
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioorthogonal protein labelling enables the study of antigen processing of citrullinated and carbamylated auto-antigens†
Tyrza
van Leeuwen‡
a,
Can
Araman‡
 a,
Linda
Pieper Pournara
a,
Arieke S. B.
Kampstra
b,
Thomas
Bakkum
a,
Mikkel H. S.
Marqvorsen
a,
Clarissa R.
Nascimento
a,
G. J. Mirjam
Groenewold
a,
Willemijn
van der Wulp
a,
Marcel G. M.
Camps
c,
George M. C.
Janssen
d,
Peter A.
van Veelen
d,
Gerard J. P.
van Westen
e,
Antonius P. A.
Janssen
f,
Bogdan I.
Florea
a,
Herman S.
Overkleeft
a,
Ferry A.
Ossendorp
c,
René E. M.
Toes
*b and
Sander I.
van Kasteren
*a
a,
Linda
Pieper Pournara
a,
Arieke S. B.
Kampstra
b,
Thomas
Bakkum
a,
Mikkel H. S.
Marqvorsen
a,
Clarissa R.
Nascimento
a,
G. J. Mirjam
Groenewold
a,
Willemijn
van der Wulp
a,
Marcel G. M.
Camps
c,
George M. C.
Janssen
d,
Peter A.
van Veelen
d,
Gerard J. P.
van Westen
e,
Antonius P. A.
Janssen
f,
Bogdan I.
Florea
a,
Herman S.
Overkleeft
a,
Ferry A.
Ossendorp
c,
René E. M.
Toes
*b and
Sander I.
van Kasteren
*a
aDivision of Bio-organic Synthesis, Leiden Institute of Chemistry and the Institute of Chemical Immunology, Leiden University, Leiden, The Netherlands. E-mail: s.i.van.kasteren@chem.leidenuniv.nl
bDepartment of Rheumatology, Leiden University Medical Center, P.O. Box 9600, 2300 RC Leiden, The Netherlands. E-mail: r.e.m.toes@lumc.nl
cDepartment of Immunology, Leiden University Medical Center, P.O. Box 9600, 2300 RC Leiden, The Netherlands
dCenter for Proteomics and Metabolomics, Leiden University Medical Center, P.O. Box 9600, 2300 RC Leiden, The Netherlands
eComputational Drug Discovery, Drug Discovery and Safety, LACDR, Leiden University, Leiden, The Netherlands
fDepartment of Molecular Physiology, Leiden Institute of Chemistry and the Oncode Institute, Leiden University, Leiden, The Netherlands
First published on 23rd February 2021
Abstract
Proteolysis is fundamental to many biological processes. In the immune system, it underpins the activation of the adaptive immune response: degradation of antigenic material into short peptides and presentation thereof on major histocompatibility complexes, leads to activation of T-cells. This initiates the adaptive immune response against many pathogens. Studying proteolysis is difficult, as the oft-used polypeptide reporters are susceptible to proteolytic sequestration themselves. Here we present a new approach that allows the imaging of antigen proteolysis throughout the processing pathway in an unbiased manner. By incorporating bioorthogonal functionalities into the protein in place of methionines, antigens can be followed during degradation, whilst leaving reactive sidechains open to templated and non-templated post-translational modifications, such as citrullination and carbamylation. Using this approach, we followed and imaged the post-uptake fate of the commonly used antigen ovalbumin, as well as the post-translationally citrullinated and/or carbamylated auto-antigen vinculin in rheumatoid arthritis, revealing differences in antigen processing and presentation.
Introduction
Proteolysis plays an essential role in many immunological processes. From pathogen degradation to the induction of apoptosis.1,2 Dysregulation of proteolysis is therefore a hallmark in a variety of immune-related pathologies, such as the growth and metastasis of tumors,3 auto-immune pathogenesis,4,5 compromised bone desorption,6 and intracellular bacterial persistence,7 to name but a few.The biology of proteases is also complex: factors such as their compartmentalized distribution,8 post-translational activation,9 functional redundancy and interplay10 complicates their study. Multiple protease and peptidase families (e.g. aspartic, serine, and cysteine proteases) are involved in the production of MHC-restricted peptides.17–19 Many of these proteases, particularly those in the endo-lysosomal system, are under tight post-translational control,9e.g. by being produced as zymogens that are only activated upon reaching the correct vesicle.9 The often-promiscuous substrate preference by virtue of their shallow binding grooves,20 attenuation of their activity by endogenous inhibitors,21 changes in pH, and radical concentrations22 are factors that further complicate the study of these enzymes.
This is particularly important during antigen processing by antigen presenting cells (APCs).11,12 APCs take up exogenous material, degrade the proteins from these sources and present specific peptides from the degraded protein on their major histocompatibility complexes (MHCs).13
Recognition of a specific peptide-MHC (pMHC) by a cognate T-cell then leads to the initiation of the adaptive immune response against the source of the peptide.13 This process is of prime importance in the clearance of exogenous pathogens and cancer, but also for the opposite: the induction of tolerance against innocuous substances and self-tissue.14–16 As such, the understanding of the proteolysis underpinning antigen presentation is essential.
The nature of the antigen (i.e. the substrate) itself also influences proteolysis23 by altering protease specificity or overall stability. For example, a point mutation in the multiple sclerosis (MS) auto-antigen myelin basic protein (MBP85-99) prevents its cleavage by asparagine endopeptidase leading to enhanced presentation to T-cells.4 (De)stabilizing a protein fold can also alter antigenicity as exemplified by the disappearance of T-cell activation by the structurally destabilized variants RNAse S and apo-HRP compared to their stably-folded counterparts24 and the increased antigenicity of a stabilised hen egg lysozyme variant.25,26
Post-translational modification of the antigen too can alter proteolysis by altering the fine protease specificity. This can, for example, lead to the appearance of ‘neo-auto-antigens’ – non-thymic peptides that can drive auto-immune diseases. This has been observed in MS where an arginine to citrulline modification in myelin oligodendrocyte glycoprotein prevented destructive epitope processing, leading to neo-T-cell activation.5
So far, the go-to approach for studying antigen-routing has been the use of detectable model proteins (e.g. lactamases,27 luciferase,28 and HRP29) or fluorophore-modified model antigens.30–32 Reporter proteins are only active (and therefore detectable) as long as their structure. Fluorescent modification of antigens changes the charge, hydrophobicity/aromaticity, shape and structure, which alters the properties of the protein.33,34 We hypothesize that this leads to significant structural alterations that strongly affect processing and antigenicity as shown for the examples above.
We here explore bioorthogonal chemistry35–38 as an approach to study antigen degradation by immune cells. The small abiotic groups, such as an azide or alkyne, incorporated into model antigens will allow the visualization even after the antigen has been extensively degraded; whilst keeping protein structural perturbations to a minimum, (Fig. 1A). Recombinant proteins containing such labels can readily be obtained using the BONCAT-approach described by the Tirrell-group40–42: The substitution of natural Met-residues by azidohomoalanine and homopropargylglycine (Aha/Hpg, Fig. 1), without loss of their activity43 or effect on viability.44,45
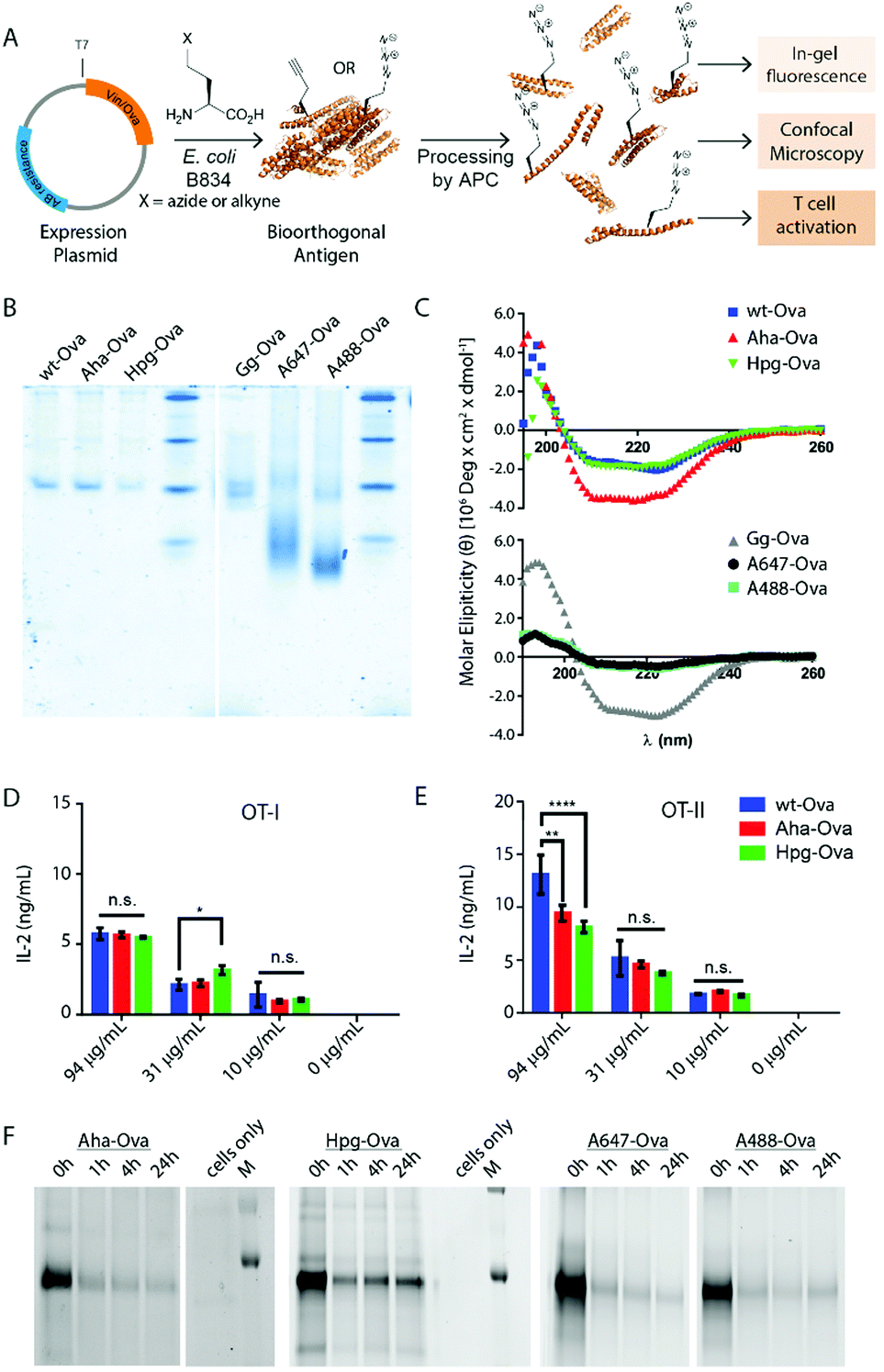 | ||
| Fig. 1 Bioorthogonal antigens closely approximate the biophysical behavior of wild type antigens. (A) Expression of antigenic proteins in the E. coli auxotrophic strain B834 allows replacement of methionines with azidohomoalanine (Aha) or homopropargylglycine (Hpg). These labels allow the in cell imaging of degradation whilst at the same time allowing the study of the antigens by T-cell activation; (B) native-PAGE analysis shows similar migration behavior of wt-Ova, Aha-Ova and Hpg-Ova, whilst showing altered migration of fluorophore modified Ova-analogues; (C) circular dichroism of bioorthogonal and fluorophore Ova-analogues; (D) activation of OT-I cells by Ova-variants as measured by IL-2 production; (E) activation of OT-II cells by these same antigens. Asterisks refer to given P values, * p < 0.05, ** p < 0.005, **** p < 0.00005. Group mean values were analyzed by two-way analysis of variance with Tukey post hoc significant difference test using GraphPad Prism 6.0. Data are represented as means ± SD; (F) tracking of degradation of antigen inside D1 dendritic cells. | ||
In this study we produce, characterize and explore bioorthogonal antigens to follow degradation and use these reagents to study the effect of pathogenic PTMs such as citrullination and carbamylation on these processes.
Results and discussion
We first produced methionine-containing and bioorthogonally labelled variants of the model antigen ovalbumin (Ova), as this protein has played a central role in the development and characterisation of many immunological tools. Ova was produced from the plasmid pMCSG7 that contains the Ova-gene fused to a Hexa-His-Tag (His6), via a TEV cleavage site (a kind gift from N. del Cid and M. Raghavan47). The plasmid was transformed into the methionine auxotrophic strain B834 of E. coli46 in a methionine-free expression medium augmented with Aha or Hpg.40,42 Ova containing Aha amino acids (Aha-Ova), homopropargylglycine (Hpg-Ova) or methionine (wt-Ova) were produced in yields of 2–3 mg L−1 culture after purification. LC-MS analysis of the bioorthogonal variants indicated quantitative incorporation of the bioorthogonal handles during expression (Fig. S1, ESI†).48–51We analysed the bioorthogonal antigens and compared them in terms of structure and stability to bacterially expressed Ova (wt-Ova), Ova isolated from hen's eggs (Gallus gallus; Gg-Ova), and fluorophore modified Ova variants. Native PAGE analysis (Fig. 1B) showed equal gel migration properties for wt-, Aha- and Hpg-Ova to Gg-Ova. Alexa Fluor 647-Ova and Alexa Fluor 488-modified Ova (A647-Ova and A488-Ova) showed multiple bands, most of which migrated faster. Circular dichroism (Fig. 1C, CD) of Hpg-, and Aha-Ova showed similar elipticity to wt- and Gg-Ova variants. A drastic drop in elipticity was observed for A647- and A488-Ova (Fig. 1C, lower graph), indicating a strong reduction in structural stability upon fluorophore modification.
To determine whether this similarity translated to similar T-cell responses,52 we tested both MHC-I and -II restricted T-cell activation. OT-I and OT-II T-cells – isolated and purified from mouse spleen – were co-cultured with murine bone marrow derived dendritic cells (BMDCs) as antigen presenting cells (APCs). The APCs were pulsed for 4h with the antigens and chased for 24 h before addition of the T-cells.54,55 Induction of T-cell activation by Aha-Ova and Hpg-Ova-induced T-cell activation was similar to that of wt-Ova (Fig. 1D and E). CD4 T-cell activation differed only at the highest Ova-concentration; with the difference in presentation dissipating at the lower concentrations (Fig. 1E). A647- and A488-Ova yielded strange results: the experimental variation between replicates varied over two orders of magnitude between repetitions (examples shown in Fig. S2, ESI†), leaving fluorophore modified Ova unreliable to study T cell responses.
Next, the bioorthogonal groups were used to monitor antigen degradation in the APCs (Fig. 1F). D1 APCs53 were pulsed with Aha-, Hpg-, A488- and A647-Ova for 2 h, followed by washing, and different chase periods (0 h, 1 h, 4 h or 24 h). After fixation, the cells were either lysed and subjected to a copper-catalyzed Huisgen cycloaddition (ccHc)-reaction with A647-azide/alkyne and SDS-PAGE (Fig. 1F), or fixed, permeabilized and imaged by confocal microscopy (Fig. 2).
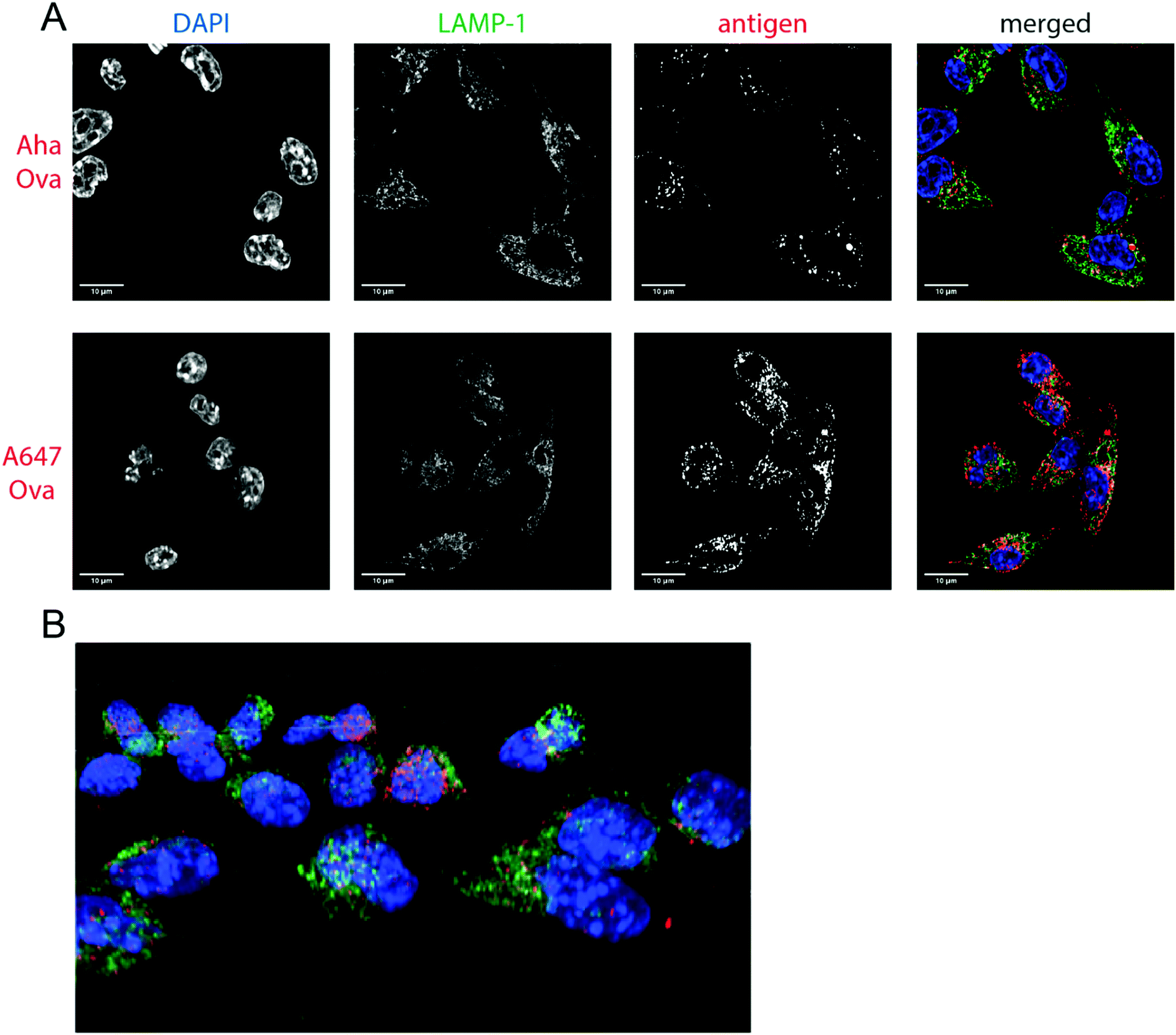 | ||
| Fig. 2 Imaging processing of bioorthogonal and fluorophore-modified Ova in DC 2.4-APCs via confocal microscopy. DCs were incubated for 2 h (pulse) with Aha or A647-Ova. Cells were fixed with 0.5% PFA and processed for immunofluorescence with LAMP-1 as a lysosomal marker (green in merged images). The nucleus was stained with DAPI (blue in merged images). Aha-Ova was stained using ccHc with A647-alkyne fluorophore (grey in single channel, red in merged images). (A) Upper panel: imaging of Aha-Ova, lower panel: imaging of A647-Ova (B) screenshot from Movie S1 (ESI†). Scale bar is 10 μm (white bar, left corner). | ||
Fluorescent SDS-PAGE analysis showed the degradation of Ova over time. No intermediate breakdown fragments were observed for this protein (the signal of the unreacted fluorophore covered the small breakdown fragments <3 kDa). Interestingly, the pre-labelled A647-Ova (for which this was not an issue), showed a disappearance of the main Ova-band, but no appearance of low M.W. fragments. This suggests the terminal degradation-fluorophore fragments diffuse away, or that the fluorophore itself does not survive the conditions in the lysosome.
Confocal microscopy of Aha-Ova and A647-Ova (Fig. 2, Fig. S3, S4 and Movie S1, ESI†) in the larger DC2.4 dendritic cell line56 (Fig. 2), as well as the D1 cells53 (Fig. S3, ESI†) showed A647-Ova localized partly to (late) LAMP-1 positive vesicles after the 2 h pulse. Aha-Ova showed no such accumulation in LAMP-1-positive vesicles, suggesting an effect of the pendant fluorophore or absent secondary protein structure to the routing of these reagents. Increased uptake of fluorophore-modified antigen was also observed in both the DC2.4 and D1-cell lines, suggesting further interference of the pendant fluorophores with routing biology (Fig. 2B).
After these studies on the model protein Ova, we next applied the technique to the study of the auto-antigen vinculin (Vin). During the pathogenesis of rheumatoid arthritis (RA), the carbamylation of lysines and the citrullination of arginines of proteins such as Vin is believed to yield neo-antigens that serve as driver of the disease. Over 60% of RA-patients have mature class-switched antibodies against citrullinated57–59 and carbamylated60,61 proteins in their blood.62 This suggests T-cell help to the auto-antibody producing B-cell has taken place. This in turn suggests that carbamylation and citrullination alters the processing and presentation of CD4 restricted neo-epitopes in this disease. Yet, very little is known about T-cell activation by these PTM-modified antigens.
We therefore expressed a truncated variant of Vin comprising amino acids 453–741 of the native sequence (containing the potential epitope DERAA63,64) as an N-terminal Deca-His-fusion (His10) via an Enterokinase cleavage site (DDDDDKH). Vin contains 8 internal Met-codons. Bioorthogonal expression yielded a mixture containing mainly 7 or 8 copies of Hpg or Aha (i.e. the N-terminal Met not cleaved). Minor variants with fewer (6, 5 and 4) copies of Aha and Hpg were also detected (combined abundance <10%; Fig. S5, ESI†).
Bioorthogonal antigens carry their label in place of Met-residues leaving the nucleophilic lysine sidechain free for post-translational modification. We therefore produced carbamylated and citrullinated variants of the proteins using methods described previously.65,66 All Vin variants used in this work contained 23 possible carbamylation sites (incl. the N-terminus). Results of SDS-PAGE and LC-MS measurements showed that >19 amines were carbamylated for all variants of Vin (Fig. S6A and B, ESI†) as determined by LC-MS. In vitro citrullination was achieved using recombinant human PAD4, an enzyme that is responsible for quantitative Arg to Cit conversion, for all wt and bioorthogonal variants of Vin (Fig. S6A and B, ESI†).
ACPAs have been shown to recognize citrullinated antigens in RA, of which DERAA containing HLA-DR2 epitopes are favoured. The introduction of bioorthogonal groups did not alter recognition by RA-associated auto-antibodies: citrullinated bioorthogonal Vin variants are recognized by ACPAs to the same degree as their wt counterparts by ELISA (Fig. S7, ESI†). Interestingly, anti-carbamylated protein antibodies (ACarPAs) did not recognize carbamylated bioorthogonal antigens as strong as they recognize wt versions (Fig. S8, ESI†). This might arise from the structural stabilizing effects of carbamylation as seen above.
We first analysed whether these antigens gave a T-cell response, and particularly what the effect of citrullination and carbamylation was on this response. For this we used a Jurkat T-cell line (J76) transduced with a T-cell receptor (TCR) restricted to a specific peptide present in vinculin (REEVFDERAANFENH; Vin-DERAA).67 The resultant reporter cells allow simultaneous determination of the activity of the transcription factors NF-κB, NFAT and AP-1 that play key roles in T-cell activation, allowing the antigen-specific activation to be measured by flow cytometry. To test the activity of this T-cell reagent, an HLA-DQA1*03:01/DQB1*03:02+ (HLA-DQ8+)-positive monocyte-derived dendritic cells, isolated from HLA-DQ8+ donors, were pulsed (overnight) with the different bioorthogonal proteins and their modified counterparts (3 μM). Vin-DERAA-specific Jurkats were then added and their activation measured by flow cytometry (Fig. 3 and Fig. S9, ESI†).
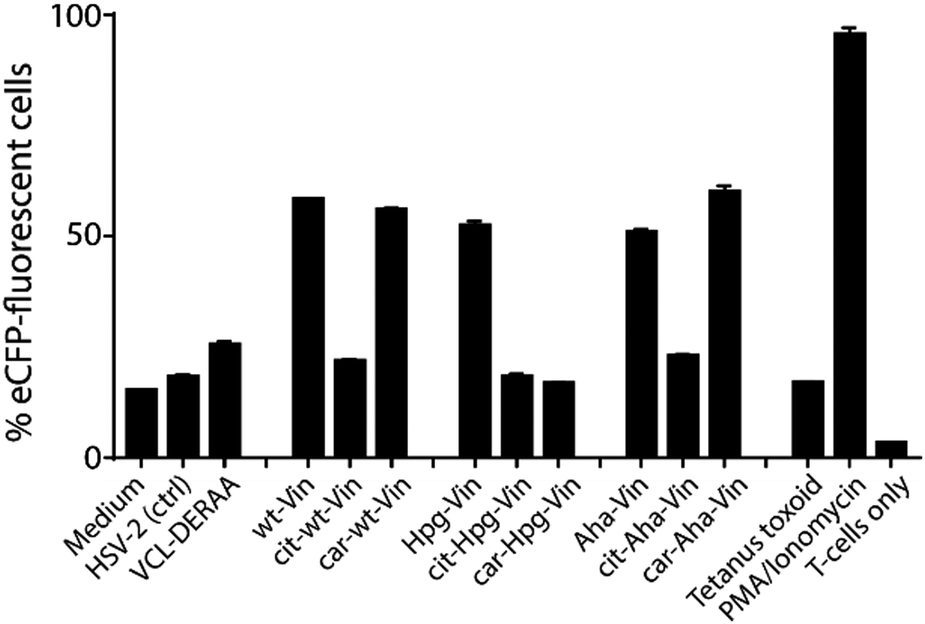 | ||
| Fig. 3 Activation of anti-DERAA T-cells by bioorthogonal vinculin derivatives. PBMC derived dendritic cells, isolated from HLA-DQ8+ donors were pulsed with peptides or Vin variants were cocultured with the Jurkat cells and used for stimulation. Activation of Vin-DERAA-specific Jurkat cells was measured with eCFP expression within the cells. The eCFP expression is dependent on NFκB and is therefore a marker of TCR activation and costimulation, the percentage of live eCFP-positive cells is plotted in bars. HSV2 (Herpes Simplex Virus 2 peptide) and Tetanus toxoid protein are negative controls. VCL-DERAA peptide and PMA-ionomycin are controls. Error bars show the SD. Data is representative of two experiments with biological triplicates. | ||
Some striking differences were observed in the ability of these peptides to activate T-cells: the citrullinated antigens showed a complete abolition of T-cell activation. This suggests the loss of two positive charges at T-cell contact sites can abrogate TCR binding. Altered proteolytic processing of the citrullinated variant likely does not play a role in this case, as this would have also led to the abolition of Carb-Vin recognition.
Wt and Carb-antigens (with the exception of Carb-Hpg-Vin) showed robust T-cell activation, yet citrullinated antigens did not. We hypothesized that this difference could either be due to the change in a TCR-contact residue, or in altered processing of the protein, resulting from the overall change in properties imbued by the citrullination or carbamylation. We therefore determined the effects of carbamylation and citrullination on protein fold stability. Carbamylation had no effect on protein fold and fold-stability (Fig. S7C, ESI†). Citrullination of the Vin on the other hand, did result in a big effect on protein structure: these proteins became high in β-sheet content and random coil-rich structure with a decrease in the content of α-helices (Fig. S7C, ESI†).
To assess whether this fold of the protein changed the rate and nature of processing, we subjected the variants to in vitro degradation with isolated lysosomes.68 Carbamylated wt- Aha- and Hpg-Vin variants all showed a greater resistance to proteolysis than their non-modified counterparts (Fig. 4A; >90% intact for Carb-Vin vs. >50% for wt-Vin at t = 18 h) Citrullination, despite the induced changes in structure, had no significant effects on the rate of degradation (Fig. S10, ESI†). Above effects could be observed with Aha-Vin in a more consistent manner than with Hpg-Vin suggesting distinct bioorthogonal group preferences for distinct antigens. Thus, we moved forward with Aha-Vin as the bioorthogonal variant for further experiments.
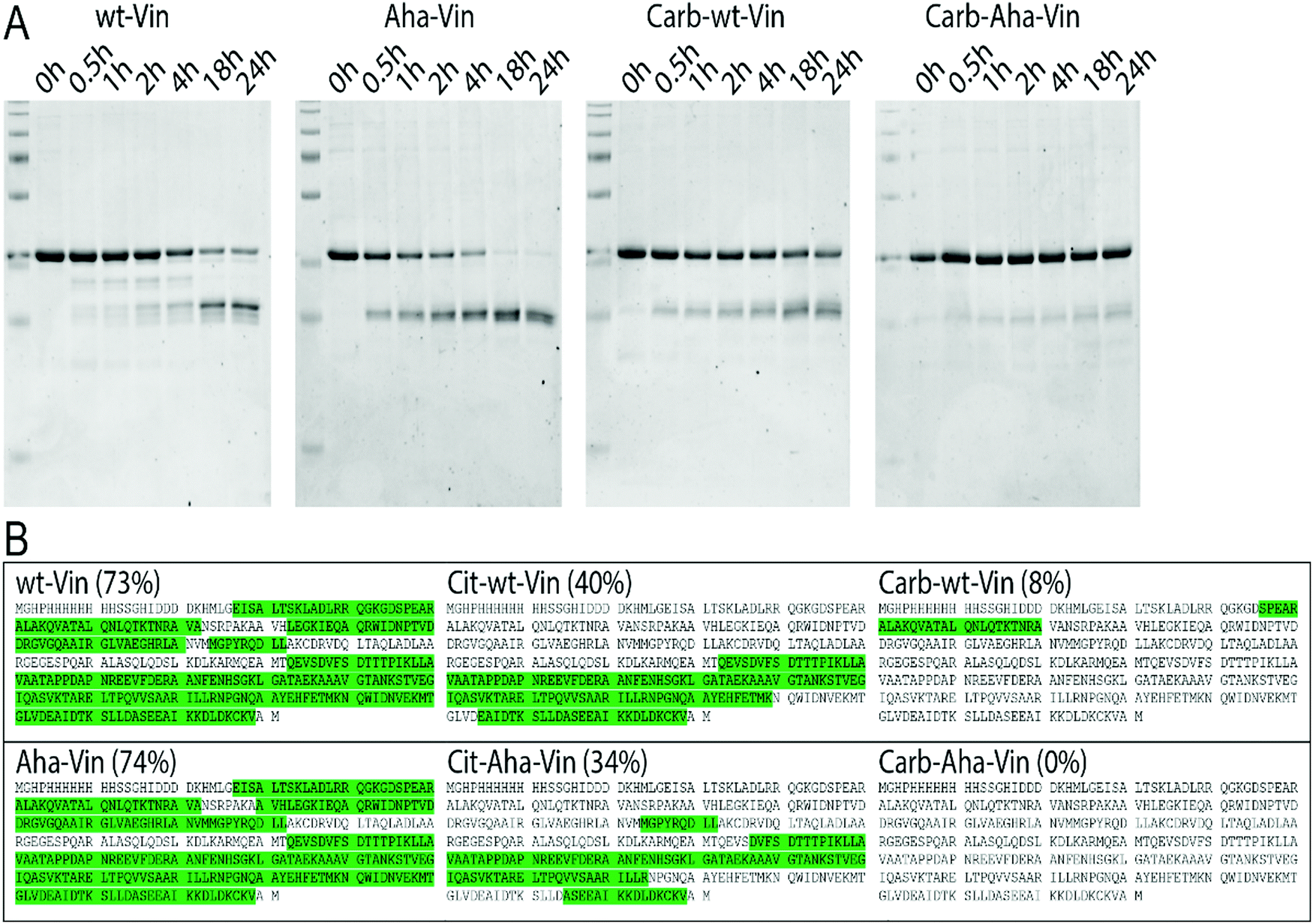 | ||
| Fig. 4 Effect of post-translational modifications on vinculin degradation. Incubation of vinculin variants with isolated lysosomes (A) or CatS (B) show decreased proteolysis of wt and Aha-Vin upon carbamylation. Vin peptides recovered after CatS digestion (n = 3 per variant) were mapped against the protein sequence. Peptide sequences were marked green, when found in at least 2/3 replicates. | ||
To see whether citrullination or carbamylation altered proteolysis at the sequence level, all Vin variants (including the bioorthogonal variants) were subjected to in vitro digestion by endolysosomal protease cathepsin S (CatS).69 The proteins were digested for 1 hour, prior to analysis by tandem MS–MS without further tryptic digest. The observed peptides were then mapped against the amino acid sequence to obtain the coverage per variant (Fig. 4B and Fig. S11, S12, ESI†). Major differences were found in processing of carbamylated, and citrullinated, variants compared to the wt. The coverage of the Carb-variants was very low compared to wt (8% compared to 73%). Citrullination yielded an intermediate decrease in coverage (40%). All bioorthogonal antigens showed similar trends to their wild-type counterparts.
To assess whether this increased resistance to proteolysis in vitro also translated to an increased stability in dendritic cells, BMDCs were pulsed with the bioorthogonal wt/PTM Vin antigens for 2 h and then chased for 1 h, 2 h, 3 h (citrullination), and/or 24 h or 48 h (carbamylation). This experiment confirmed the in vitro results: Cit-Aha-Vin, and wt-Aha-Vin were fully degraded within 24 h, yet Carb-Vin variants remained detectable even at the 48 h time point (Fig. S13, ESI†). We performed an MTT assay to verify that the used amounts of Vin variants were nontoxic to DC2.4 cells (Fig. S14, ESI†). Additionally, confocal microscopy of the PTM-modified Aha-Vin variants on DC2.4 cells showed that neither carbamylation nor citrullination had a major effect on antigen localization at the 3 h and 6 h time points (Fig. 15, ESI†).
These data suggest that alterations in processing are not responsible for the changes observed in T-cell activation upon citrullination and that – despite the change of only two atoms in the epitope – the Arg → Cit modification is responsible for the abolition of T-cell activation. To provide further evidence in support of this hypothesis, the presentation of minimal epitopes (REEVFDERAANFENH) to the Jurkat T-cells was assessed (Fig. 5), and indeed no responses were observed for the citrullinated minimal epitope, compared to the wt, further supporting this hypothesis. Finally, we performed template guided molecular docking simulations on the minimal Vin-DERAA epitope (Fig. 6). The results clearly indicated that both Arg residues are exposed on the extracellular face of the protein. Interestingly, docking the Cit-Vin-DERAA in a similar manner seems to indicate that the citrullinated DERAA-Arg can be buried in the binding site, instead exposing the negatively charged glutamic acid residue. This provides a structural rationale for the observed suppression of T-cell activation upon citrullination.
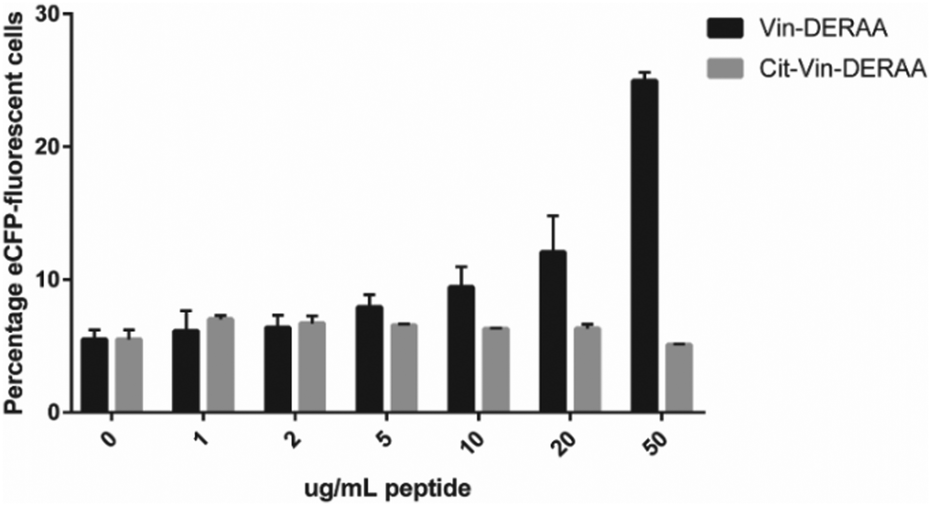 | ||
| Fig. 5 FACS read-out for T-cell activation with titration of wt- and Cit-VCL-DERAA peptide. Percentage of eCFP positive cells was read out as a measure for T-cell activation (n = 3). | ||
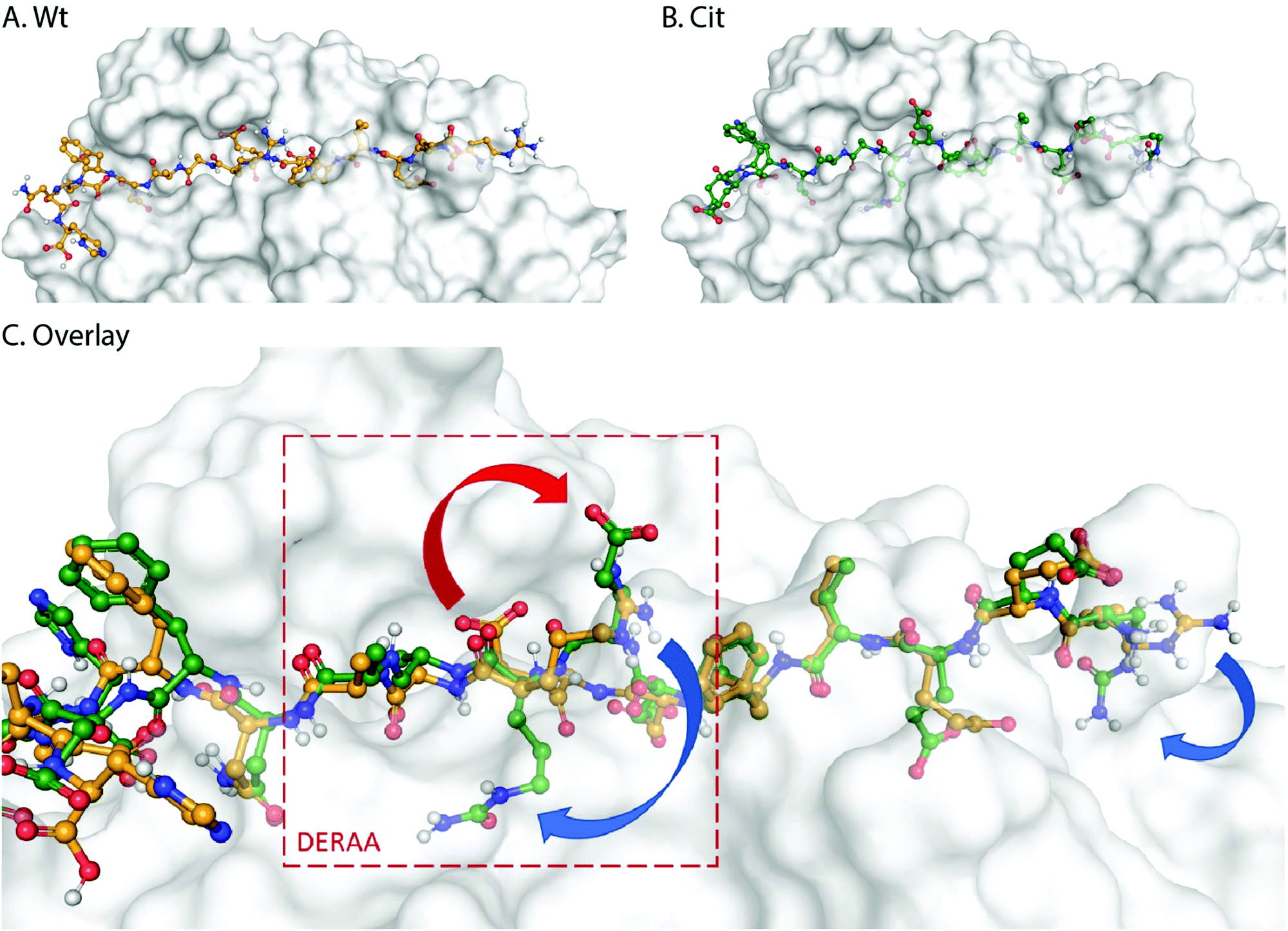 | ||
| Fig. 6 Molecular docking of wt- and Cit-VCL-DERAA in HLA-DQA1*03:01/DQB1*03:02 (HLA-DQ8, PDB: 6DFX). Arg residues in wt-VCL-DERAA (A) are exposed extracellularly whereas for Cit-VCL-DERAA (B) citrullinated arginines are buried (C, blue arrows) and Glu residue becomes exposed extracellularly (C, red arrow). | ||
Conclusions
In summary, we were able to show that our bioorthogonal antigen variants are powerful tools to track the fate of antigens upon processing by APCs. The antigens – when expressed with Aha or Hpg in place of Met – behave similarly to wild type antigens. Additionally, the bioorthogonal groups are stable to the harsh conditions found in the lysosome, the handles are not cleaved/destroyed by proteolysis, and lysine PTMs can be studied. One limitation of this approach (using azides/alkynes with the Cu-catalyzed Huisgen cycloaddition reaction for labelling) is the need for fixation/lysis of the cells prior to performing this reaction. Despite some recent efforts to combat this, the Cu-catalyst is still too toxic to immune cells to allow meaningful live cell imaging.The use of live-cell compatible click reactions would be a great future direction of this field.70 Reactions, such as the strain-promoted [3+2] cycloadditions,71 and the various guises of the inverse electron demand Diels–Alder reaction,72 have proven their merit in live-cell labelling experiments.73–75 Unfortunately, they are not yet suitable in the context described in this article: first, the non-homogeneous subcellular distribution of the fluorophores used in these reactions can result in ‘distribution artefacts’, thus the non-homogeneous biodistribution to given subcellular structures, and their preferential reactions there.76 There is also a conundrum relating to the rates of these reactions: those reagents that can react on the timescales needed to illuminate the antigen presentation pathway, are of limited stability to the conditions found in the endo-lysosome.39 Once these issues are addressed, the live cell imaging of antigens during presentation remains a tantalising prospect.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Reuma Nederland (Netherlands Rheumatoid Arthritis Fund) with grant number 15-2-202 and the European Research Council with grant ERC-CoG-865175. The authors acknowledge the assistance of Hans van den Elst with LCQ OrbiTrap and proteomics experiments.References
- C. Cho, D. O'Dell Bunch, J.-E. Faure, E. H. Goulding, E. M. Eddy, P. Primakoff and D. G. Myles, Science, 1998, 281, 1857–1859 CrossRef CAS PubMed.
- I. Budihardjo, H. Oliver, M. Lutter, X. Luo and X. Wang, Annu. Rev. Cell Dev. Biol., 1999, 15, 269–290 CrossRef CAS PubMed.
- J. E. Koblinski, M. Ahram and B. F. Sloane, Clin. Chim. Acta, 2000, 291, 113–135 CrossRef CAS.
- B. Manoury, D. Mazzeo, L. Fugger, N. Viner, M. Ponsford, H. Streeter, G. Mazza, D. C. Wraith and C. Watts, Nat. Immunol., 2002, 3, 169–174 CrossRef CAS PubMed.
- S. A. Jagessar, I. R. Holtman, S. Hofman, E. Morandi, N. Heijmans, J. D. Laman, B. Gran, B. W. Faber, S. I. van Kasteren, B. J. Eggen and B. A. t Hart, J. Immunol., 2016, 197, 1074–1088 CrossRef CAS PubMed.
- B. Turk, Nat. Rev. Drug. Discovery, 2006, 785 CrossRef CAS PubMed.
- O. Steele-Mortimer, Curr. Opin. Microbiol., 2008, 11, 38–45 CrossRef CAS PubMed.
- V. Turk, V. Stoka, O. Vasiljeva, M. Renko, T. Sun, B. Turk and D. Turk, Biochim. Biophys. Acta Prot. Proteomics, 2012, 1824, 68–88 CrossRef CAS PubMed.
- M. Rosenfeld, G. Kreibich, D. Popov, K. Kato and D. Sabatini, J. Cell Biol., 1982, 93, 135–143 CrossRef CAS PubMed.
- S. I. van Kasteren and H. S. Overkleeft, Curr. Opin. Chem. Biol., 2014, 23, 8–15 CrossRef CAS PubMed.
- K. L. Rock, D. J. Farfán-Arribas and L. Shen, J. Immunol., 2010, 184, 9–15 CrossRef CAS PubMed.
- J. A. Villadangos, R. A. Bryant, J. Deussing, C. Driessen, A. M. Lennon-Dumenil, R. J. Riese, W. Roth, P. Saftig, G. P. Shi, H. A. Chapman, C. Peters and H. L. Ploegh, Immunol. Rev., 1999, 172, 109–120 CrossRef CAS PubMed.
- K. L. Rock, E. Reits and J. Neefjes, Trends Immunol., 2016, 37, 724–737 CrossRef CAS PubMed.
- E. R. Unanue, V. Turk and J. Neefjes, Annu. Rev. Immunol., 2016, 34, 265–297 CrossRef CAS PubMed.
- J. Yi, L. Balagopalan, T. Nguyen, K. M. McIntire and L. E. Samelson, Nat. Commun., 2019, 10, 277 CrossRef PubMed.
- M. D. Rosenblum, K. A. Remedios and A. K. Abbas, J. Clin. Invest., 2015, 125, 2228–2233 CrossRef PubMed.
- P. Guermonprez, J. Valladeau, L. Zitvogel, C. Théry and S. Amigorena, Annu. Rev. Immunol., 2002, 20, 621–667 CrossRef CAS PubMed.
- R. Unanue, V. Turk and J. Neefjes, Annu. Rev. Immunol., 2016, 34, 265–297 CrossRef PubMed.
- J. S. Blum, P. A. Wearsch and P. Cresswell, Annu. Rev. Immunol., 2013, 31, 443–473 CrossRef CAS PubMed.
- Y. Choe, F. Leonetti, D. C. Greenbaum, F. Lecaille, M. Bogyo, D. Brömme, J. A. Ellman and C. S. Craik, J. Biol. Chem., 2006, 281, 12824–12832 CrossRef CAS PubMed.
- S. Magister and J. Kos, J. Cancer, 2012, 4, 45–56 CrossRef PubMed.
- A. Savina, C. Jancic, S. Hugues, P. Guermonprez, P. Vargas, I. C. Moura, A. M. Lennon-Dumenil, M. C. Seabra, G. Raposo and S. Amigorena, Cell, 2006, 126, 205–218 CrossRef CAS PubMed.
- S. Scheiblhofer, J. Laimer, Y. Machado, R. Weiss and J. Thalhamer, Expert Rev. Vaccines, 2017, 16, 479–489 CrossRef CAS PubMed.
- L. Delamarre, R. Couture, I. Mellman and E. S. Trombetta, J. Exp. Med., 2006, 203, 2049–2055 CrossRef CAS PubMed.
- T. Ohkuri, S. Nagatomo, K. Oda, T. So, T. Imoto and T. Ueda, J. Immunol., 2010, 185, 4199–4205 CrossRef CAS PubMed.
- T. So, H. O. Ito, T. Koga, S. Watanabe, T. Ueda and T. Imoto, J. Biol. Chem., 1997, 272, 32136–32140 CrossRef CAS PubMed.
- I. Cebrian, G. Visentin, N. Blanchard, M. Jouve, A. Bobard, C. Moita, J. Enninga, L. F. Moita, S. Amigorena and A. Savina, Cell, 2011, 147, 1355–1368 CrossRef CAS PubMed.
- A. Giodini and P. Cresswell, EMBO J., 2008, 27, 201–211 CrossRef CAS PubMed.
- C. C. Norbury, B. J. Chambers, A. R. Prescott, H. G. Ljunggren and C. Watts, Eur. J. Immunol., 1997, 27, 280–288 CrossRef CAS PubMed.
- N. van Montfoort, M. G. Camps, S. Khan, D. V. Filippov, J. J. Weterings, J. M. Griffith, H. J. Geuze, T. van Hall, J. S. Verbeek, C. J. Melief and F. Ossendorp, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6730–6735 CrossRef CAS PubMed.
- M. F. Princiotta, D. Finzi, S. B. Qian, J. Gibbs, S. Schuchmann, F. Buttgereit, J. R. Bennink and J. W. Yewdell, Immunity, 2003, 18, 343–354 CrossRef CAS PubMed.
- N. Zaidi, T. Herrmann, D. Baechle, S. Schleicher, J. Gogel, C. Driessen, W. Voelter and H. Kalbacher, FEBS J., 2007, 274, 3138–3149 CrossRef CAS PubMed.
- E. W. Voss, Jr., C. J. Workman and M. E. Mummert, Biotechniques, 1996, 20, 286–291 CrossRef PubMed.
- A. M. Pickering and K. J. Davies, Free Radical Biol. Med., 2012, 52, 239–246 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS PubMed.
- L.-H. Qin, W. Hu and Y.-Q. Long, Tetrahedron Lett., 2018, 59, 2214–2228 CrossRef CAS.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007–11022 RSC.
- T. Bakkum, T. van Leeuwen, A. J. Sarris, D. M. van Elsland, D. Poulcharidis, H. S. Overkleeft and S. I. van Kasteren, ACS Chem. Biol., 2018, 13, 1173–1179 CrossRef CAS PubMed.
- K. L. Kiick and D. A. Tirrell, Tetrahedron, 2000, 56, 9487–9493 CrossRef CAS.
- K. L. Kiick, R. Weberskirch and D. A. Tirrell, FEBS Lett., 2001, 502, 25–30 CrossRef CAS PubMed.
- J. C. van Hest, K. L. Kiick and D. A. Tirrell, J. Am. Chem. Soc., 2000, 122, 1282–1288 CrossRef CAS.
- S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J. Kirkpatrick, N. J. Oldham, D. C. Anthony and B. G. Davis, Nature, 2007, 446, 1105 CrossRef CAS PubMed.
- D. M. van Elsland, E. Bos, W. de Boer, H. S. Overkleeft, A. J. Koster and S. I. van Kasteren, Chem. Sci., 2016, 7, 752–758 RSC.
- D. Poulcharidis, K. Belfor, A. Kros and S. I. van Kasteren, Chem. Sci., 2017, 8, 5585–5590 RSC.
- W. B. Wood, J. Mol. Biol., 1966, 16, 118–133 CrossRef CAS PubMed.
- N. Del Cid, L. Shen, J. Belleisle and M. Raghavan, PLoS One, 2012, 7, e41727 CrossRef CAS PubMed.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS PubMed.
- S. I. van Kasteren, H. B. Kramer, D. P. Gamblin and B. G. Davis, Nat. Protoc., 2007, 2, 3185–3194 CrossRef CAS PubMed.
- D. J. Harvey, D. R. Wing, B. Kuster and I. B. Wilson, J. Am. Soc. Mass Spectrom, 2000, 11, 564–571 CrossRef CAS PubMed.
- M. W. Walker and E. Lloyd-Evans, Methods Cell Biol., 2015, 126, 21–43 CAS.
- L. Delamarre, M. Pack, H. Chang, I. Mellman and E. S. Trombetta, Science, 2005, 307, 1630–1634 CrossRef CAS PubMed.
- C. Winzler, P. Rovere, M. Rescigno, F. Granucci, G. Penna, L. Adorini, V. S. Zimmermann, J. Davoust and P. Ricciardi-Castagnoli, J. Exp. Med., 1997, 185, 317–328 CrossRef CAS PubMed.
- S. Gillis, M. M. Ferm, W. Ou and K. A. Smith, J. Immunol., 1978, 120, 2027–2032 CAS.
- B. L. Levine, C. H. June, Y. Ueda, M. L. Huang, G. J. Freeman, L. M. Nadler and S. G. Ward, Int. Immunol., 1995, 7, 957–966 CrossRef PubMed.
- Z. Shen, G. Reznikoff, G. Dranoff and K. L. Rock, J. Immunol., 1997, 158, 2723–2730 CAS.
- Z. Baka, B. Gyorgy, P. Geher, E. I. Buzas, A. Falus and G. Nagy, Joint Bone Spine, 2012, 79, 431–436 CrossRef CAS PubMed.
- W. R. Fearon, Biochem. J., 1939, 33, 902–907 CrossRef CAS PubMed.
- E. R. Vossenaar, A. J. Zendman, W. J. van Venrooij and G. J. Pruijn, BioEssays, 2003, 25, 1106–1118 CrossRef CAS PubMed.
- C. J. van Dalen, M. W. Whitehouse, C. C. Winterbourn and A. J. Kettle, Biochem. J., 1997, 327(Pt 2), 487–492 CrossRef CAS PubMed.
- J. Shi, R. Knevel, P. Suwannalai, M. P. van der Linden, G. M. Janssen, P. A. van Veelen, N. E. Levarht, A. H. van der Helm-van Mil, A. Cerami, T. W. Huizinga, R. E. Toes and L. A. Trouw, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17372–17377 CrossRef CAS PubMed.
- G. S. Firestein and I. B. McInnes, Immunity, 2017, 46, 183–196 CrossRef CAS PubMed.
- P. A. Weller, E. P. Ogryzko, E. B. Corben, N. I. Zhidkova, B. Patel, G. J. Price, N. K. Spurr, V. E. Koteliansky and D. R. Critchley, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5667–5671 CrossRef CAS PubMed.
- J. van Heemst, D. T. S. L. Jansen, S. Polydorides, A. K. Moustakas, M. Bax, A. L. Feitsma, D. G. Bontrop-Elferink, M. Baarse, D. van der Woude, G.-J. Wolbink, T. Rispens, F. Koning, R. R. P. de Vries, G. K. Papadopoulos, G. Archontis, T. W. Huizinga and R. E. Toes, Nat. Commun., 2015, 6, 6681 CrossRef CAS PubMed.
- J. Fando and S. Grisolia, Eur. J. Biochem., 1974, 47, 389–396 CrossRef CAS PubMed.
- X. Chang, R. Yamada, A. Suzuki, Y. Kochi, T. Sawada and K. Yamamoto, Rheumatology, 2005, 44, 1374–1382 CrossRef CAS PubMed.
- S. Rosskopf, J. Leitner, W. Paster, L. T. Morton, R. S. Hagedoorn, P. Steinberger and M. H. M. Heemskerk, Oncotarget, 2018, 9, 17608–17619 CrossRef PubMed.
- M. W. Walker and E. A. Lloyd-Evans, Meth. Cell Biol., 2015, 126, 21–43 CAS.
- J. D. Colbert, S. P. Matthews, G. Miller and C. Watts, Eur. J. Immunol., 2009, 39, 2955–2965 CrossRef CAS PubMed; K. Porte, M. Riberaud, R. Châtre, D. Audisio, S. Papot and F. Taran, ChemBioChem, 2020, 22, 100–113 CrossRef PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- H. E. Murrey, J. C. Judkins, C. W. Am Ende, T. E. Ballard, Y. Fang, K. Riccardi, L. Di, E. R. Guilmette, J. W. Schwartz, J. M. Fox and D. S. Johnson, J. Am. Chem. Soc., 2015, 137, 11461–11475 CrossRef CAS PubMed.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- M. Baalmann, M. J. Ziegler, P. Werther, J. Wilhelm and R. Wombacher, Bioconjugate Chem., 2019, 30, 1405–1414 CrossRef CAS PubMed.
- C. W. Cunningham, A. Mukhopadhyay, G. H. Lushington, B. S. Blagg, T. E. Prisinzano and J. P. Krise, Mol. Pharmaceutics, 2010, 7, 1301–1310 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00009h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |