
Guest–host interlinked PEG-MAL granular hydrogels as an engineered cellular microenvironment†
Adrienne E.
Widener
a,
Mallika
Bhatta
a,
Thomas E.
Angelini
 ba and
Edward A.
Phelps
*a
ba and
Edward A.
Phelps
*a
aJ. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Gainesville, FL, USA. E-mail: ephelps@bme.ufl.edu
bDepartment of Mechanical and Aerospace Engineering, University of Florida, Gainesville, FL, USA
First published on 28th December 2020
Abstract
We report the development of a polyethylene glycol (PEG) hydrogel scaffold that provides the advantages of conventional bulk PEG hydrogels for engineering cellular microenvironments and allows for rapid cell migration. PEG microgels were used to assemble a densely packed granular system with an intrinsic interstitium-like negative space. In this material, guest–host molecular interactions provide reversible non-covalent linkages between discrete PEG microgel particles to form a cohesive bulk material. In guest–host chemistry, different guest molecules reversibly and non-covalently interact with their cyclic host molecules. Two species of PEG microgels were made, each with one functional group at the end of the four arm PEG-MAL functionalized using thiol click chemistry. The first was functionalized with the host molecule β-cyclodextrin, a cyclic oligosaccharide of repeating D-glucose units, and the other functionalized with the guest molecule adamantane. These two species provide a reversible guest–host interaction between microgel particles when mixed, generating an interlinked network with a percolated interstitium. We showed that this granular configuration, unlike conventional bulk PEG hydrogels, enabled the rapid migration of THP-1 monocyte cells. The guest–host microgels also exhibited shear-thinning behavior, providing a unique advantage over current bulk PEG hydrogels.
Introduction
Synthetic analogs of natural extracellular matrices have emerged that are well-suited for basic science and regenerative medicine applications.1 Polyethylene glycol (PEG) is one of the most commonly implemented synthetic polymers to model extracellular matrix (ECM) with useful properties for regenerative therapeutics. PEG has a well-established chemistry and a long history of safety in vivo.2–5 Addition of reactive groups flanking the PEG chain, such as vinyl-sulfone, norbornene, acrylate, or maleimide (MAL), allows for crosslinking and conjugation of biomolecules in aqueous solution at physiological pH.6–11 A major strength of these strategies is the modular “plug-and-play” design of the base hydrogel system, which allows bioactivity and mechanical properties to be independently tailored. PEG is intrinsically resistant to protein adsorption and cell adhesion, providing low background interference with incorporated biofunctionalities. PEG hydrogel systems are made susceptible to cellular invasion by crosslinking with protease (MMP)-cleavable peptide sequences,7,12 domains for cell adhesion (e.g., RGD peptide),13 and tethered growth factors.14–18 Due to its modular nature and excellent in vivo properties, PEG is an appealing platform for the fabrication of regenerative therapies.19Despite successful tissue engineering applications and the attractive flexibility of the crosslinking chemistry toolboxes,20 bulk PEG hydrogels remain inadequate systems for modeling certain types of rapid and dynamic cell behavior at the tissue scale. This is because cell movement in the dense bulk PEG hydrogel depends on proteolysis of degradable crosslinks,21 and is thus limited to a maximal rate of approximately 75–170 μm per day.5,22 In contrast, immune cells migrate in biological tissues via proteolytically-independent mechanisms at speeds that can exceed 25 μm per minute,23 which constitutes a 500-fold difference in cell migration velocity between PEG hydrogels and biological tissues. Thus, there is a significant unmet need to engineer a solution to this cell migration speed limit if synthetic ECMs are to meet the needs of demanding biological experiments where tissue-like cell migration is a fundamental aspect. We propose that the generation of hydrogels with narrow cell-scale negative spaces, akin to the tissue interstitium, is a robust strategy to increase cell migration speeds in PEG hydrogels.
Hydrogels containing highly interconnected macroporous structures have been investigated to improve cell migration from surrounding tissues into the interior of implanted scaffolds.24–28 Methods to generate macroporous PEG hydrogels include salt-particle leaching,27,29 cryogels,30–33 and sacrificial negative molds.25,26,34 However, these strategies depend on tissue remodeling of the large void spaces. In contrast, T lymphocyte migration through native tissues occurs largely through amoeboid-type mechanisms without structurally changing the local matrix architecture.35 Further, in each of these methods, the pore-forming process is incompatible with living cells, which must typically be later seeded into the scaffold after fabrication. It could be beneficial for seeding the negative space of porous scaffolds with cells, if the process could be made more friendly to cells so that they can be incorporated at the time of scaffold formation instead of requiring an additional seeding step.
Granular hydrogels are an emerging paradigm in the field of biosynthetic hydrogels that constitute a starting point from which to create an artificial interstitium. Landmark examples of granular hydrogels include: (1) enzymatic and click-chemistry annealing of hyaluronic acid microgels36,37 or PEG hydrogel microgels38–40 (2) physically interacting granular hydrogels composed of tightly-packaged or “jammed” microgels,41 and (3) reversible interactions between discrete gel components based on hyaluronic acid microgels crosslinked by β-cyclodextrin/adamantane guest–host interactions.42
Here we demonstrate a granular hydrogel scaffold composed of polyethylene glycol maleimide (PEG-MAL) microgels that have been functionalized with guest–host molecules. These guest–host molecules provide an interlinking reversible interaction between the PEG-MAL microgels. The PEG-MAL microgels introduce a porous void space that enables rapid cell migration which can be visualized to study immune cell interactions within the matrix. In addition, reversible interlinks between the microgels give the material self-healing and shear-thinning properties. Bulk bio-synthetic hydrogels such as those based on PEG have been implemented successfully for a wide arrange of regenerative applications. Yet one important area in which these materials perform poorly is in applications where rapid cell invasion/migration would be beneficial. Rapid cell invasion is particularly important to the behavior of immune cells. Examples of therapeutic applications that could benefit from rapid invasion of immune and other cell types include re-vascularization that depends on macrophage regulatory activity, antigen depots for cancer vaccines or tolerance treatments that depend on antigen-presenting cell migration, and in vitro models of lymph nodes and invasive tumor organoids for organ-on-a-chip devices.
Results
PEG-MAL has been successfully used for a wide range of biological applications. However, as maleimide reacts quickly with thiols, it can potentially lead to inhomogeneously crosslinked gels when reacted at physiological pH.43 To generate large quantities of homogenous PEG-MAL microgels, we sought to slow down the crosslinking reaction, enabling more time for handling of the pre-gel solution. We studied the effect of pH, crosslinker, and the number of PEG arms to optimize the PEG-MAL gelation parameters. Due to the very short gelation time of PEG-MAL at neutral pH (<10 seconds), rheometric measurement of gelation point was not practical to perform. Instead, we performed manual estimation of gelation point using the point when increasing viscosity inhibited pipetting as an indicator for gelation. Bulk gels were formed by rapidly mixing 4-arm PEG-MAL and 8-arm PEG-MAL macromers with PEG-dithiol or dithiothreitol (DTT) over a range of pH. We found that 4-arm PEG-MAL took twice as long to gel than 8-arm PEG-MAL (ESI Fig. 1A†) while crosslinking with PEG-dithiol resulted in slower gelation than DTT. Solutions of macromer and crosslinker colored with cyan and magenta-colored food dyes under a stereoscope were used to observe gel homogeneity upon mixing of macromer with crosslinker. PEG-MAL macromer was pipetted between two glass slides separated by a spacer, followed by PEG-dithiol crosslinker. The solutions were mixed by pipetting up and down until gelation occurred (ESI Fig. 1B†). We observed that decreasing pH allowed for more homogeneously mixed gels (ESI Fig. 1C†). At pH 7.0, 4-arm PEG-MAL had a rapid gelation time of approximately 4 seconds that made it difficult to obtain homogeneous mixing of gel components, leaving microdomains of highly crosslinked areas and low crosslinked areas, as seen by the separate cyan and magenta regions (ESI Fig. 1C†). At pH 5.6, the gelation time of 4-arm PEG-MAL was 34 seconds, and at pH 4.0, the gelation time was 215 seconds. By using a pH with extended gelation times (pH 5.6), a more homogeneous mixture of gel components was achieved.Water-soluble macrocyclic hosts that have proven useful for the generation of reversible bonds in biomaterials design include cyclodextrins and cucurbit[n]urils.44 In guest–host chemistry, different guest molecules reversibly and non-covalently interact with their cyclic host molecules through hydrophobic and hydrophilic interactions. The host molecule we used is β-cyclodextrin, a cyclic oligosaccharide of seven repeating D-glucose units. Cyclodextrin molecules are shaped like a truncated cone with a hydrophobic inner core and hydrophilic outer surface. Cyclodextrins are available at low cost, are water-soluble, non-toxic, and included on the US FDA list of generally recognized safe molecules due to their long history of safe usage in pharmaceutical, food, and cosmetic products. There are many potential guest molecules for complexation with cyclodextrin. The roughly spherical hydrophobic adamantane group has been extensively characterized for β-cyclodextrin complexation in biomaterials research,44 and it will be used here. However, other guest molecules with higher or lower affinities can be substituted if needed.
Here, guest–host molecular interactions45,46 were used to provide reversible non-covalent linkages between densely-packed PEG-MAL hydrogel microgels to generate a granular system with an intrinsic interstitium-like microarchitecture. Synthesis of β-cyclodextrin and adamantane functionalized hydrogel precursor molecules was performed using standard Michael-type addition to multi-arm PEG-MAL before crosslinking similarly to our published methods of functionalization of PEG-MAL macromer with the G![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) SPC peptide for cell-adhesion.9,47 Characterization of tethering of thiolated ligands such as GRGDSPC peptide to PEG-MAL macromer was previously demonstrated to be highly efficient.9,15,47,48 The functionalized PEG-MAL macromer solutions were homogeneously mixed with a stoichiometrically-balanced amount of PEG-dithiol crosslinker at pH 5.6, and a trace amount of Alexa Fluor 488 or 568 maleimide was added to allow for visualization. Similarly to PEG microgel fabrication methods described by the Anseth group,40 this mixture was added to a larger volume of mineral oil stabilized with SPAN80 surfactant, and immediately vortexed at full speed to form an emulsion (Fig. 1A). The emulsion was maintained with gentle rocking agitation for 30 minutes while the microgels crosslinked. PEG microgels were purified from the emulsion by centrifugation, and multiple washing steps (3× Triton-X 100, 1× 50% acetone in deionized water, 1× DI water, 2× PBS) removed residual mineral oil and surfactant. This emulsion method generated microgels that ranged from 5 μm to 200 μm in diameter, depending on the vortexing time. We found that vortexing the solution for a more extended period (90 s) generated smaller microgels (average = 5.6 μm) and for shorter periods (15 s) generated larger microgels (average = 106.7 μm) (Fig. 1B and C). By measuring the polydispersity index of the resulting microgels vortexed for different times, we found that the polydispersity decreased with increasing vortexing time, indicating that the longer the vortex time, the more homogeneous the material (Fig. 1C). When the two species of microgel were mixed in equal proportions and compacted via centrifugal filtration, an interlinked granular network formed (Fig. 1D).
SPC peptide for cell-adhesion.9,47 Characterization of tethering of thiolated ligands such as GRGDSPC peptide to PEG-MAL macromer was previously demonstrated to be highly efficient.9,15,47,48 The functionalized PEG-MAL macromer solutions were homogeneously mixed with a stoichiometrically-balanced amount of PEG-dithiol crosslinker at pH 5.6, and a trace amount of Alexa Fluor 488 or 568 maleimide was added to allow for visualization. Similarly to PEG microgel fabrication methods described by the Anseth group,40 this mixture was added to a larger volume of mineral oil stabilized with SPAN80 surfactant, and immediately vortexed at full speed to form an emulsion (Fig. 1A). The emulsion was maintained with gentle rocking agitation for 30 minutes while the microgels crosslinked. PEG microgels were purified from the emulsion by centrifugation, and multiple washing steps (3× Triton-X 100, 1× 50% acetone in deionized water, 1× DI water, 2× PBS) removed residual mineral oil and surfactant. This emulsion method generated microgels that ranged from 5 μm to 200 μm in diameter, depending on the vortexing time. We found that vortexing the solution for a more extended period (90 s) generated smaller microgels (average = 5.6 μm) and for shorter periods (15 s) generated larger microgels (average = 106.7 μm) (Fig. 1B and C). By measuring the polydispersity index of the resulting microgels vortexed for different times, we found that the polydispersity decreased with increasing vortexing time, indicating that the longer the vortex time, the more homogeneous the material (Fig. 1C). When the two species of microgel were mixed in equal proportions and compacted via centrifugal filtration, an interlinked granular network formed (Fig. 1D).
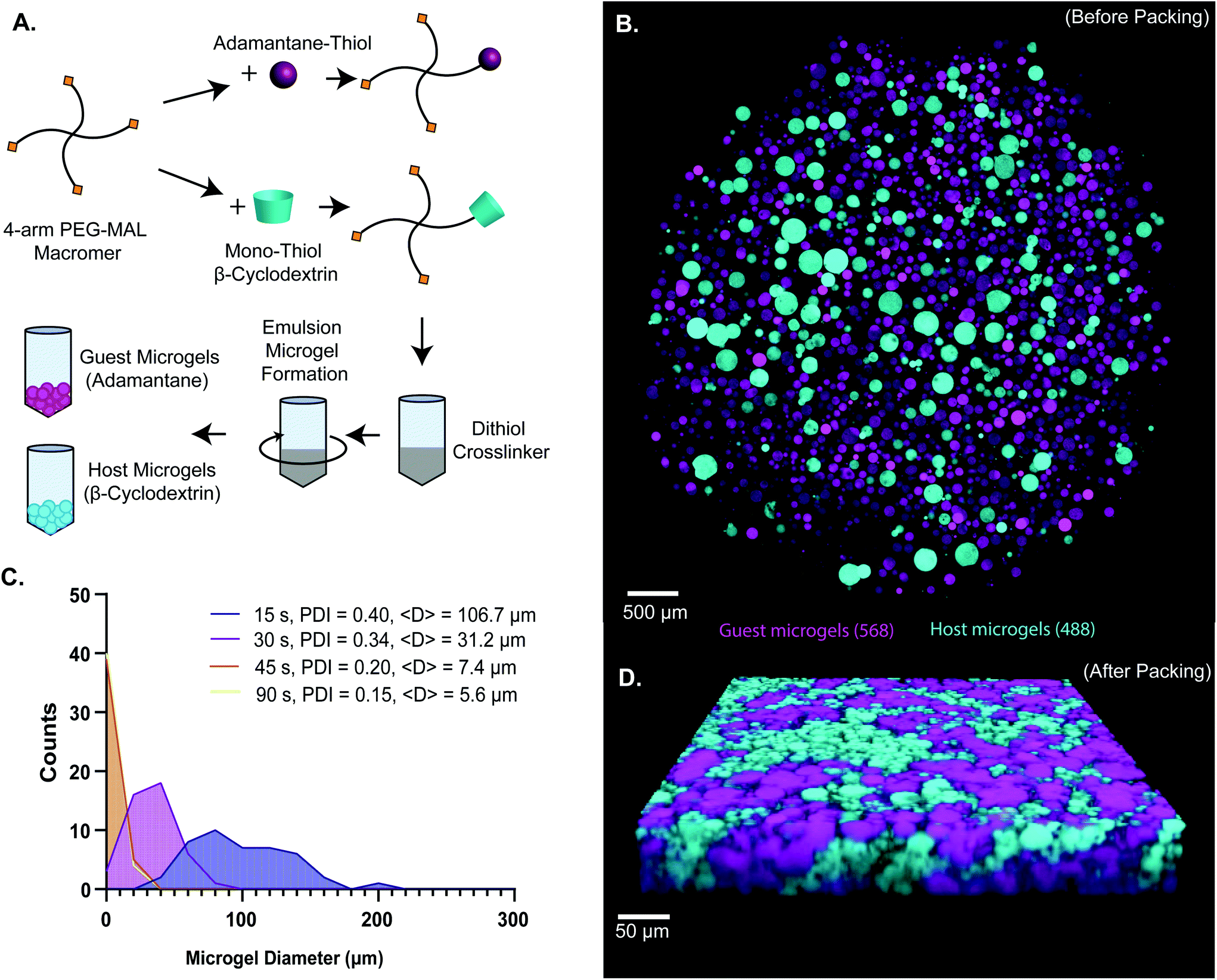 | ||
| Fig. 1 (a) Scheme of 4-arm PEG-MAL microgel synthesis. 4-arm PEG-MAL macromer is first functionalized with 1-adamantane-thiol and mono-thiol-β-cyclodextrin through michael-type addition chemistry. The pegylated adamantane (top) and pegylated β-cyclodextrin (bottom) are then added to a crosslinker, PEG-dithiol, quickly transferred into mineral oil, and vortexed to create an emulsion. On the right is an example schematic of the two different types of microgels (guest microgels and host microgels). (b) Confocal image of microgels before packing. (c) Frequency histogram of size distributions of microgels made at 15, 30, 45 and 90 s vortex times. Polydispersity index (pdi) decreased with increasing vortex time. Average diameter, <d>, also decreased with increasing vortex time: 106.7 μm for 15 s, 31.2 μm for 30 s, 7.4 μm for 45 s, and 5.6 μm for 90 s.(d) confocal microscopy z-stack volume rendering of packed guest-microgels and host-microgels. The percolated interstitium is shown between the different species of microgels, allowing for cellular infiltration and uninhibited movement. | ||
We next sought to determine the relationship microgel diameter and the size of the void spaces of the interstitium. To demonstrate this interstitium, we added a high-molecular-weight TRITC-dextran that was too large to penetrate the microgels to fill the void space of the granular hydrogel scaffold. Confocal microscopy z-stacks were rendered in three dimensions (3D) to visualize the void spaces between the microgels (Fig. 2A), demonstrating a continuous network of pores between the microgels. We quantified the inter-microgel distance and average pore area in scaffolds formed from different size microgels generated by varying the emulsion vortexing conditions (Fig. 2B). We found that all microgel sizes created scaffolds with similar pore structures that varied over a range of length scales due to the heterogeneity of microgel sizes. There was a trend of decreasing interparticle distance and pore area with decreasing microgel size, as expected. However the average interparticle distance was around 10 μm, on the same length scale as cells. From here, we chose to fix the average particle size to be between 10 and 100 μm (vortexing time of 30 s), as microgels smaller than 10 μm tended to self-aggregate through colloidal interaction,49 which was detrimental to scaffold formation, while microgels larger than 100 μm created scaffolds with a greater percentage of pores larger than cell-length scales (10 μm diameter). Based on these studies, we vortexed our microgels for 30 s to avoid aggregation of the particles due to colloidal interactions and to maintain the interstitium size ideal for cell invasion.
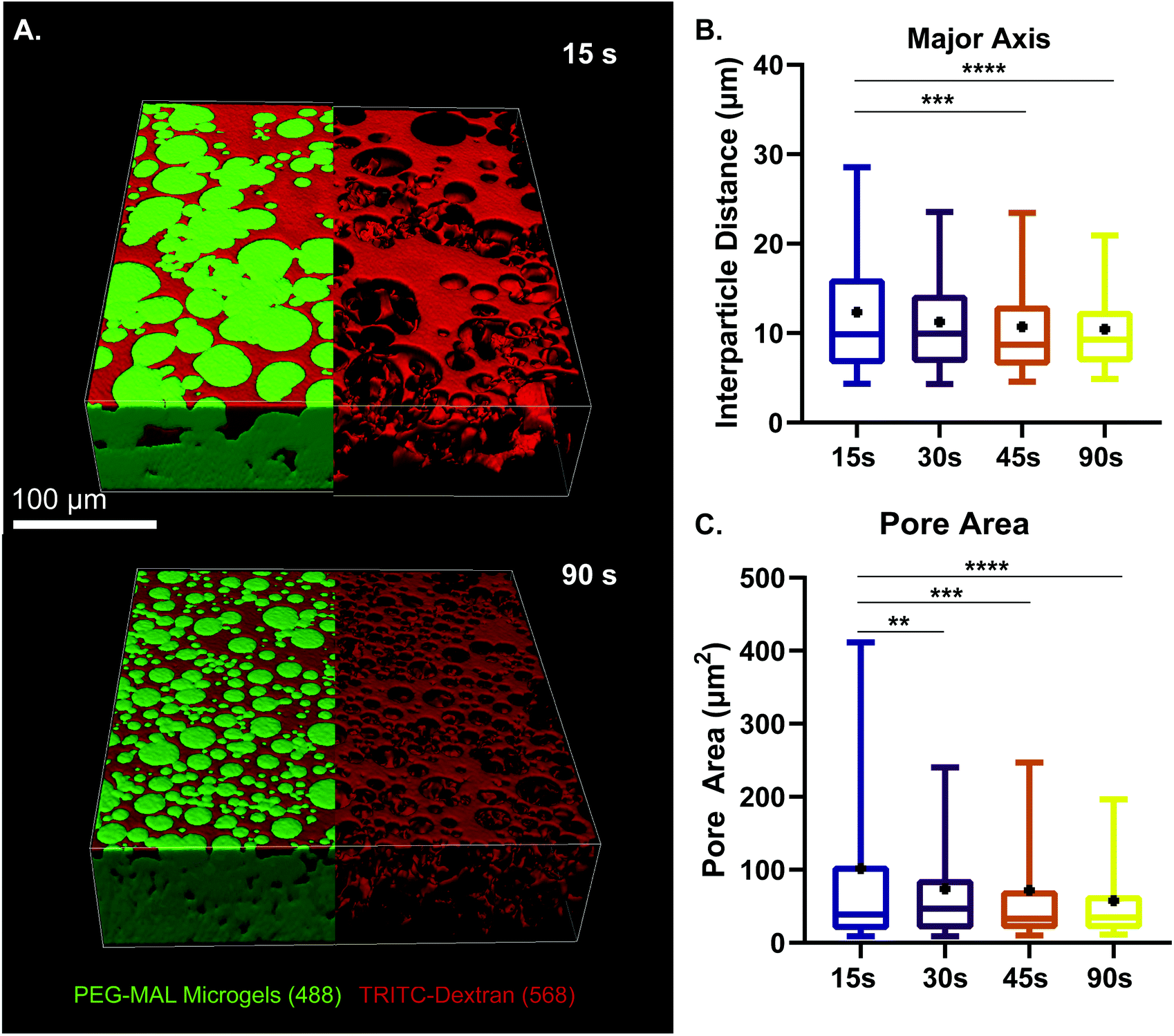 | ||
| Fig. 2 Characterization of microgel diameter and pore size. (a) z-Stack projection of PEG-MAL microgels vortexed for 15 and 90 s. Visualization of void space by using high-molecular-weight tritc dextran to fill pores within the granular hydrogel scaffold. Scale bar = 100 μm. (b) Interparticle distance between pores of scaffold at different vortexing times. (c) Pore area analysis at different vortexing times. Data is displayed as a box and whisker plot. The box extends from the 25th to 75th percentile. The median is denoted the middle line. The whiskers extend to the 5th and 95th percentile, mean is denoted by the black dot. Significance determined by one-way anova, p < 0.0001. | ||
We next validated the ability of adamantane-functionalized microgels to bind β-cyclodextrin by incubation with soluble Alexa Fluor 488-labeled β-cyclodextrin, made by reacting Alexa-Fluor 488 maleimide with mono-thiol-β-cyclodextrin (Fig. 3A). We found adamantane functionalized PEG-MAL bound significantly more β-cyclodextrin-488 than unfunctionalized microgels (Fig. 3B), indicating that the guest–host molecules were able to interact. The reverse experiment to determine binding of monomeric adamantane to β-cyclodextrin functionalized microgels was not feasible as monomeric adamantane has low water solubility. Macroscopic videos were taken to demonstrate the bulk properties of the microgel scaffold with and without the guest–host interactions. In the first example (Fig. 4A and ESI Videos 1 and 2†), scaffolds of packed microgels were placed onto a glass slide and manually sheared by a spreading motion with a spatula. In the guest–host gels, the scaffolds retained their shape, whereas the unfunctionalized PEG-MAL microgels without guest–host interactions dissociated and broke apart. In the second example, a 100 μL volume of water was dripped onto the microgel scaffolds and the scaffolds were observed for the ability to retain shape (Fig. 4B and ESI Videos 3 and 4†). In the guest–host gels, the scaffolds did not dissociate with the addition of water droplets, whereas the unfunctionalized PEG-MAL microgels were rapidly disrupted by the force of the water droplets.
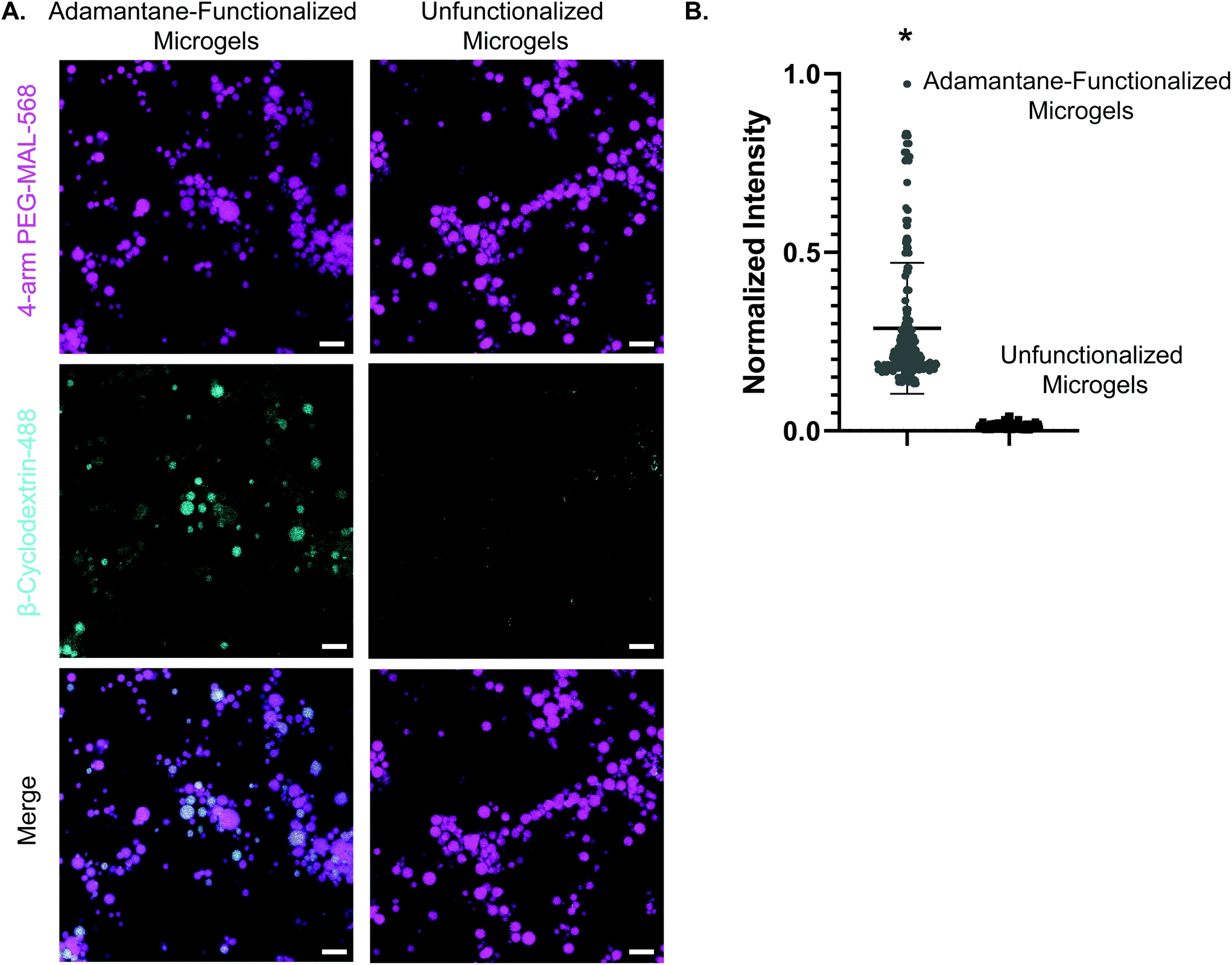 | ||
| Fig. 3 Confirmation of guest–host interactions through colocalization. (a) Panel 1: adamantane-functionalized PEG-MAL microgels and unfunctionalized microgels labeled with AlexaFluor-568. Panel 2: AlexaFluor-488-β-cyclodextrin fluorescence in the gels after incubation with soluble AlexaFluor-488-β-cyclodextrin. Panel 3: merge of PEG-MAL microgels and AlexaFluor-488-β-cyclodextrin. Scale bar = 50 μm for all panels. (c) Normalized fluorescence intensity for soluble AlexaFluor-488-β-cyclodextrin with adamantane-functionalized microgels and unfunctionalized microgels. Data is displayed as mean + s.e.m. Significance calculated by students t-test, p < 0.0001. | ||
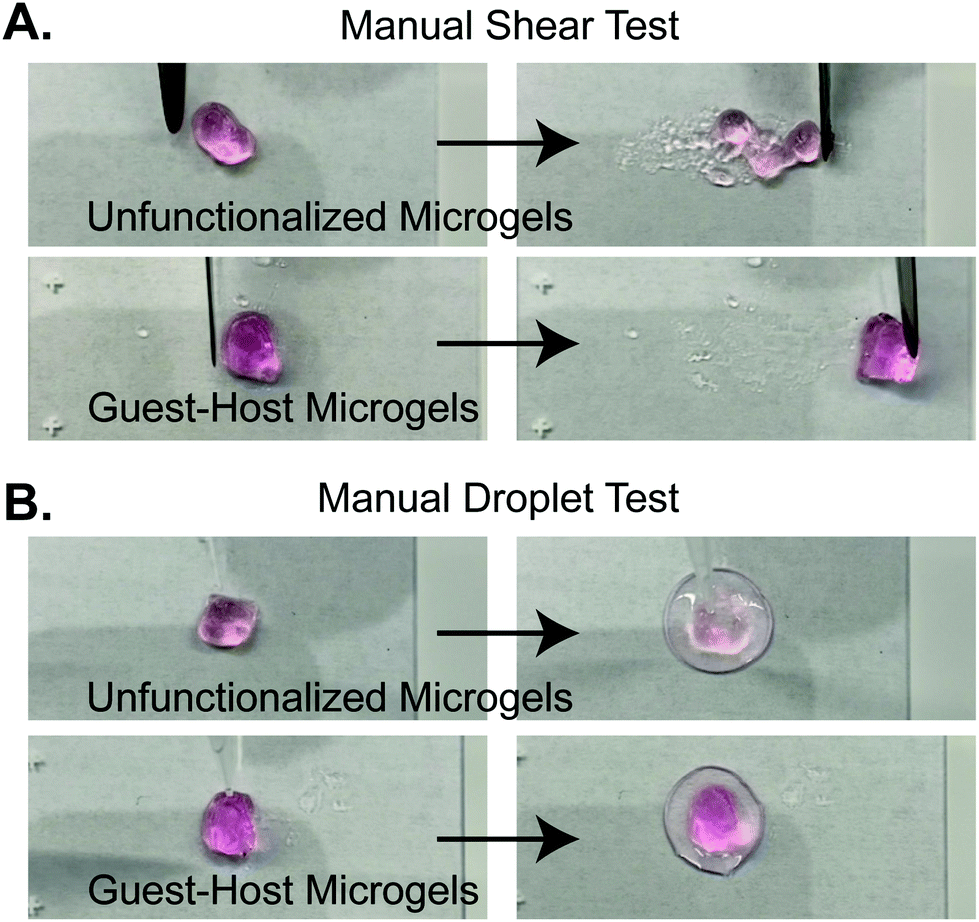 | ||
| Fig. 4 Stills from supplementary videos demonstrating macroscopic properties of guest-microgels and host-microgels mixed microgel scaffolds. (a) Manual shearing of scaffold by spatula. Unfunctionalized microgels dissociate upon shearing. Guest–host microgels maintain shape during manipulation. (b) Unfunctionalized microgels are dissociated by wetting with a water droplet. Guest–host microgels do not dissociate upon wetting. | ||
To confirm guest–host interactions between the two gel species, we also tested macroscopic adamantane- and β-cyclodextrin-functionalized bulk PEG-MAL gels (ESI Fig. 2 and ESI Videos 5–8†). When two guest or two host bulk PEG-MAL gels were brought into contact, the materials did not adhere and were easily separable. When a guest bulk PEG-MAL gel was brought into contact with a host bulk PEG-MAL gel, the gels bonded strongly enough to resist gravity and formed a contiguous interface visible at the micro-scale by confocal microscopy (ESI Fig. 2†).
To quantify how the addition of guest–host interactions to packed microgels influenced their material properties, we performed a series of rheological tests. To form macroscopic granular gels, equal amounts of β-cyclodextrin-functionalized and adamantane-functionalized PEG-MAL microgels (or unfunctionalized microgels for the control group) were combined in suspension and packed by centrifugal filtration (Fig. 1D and 5A). In a low amplitude (1%) oscillatory frequency sweep from 0.1 to 10 Hz (Fig. 5B), both groups exhibited rheological behaviors like those of a damped elastic solid, having a relatively frequency-independent elastic modulus (G′) that is larger than a weakly frequency-dependent viscous modulus (G′′) over the entire frequency range tested. An oscillatory strain sweep at a frequency of 1 Hz from 0.01 to 500% was conducted to determine the approximate yield stress for both guest–host microgels and unfunctionalized microgels (Fig. 5C). The approximate yield stress as defined by the threshold stress at which the elastic shear modulus begins to drop50 was determined to be 90 Pa for guest–host microgels and 150 Pa for unfunctionalized microgels. To further investigate how guest–host interactions influence the yielding and shear thinning of packed microgels, we conducted a unidirectional shear rate sweep. While these flow profiles have complex time-dependent shapes, they both exhibit weakly-varying regions at low shear-rates, where the shear stresses lay close to the yield stresses determined above (Fig. 5D). We note that at the highest shear rates, the unfunctionalized microgels had the tendency to expel from the instrument so we do not focus on this flow regime here.
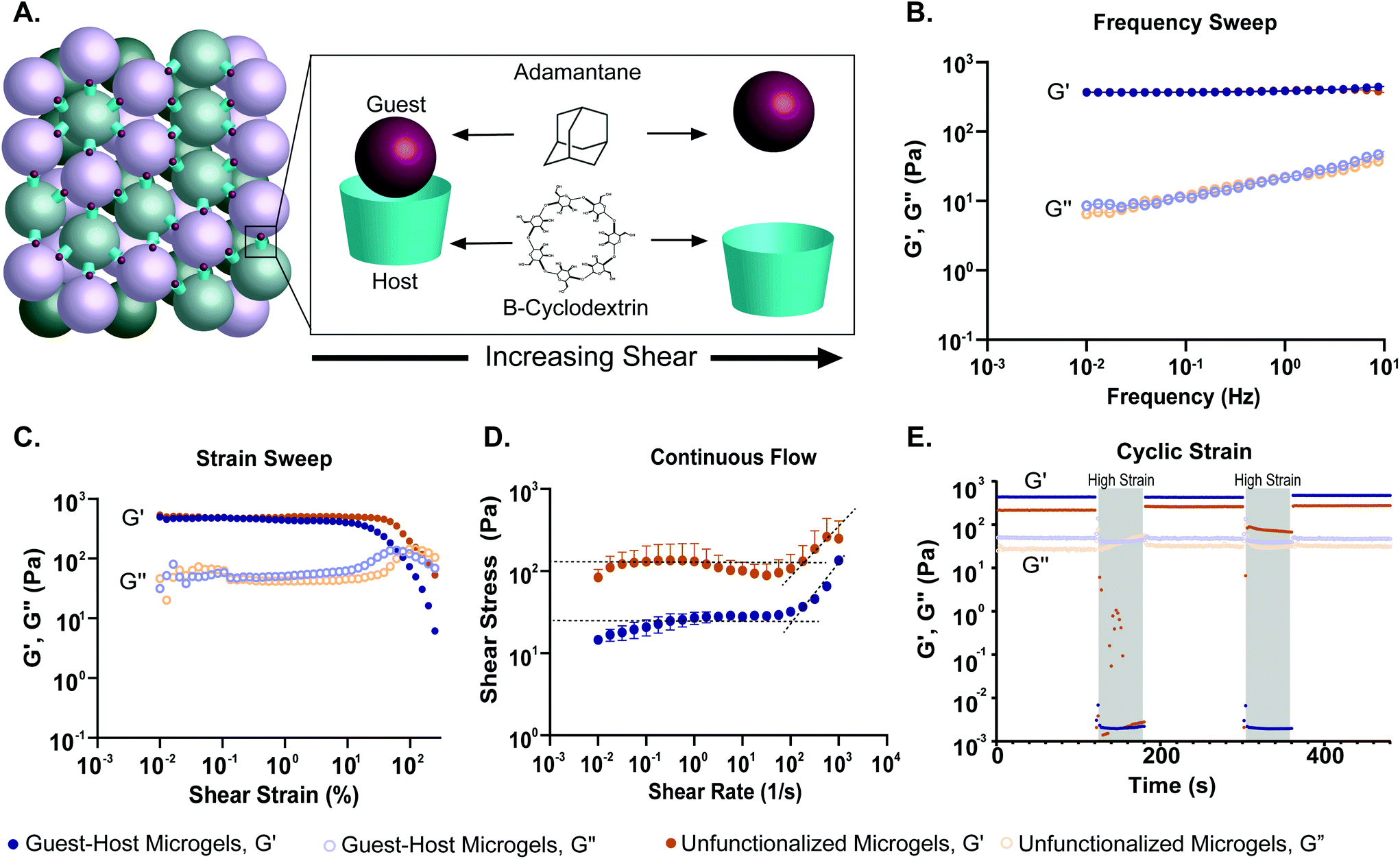 | ||
| Fig. 5 Oscillatory shear rheological properties of PEG-MAL granular hydrogels. (a) Schematic showing PEG-MAL microgels functionalized with adamantane and β-cyclodextrin dissociating under increasing shear. (b) Storage (G′) and loss (G′′) modulus over a frequency sweep from 0.1 to 10 Hz of guest–host modified PEG-MAL microgels (blue) and unfunctionalized PEG-MAL microgels (orange). The frequency sweep indicates that both hydrogel systems are viscoelastic systems, transitioning from a primarily liquid to a primarily solid state with increasing frequency. (c) The strain sweep demonstrates that both microgel systems experience a drop in storage and loss moduli after a certain yield strain, indicative of the material yielding. The yield strain for guest–host microgels is lower (90 pa) than that of the unfunctionalized microgels (150 pa), indicating that guest–host microgels may be easier to inject. (d) Shear-thinning was identified in both the microgel systems via a continuous flow experiment. The dotted lines represent the linear regression at the zero-frequency limit. The curvature of the graph was determined to be due to the time-dependent nature of restruction within the material. (e) A cyclic strain experiment was conducted between low (1%) and high (500%) strain over 120 and 60 s, respectively. The high strain regions are indicated by a grey overlay. Both demonstrated thixotropic behavior, however after the first high strain region, the unfunctionalized microgels were expelled from the instrument on all attempted runs. This is indicative of the inability of the material to retain shape and reform after high strain. The guest–host microgels were able to retain shape and reform after both high strain regions, demonstrating the ability of guest–host molecules to self-heal within the material. | ||
To test the shear-thinning and rapid post-shear reassembly of the guest–host microgels, oscillatory strain tests were conducted at alternating low (1%) and high (500%) strain (1 Hz) (Fig. 4E). In both the unfunctionalized microgels and the guest–host microgels, the material showed shear-thinning properties. However, the unfunctionalized microgels were unable to recover after the first period of high strain due to the material being expelled from the instrument at high strain. This is speculated to be due to the lack of interlinking molecules. However, the guest–host microgels showed self-healing and recovery characteristics compared to the failed unfunctionalized microgel test. The ability of the material to recover after high strain is indicative of the reassembly of the guest–host interactions. This is applicable in a clinical setting where a scaffold may need to be administered by injection while remaining a single continuous structure. As expected for non-adhesive microgels, to maintain the shape of a packed unfunctionalized microgel sample, some form of container is needed. By contrast, samples of guest–host microgels maintained their shapes without the need for containers, since they are held together by reversible guest–host interactions. Both materials, however, were shear reversible and recovered mechanical integrity after high shear.
Despite advances in the use of PEG hydrogel to model complex biological processes such as organoid formation,51,52 bulk PEG-MAL hydrogel remains an inadequate system for modeling certain types of rapid and dynamic cell migration. Cell migration is inhibited due to the nanoscale crosslinking mesh network of these hydrogels. Instead, it depends on the proteolysis of degradable peptide crosslinks, while non-degradable PEG hydrogels are not expected to support cell migration at all. Granular hydrogels provide a percolated interstitium created by the negative space between packed microgels. Here, we studied the rapid cell invasion of THP-1 monocytes through guest–host interlinked microgels. THP-1 is a monocyte cell line that has a migratory phenotype and exhibits adhesion-independent growth. Certainly, this is not the only cell model that could be used, but it is an appropriate choice to study interstitial cell motility by analogy to peripheral blood monocyte homing to target cells in the periphery.
The groups tested were guest–host microgels with and without RGD cell adhesion peptide, unfunctionalized microgels with and without RGD, Matrigel as a positive control for cell migration, and bulk non-degradable PEG-MAL hydrogels as a negative control for cell migration. We did not include bulk PEG-MAL + RGD as a control in the transwell migration study because the tight crosslinking structure of the non-degradable bulk PEG-MAL + RGD would remain on the nanoscale, we do not expect cells to be able to transit bulk PEG-MAL. We did not include an enzyme-degradable version of the bulk gel because we wanted to compare granular scaffolds to bulk scaffolds composed to the same base material.
We used a transwell invasion assay to understand THP-1 monocyte invasion through each species of gel towards a 10% FBS chemoattractant (Fig. 6A–D). After a 24-hour incubation period, we found that cells had invaded through the gel interior, and many cells reached the lower membrane for all groups except for bulk PEG-MAL hydrogels (Fig. 6D). The distance of each cell from the lower membrane was captured by confocal microscopy and plotted as a histogram for each material (Fig. 6B). We found that more cells were localized closer to the membrane for guest–host microgels than unfunctionalized microgels or Matrigel, suggesting that the scaffold invasion speed was highest for the guest–host microgel matrix. For bulk PEG-MAL, all cells remained on the surface of the gel and did not penetrate the gel interior. The cells on the bulk gel surface were not able to be visualized in 3D by confocal due to the limited working distance of our microscope objectives, but we confirmed the cells’ location on top of the bulk gels using our widefield tissue culture microscope. Guest–host microgels functionalized with RGD had the highest percentage of invading cells per unit of volume within the gel interior. Significantly higher numbers of cells invaded guest–host microgels with RGD than unfunctionalized microgels with RGD (Fig. 6C). This may be due to a lack of inter-microgel mechanics in the unfunctionalized group, making it more likely for cells to interact with single microgel spheres rather than ‘flowing and squeezing’ through the interlinked guest–host material.
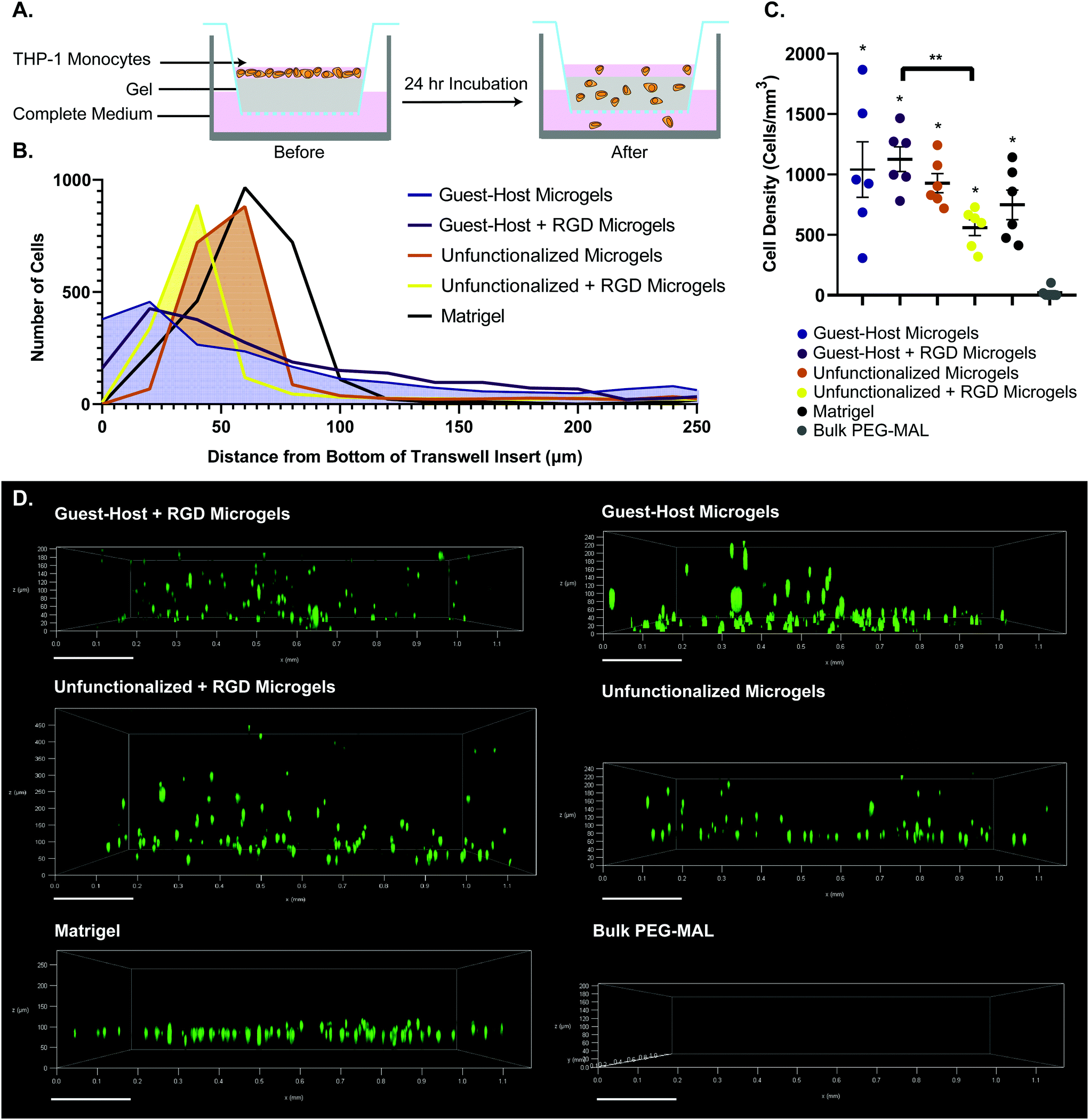 | ||
| Fig. 6 Transwell invasion assay of THP-1 monocytes through various gel species. (a) Schematic of the transwell insert assay before and after 24 hours invasion. (b) Histogram showing accumulated positions of all cells imaged, relative to the bottom of the transwell insert. (c) Number of cells per unit volume invaded into the interior of each gel species. n = 6. Data is displayed as mean ± s.e.m. significance determined by one-way anova, p < 0.05.(d) representative cell position at 24 hours after invasion. 0 μm on each sample indicates the location of bottom of the transwell insert. THP-1 monocytes were labeled with celltracker deep red. For guest–host + RGD, guest–host, unfunctionalized + RGD, and unfunctionalized groups, labeled microgels were captured in the raw microscopy data, but are omitted from the figure to visualize the cells unobstructed. | ||
Lastly, we captured a 3D time lapse recording of THP-1 cells moving in guest–host + RGD microgel scaffolds by confocal microscopy to observe, qualitatively, how the cells move in the material in real time (Fig. 7 and ESI Videos 9 and 10†). While some cells moved within a small local area resembling more Brownian-type motion, other cells engaged in directionally persistent walks. Cells were also observed to readily move across the interface between adamantane- and β-cyclodextrin functionalized microgels, indicating that guest–host interactions do not appear to impede cell motility in the material. We tracked some cells as moving at speeds of 30 μm per hour. While this is slower than immune cell migration through natural matrices, it is approximately ten times faster than cell migration rates that have been achieved in protease-degradable bulk PEG gels.5,22 In summary, these results are consistent with interlinked guest–host PEG-MAL microgels being a promising addition to the emerging field of granular hydrogel biomaterials and warrant further investigation to continue characterizing cellular behavior and in vivo responses of these unique materials.
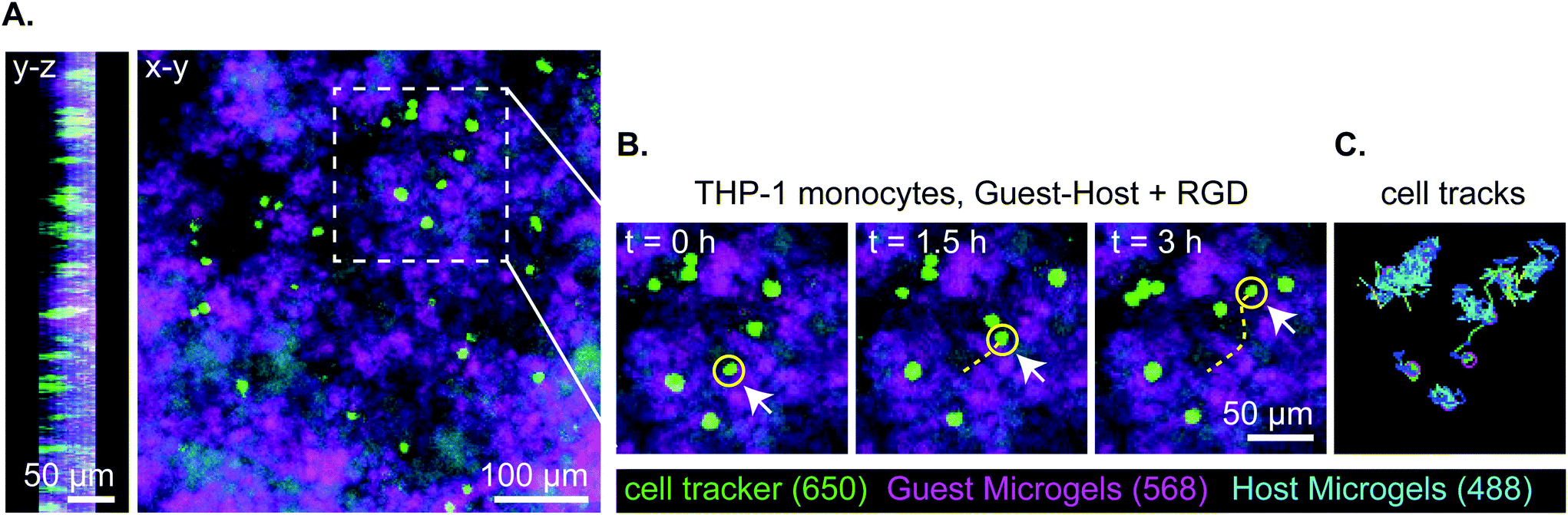 | ||
| Fig. 7 Representative stills of 3 hours of total 18-hour transwell invasion assay of THP-1 monocytes through guest–host + RGD microgels. Total cell tracks for 18 hours were omitted for clarity but can be seen in supplement video 5. (a) Orthogonal views of z-stack in y–z and x–y. Cells are located within the material and distributed throughout. (b) Still images of THP-1 migration at t = 0 h, 1.5 h, 3 h. THP-1 monocytes are labeled with celltracker deep red, the guest microgels are labeled with AlexaFluor-568, and the host microgels are labeled with AlexaFluor-488. The yellow line denotes the track of a representative cell moving through the interstitium of the microgels. (c) The representative cell tracks over a 3 hour time period. | ||
Discussion
In this study, we engineered guest–host microgels that maintain the biomaterial advantages of bulk PEG hydrogels while allowing for shear-thinning and rapid cell invasion. Previously, bulk PEG-MAL hydrogels have been limited by fast maleimide reaction kinetics that made it difficult to thoroughly mix the macromer with the crosslinker before gelation occurred.43 However, by optimizing the pH, crosslinker, and number of PEG arms, the reaction time of the hydrogel crosslinking was slowed down to a manageable rate, and the solution was mixed homogeneously before gelation. We found that 4-arm PEG-MAL has a significantly longer gelation time than 8-arm PEG-MAL over all pH tested. This is likely due to the availability of arms in 8-arm PEG-MAL compared to 4-arm PEG-MAL. Following LeChatelier's principle: with the increase in the availability of reaction sites, the rate of reaction increases.53 Hydrogels crosslinked with DTT had a faster gelation time than those crosslinked with PEG-dithiol, perhaps due to the length of the PEG-dithiol molecule inhibiting rapid reaction kinetics. With these modifications, and at lower pH, the hydrogel was able to be homogeneously mixed before gelation occurred (ESI Fig. 1†).Microgels were made via a water-in-oil emulsion to create large batches of microgels. Microfluidic devices offer an alternative approach to generate PEG hydrogel microgels,40,54 and can create spheres of uniform diameter, but require parallelization of devices to increase batch yields.55 Because PEG-MAL gels quickly, microfluidics require the addition of the crosslinker at the point of droplet formation or a second curing step such as UV light after droplet formation. Here, we generated large quantities of PEG-MAL microgels by thoroughly mixing the PEG macromer and crosslinker under conditions that slowed down the gelation speed, then quickly vortexed the solution to create an emulsion. We found that by altering the time of vortexing, we could control the average size of the microgels (Fig. 1C). This approach was used to control the length scale of the overall interstitium made by the negative space of packed microgels. By controlling the size of the microgels, we were also able to avoid colloidal behavior of small microgels, due to the strength of intermolecular forces interacting between particles of less than 10 μm diameter.49
We determined that guest–host interactions were occurring between microgels by incubating adamantane-functionalized 4-arm PEG-MAL microgels with soluble Alexa Fluor 488-labeled β-cyclodextrin. Microgels functionalized with adamantane showed more significant colocalization with Alexa Fluor 488-labeled β-cyclodextrin than unfunctionalized microgels, indicating that guest–host interactions occur on microgels (Fig. 3). The reverse experiment was omitted due to the lower water solubility of adamantane-thiol. Future studies should include NMR spectroscopy to quantify guest–host interactions between adamantane-functionalized microgels and β-cyclodextrin-functionalized microgels.
We found that the Adamantane-microgel species tended to self-associate at the microscale when mixed with β-cyclodextrin microgels (Fig. 1B and D). We speculate that this behavior is due to the hydrophobicity of the adamantane, creating microscopic pockets of hydrophobic adamantane-microgels surrounded by the hydrophilic β-cyclodextrin-microgels. Furthermore, at the nanoscale, the adamantane molecules could create a micellar effect within the microgel, forming a hydrophobic core surrounded by hydrophilic PEG chains when in aqueous solution. Although the guest–host interactions have been shown to interact and enhance the mechanical properties of the unfunctionalized PEG-MAL microgels (Fig. 3–5), a micellular or self-association effect could limit the guest–host interactions to some extent, as it decreases the availability of the guest molecule. In contrast, when bulk PEG-MAL hydrogels are functionalized with the same concentration of adamantane and β-cyclodextrin and brought into contact, the gels become inseparable (ESI Fig. 2, ESI Videos 5–8†). This behavior suggests that due to the greater surface area brought into contact by the bulk gels, the guest–host interactions have an additive effect that substantially increases the strength of the interaction.
The role of guest–host interactions on the material properties of packed microgels was investigated through rheological studies. We found that all groups exhibited dominantly solid-like behaviors (Storage modulus, G′ > Loss modulus, G′′) at low strains, having nearly frequency-independent elastic moduli, even in the low-frequency limit. Interestingly, the storage and loss moduli of guest–host microgels were not significantly different from PEG-MAL microgel controls. This result is in contrast to a prior study in which guest–host granular hydrogels displayed greater mechanics than unfunctionalized controls.42 The difference between our system and that published by Mealy et al. is that in our material, the guest–host interactions occur between the two species of adamantane and β-cyclodextrin functionalized microgel particles, leaving an interstitial void space. Mealy et al. filled the interstitial void space, formed by a single-species of adamantane-functionalized hyaluronic acid microgels, with β-cyclodextrin functionalized hyaluronic acid, thus creating a denser brick-and-mortar like structure with higher mechanical properties (G′ ∼ 6 kPa). The mechanical properties of the guest–host granular matrix we developed are similar to the modulus of soft tissues such as the liver and pancreas (G′ < 1 kPa).56–58 Our results, by contrast, indicate that the low-strain linear rheology of packed microgels with guest–host interactions is dominated by the elastic properties of the PEG-MAL microgels, and not their adhesive interactions. However, we find that the guest–host interactions begin to contribute to the material properties at high strains, where particles begin to rearrange. In this regime of behavior, we see that the added adhesive from guest–host interactions reduces the material's yield stress. While one may expect that adding adhesions would make a granular material stronger, it has been shown in other microgel systems that yield strain decreases with increasing adhesion strength.59 This behavior arises from heterogeneities that emerge in packed systems with adhesion. Our fluorescence micrographs show that guest–host interactions occur through large-scale network-like structures, rather than through a perfectly alternating guest–host pairs at the single-microgel level. This structural heterogeneity likely serves to concentrate tensile-stresses along guest–host interfaces. Thus, while the averaged-out stress in the guest–host system may be low, the stresses at these interfaces can be very high, creating the opportunity for yielding. The ability for our guest–host microgel scaffold to yield when under low stresses, while possessing a fairly high elastic modulus before yielding, can be leveraged in applications to achieve superior performance as discussed below.
Previously demonstrated guest–host hydrogel systems exhibited shear-thinning and self-healing properties, rendering the materials injectable.42,44,60–62 Here, we investigated the shear-thinning and recovery characteristics of the granular hydrogel by conducting a strain sweep, unidirectional shear-rate sweeps, and cycling between low (1%) and high (500%) strains to simulate injection conditions. We found that both granular hydrogel systems displayed thixotropic behavior. We determined the approximate yield stress of the guest–host and PEG-MAL microgel system by observing the threshold stress at which the elastic shear modulus began to drop. We found the guest–host microgel system to have a yield stress of 90 Pa and 150 Pa for the PEG-MAL control. This indicates that guest–host microgels may be easier to inject compared to their PEG-MAL controls. As mentioned before, the PEG-MAL control material without guest–host interactions failed to hold together at high shear-rates, whereas guest–host microgels were able to undergo the tests and recover. This provides evidence to the self-healing capabilities of guest–host microgels and indicates that the stabilizing action of the guest–host bond will help to ensure that the microgel scaffold's material properties will not change as a result of injection. By contrast, unfunctionalized microgels are likely to disperse upon injection, changing the scaffold's material properties in an uncontrolled way.
Finally, we then sought to understand if guest–host microgels were permissive to cell invasion. We functionalized guest–host microgels with RGD peptide to provide adhesion sites for cells to migrate towards a chemoattractant. THP-1 monocytes were used as a model for immune cell migration due to their migratory phenotype. We found that the granular microgel systems permitted rapid cell invasion into the interior of the gel. All groups of microgels and Matrigel showed significantly more cell invasion into the interior of the gel than bulk PEG-MAL hydrogel, which did not permit cell invasion within the timeframe analyzed. This is due to the nanoscale crosslinking in bulk PEG hydrogels, ultimately inhibiting rapid cell migration. These results show that both guest–host and unfunctionalized PEG microgel systems provide a scaffold in which cells can migrate rapidly. As we plan to conduct future detailed studies of cell migration in the granular guest–host gels, it would be premature to assign a mechanism responsible for the observed migration behavior. However, we speculate that the THP-1 cells utilized a non-adhesive mode of migration due to similar rates of cell invasion for guest–host gels with and without RGD. It has been well-established that leukocytes can migrate via adhesion-independent mechanisms in 3D matrices.63,64 For example, it has been shown that genetic depletion of all 24 integrin heterodimers does not alter migration velocities for many immune cell types.65 In such models, amoeboid movement in confined environments can be driven entirely by actin polymerization rather than force coupling. For a review see Lämmermann and Sixt.66 Integrins are dispensable for interstitial immune cell migration but can be employed when the cells are confined to 2D environments. It could be that migration speed is RGD-independent in guest–host gels because the cells perform integrin-independent ‘chimneying’ or ‘flowing and squeezing’ between closely adjacent and mechanically stable surfaces. Conversely, immune cells may have more difficulty squeezing between unfunctionalized gels where the adjacent surfaces are unstable. In the unfunctionalized gels, attachment to RGD may slow down migration because the cells adhere to and ‘treadmill’ on individual microgels, unable to generate as much forward movement as in an interconnected network.
Conclusions
Here we report the development of a polyethylene glycol (PEG) hydrogel scaffold stabilized with guest–host interactions between microgels. The granular system provided by packing microgels together created an open interstitium that enabled starkly faster cell migration than conventional bulk PEG hydrogels. The granular system showed shear-thinning capabilities that suggest the guest–host microgels would be injectable. Due to batch emulsion techniques, these microgels can be made in large volumes using off-the-shelf chemistries, making it available to adopt in many lab settings. These results warrant next steps to investigate further and fine-tune the guest–host molecular interactions occurring within the material system and explore the in vivo response to implanted guest–host microgels. In the future, guest–host microgels could provide a modular rapid cell migration platform while preserving the engineering toolbox that has made bulk PEG-MAL a higher versatile synthetic analog of the natural ECM.Experimental section
Chemicals and reagents
4-Arm polyethylene glycol maleimide (20 kDa) was purchased from Laysan Bio. 8-Arm polyethylene glycol maleimide (40 kDa) and PEG-dithiol(3.5 kDa) were purchased from Jenkem Technology. 1-Adamantane-Thiol, Span80, Mineral Oil, and Triton-X 100 were obtained from Sigma Aldrich. Mono-(6-mercapto-6-deoxy)-β-cyclodextrin was obtained from Zhiyuan Biotechnology. GRGDSPC peptide was purchased from Genscript Biotech.Hydrogel gelation characterization
Four-arm polyethylene glycol maleimide (4-arm PEG-MAL) (20 kDa) macromer and PEG-dithiol were dissolved in 1× PBS with 1% HEPES at varying pH (4.0, 5.0 5.2, 5.4 5.6, 5.8, 6.0, 7.0, 7.4). The 4-arm PEG-MAL macromer was mixed with solubilized magenta food coloring, and the PEG-dithiol crosslinker was mixed with cyan food coloring. 100 μL PEG-MAL macromer was pipetted between two glass microscope slides separated by 2 mm spacers. 100 μL of dyed PEG-dithiol crosslinking solution was then added between the two slides, pipetting up and down until the viscosity of the solution was too great for further mixing. The mixing was recorded macroscopically on an iPhone 11 camera, and still images were extracted from the video at the point of gelation for each pH sample.Microgel synthesis and preparation
Guest–host PEG-MAL microgels were generated by close-packing two species of PEG microgels functionalized with guest/host molecules. 120 mg mL−1 of 4-arm PEG-MAL macromer dissolved in 1× PBS with 1% HEPES at pH 5.6, was reacted with adamantane-thiol in dimethyl sulfoxide (DMSO) or mono-6-mercapto-β-cyclodextrin at approximately a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio, one functional group to every 4 arms, with PEG-MAL for 30 minutes to achieve a theoretical concentration of 0.45 mg mL−1 for Ada and 3.0 mg mL−1 for β-CD. The functionalized PEG-MAL macromers were labeled with a trace amount of Alexa Fluor 568-maleimide or Alexa Fluor 488-maleimide and allowed to incubate for an additional 30 minutes (fluorophore labeling was conducted to enable visualization of the gels but was of negligible impact to the network structure). 29.2 mg mL−1 of PEG-dithiol in 1× PBS with 1% HEPES at pH 5.6 was added in 1:1 volume ratio to each solution, quickly pipetted up and down several times to mix thoroughly, then transferred to a 30× volume of mineral oil with 2% vol/vol SPAN80 surfactant (to stabilize the emulsion) in a 50 ml conical tube. The tube was vortexed to generate an emulsion, then allowed to finish gelation for 30 minutes while gently rocking for a final 6 wt% gel. Crosslinked microgels were collected by centrifugation at 3000g for 5 minutes and washed with 0.3% Triton X-100 in deionized (DI) water, 50% acetone in DI water, DI water, and 1× PBS. To form networks, equal amounts adamantane-microgels and β-cyclodextrin-microgels were mixed and packed together by centrifugation using Costar 0.45 μm microcentrifuge filters at 4000g for 10 minutes. Microgel size distribution and guest–host interactions were characterized by optical microscopy and quantified using the particle tracker plugin of the FIJI distribution of ImageJ.67 Unfunctionalized microgels were generated as described above but excluding the functionalization of the guest host molecules, and increasing the crosslinking concentration to 42.4 mg mL−1 of PEG-dithiol. RGD functionalized microgels were generated similarly to the guest–host microgels, but the ratio of functional group to PEG-MAL arms changed to 1:16 to achieve the optimal concentration of adhesion sites. 0.49 mg mL−1 of GRGDSPC peptide was reacted for 30 minutes with PEG-MAL after functionalization with individual guest–host molecules to achieve a final concentration of 0.01 mg mL−1.
:4 ratio, one functional group to every 4 arms, with PEG-MAL for 30 minutes to achieve a theoretical concentration of 0.45 mg mL−1 for Ada and 3.0 mg mL−1 for β-CD. The functionalized PEG-MAL macromers were labeled with a trace amount of Alexa Fluor 568-maleimide or Alexa Fluor 488-maleimide and allowed to incubate for an additional 30 minutes (fluorophore labeling was conducted to enable visualization of the gels but was of negligible impact to the network structure). 29.2 mg mL−1 of PEG-dithiol in 1× PBS with 1% HEPES at pH 5.6 was added in 1:1 volume ratio to each solution, quickly pipetted up and down several times to mix thoroughly, then transferred to a 30× volume of mineral oil with 2% vol/vol SPAN80 surfactant (to stabilize the emulsion) in a 50 ml conical tube. The tube was vortexed to generate an emulsion, then allowed to finish gelation for 30 minutes while gently rocking for a final 6 wt% gel. Crosslinked microgels were collected by centrifugation at 3000g for 5 minutes and washed with 0.3% Triton X-100 in deionized (DI) water, 50% acetone in DI water, DI water, and 1× PBS. To form networks, equal amounts adamantane-microgels and β-cyclodextrin-microgels were mixed and packed together by centrifugation using Costar 0.45 μm microcentrifuge filters at 4000g for 10 minutes. Microgel size distribution and guest–host interactions were characterized by optical microscopy and quantified using the particle tracker plugin of the FIJI distribution of ImageJ.67 Unfunctionalized microgels were generated as described above but excluding the functionalization of the guest host molecules, and increasing the crosslinking concentration to 42.4 mg mL−1 of PEG-dithiol. RGD functionalized microgels were generated similarly to the guest–host microgels, but the ratio of functional group to PEG-MAL arms changed to 1:16 to achieve the optimal concentration of adhesion sites. 0.49 mg mL−1 of GRGDSPC peptide was reacted for 30 minutes with PEG-MAL after functionalization with individual guest–host molecules to achieve a final concentration of 0.01 mg mL−1.
Pore size analysis was determined by incubating the assembled microgel scaffold with high molecular weight TRITC-Dextran (500 kDa, Sigma-Aldrich) for 1 hour and were then imaged using confocal microscopy. The high molecular weight prevents dextran from diffusing into the microgels and instead labels the pores around the individual microgels. The images were binarized, watershed and particles were analyzed using the FIJI distribution of ImageJ. The area and major axis lengths for each pore were then averaged across each condition.
Guest–host interaction confirmation
Adamantane-functionalized microgels and unfunctionalized microgels were made as described above. Mono-thiol-β-cyclodextrin was dissolved in 1× PBS at pH 7.2 at 3 mM and incubated with an equimolar amount of Alexa Fluor 488 maleimide for 30 min. Microgels were incubated with the Alexa Fluor-488-β-cyclodextrin for 24 hours. Post-incubation, the microgels were washed 2 times with 1× PBS and imaged using confocal microscopy. Manual droplet tests were performed by slowly dropping 100 μL of deionized water onto approximately 200 μL of gel. Manual shear and droplet tests were captured macroscopically using an iPhone 11 camera. Bulk gels were made from equal parts functionalized PEG-MAL and PEG-dithiol in 1X PBS, pH 7.4 and captured macroscopically using an iPhone 11 camera.Microscopy
Microgels were imaged on a Leica SP8 confocal laser-scanning microscope using 10×/0.3 and 20×/0.8 numerical aperture Plan-Apochromat air objectives at 1024 × 1024-pixel resolution. Images were processed and quantified using the FIJI distribution of ImageJ.67 ROIs were determined using CellMagicWand Plug-In for ImageJ68 and Alexa Fluor 488 β-cyclodextrin fluorescence intensities were normalized to Alexa Fluor 568 fluorescence intensities.69Rheology
All rheological measurements were performed on either an Anton Paar MCR 302 rheometer or an Anton Paar MCR 702 rheometer, fitted with a 20 mm roughened plate on plate configuration with a 1 mm gap height at 25 °C. To load samples between the geometries, about 1 mL of the microgels were placed on the bottom plate at room temperature, the plate geometry was then gradually lowered, filling the gap with the microgel sample. Excess sample was trimmed from the periphery. Oscillatory shear strain amplitude sweeps were performed at 1 Hz between strains of 0.01 and 500%. Storage modulus (G′) and loss modulus (G′′) were determined from frequency sweeps performed at 1% strain from 10 to 0.01 Hz using the same geometric configuration. Unidirectional shear-rate sweeps were performed by shearing the sample at a chosen shear-rate (γ) while measuring shear stress (σ). An effective viscosity is determined from the ratio of shear-stress to shear-rate in these measurements. A linear regression was performed to determine the zero-frequency limit and result yield stress of the microgel systems on GraphPad. Strain cycle and recovery experiments were conducted by alternating between 1% strain for 120 s and 500% strain for 60 s over three periods at 1 Hz.Transwell invasion assay
The THP-1 Monocyte cell line was maintained in RPMI 1640+Glutamax between P6–P10. Cells were stained with CellTracker Deep Red prior to the invasion assay and starved in serum-free media for 24 hours. All groups of microgels were sterilized for 48 hours in 70% isopropyl alcohol. Approximately 100 μl of microgels were placed into the bottom of the transwell insert and spun down to create an even layer by rotating bucket centrifugation at 180g for 5 min. For the bulk gels, reagents were dissolved in RPMI 1640 at pH 7.4 and filtered through Costar 0.45 μm microcentrifuge filters. 50 μl of PEG-MAL macromer was placed at the bottom of the transwell insert, then 50 μl of PEG-dithiol was added to each of the inserts. The Matrigel was thawed on ice for 24 hours at 4 °C, 100 μl of Matrigel was pipetted with a chilled pipette tip into a chilled transwell insert. All gels were then incubated at 37 °C for 30 minutes before adding cells. 100 μl of stained cells at 1E6 cells per ml in serum-free media was added on top of the gels. 600 μl of serum positive media was placed in the well below the transwell insert and allowed to incubate for 24 hours. After 24 hours, the transwell inserts were removed and placed into a new plate. Inserts were imaged on a confocal laser-scanning microscope (Leica SP8) with ×10/0.3 and ×20/0.8 numerical aperture Plan-Apochromat air-objectives at 1024 × 1024-pixel resolution. Z-Stacks were rendered from 5 μm slices over 200–500 μm of the sample, starting from the bottom of the transwell insert. Images were processed and quantified in ImageJ. Volume and center of mass were calculated using the 3D Object Counter plugin for ImageJ.69 Time lapse migration of cells through the guest–host microgels matrix was taken over 18 hours, with an imaging rate of 5 min. The files were then transferred to Image J for analysis of cell tracks using the TrackMate tool.70Statistical analysis
Means among three or more groups were compared by a one-way analysis of variance (ANOVA) in GraphPad Prism 8 software. If deemed significant, Tukey's post hoc pairwise comparisons were performed. Means between two groups were compared by two-tailed Student's t-test. A confidence level of 95% was considered significant. The statistical test used, exact P values, and definition of n are all indicated in the individual figure legends. All error bars in the figures display the mean ± s.e.m.Author contributions
A. W. performed the experiments and analyzed the data. M. B. conducted initiating studies to establish the emulsion technique for microgel fabrication. T. A. provided access to mechanical testing instrumentation, helped perform rheological measurements, and analyzed and interpreted the rheometry data. E. P. conceived the project and supervised the work. A. W., T. A., and E. P. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was funded by the Intramural Research Program of UF's Wertheim College of Engineering (E.A.P.), the J. Crayton Pruitt Family Department of Biomedical Engineering (E.A.P.), and by a JDRF Agreement Award (no. 2-SRA-2019-781-S-B). The authors thank Anton Paar for the use of the Anton Paar 702 rheometer through their VIP academic research program (T.A.). This material is based on work supported by the National Science Foundation under grant no. DMR-1352043 (T.A.). We also thank Drs. Christine Schmidt and Jorge Mojica Santiago (University of Florida) for assistance with their Anton Paar MCR 302 Rheometer.References
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS
.
- S. C. Rizzi and J. A. Hubbell, Biomacromolecules, 2005, 6, 1226–1238 CrossRef CAS
- T. P. Kraehenbuehl, P. Zammaretti, A. J. Van Der Vlies, R. G. Schoenmakers, M. P. Lutolf, M. E. Jaconi and J. A. Hubbell, Biomaterials, 2008, 29, 2757–2766 CrossRef CAS
- C.-C. Lin and K. S. Anseth, Pharm. Res., 2008, 26, 631–643 CrossRef
- G. P. Raeber, M. P. Lutolf and J. A. Hubbell, Biophys. J., 2005, 89, 1374–1388 CrossRef CAS
- T. P. Kraehenbuehl, L. S. Ferreira, P. Zammaretti, J. A. Hubbell and R. Langer, Biomaterials, 2009, 30, 4318–4324 CrossRef CAS
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef
- E. A. Phelps, N. O. Enemchukwu, V. F. Fiore, J. C. Sy, N. Murthy, T. A. Sulchek, T. H. Barker and A. J. Garcia, Adv. Mater., 2012, 24, 64–70 CrossRef CAS
- J. J. Moon, J. E. Saik, R. A. Poché, J. E. Leslie-Barbick, S.-H. Lee, A. A. Smith, M. E. Dickinson and J. L. West, Biomaterials, 2010, 31, 3840–3847 CrossRef CAS
- A. S. Gobin and J. L. West, FASEB J., 2002, 16, 751–753 CrossRef CAS
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS
- D. L. Hern and J. A. Hubbell, J. Biomed. Mater. Res., 1998, 39, 266–276 CrossRef CAS
- B. K. Mann, R. H. Schmedlen and J. L. West, Biomaterials, 2001, 22, 439–444 CrossRef CAS
- E. A. Phelps, K. L. Templeman, P. M. Thule and A. J. Garcia, Drug Delivery Transl. Res., 2015, 5, 125–136 CrossRef CAS
- E. A. Phelps, N. Landazuri, P. M. Thule, W. R. Taylor and A. J. Garcia, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3323–3328 CrossRef CAS
- E. A. Phelps and A. J. Garcia, Curr. Opin. Biotechnol, 2010, 21, 704–709 CrossRef CAS
- E. A. Phelps and A. J. Garcia, Regener. Med., 2009, 4, 65–80 CrossRef
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS
- M. Hahn, L. Taite, J. Moon, M. Rowland, K. Ruffino and J. West, Biomaterials, 2006, 27, 2519–2524 CrossRef CAS
- J. J. Rice, M. M. Martino, L. De Laporte, F. Tortelli, P. S. Briquez and J. A. Hubbell, Adv. Healthcare Mater., 2013, 2, 57–71 CrossRef CAS
- J. A. Shepard, A. Huang, A. Shikanov and L. D. Shea, J. Controlled Release, 2010, 146, 128–135 CrossRef CAS
- M. J. Miller, Science, 2002, 296, 1869–1873 CrossRef CAS
- B. D. Ratner and S. J. Bryant, Annu. Rev. Biomed. Eng., 2004, 6, 41–75 CrossRef CAS
- M. C. Ford, J. P. Bertram, S. R. Hynes, M. Michaud, Q. Li, M. Young, S. S. Segal, J. A. Madri and E. B. Lavik, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2512–2517 CrossRef CAS
- R. M. Namba, A. A. Cole, K. B. Bjugstad and M. J. Mahoney, Acta Biomater., 2009, 5, 1884–1897 CrossRef CAS
- Y.-C. Chiu, M.-H. Cheng, H. Engel, S.-W. Kao, J. C. Larson, S. Gupta and E. M. Brey, Biomaterials, 2011, 32, 6045–6051 CrossRef CAS
- P. D. Rios, M. Skoumal, J. Liu, R. Youngblood, E. Kniazeva, A. J. Garcia and L. D. Shea, Biotechnol. Bioeng., 2018, 115, 2356–2364 CrossRef CAS
- B. Jiang, T. M. Waller, J. C. Larson, A. A. Appel and E. M. Brey, Tissue Eng., Part A, 2013, 19, 224–234 CrossRef CAS
- S. Ng, S. March, A. Galstian, N. Gural, K. R. Stevens, M. M. Mota and S. N. Bhatia, Sci. Rep., 2017, 7, 45424 CrossRef CAS
- R. Aliperta, P. B. Welzel, R. Bergmann, U. Freudenberg, N. Berndt, A. Feldmann, C. Arndt, S. Koristka, M. Stanzione, M. Cartellieri, A. Ehninger, G. Ehninger, C. Werner, J. Pietzsch, J. Steinbach, M. Bornhäuser and M. P. Bachmann, Sci. Rep., 2017, 7, 42855 CrossRef CAS
- P. B. Welzel, M. Grimmer, C. Renneberg, L. Naujox, S. Zschoche, U. Freudenberg and C. Werner, Biomacromolecules, 2012, 13, 2349–2358 CrossRef CAS
- Y. Hwang, N. Sangaj and S. Varghese, Tissue Eng., Part A, 2010, 16, 3033–3041 CrossRef CAS
- S. I. Somo, B. Akar, E. S. Bayrak, J. C. Larson, A. A. Appel, H. Mehdizadeh, A. Cinar and E. M. Brey, Tissue Eng., Part C, 2015, 21, 773–785 CrossRef CAS
- P. Friedl and E. B. Brocker, Cell. Mol. Life Sci., 2000, 57, 41–64 CrossRef CAS
- L. R. Nih, E. Sideris, S. T. Carmichael and T. Segura, Adv. Mater., 2017, 29, 1606471 CrossRef
- N. J. Darling, W. Xi, E. Sideris, A. R. Anderson, C. Pong, S. T. Carmichael and T. Segura, Adv. Healthcare Mater., 2020, 9, 1901391 CrossRef CAS
- S. Xin, O. M. Wyman and D. L. Alge, Adv. Healthcare Mater., 2018, 7, 1800160 CrossRef
- S. Xin, D. Chimene, J. E. Garza, A. K. Gaharwar and D. L. Alge, Biomater. Sci., 2019, 7, 1179–1187 RSC
- A. S. Caldwell, G. T. Campbell, K. M. T. Shekiro and K. S. Anseth, Adv. Healthcare Mater., 2017, 6, 1700254 CrossRef
- L. Riley, L. Schirmer and T. Segura, Curr. Opin. Biotechnol, 2018, 60, 1–8 CrossRef
- J. E. Mealy, J. J. Chung, H. H. Jeong, D. Issadore, D. Lee, P. Atluri and J. A. Burdick, Adv. Mater., 2018, 30, e1705912 CrossRef
- N. J. Darling, Y.-S. Hung, S. Sharma and T. Segura, Biomaterials, 2016, 101, 199–206 CrossRef CAS
- S. M. Mantooth, B. G. Munoz-Robles and M. J. Webber, Macromol. Biosci., 2019, 19, e1800281 CrossRef
- T. Xiao, L. Xu, L. Zhou, X.-Q. Sun, C. Lin and L. Wang, J. Mater. Chem. B, 2019, 7, 1526–1540 RSC
- C. B. Rodell, J. E. Mealy and J. A. Burdick, Bioconjugate Chem., 2015, 26, 2279–2289 CrossRef CAS
- E. A. Phelps, D. M. Headen, W. R. Taylor, P. M. Thule and A. J. Garcia, Biomaterials, 2013, 34, 4602–4611 CrossRef CAS
- N. O. Enemchukwu, R. Cruz-Acuna, T. Bongiorno, C. T. Johnson, J. R. Garcia, T. Sulchek and A. J. Garcia, J. Cell Biol., 2016, 212, 113–124 CrossRef CAS
-
E. R. Weeks, in Statistical Physics of Complex Fluids, ed. S. Maruyama and M. Tokuyama, Tohoku University Press, Sendai, Japan, 2007, ch. 2, vol. 6, pp. 1–87 Search PubMed
- C. S. O'Bryan, T. Bhattacharjee, S. R. Niemi, S. Balachandar, N. Baldwin, S. T. Ellison, C. R. Taylor, W. G. Sawyer and T. E. Angelini, MRS Bull., 2017, 42, 571–577 CrossRef
- R. Cruz-Acuña, M. Quirós, A. E. Farkas, P. H. Dedhia, S. Huang, D. Siuda, V. García-Hernández, A. J. Miller, J. R. Spence, A. Nusrat and A. J. García, Nat. Cell Biol., 2017, 19, 1326–1335 CrossRef
- N. Gjorevski, N. Sachs, A. Manfrin, S. Giger, M. E. Bragina, P. Ordóñez-Morán, H. Clevers and M. P. Lutolf, Nature, 2016, 539, 560–564 CrossRef CAS
-
C. G. Olivera-Fuentes and C. M. Colina, presented in part at the International Conference on Engineering Education Coimbra, Portugal, September 3–7, 2007, 2007
- L. P. B. Guerzoni, J. Bohl, A. Jans, J. C. Rose, J. Koehler, A. J. C. Kuehne and L. De Laporte, Biomater. Sci., 2017, 5, 1549–1557 RSC
- D. M. Headen, J. R. García and A. J. García, Microsyst. Nanoeng., 2018, 4, 17076 CrossRef CAS
- M. Ayyildiz, R. G. Aktas and C. Basdogan, Biorheology, 2014, 51, 47–70 CAS
- A. Rubiano, D. Delitto, S. Han, M. Gerber, C. Galitz, J. Trevino, R. M. Thomas, S. J. Hughes and C. S. Simmons, Acta Biomater., 2018, 67, 331–340 CrossRef
- C. Wex, M. Frohlich, K. Brandstadter, C. Bruns and A. Stoll, J. Mech. Behav. Biomed. Mater., 2015, 41, 199–207 CrossRef CAS
- Z. Shao, A. S. Negi and C. O. Osuji, Soft Matter, 2013, 9, 5492 RSC
- C. B. Rodell, A. L. Kaminski and J. A. Burdick, Biomacromolecules, 2013, 14, 4125–4134 CrossRef CAS
-
A. C. Yu, L. M. Stapleton, J. L. Mann and E. A. Appel, in Self-assembling Biomaterials, 2018, pp. 205–231, DOI:10.1016/b978-0-08-102015-9.00010-1
- L. Zou, A. S. Braegelman and M. J. Webber, ACS Appl. Mater. Interfaces, 2019, 11, 5695–5700 CrossRef CAS
- P. Kameritsch and J. Renkawitz, Trends Cell Biol., 2020, 30, 818–832 CrossRef CAS
- K. M. Yamada and M. Sixt, Nat. Rev. Mol. Cell Biol., 2019, 20, 738–752 CrossRef CAS
- T. Lämmermann, B. L. Bader, S. J. Monkley, T. Worbs, R. Wedlich-Söldner, K. Hirsch, M. Keller, R. Förster, D. R. Critchley, R. Fässler and M. Sixt, Nature, 2008, 453, 51–55 CrossRef
- T. Lämmermann and M. Sixt, Curr. Opin. Cell Biol., 2009, 21, 636–644 CrossRef
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS
- D. E. Wilson, G. B. Smith, A. L. Jacob, T. Walker, J. Dimidschstein, G. Fishell and D. Fitzpatrick, Neuron, 2017, 93, 1058–1065 CrossRef CAS
- S. Bolte and F. P. Cordelieres, J. Microsc., 2006, 224, 213–232 CrossRef CAS
- J.-Y. Tinevez, N. Perry, J. Schindelin, G. M. Hoopes, G. D. Reynolds, E. Laplantine, S. Y. Bednarek, S. L. Shorte and K. W. Eliceiri, Methods, 2017, 115, 80–90 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01499k |
| This journal is © The Royal Society of Chemistry 2021 |