, L. Gremer, C. S. R. Grüning, G. Matthews, G. Meisl, T. P. J. Knowles, D. Willbold, P. Neudecker, H. Heise, G. Ullah, W. Hoyer and M. Muschol, Chem. Sci., 2018, 9, 5937–5948 RSC.
- S. Higashi, E. Iseki, R. Yamamoto, M. Minegishi, H. Hino, K. Fujisawa, T. Togo, O. Katsuse, H. Uchikado, Y. Furukawa, K. Kosaka and H. Arai, Brain Res., 2007, 1184, 284–294 CrossRef CAS PubMed.
- M. J. Guerrero-Muñoz, D. L. Castillo-Carranza, S. Krishnamurthy, A. A. Paulucci-Holthauzen, U. Sengupta, C. A. Lasagna-Reeves, Y. Ahmad, G. R. Jackson and R. Kayed, Neurobiol. Dis., 2014, 71, 14–23 CrossRef PubMed.
- T. L. Spires-Jones, J. Attems and D. R. Thal, Acta Neuropathol., 2017, 134, 187–205 CrossRef CAS.
- F. Hasecke, C. Niyangoda, G. Borjas, J. Pan, G. Matthews, M. Muschol and W. Hoyer, Angew. Chem., Int. Ed., 2021, 60, 3016–3021 CrossRef CAS PubMed.
- D. Puzzo, R. Piacentini, M. Fá, W. Gulisano, D. D. Li Puma, A. Staniszewski, H. Zhang, M. R. Tropea, S. Cocco, A. Palmeri, P. Fraser, L. D'Adamio, C. Grassi and O. Arancio, eLife, 2017, 6, e26991 CrossRef PubMed.
- S. I. A. Cohen, M. Vendruscolo, M. E. Welland, C. M. Dobson, E. M. Terentjev and T. P. J. Knowles, J. Chem. Phys., 2011, 135, 065105 CrossRef PubMed.
- T. P. J. Knowles, C. A. Waudby, G. L. Devlin, S. I. A. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, Science, 2009, 326, 1533–1537 CrossRef CAS PubMed.
- S. I. A. Cohen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, in Amyloid Fibrils and Prefibrillar Aggregates, ed. D. E. Otzen, Wiley-VCH, Weinheim, Germany, 2013, pp. 183–209 Search PubMed.
- U. Sengupta, N. Puangmalai, N. Bhatt, S. Garcia, Y. Zhao and R. Kayed, Mol. Neurobiol., 2020, 57, 2741–2765 CrossRef CAS PubMed.
- S. Massimo, Curr. Protein Pept. Sci., 2010, 11, 343–354 CrossRef PubMed.
- L. Breydo and V. N. Uversky, FEBS Lett., 2015, 589, 2640–2648 CrossRef CAS PubMed.
- G. M. Shankar, S. Li, T. H. Mehta, A. Garcia-Munoz, N. E. Shepardson, I. Smith, F. M. Brett, M. A. Farrell, M. J. Rowan, C. A. Lemere, C. M. Regan, D. M. Walsh, B. L. Sabatini and D. J. Selkoe, Nat. Med., 2008, 14, 837–842 CrossRef CAS PubMed.
- J. Lee, E. K. Culyba, E. T. Powers and J. W. Kelly, Nat. Chem. Biol., 2011, 7, 602–609 CrossRef CAS PubMed.
- T. R. Serio, A. G. Cashikar, A. S. Kowal, G. J. Sawicki, J. J. Moslehi, L. Serpell, M. F. Arnsdorf and S. L. Lindquist, Science, 2000, 289, 1317–1321 CrossRef CAS PubMed.
- D. L. Brody, H. Jiang, N. Wildburger and T. J. Esparza, Alzheimer's Res. Ther., 2017, 9, 62 CrossRef PubMed.
- W. F. Goure, G. A. Krafft, J. Jerecic and F. Hefti, Alzheimer's Res. Ther., 2014, 6, 42 CrossRef PubMed.
- T. Miti, M. Mulaj, J. D. Schmit and M. Muschol, Biomacromolecules, 2015, 16, 326–335 CrossRef CAS PubMed.
- J. Foley, S. E. Hill, T. Miti, M. Mulaj, M. Ciesla, R. Robeel, C. Persichilli, R. Raynes, S. Westerheide and M. Muschol, J. Chem. Phys., 2013, 139, 121901 CrossRef PubMed.
- S. E. Hill, T. Miti, T. Richmond and M. Muschol, PLoS One, 2011, 6, e18171 CrossRef CAS PubMed.
- S. E. Hill, J. Robinson, G. Matthews and M. Muschol, Biophys. J., 2009, 96, 3781–3790 CrossRef CAS PubMed.
- T. C. Dickson and J. C. Vickers, Neuroscience, 2001, 105, 99–107 CrossRef CAS PubMed.
- S. Han, M. Kollmer, D. Markx, S. Claus, P. Walther and M. Fändrich, Sci. Rep., 2017, 7, 43577 CrossRef PubMed.
- J. C. Morris, M. Storandt, D. W. J. McKeel, E. H. Rubin, J. L. Price, E. A. Grant and L. Berg, Neurology, 1996, 46, 707–719 CrossRef CAS PubMed.
- C. A. Lemere, J. K. Blusztajn, H. Yamaguchi, T. Wisniewski, T. C. Saido and D. J. Selkoe, Neurobiol. Dis., 1996, 3, 16–32 CrossRef CAS PubMed.
- E. Head, I. T. Lott, D. M. Wilcock and C. A. Lemere, Curr. Alzheimer Res., 2016, 13, 18–29 CrossRef CAS PubMed.
- D. P. Perl, Mt. Sinai J. Med., 2010, 77, 32–42 CrossRef PubMed.
- D. Hayne, S. Lim and P. Donnelly, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC.
- A. Abedini, A. Plesner, P. Cao, Z. Ridgway, J. Zhang, L.-H. Tu, C. T. Middleton, B. Chao, D. J. Sartori, F. Meng, H. Wang, A. G. Wong, M. T. Zanni, C. B. Verchere, D. P. Raleigh and A. M. Schmidt, eLife, 2016, 5, e12977 CrossRef PubMed.
- R. Mishra, K. Sörgjerd, S. Nyström, A. Nordigården, Y.-C. Yu and P. Hammarström, J. Mol. Biol., 2007, 366, 1029–1044 CrossRef CAS PubMed.
- E. Frare, P. Polverino de Laureto, J. Zurdo, C. M. Dobson and A. Fontana, J. Mol. Biol., 2004, 340, 1153–1165 CrossRef CAS PubMed.
- G. Zandomeneghi, M. R. H. Krebs, M. G. McCammon and M. Fändrich, Protein Sci., 2004, 13, 3314–3321 CrossRef CAS PubMed.
- E. Cerf, R. Sarroukh, S. Tamamizu-Kato, L. Breydo, S. Derclaye, Y. F. Dufrêne, V. Narayanaswami, E. Goormaghtigh, J.-M. Ruysschaert and V. Raussens, Biochem. J., 2009, 421, 415–423 CrossRef CAS PubMed.
- J.-M. Ruysschaert and V. Raussens, Methods Mol. Biol., 2018, 1777, 69–81 CrossRef CAS PubMed.
- J. C. Stroud, C. Liu, P. K. Teng and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7717–7722 CrossRef CAS PubMed.
- L. Zhou and D. Kurouski, Anal. Chem., 2020, 92, 6806–6810 CrossRef CAS PubMed.
- A. Laganowsky, C. Liu, M. R. Sawaya, J. P. Whitelegge, J. Park, M. Zhao, A. Pensalfini, A. B. Soriaga, M. Landau, P. K. Teng, D. Cascio, C. Glabe and D. Eisenberg, Science, 2012, 335, 1228–1231 CrossRef CAS PubMed.
- M. I. Apostol, K. Perry and W. K. Surewicz, J. Am. Chem. Soc., 2013, 135, 10202–10205 CrossRef CAS PubMed.
- R. Kayed, E. Head, F. Sarsoza, T. Saing, C. Cotman, M. Necula, L. Margol, J. Wu, L. Breydo, J. Thompson, S. Rasool, T. Gurlo, P. Butler and C. Glabe, Mol. Neurodegener., 2007, 2, 18 CrossRef PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- C. Condello, T. Lemmin, J. Stöhr, M. Nick, Y. Wu, A. M. Maxwell, J. C. Watts, C. D. Caro, A. Oehler, C. D. Keene, T. D. Bird, S. G. van Duinen, L. Lannfelt, M. Ingelsson, C. Graff, K. Giles, W. F. DeGrado and S. B. Prusiner, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E782–E791 CrossRef CAS PubMed.
- B. Bolognesi, J. R. Kumita, T. P. Barros, E. K. Esbjorner, L. M. Luheshi, D. C. Crowther, M. R. Wilson, C. M. Dobson, G. Favrin and J. J. Yerbury, ACS Chem. Biol., 2010, 5, 735–740 CrossRef CAS PubMed.
- M. Lindgren and P. Hammarström, FEBS J., 2010, 277, 1380–1388 CrossRef CAS PubMed.
- N. D. Younan and J. H. Viles, Biochemistry, 2015, 54, 4297–4306 CrossRef CAS PubMed.
- J. Barton, D. S. Arias, C. Niyangoda, G. Borjas, N. Le, S. Mohamed and M. Muschol, Biomolecules, 2019, 9, 539 CrossRef PubMed.
- N. P. Harte, I. Klyubin, E. K. McCarthy, S. Min, S. A. Garrahy, Y. Xie, G. P. Davey, J. J. Boland, M. J. Rowan and K. H. Mok, J. Biol. Chem., 2015, 290, 28343–28352 CrossRef CAS PubMed.
- M. N. N. Vieira, L. Forny-Germano, L. M. Saraiva, A. Sebollela, A. M. B. Martinez, J.-C. Houzel, F. G. De Felice and S. T. Ferreira, J. Neurochem., 2007, 103, 736–748 CrossRef CAS PubMed.
- G. Fusco, S. W. Chen, P. T. F. Williamson, R. Cascella, M. Perni, J. A. Jarvis, C. Cecchi, M. Vendruscolo, F. Chiti, N. Cremades, L. Ying, C. M. Dobson and A. De Simone, Science, 2017, 358, 1440–1443 CrossRef CAS PubMed.
- S. A. Kotler, J. R. Brender, S. Vivekanandan, Y. Suzuki, K. Yamamoto, M. Monette, J. Krishnamoorthy, P. Walsh, M. Cauble, M. M. B. Holl, E. N. G. Marsh and A. Ramamoorthy, Sci. Rep., 2015, 5, 11811 CrossRef PubMed.
- B. Mannini, E. Mulvihill, C. Sgromo, R. Cascella, R. Khodarahmi, M. Ramazzotti, C. M. Dobson, C. Cecchi and F. Chiti, ACS Chem. Biol., 2014, 9, 2309–2317 CrossRef CAS PubMed.
- A. R. A. Ladiwala, J. Litt, R. S. Kane, D. S. Aucoin, S. O. Smith, S. Ranjan, J. Davis, W. E. Van Nostrand and P. M. Tessier, J. Biol. Chem., 2012, 287, 24765–24773 CrossRef CAS PubMed.
- D. R. Booth, M. Sunde, V. Bellotti, C. V. Robinson, W. L. Hutchinson, P. E. Fraser, P. N. Hawkins, C. M. Dobson, S. E. Radford, C. F. F. Blake and M. B. Pepys, Nature, 1997, 385, 787–793 CrossRef CAS PubMed.
- K. W. Tipping, T. K. Karamanos, T. Jakhria, M. G. Iadanza, S. C. Goodchild, R. Tuma, N. A. Ranson, E. W. Hewitt and S. E. Radford, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5691–5696 CrossRef CAS PubMed.
- L. R. Wilken and Z. L. Nikolov, Biotechnol. Prog., 2006, 22, 745–752 CrossRef CAS PubMed.
- H. M. Ellerby, W. Arap, L. M. Ellerby, R. Kain, R. Andrusiak, G. D. Rio, S. Krajewski, C. R. Lombardo, R. Rao, E. Ruoslahti, D. E. Bredesen and R. Pasqualini, Nat. Med., 1999, 5, 1032–1038 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |