
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revision in the first steps of the biosynthesis of the red antibiotic prodigiosin: use of a synthetic thioester to validate a new intermediate†
Maxime
Couturier
a,
Hiral D.
Bhalara
a,
Rita E.
Monson
b,
George P. C.
Salmond
b and
Finian J.
Leeper
 *a
*a
aYusuf Hamied Dept. of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: fjl1@cam.ac.uk
bDept. of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge CB2 1QW, UK
First published on 15th January 2021
Abstract
A biosynthetic pathway for the red-antibiotic, prodigiosin, was proposed over a decade ago but not all the suggested intermediates could be detected experimentally. Here we show that a thioester that was not originally included in the pathway is an intermediate. In addition, the enzyme PigE was originally described as a transaminase but we present evidence that it also catalyses the reduction of the thioester intermediate to its aldehyde substrate.
Introduction
Prodigiosin 11 is a tripyrrolic natural product produced by a wide range of bacteria.1 It originally caught researchers’ attention because of its bright red colour, but its various biological activities have renewed interest in this molecule in recent decades.2The biosynthetic pathway to prodigiosin was established by Williamson et al. via cross-feeding experiments of various knock-out mutants of Serratia ATCC sp.39006 (S39006).3 They showed that prodigiosin resulted from a bifurcated pathway (Fig. 1) with the independent synthesis of monopyrrole 2-methyl-3-amylpyrrole (MAP) 7 and bipyrrole 4-methoxy-2,2′-bipyrrole-5-carbaldehyde (MBC) 9 before a final condensation step catalysed by the enzyme PigC.
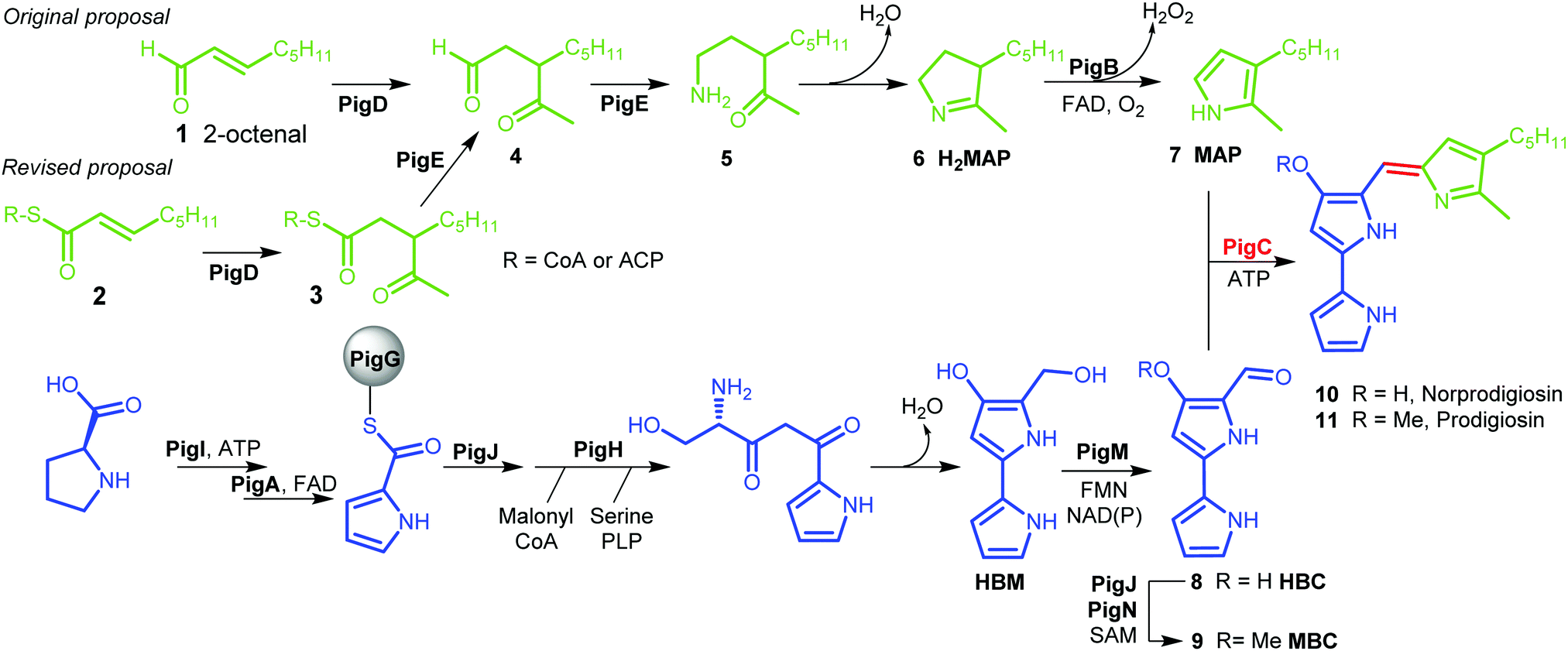 | ||
| Fig. 1 Biosynthesis of prodigiosin; green; formation of MAP, blue: formation of MBC, red: condensation. HBM is 4-hydroxy-2,2′-bipyrrole-5-methanol and HBC is 4-hydroxy-2,2′-bipyrrole-5-carbaldehyde. | ||
MBC is an intermediate in the biosynthesis of other prodiginines, natural products which share the tripyrrolic structure of prodigiosin 11. In particular, the enzymes involved in the biosynthesis of MBC in Streptomyces coelicolor A(3) have been well characterised.4 The biosynthesis of MAP, on the other hand, is specific to the formation of prodigiosin 11. 2-Octenal 1 was originally proposed to be the starting point of the pathway.3 Addition of an acetyl group from pyruvate by PigD was proposed to form 3-acetyloctanal 4, which then underwent a transamination catalysed by PigE,5 leading to amino ketone 5 which spontaneously cyclises to form dihydroMAP (H2MAP) 6. H2MAP is then oxidised to MAP 7 by PigB.6
However, Dresen et al. showed that, when presented with an aldehyde such as 2-octenal 1, PigD catalysed the direct addition of the acetyl group onto the aldehyde carbon, whereas, with less reactive α,β-unsaturated ketones and thioesters, attack on the β position was preferred.7 Kasparyan et al. extended this work to some homologues of PigD and suggested that thioester 3 may be the true natural product of PigD.8
In this work, we show that thioester 3 is indeed an intermediate in the MAP biosynthetic pathway. In addition, we provide evidence that PigE catalyses the reduction of the thioester intermediate to aldehyde 4 as well as catalysing the transamination of that aldehyde. As a result, we propose an alternative biosynthetic pathway for the formation of MAP which uses 2-octenoyl thioester 2, either with coenzyme A (CoA) or an acyl carrier protein (ACP), as its starting point (Fig. 1).
Results and discussion
Preparation of potential substrates
CoA and ACP thioesters are common intermediates in biosynthesis. In particular, straight-chain thioesters with even numbers of carbons are readily available from fatty acid biosynthesis (or degradation). In addition, it has been shown that PigD catalyses the transfer of an acetyl group from pyruvate to N-acetylcysteamine (NAC) thioester 12 to give thioester 13 (Fig. 2).7,8 Considering that NAC thioesters mimic CoA or ACP thioesters, we synthesised 13 to investigate if it could be converted to prodigiosin by S39006.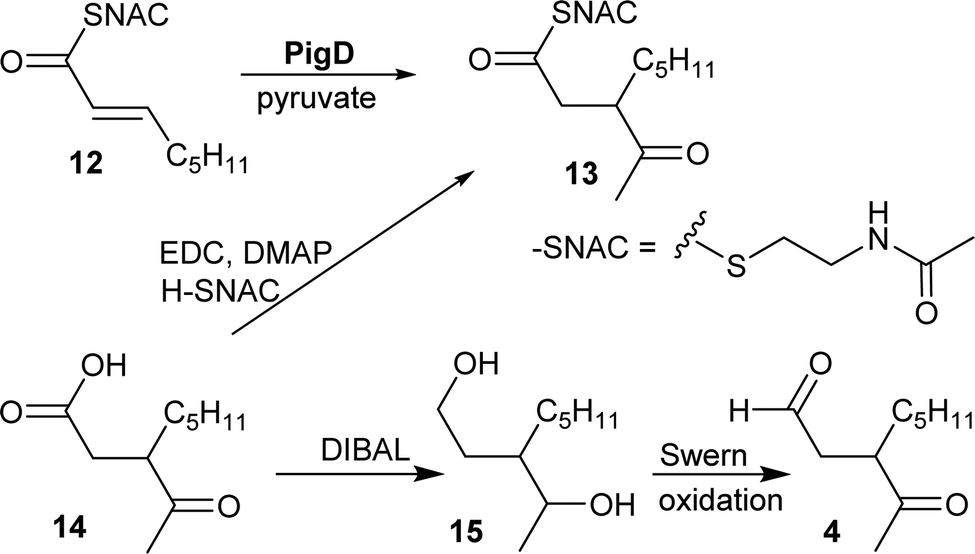 | ||
| Fig. 2 Syntheses of potential substrates of PigE. | ||
As thioesters are prone to hydrolysis and all testing of 13 would be done in aqueous solution, its hydrolysis product, 3-acetyloctanoic acid 14, was also synthesised, as a negative control, and the known PigE substrate 3-acetyloctanal 4 was synthesised as the positive control.
Acid 14 was obtained in three steps from 2-octanone (see ESI†): bromination on C-3 was followed by displacement of bromide by dimethyl malonate and then hydrolysis/decarboxylation. Acid 14 was then converted to 13via thioesterification with N-acetylcysteamine. Aldehyde 4 was obtained by reduction of 14 to the diol 15 followed by Swern oxidation.
Chemical complementation assays
S39006 ΔpigD has an in-frame deletion in pigD, avoiding any downstream polarity in the prodigiosin biosynthetic operon, and presents a white phenotype.3 However, when fed with an intermediate accepted by one of the subsequent enzymes in the MAP biosynthetic pathway this strain produces 11 and therefore presents a red phenotype.3,6In a qualitative assay, S39006 ΔpigD was streaked on agar plates and spots (2 μl, 1 M) of aldehyde 4 and thioester 13 were placed next to the streaks. After 12 h, a red colour was visible in the streak next to the spot in both cases (Fig. 3), suggesting that both compounds could restore prodigiosin production. The colour was less intense in the case of the thioester, perhaps suggesting less efficient uptake into the cell or less efficient utilisation of the NAC thioester than the true thioester intermediate.
 | ||
| Fig. 3 Chemical complementation of S39006 ΔpigD with aldehyde 4 (left) and thioester 13 (right). | ||
These results were confirmed by a quantitative complementation assay in liquid culture. After optimisation to overcome low levels of production of 11 due to the toxicity of 4, S39006 ΔpigD was treated with 4, 13 or 14 (1 mM) for 16 h and prodigiosin was then extracted from cell pellets with acidified EtOH and quantified by its absorbance at 535 nm.9 Again the results (Fig. 4) were that pigmentation was observed with aldehyde 4 and thioester 13 but not with carboxylic acid 14. The UV spectrum of the extract of the culture treated with 13 showed a maximum at 535 nm (Fig. 4c), consistent with prodigiosin.
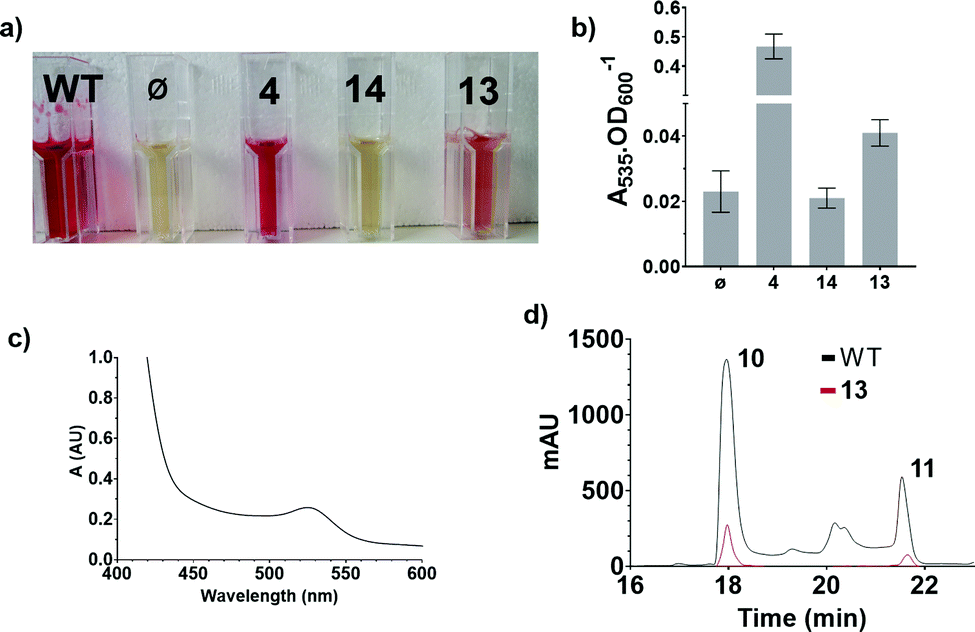 | ||
| Fig. 4 Results of the quantitative chemical complementation assays; (a) ethanolic extract of cultures of S39006 WT and S39006 ΔpigD incubated with no additive (ϕ), aldehyde 4, carboxylic acid 14 or thioester 13; (b) Absorbance at 535 nm normalised by OD600 for the specified extracts; (c) UV spectrum of the extract of S39006 ΔpigD inoculated with thioester 13; (d) Comparison of the HPLC traces (535 nm) of the extracts of S39006 WT and S39006 ΔpigD incubated with thioester 13. | ||
Extracts from cultures of S39006 ΔpigD not treated with any substrate have some absorption at 535 nm but this is a sloping baseline and no peak is seen. Nevertheless, to confirm that the increase in absorbance during the chemical complementation experiments was due to prodigiosin and not some other metabolite, an ethanolic extract from a culture of wild-type S39006 was analysed by HPLC. Two peaks absorbing at 535 nm were observed and LCMS (see ESI†) showed they were prodigiosin 11 and norprodigiosin 10. Norprodigiosin results from the condensation of the unmethylated bipyrrole HBC 8 with MAP 7 (see Fig. 1). The peaks corresponding to 11 and 10 were also observed in the HPLC trace of the extract from S39006 ΔPigD treated with 13 (Fig. 4d). This proves that thioester 13 can restore production of prodigiosin in S39006 ΔpigD. Thus it is very likely that a related thioester is an intermediate in the biosynthesis of MAP.
Bioinformatic analysis of PigE
The presence of a thioester intermediate in MAP biosynthesis implies that a reduction step converting the thioester to an aldehyde needs to be added to the original pathway. The possibility that PigE had thioester reductase activity as well as the established aminotransferase activity was investigated.The sequence of PigE was analysed using the NCBI BLAST software.10 This showed that the C-terminal region of PigE (residues 394 to 848) had strong similarity with pyridoxal phosphate (PLP)-dependent aminotransferases, particularly class III aminotransferases that generally act on or produce terminal alkylamines.11 A crystal structure of the C-terminal domain with PLP bound has been published.5 The N-terminal region (residues 2 to 341) showed homology with acyl-ACP reductases, which catalyse the reduction of fatty acyl ACP thioesters to the corresponding aldehydes.12 The majority of proteins that showed good similarity with full length PigE were homologues in other organisms known to produce prodigiosin or related compounds (e.g. TamH in tambjamine biosynthesis13).
PHYRE2 homology modelling software14 generated a structural model for the N-terminal domain (see ESI†) using the recently published structure of the acyl-ACP reductase (AAR) involved in cyanobacterial biosynthesis of hydrocarbons (26% identity).15 The second best hit was glutamyl tRNA reductase (GluTR) from Methanopyrus kandleri (17% identity), which catalyses the transformation of glutamyl tRNA into glutamate-1-semialdehyde, the first committed step of heme biosynthesis in plants, algae and most bacteria. Both these enzymes initially transfer the acyl group onto an active site cysteine residue and it is this thioester intermediate that is subsequently reduced by NADPH.16 This cysteine residue is conserved in PigE (Cys296) and is in the same position in the homology model as Cys294 in AAR when the two structures are overlaid. Interestingly both AAR and GluTR have been shown to channel their product aldehydes directly to the next enzyme in the pathway via an exit tunnel that is distinct from the substrate entry route. For AAR the next enzyme is a di-iron-dependent aldehyde-deformylating oxygenase (ADO),15 but for GluTR it is a PLP-dependent aminotransferase, glutamate-1-semialdehyde aminomutase (GSAM).17 It seems likely, therefore, that the reductase and aminotransferase domains of PigE form a structure similar to the GluTR-GSAM complex and substrate channelling of aldehyde 4 may occur.
The bioinformatic analysis, therefore, reveals the presence of two domains in PigE: a C-terminal aminotransferase domain, and a N-terminal thioester reductase domain. This strongly suggests that the N-terminal domain of PigE is the protein that catalyses the reduction of the thioester intermediate.
In vitro characterisation of PigE
Plasmid pQE80-L::oriT containing pigE from S39006 was transformed into E. coli BL21, giving strain E. coli pPigE. PigE protein was purified from cultures of this strain. A UV spectrum of the purified PigE showed a peak at 410 nm, consistent with PLP bound as its Schiff's base with the active-site lysine residue. (Fig. 5).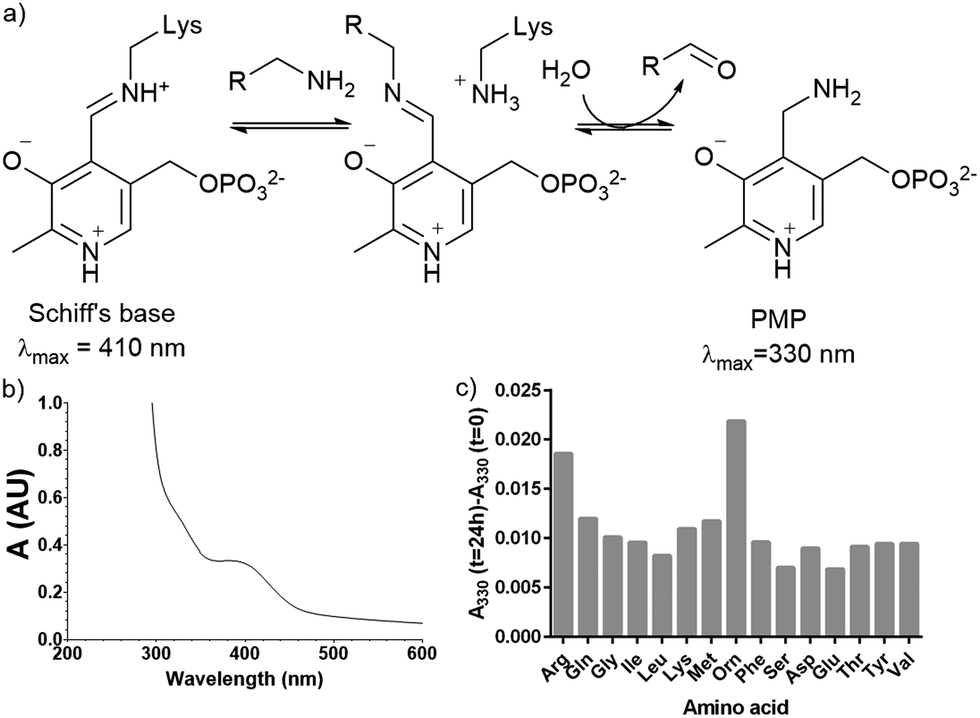 | ||
| Fig. 5 (a) Intermediates in PLP-catalysed transamination; (b) UV spectra of isolated PigE; (c) change in the A330 of PigE after 24 h in the presence of various amino acids. | ||
To determine the likely amino donor in the transaminase reaction PigE was incubated with various amino acids individually and the formation of the pyridoxamine phosphate (PMP) intermediate was monitored by recording the absorbance at 330 nm. The most significant increase was observed with ornithine and arginine (Fig. 5c) suggesting that PigE is a transaminase dependent on these amino acids.
Lysates prepared from E. coli BL21 and E. coli pPigE were supplemented with thioester 13, ornithine and PLP and incubated at 30 °C for 16 h. After removing cell debris, the resulting mixtures were analysed by LCMS and compared with synthetic samples of H2MAP 6 and thioester 13 (Fig. 6). In the extract from E. coli BL21, only the peak corresponding to the thioester 13 was visible. In the extract from E. coli pPigE, the main peak corresponded to H2MAP 6 and only a very small peak corresponding to 13 was visible. This is a strong indication that the presence of PigE was necessary for 13 to be reduced in addition to the transamination to give 6.
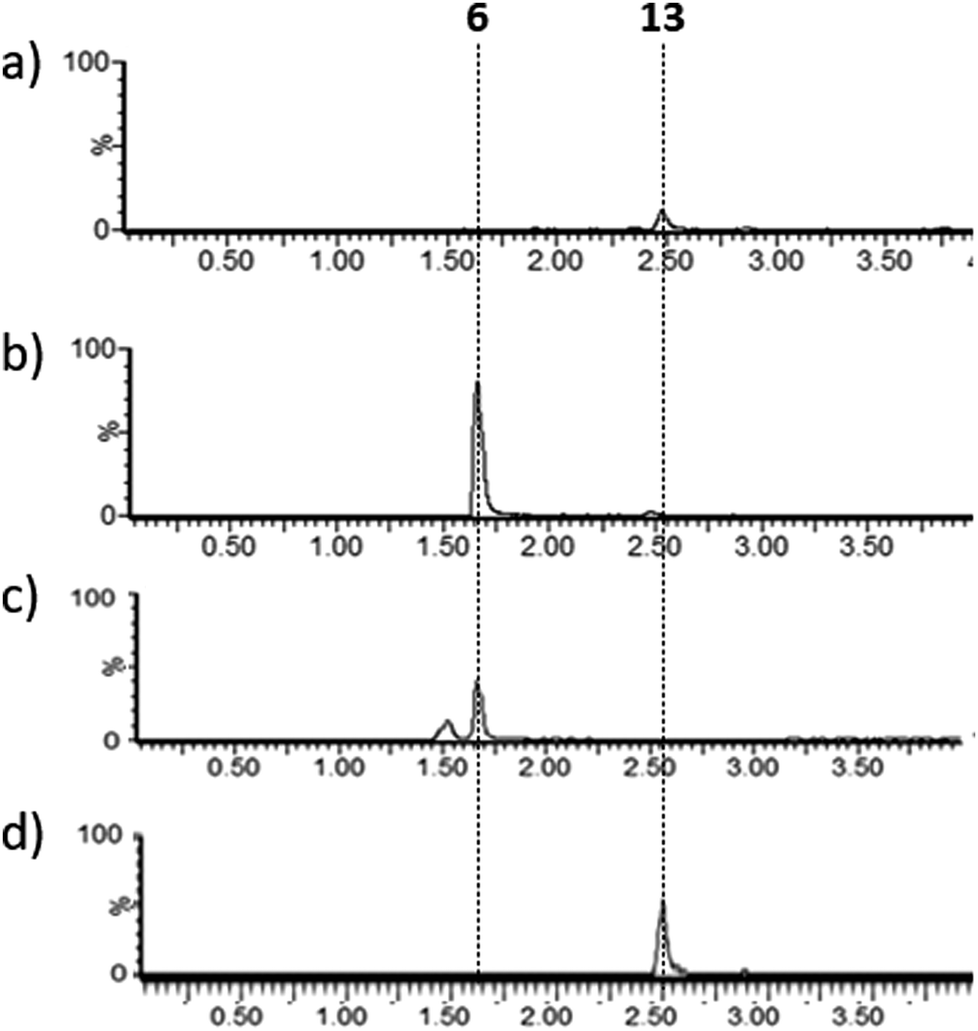 | ||
| Fig. 6 Extracted ion chromatograms (m/z 154.2 + 288.2) (a) and (b) after incubation of 13 with cell lysates of (a) E. coli BL21 or (b) E. coli pPigE, (c) synthetic H2MAP 6 (m/z 154.16, 6 + H+), (d) synthetic 13 (m/z 288.16, 13 + H+). Retention times corresponding to 6 and 13 are shown. | ||
When purified PigE was incubated with thioester 13, ornithine and NAD(P)H at 30 °C for 16 h in phosphate buffer, no peak corresponding to H2MAP could be detected by LCMS. Similarly experiments with the same substrates in the presence of a variety of metal ions failed to show any significant change in absorbance of NAD(P)H at 340 nm over time resulting from the presence of PigE. Taken together these experiments suggest that additional cofactors and/or metal ions contained in the cell lysates may be necessary for rapid reaction to occur. It is noteworthy that the activity of GluTR, to which the N-terminal domain of PigE may be related, is affected by several regulatory proteins and by the presence of the tRNA18 and activity can easily be lost by oxidation of the active site cysteine.16
Conclusions
We have shown that thioester 13 can restore production of prodigiosin in the deficient strain, S39006 ΔpigD. In the presence of PigE, the thioester can be reduced, and the resulting aldehyde undergoes transamination, leading to the next biosynthetic intermediate, H2MAP.We therefore propose the revised biosynthetic pathway for prodigiosin shown in Fig. 1. Our results, along with the bioinformatic analysis, support the notion that PigE is a bifunctional enzyme, with a thioester reductase domain in its N-terminal region and a transaminase domain in its C-terminal region. We also propose that the amino donor for the transaminase activity is mostly likely ornithine or arginine.
Experimental
Prodigiosin production and quantification
Cell extract assays
E. coli BL21 or E. coli pPigE were cultured in 200 ml of LB until and OD600 = 0.6. Cultures were then induced with 1 mM IPTG at 16 °C for 14–16 h. Cells were harvested by centrifugation at 4500 rpm for 20 min at 4 °C. The cells were held on ice for 15 min and then resuspended in 20 ml of phosphate buffer. After a further 30 min on ice, the cell suspensions were sonicated (6 × 20 s with 30 s breaks). Eppendorf tubes were charged with 13 (1 mM), and ornithine and PLP (1.25 mM final concentrations), then cell lysate was added to a total volume of 0.5 ml. The resulting mixture was gently shaken overnight at 30 °C, then centrifuged and the supernatant was analysed directly by LCMS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the BBSRC, UK (awards BB/N008081/1 and BB/K001833/1). MC was supported by the Frances and Augustus Newman foundation, and HDB by the Cambridge Commonwealth Trust and Emmanuel College, Cambridge.Notes and references
- N. R. Williamson, P. C. Fineran, F. J. Leeper and G. P. C. Salmond, Nat. Rev. Microbiol., 2006, 4, 887–899 CrossRef CAS.
- A. Fürstner, Angew. Chem., Int. Ed., 2003, 42(31), 3582–3603 CrossRef.
- N. R. Williamson, H. T. Simonsen, R. A. A. Ahmed, G. Goldet, H. Slater, L. Woodley, F. J. Leeper and G. P. C. Salmond, Mol. Microbiol., 2005, 56(4), 971–989 CrossRef CAS.
- A. M. Cerdeño, M. J. Bibb and G. L. Challis, Chem. Biol., 2001, 8(8), 817–829 CrossRef.
- X. Lou, T. Ran, N. Han, Y. Gao, J. He, L. Tang, D. Xu and W. Wang,, Biochem. Biophys. Res. Commun., 2014, 447(1), 178–183 CrossRef CAS.
- M. Couturier, H. D. Bhalara, S. R. Chawrai, R. Monson, N. R. Williamson, G. P. C. Salmond and F. J. Leeper, ChemBioChem, 2020, 21(4), 523–530 CrossRef CAS.
- C. Dresen, M. Richter, M. Pohl, S. Lüdeke and M. Müller, Angew. Chem., Int. Ed., 2010, 49(37), 6600–6603 CrossRef CAS.
- E. Kasparyan, M. Richter, C. Dresen, L. S. Walter, G. Fuchs, F. J. Leeper, T. Wacker, S. L. A. Andrade, G. Kolter, M. Pohl and M. Müller, Appl. Microbiol. Biotechnol., 2014, 98(23), 9681–9690 CrossRef CAS.
- H. Slater, M. Crow, L. Everson and G. P. C. Salmond, Mol. Microbiol., 2003, 47(2), 303–320 CrossRef CAS.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215(3), 403–410 CrossRef CAS.
- H. J. Cha, J. H. Jeong, C. Rojviriya and Y. G. Kim, PLoS One, 2014, 9(11), e113212 CrossRef CAS; D. Schiroli and A. Peracchi, Biochim. Biophys. Acta, Gen. Subj., 2015, 1854(9), 1200–1211 CrossRef.
- A. Schirmer, M. A. Rude, X. Li, E. Popova and S. B. del Cardayre, Science, 2010, 329(5991), 559–562 CrossRef CAS.
- C. Burke, T. Thomas, S. Egan and S. Kjelleberg, Environ. Microbiol., 2007, 9(3), 814–818 CrossRef CAS.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg,, Nat. Protoc., 2015, 10(6), 845–858 CrossRef CAS.
- Y. Gao, H. Zhang, M. Fan, C. Jia, L. Shi, X. Pan, P. Cao, X. Zhao, W. Chang and M. Li, Nat. Commun., 2020, 11(1), 1525 CrossRef CAS.
- J. Moser, W. D. Schubert, V. Beier, I. Bringemeier, D. Jahn and D. W. Heinz, EMBO J., 2001, 20(23), 6583–6590 CrossRef CAS.
- C. Lüer, S. Schauer, K. Möbius, J. Schulze, W.-D. Schubert, D. W. Heinz, D. Jahn and J. Moser, J. Biol. Chem., 2005, 280(19), 18568–18572 CrossRef CAS; L. A. Nogaj and S. I. Beale, J. Biol. Chem., 2005, 280(26), 24301–24307 CrossRef.
- A. Zhao, Y. Fang, X. Chena, S. Zhao, W. Dong, Y. Lin, W. Gong and L. Liu, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(18), 6630–6635 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00173b |
| This journal is © The Royal Society of Chemistry 2021 |