
Black BiVO4: size tailored synthesis, rich oxygen vacancies, and sodium storage performance†
Xiaosa
Xu‡
a,
Youxun
Xu‡
a,
Fei
Xu
 *ab,
Guangshen
Jiang
a,
Jie
Jian
a,
Huiwu
Yu
a,
Enming
Zhang
a,
Dmitry
Shchukin
c,
Stefan
Kaskel
b and
Hongqiang
Wang
*a
*ab,
Guangshen
Jiang
a,
Jie
Jian
a,
Huiwu
Yu
a,
Enming
Zhang
a,
Dmitry
Shchukin
c,
Stefan
Kaskel
b and
Hongqiang
Wang
*a
aState Key Laboratory of Solidification Processing, Center for Nano Energy Materials, School of Materials Science and Engineering, Northwestern Polytechnical University, Shaanxi Joint Laboratory of Graphene (NPU), Xi'an, 710072, P. R. China. E-mail: feixu@nwpu.edu.cn; hongqiang.wang@nwpu.edu.cn
bDepartment of Inorganic Chemistry, Technische Universität Dresden, Bergstrasse 66, 01062 Dresden, Germany
cDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK
First published on 6th January 2020
Abstract
BiVO4 nanoparticles with tailored sizes and rich oxygen vacancies were facilely synthesized via pulsed laser irradiation of colloidal nanoparticles (PLICN). As anode materials for Na-ion batteries, the revolutionized sodium storage process found in sub-10 nm BiVO4 nanocrystals results in outstanding properties including extraordinary rate capability and remarkable cycling stability.
Introduction
Over the last decades, the synthesis of well-defined inorganic colloids with the size ranging from submicrometer to nanometer has been intensively pursued from both scientific and technological perspectives.1–3 Control over the size evolution has been thus of significance in the synthesis of a variety of colloidal particles,4–6 owing to their size-dependent properties that vary a lot from their bulk counterparts. Meanwhile, the size-uniformity of the synthesized colloids has also played an important role for many applications owing to their advanced collective behaviors.7,8 Mechanistic studies have shown that uniform sized colloids can be produced by wet chemical strategies when the burst of nucleation is combined with the subsequent diffusion-controlled growth.9,10 For example, uniform-sized metal nanocrystals such as gold, silver, platinum, and palladium can be synthesized by reducing the metal salts in alcohols in the presence of surfactants;3 monodisperse nanocrystals of metal oxides can be fabricated by thermal decomposition of metal-surfactant complexes;11 monodisperse nanocrystals of chalcogenide materials can be obtained in surfactant solution by hot injection at high temperature.12 As promising materials, multinary metal oxides have much potential to advance their electrochemical,13 optoelectronic,14–16 photochemical17 and catalytic applications,18 owing to the inherent compositional complexity for regulating their physical and chemical characteristics. Thus many efforts have been devoted to preparing multinary metal oxides either by solid phase reaction19 or soft chemical methods.20,21 However, in contrast to the successes achieved on the judicious size-manipulation of elementals, binary metal oxides and chalcogenides, there have been unfortunately rare reports on the synthesis of size-tailored and uniform-sized multinary metal oxide based colloids. Seeking a strategy that enables the general synthesis of uniform-sized multinary metal oxides with well-defined chemical features has remained a great challenge.Herein, we demonstrate a general synthetic method of pulsed laser irradiation of colloidal nanoparticles (PLICN) for constructing size-tailored and uniform colloids of multinary metal oxides. As an example, we show the room temperature synthesis of bismuth vanadium oxides (BiVO4) with uniform sizes ranging from submicrometer down to ultrafine several nanometers, by pulse laser irradiation of bulk BiVO4 particles in desired solvents but without any other additives. By judiciously varying the laser fluence and irradiation time, the sizes of BiVO4 colloids can be readily modulated. Meanwhile, such strategy enriches the family of black materials with a new member of black BiVO4 owing to the formation of oxygen vacancies in BiVO4 upon laser irradiation. The described synthetic platform for size-tailored multinary metal oxides has significant implications and applications. The proof-of-concept application is demonstrated here as anode material for sodium storage, where nanosize-dependent fast kinetic characteristics and oxygen vacancy defects contribute together to the outstanding performances. Our primary results show that sodium storage performances found in sub-10 nm BiVO4 nanocrystals renders the electrodes with high reversible capacity (565 mA h g−1 at 0.1 A g−1), extraordinary rate capability (297 mA h g−1 at 2.0 A g−1) and remarkable cycling stability (470 mA h g−1 at 0.1 A g−1 after 200 cycles), which is the best in the performance records of BiVO4 based anodes, and also comparable or even among the top values if compared with many other multinary metal oxides anode materials. We thus believe these findings could open a new research domain in the field of multinary metal oxide inorganics and shed a new light on the pursuit of high-performance but not limited to energy storage.
Results and discussion
Size-tailoring of the BiVO4 colloids
Fig. 1a schematically depicts the laser generation of BiVO4 colloidal particles with tailored sizes by PLICN at room temperature. In brief, bulk BiVO4 with irregular shapes (Fig. 1b) was first dispersed in acetone by sonication, and then was subjected for pulsed laser irradiation. The laser beam of 355 nm wavelength was chosen to irradiate the BiVO4 suspensions, owing to its absorption cut-off wavelength of 520 nm. After laser irradiation for 10 minutes, the pristine monoclinic BiVO4 microplates were found transformed to a large number of uniform spherical particles with an average size of 400 nm (BVO-400) (Fig. 1c) with laser fluence of 78 mJ per pulse per cm2. Upon increasing the laser fluence to 156 mJ per pulse per cm2, spherical particles with an average size of 50 nm (BVO-50) were obtained (Fig. 1d). | ||
| Fig. 1 Synthesis of size-tailored BiVO4 colloids. (a) Schematic illustration of the pulsed laser irradiation of BiVO4 particles. (b) SEM image of pristine BiVO4 particles (BVO). Inset: the optical image of BiVO4 prinstine particles dropping on a glass slide. (c) FESEM image of submicrometer spheres (BVO-400), (d) TEM image of nanospheres (BVO-50), and (e) TEM image of sub-10 nm BiVO4 nanocrystals (BVO-5) obtained by pulsed laser irradiation of pristine BiVO4. Insets: optical images of them dropping on the glass slides. (f) SEM and (g) TEM mappings of BVO-400 and BVO-50. (h) HRTEM image of BVO-5. | ||
X-Ray Diffraction (XRD) analysis of the samples BVO-400 and BVO-50 (Fig. S1a†) reveals both have typical diffraction patterns of monoclinic BiVO4 (JCPDF#00-014-0688). As shown in the energy dispersive X-ray fluorescence spectroscopy (EDX) mapping (Fig. 1f and g), elements of Bi, V and O homogeneously distributed in the obtained spherical particles, further confirming BiVO4 is stable upon pulsed laser irradiation. When the laser fluence is increased to 260 mJ per pulse per cm2, the size of the obtained product dramatically decreases to below 10 nm (with an average size of 5 nm, BVO-5). The high-resolution TEM (HRTEM) images (Fig. 1h) reveal its crystalline nature with lattice spacing of 0.308, 0.312, 0.213 and 0.237 nm, corresponding respectively to the interplanar d-spacing of monoclinic BiVO4 planes of (121), (130), (150) and (220). The phase stability of BVO-5 upon laser irradiation was further confirmed by the XRD pattern (Fig. S1b†), showing the similar peaks to that of raw material. The peak widening in the XRD pattern around 18.7°, 28.8° and 30.5° indicates the occurring of crystal size decrease after laser irradiation, which agrees with the result from HRTEM observation (Fig. 1e).22 It should be noted that the size-tailored synthesis of BiVO4 colloidal particles has so far been rarely reported, especially with the size down to less than 10 nm, which could be promising to induce unique size-related physical/chemical characteristics. As the typical demonstration, we found that colloidal particles of BVO-400, and BVO-50 fail to emit any photoluminescence (PL) when illuminated by a 365 nm UV lamp, while the colloids of BVO-5 demonstrates a shining blue emission under the same condition (Fig. S2a†). A further characterization reveals the absolute PL quantum yields of BVO-5 could reach up to 67% (Fig. S2b†), which might be due to the quantum confinement of BVO-5.
Compared with ‘bottom-up’ wet chemistry methods for monodisperse colloidal particles, PLICN is a special and environmentally friendly ‘top-down’ strategy towards colloids with controllable sizes, by utilizing the unique heating–melting–evaporation mechanism. In such an approach, a nanoparticle will melt or evaporate if absorbing enough laser beam energy. To quantitatively define the specific fluence that results in the melting or evaporation of particles with given sizes, the Mie theory can be used to calculate the absorption cross-section σabsλ, for particles of different sizes dp, and then to determine the energy absorbed by a particle irradiated by a single laser pulse [eqn (1)]:
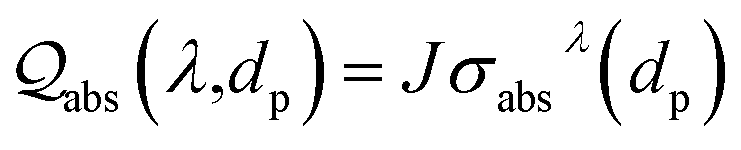 | (1) |
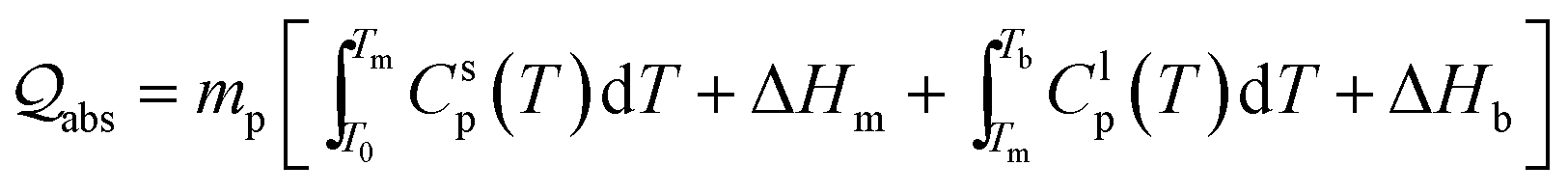 | (2) |
The required laser fluence J* for particles of different sizes that will heat a particle from T0 (298.15 K) to the melting point Tm, to complete melting, to the boiling point Tb, and to full evaporation, can be determined. Therefore, by using such equation, the size of the produced colloids can be estimated. The physical and thermodynamic constants used in eqn (2) is, the density ρp, the heat capacities Csp for solids and Clp for liquids, the melting heat ΔHm, and the boiling heat ΔHb.
Generally, particles with submicrometer sizes start to melt with the increase of laser fluence during the PLICN.24 At the beginning, the bulk BiVO4 particles absorbed the laser energy and started to melt, forming many small particles (Fig. S3a†). Then melted larger particles can take up smaller particles (Fig. S3b†) until all the smaller particles are consumed to create homogenous sizes of submicrometer spheres (Fig. S3c and d†),25 which could explain the acquiring of uniform BVO-400 colloids (Fig. 1c). When the laser fluence increases beyond 260 mJ per pulse per cm2, the full evaporation of particles leads to a dramatic size decrease (Fig. 1e). For the laser irradiation with fluences in between, the partial evaporation could lead to the detachment of smaller fragments from molten BiVO4 particle. Such partial evaporation could create a liquid that is supersaturated with nanoparticle fragments, which significantly increased the collision frequency of the detached fragments. This allows them to coalesce into larger particles, which could explain the formation of BiVO4 nanospheres with the size of 50 nm (Fig. 1d). Usually larger particles need higher energy to completely melt or evaporate owing to the large heat capacity of particles, whereas smaller particles also require much higher energy owing to the low optical cross-section of smaller particles (see eqn (1)). Therefore, the sub-10 nm BiVO4 nanocrystals (Fig. 1e) acquired by the laser evaporation is usually stable against the laser irradiation at the same laser fluence and thus shows no further coalesce.
Black nature of the BiVO4 colloids
In addition to the size evolution of BiVO4 shown above, we found another interesting phenomenon that the original yellow colour of BiVO4 turned to black during laser irradiation for different sized BiVO4 colloids (Fig. S4 and ESI Video†). As shown in the insets of Fig. 1b–e, we coated the pristine BiVO4 as well as the laser generated BiVO4 colloids on the glass substrates. It can be observed that all these laser-generated BiVO4 colloids demonstrate black colour. Further, the diffuse reflectance UV-vis spectra (Fig. S5†) reveals that the pristine BiVO4 exhibits an absorption onset at around 520 nm, while laser generated samples demonstrated greatly enhanced light absorption especially at the long-wavelength range (>500 nm). This is in good agreement with the phenomenon of the visually black BiVO4, which is as far as we know a new type of artificial black material.The phase stability of BiVO4 has been evidenced by above structural characterizations (Fig. 1g and h and XRD in Fig. S1†), and the possibility of BiVO4 decomposition during laser irradiation that contains transient extreme conditions was thus excluded. We therefore tried to understand the reason behind the blackening of BiVO4. It was found that the colour of laser generated BiVO4 colloids turns back to yellow when annealed in air, while the colour remained black when annealed in nitrogen (Fig. S6†). This implies that the blackening of BiVO4 could be related to oxygen related defects. Raman spectra of pristine BiVO4 and the size-tailored black BiVO4 colloids were further analysed to detect any difference of the bonding vibrational states associated with oxygen related defects. As shown in Fig. 2a, typical vibrational bands of BiVO4 around 210, 324, 366, and 826 cm−1 are observed for all samples. After laser irradiation, the Raman peaks at 324 and 366 cm−1 assigned to the asymmetric and symmetric deformation modes of the VO43− tetrahedron26 become weaker and broader (BVO-400, BVO-50). The peaks broadening related to the formation of defects27 becomes more substantial with the increase of laser fluence (BVO-5). Furthermore, the Raman peaks of BVO-400, and BVO-50 at 826 cm−1 (attributed to the stretching modes of V–O bonds) shift to lower frequencies in comparison to that of pristine BiVO4, and the shifts observed for BVO-5 is especially pronounced, indicating oxygen vacancies may exist in the lattice of BiVO4.27,28
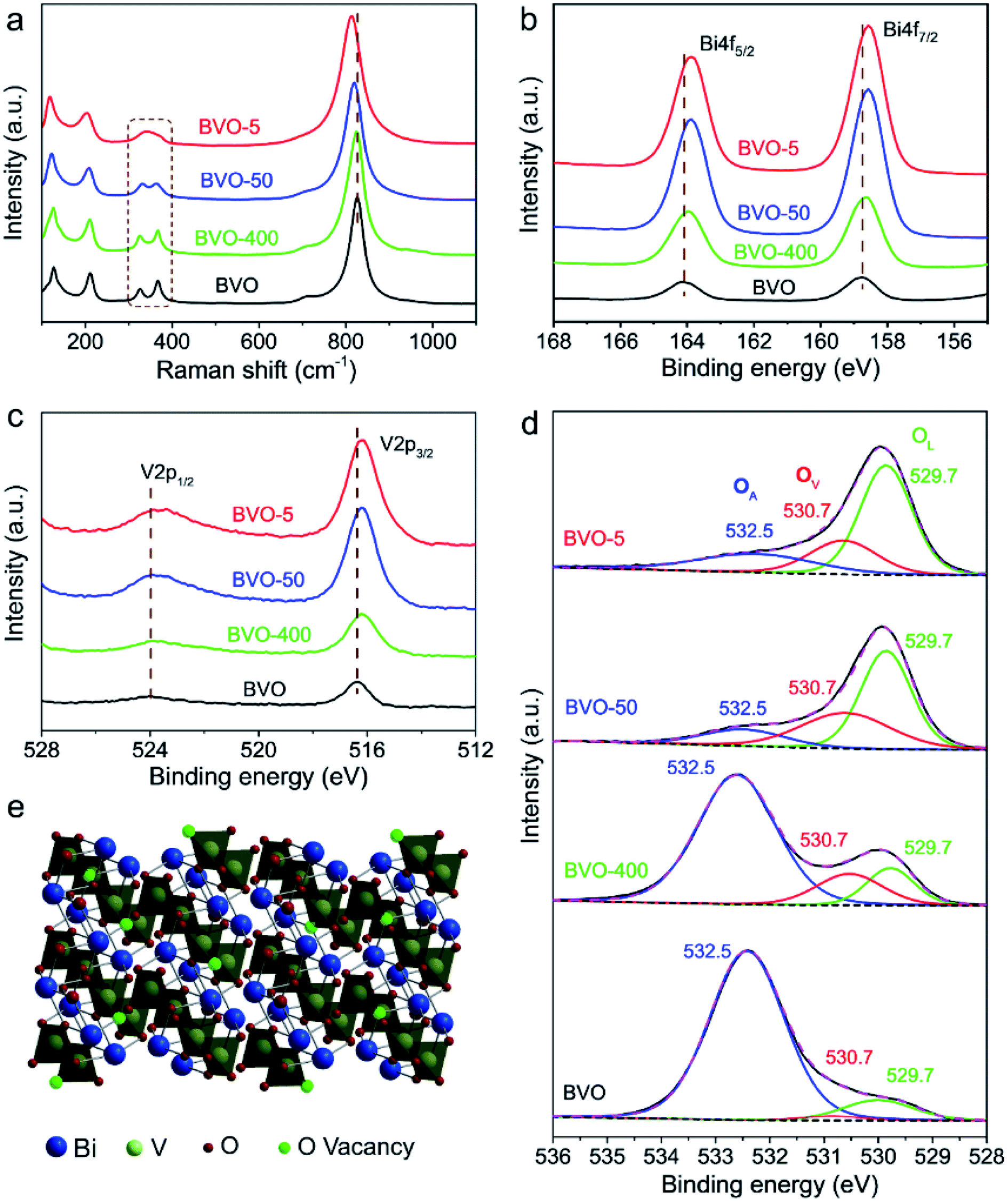 | ||
| Fig. 2 Composition and structural analysis. (a) Raman spectra, (b–d) XPS spectra of pristine BiVO4, BVO-400, BVO-50 and BVO-5. (e) Crystal structure of the monoclinic scheelite polymorph of BiVO4 with rich oxygen vacancies. | ||
X-ray photoelectron spectroscopy (XPS) analysis was performed to further understand the laser induced defects evolution in BiVO4 colloids. The binding energy of V 2p and Bi 4f (Fig. 2b and c) slightly shift to lower binding energy after laser irradiation, which is probably due to partial reduction of V5+ and Bi3+ ions and the migration of electrons bound to oxygen and metal ions towards oxygen vacancies.29 The O 1s XPS spectra of the BiVO4 samples (Fig. 2d) could be deconvoluted into three component peaks at 529.7, 530.7, and 532.5 eV, which correspond to respectively the lattice oxygen (OL), oxygen vacancies (OV) and adsorbed oxygen (OA).29,30 It can be seen that the intensity of peaks of Bi 4f, V 2p and OL are significantly enhanced, which could be due to the increased laser-induced-annealing effect upon the laser fluence increasing. The OA is found drastically reduced after laser treatment, and this is because the oxygen-containing functional group on the particles surface could be easily removed by laser.31 The creation of oxygen vacancies during laser irradiation could be confirmed by the OV peak signals evolution shown in Fig. 2c, which demonstrates that the OV peaks of black BiVO4 (BVO-400, BVO-50, BVO-5) are much stronger in comparison to that of pristine BiVO4. This is in agreement with the cases in binary metal oxides, such as TiO2.32 The following process could explain the oxygen vacancies formation. BiVO4 particles melt or evaporate once it absorbs enough laser energy. The particles surface temperature could be high enough (thousands of degrees Celsius) to decompose the surrounding liquid medium such as acetone.33 The produced reductive gas from the decomposition of liquid medium could partially reduce the BiVO4 colloids, generating black BiVO4 with rich oxygen vacancies.
Sodium-ion storage performance
The laser-based ‘top-down’ strategy towards BiVO4 nanocrystals with tailored size down to sub-10 nm sizes and rich oxygen vacancies (depicted in Fig. 2e) is expected to offer outstanding performance in applications such as photocatalysis, optoelectronic conversion as well as energy storage. The first of these potentials is confirmed here by using the BiVO4 for sodium ion storage as an example. Our interest in employing these size-tailored black BiVO4 as anode for sodium ions batteries (SIBs) is encouraged by their following structural features that oxygen vacancies can change the electronic structures for improved conductivity;34–36 The reduced dimensions of BiVO4 nanoparticles, especially to several nanometers would help to shorten transport paths and accelerate fast kinetics, offer large interfaces for sufficient Na+ insertion/extraction reaction, and accommodate the strain with volume change,37,38 which are the major haunted problems on metal oxide anode in SIBs.Reduced graphene oxide (rGO) was employed as a platform to anchor black BiVO4 colloids (Fig. 3a) by a facile hydrothermal method, which resulted in the uniform decoration of colloids of BVO-400, BVO-50 and BVO-5 on rGO (SEM of Fig. S7, STEM with element mappings of Fig. S8a–f, and Raman spectra of Fig. S9a†) and the BiVO4 content is determined by thermogravimetric analysis (TGA) in air (Fig. S9b†). The Na-storage performance of the as-obtained BVO@rGO was investigated in half-cells configuration with Na foil as the counter electrode. It would be significant to investigate if the black feature plays a positive role in the Na-storage performance. We thus compared the performance of black BVO-400 and the yellow BVO-400. The yellow one was prepared by annealing the black BVO-400 in air (designated as yellow-BVO@rGO-400) while keeping other procedures unchanged (Fig. 3b). Compared with BVO@rGO-400, the yellow-BVO@rGO-400 exhibits lower capacity with only a capacity of 241 mA h g−1 after 20 cycles at 0.1 A g−1 and faster capacity decay, confirming oxygen vacancies play a critical role in enhancing Na+ storage, which is in agreement with the recent reports.39,40 In addition, the pristine BiVO4 microplates without laser irradiation were also loaded on rGO (designated as BVO@rGO-bulk), while the electrochemical tests show that BVO@rGO-bulk suffers from dramatic capacity fading (Fig. S10a†) and inferior rate capability (Fig. S10b†).
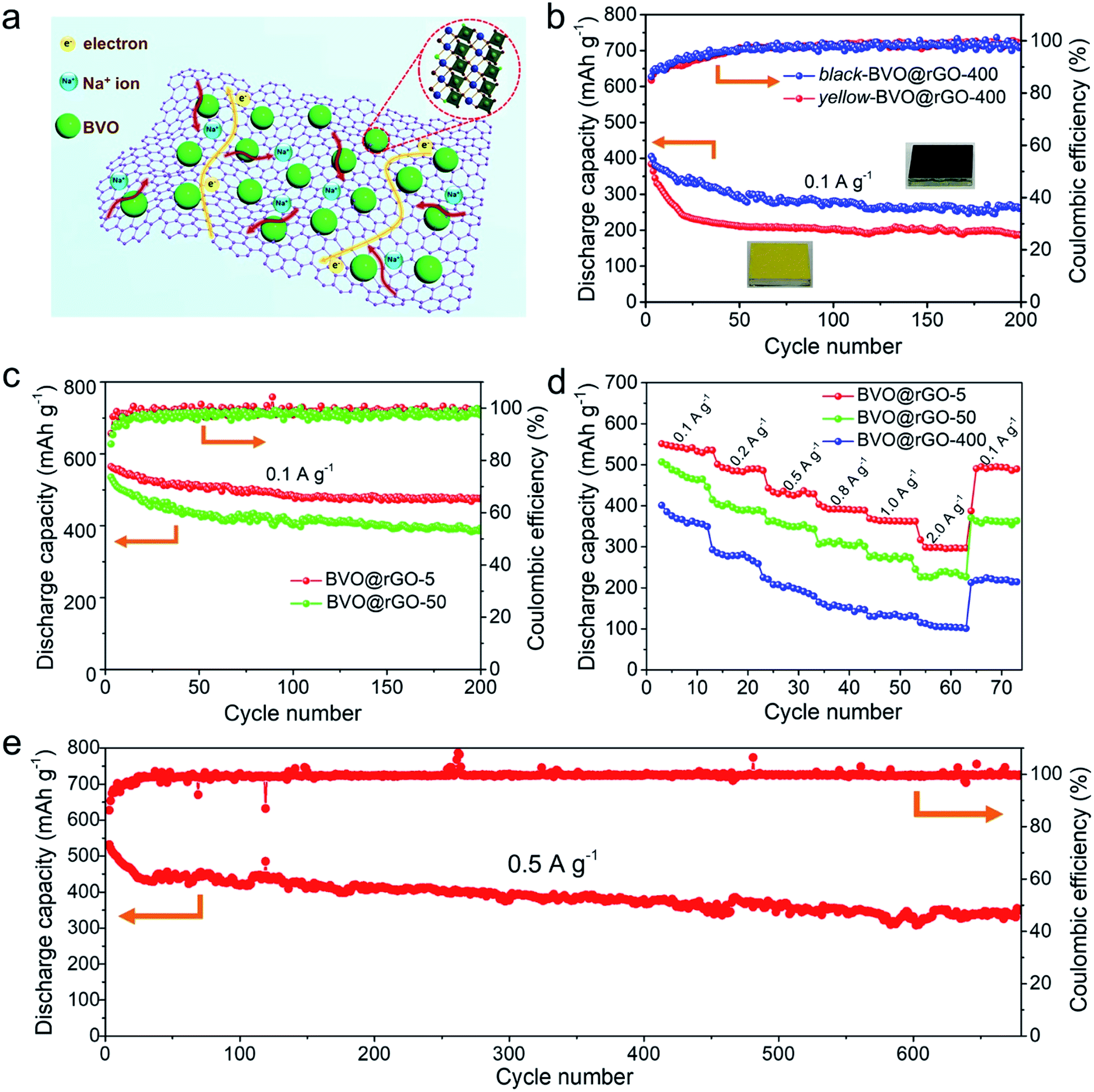 | ||
| Fig. 3 Electrochemical performance of BVO@rGO electrodes. (a) Schematic illustration of sodiation processes for the BVO@rGO. (b) Cycling performance of black-BVO@rGO-400 and yellow-BVO@rGO-400 at 0.1 A g−1 with the voltage range of 0.01–3.0 V. (c) Cycling performance of BVO@rGO-5 and BVO@rGO-50 at 0.1 A g−1. (d) Rate capabilities of BVO@rGO-5, BVO@rGO-50, and BVO@rGO-400 electrodes from 0.1 to 2.0 A g−1. (e) Long-term cycling performance of BVO@rGO-5 electrode at 0.5 A g−1. Note: the specific capacity contribution of BiVO4 in the electrodes were calculated according to the eqn. S1 and S2‡ in Experimental methods. | ||
To further explore the Na-storage performance of the black BiVO4 colloids with tailored size, BVO@rGO-50 and BVO@rGO-5 were also prepared. The BVO@GO-5 electrode showed an initial coulombic efficiency of 60.1% (Fig. S11†), which is much higher than that of BVO@GO-50 (56.5%), black-BVO@rGO-400 (47.3%) and yellow-BVO@rGO-400 (46.9%). In addition, as shown in Fig. 3c, both the capacity and their retention ratio improve steadily as the BiVO4 particle size reduces. In comparison, BVO@rGO-5 always delivers highest capacities throughout the whole cycles and obtains a stable reversible capacity of 470 mA h g−1 after 200 cycles. Meanwhile, BVO@rGO-5 has the highest capacity retention ratio of 85.0%, indicative of a decay of only 0.076% per cycle. Another striking performance is the rate capability. BVO@rGO-5 can deliver highest capacities of 535, 491, 329, 392, 362 and 297 mA h g−1 at every gradient current density of 0.1, 0.2, 0.5, 0.8, 1.0 and 2.0 A g−1, respectively, in comparison to BVO@rGO-50 and BVO@rGO-400 (Fig. 3d). Remarkably, when the current density goes back to 0.1 A g−1, the capacity of BVO@rGO-5 quickly recovers to 493 mA h g−1, demonstrating excellent reversibility. To the best of our knowledge, the maximum capacity achieved in BVO@rGO-5 hits a new record among all reports BiVO4-based anode materials thus far (Fig. S12†),41,42 and is also even higher comparing with other multinary metal oxide anode materials (Table S1†). To further certify the black BiVO4 a promising anode material, we investigated its cycling performance at 0.01–1.0 V (Fig. S13a†). Obviously, a reversible capacity of 316 mA h g−1 was obtained after 100 cycles and the capacity retention ratio reached 76% comparing with that at 0.01–3.0 V. Also, the high capacity proportion of 75% at different current densities indicates its low operating voltage as anode material for SIBs (Fig. S13b and c†). It can be concluded that the improved capacity, cycling performance and rate capability is closely associated with the reduced particle size, which permits a high contact area for high utilization of BiVO4 particles, shortened diffusion path of sodium ions. Furthermore, it is found that a high reversible capacity of 340 mA h g−1 is still achieved after 680 cycles at 0.5 A g−1 (Fig. 3e), despite capacity decay in the initial cycles, implying the superior high-rate cycling stability. Further increasing the BiVO4 content leads to reduced cyclic capacity (Fig. S14†), probably due to the aggregation of excess BiVO4.
Cyclic voltammetry (CV) was employed to scrutinize the Na-ion storage behavior of BVO@rGO-5 (Fig. S15†). During the first reduction process, two broad reduction peaks appeared at around 0.89–1.42 V, which might be assigned to the conversion reaction of BiVO4 (3Na + BiVO4 → Bi + Na3VO4) as well as the irreversible formation of a solid electrolyte interface (SEI) film,41 disappearing in the subsequent scans. The peaks at 0.46 V and 0.19 V imply the reversible alloying reaction of Bi ↔ NaBi ↔ Na3Bi. With repeated cycling, the cathodic peak corresponding to alloying reaction at 0.46 V is divided into two peaks at 0.56 V and 0.34 V. In the anodic scan, the peaks at 0.68 V and 0.80 V are related to the desodiation process of Na3Bi. The feeble peaks located at 1.58 V and 2.17 V might be defined as the Na+ extraction reaction from the host originating from the valence change of V.43,44 Such a result is further certified by the ex situ XRD patterns of BVO@rGO-5 (Fig. S16†), in which x-value varies with the charged/discharged state, for example, NaVO2 was detected when discharging to 0.6 V. The CV curves almost overlap in the following cycles, reflecting its good electrochemical reversibility.
Effects of reduced colloids dimension on sodium storage kinetics
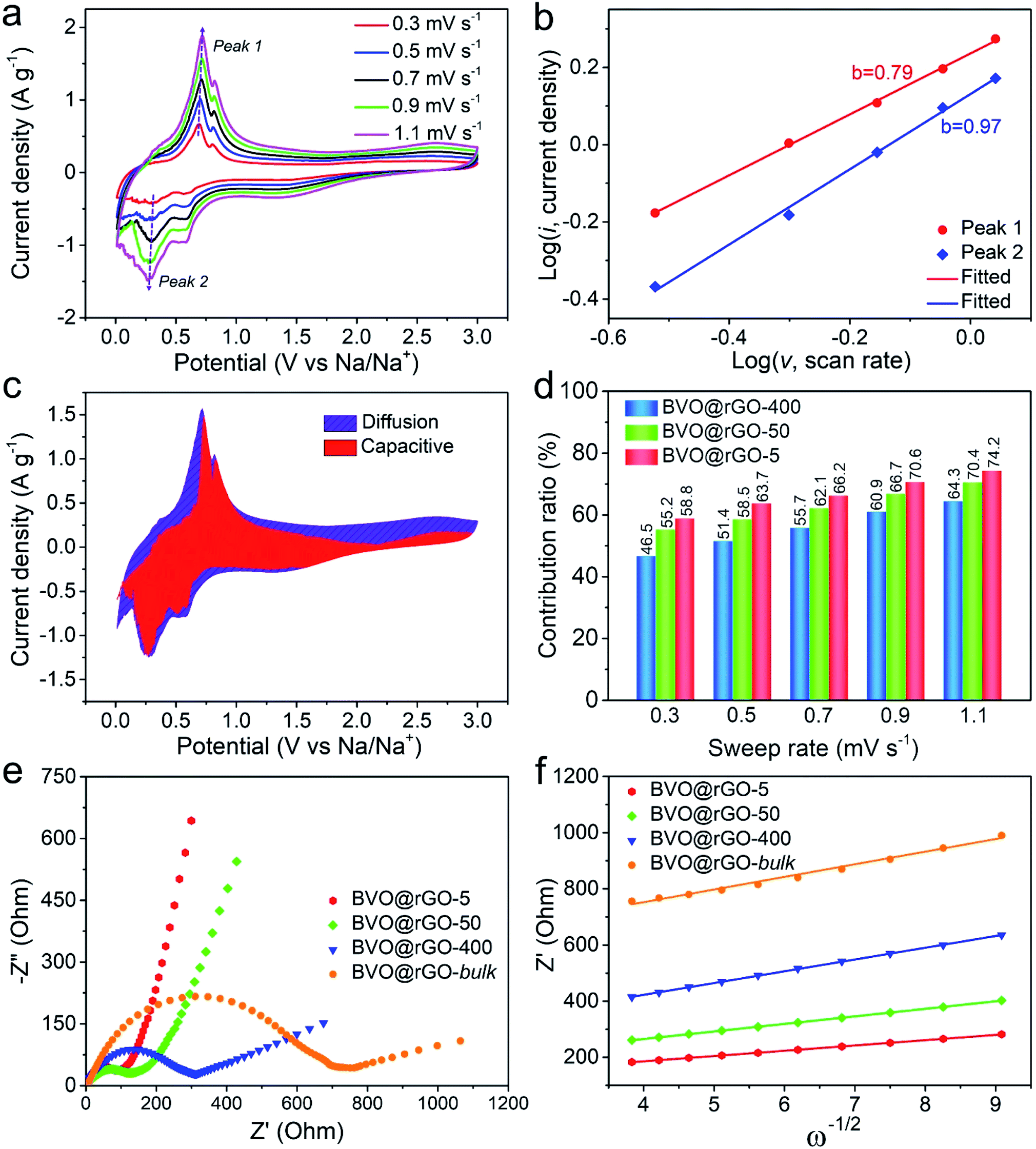
We further conducted CV measurements for these size-tailored BVO@rGO electrodes at different scan rates to reveal the size-dependent fast kinetics (Fig. 4a, S17a and S18a†). The charge storage mechanism was qualitatively analysed according to the eqn (3).45log(i) = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(v) + log(a) log(v) + log(a) | (3) |
It has been established that b value has two well-defined conditions: the value of 0.5 indicates a diffusion-dominated process and the value of 1.0 represents the capacitive-controlled one. For BVO@rGO-5, the b value for the cathodic Peak 1 (sodiation process) is as high as 0.97 (Fig. 4b), very close to 1, strongly suggesting that majority of kinetics are surface controlled. In sharp contrast, the b value of the same sodiation process decreases to 0.75 and 0.55 with the size increases to 50 nm and 400 nm (Fig. S17b and S18b†), indicating the gradual enhancement of diffusion-dominated process. Likewise, for the desodiation process (Peak 2), the b value follows the same trend. These results clearly demonstrate the size-dependent kinetics for the sodium storage behavior of the size-tailored BiVO4 colloids.
 | ||
| Fig. 4 Na+ Storage mechanism and kinetics analysis. (a) CV curves of BVO@rGO-5 electrode at different scan rates. (b) log(i) versus log(v) plots at different oxidation and reduction states. (c) CV curve with the pseudocapacitive contribution shown in the red region at a scan rate of 0.9 mV s−1. (d) The comparison of capacitive contribution at different scan rates in BVO@rGO electrodes. (e) Electrochemical impedance spectra for BVO@rGO electrodes after 200th cycle at 0.1 A g−1. (f) Corresponding fitting of the coefficient of Warburg impedance (σw) from the linear fitting line of Z′ versus ω−1/2. | ||
To quantify the ratio of the Na-ion capacitive contribution, the current response i at a fixed potential V is separated into capacitive effects (k1v) and diffusion-controlled contribution (k2v1/2) according to the eqn (4).46
| i(V) = k1v + k2v1/2 | (4) |
Fig. 4c clearly shows the typical contribution from capacitive process (red region) and diffusion process (blue region) for BVO@rGO-5. A dominating capacitive contribution up to 70.6% is achieved for BVO@rGO-5 at 0.9 mV s−1. Moreover, the role of capacitive contribution further improves with the increasing the sweep rates and reaches a maximum value of 74.2% at 1.1 mV s−1 (Fig. 4d). These results show that the capacitive contribution always dominates the total capacity during charge–discharge processes for BVO@rGO-5. By similar analysis of BVO@rGO-50 and BVO@rGO-400 (Fig. S17c, d, S18c and d†), the capacitive contributions at various sweep rates are summarized in Fig. 4d. It is obvious that the capacitive contribution decreases monotonically as the particle increases from 5, 50 to 400 nm.
Electrochemical impedance spectroscopy (EIS) measurements were used to clarify the electron- and sodium-ion transport property of these BiVO4 (Fig. 4e and f). The charge transfer resistance (Rct) reflected from the semicircle in the high frequency range and the ions diffusion resistance associated with a sloping straight line in the low frequency range are shown in Nyquist plots. Obviously, the Rct increases in a sequence of the BVO@rGO-5 (87 Ω), BVO@rGO-50 (128 Ω), BVO@rGO-400 (313 Ω) and BVO@rGO-bulk (729 Ω), indicating an elevated charge transfer with reduced size. Meanwhile, the Na+ diffusion coefficient (DNa) can be calculated by the eqn (5).47
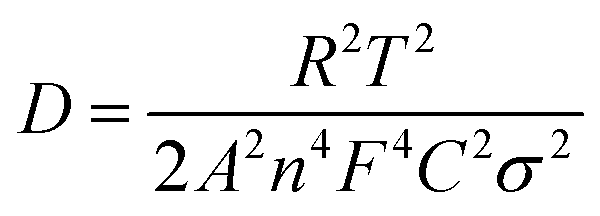 | (5) |
It can be found that BVO@rGO-5 still possesses a higher DNa of 6.3 × 10−15 cm2 s−1 than BVO@rGO-50 and BVO@rGO-400 (3.1 × 10−15 and 1.3 × 10−15 cm2 s−1, respectively) after 200 cycles, which is almost 6 times as much as that of BVO@rGO-bulk (1.1 × 10−15 cm2 s−1), indicating a lower ion diffusion resistance and accelerated Na+ transportation. Therefore, reducing particle size and introducing oxygen vacancies are thus highly promising methods to improve electrochemical performance of materials. The pulsed laser irradiation presented here provides a new strategy for facile synthesis of size-tailored multinary metal oxide particles ranging from submicrometer to nanometer and introduces oxygen vacancies in them, simultaneously.
The unique feature observed in these BVO@rGO indicates the following crucial aspects. First, the ultrafine BiVO4 particles with large surface area in BVO@rGO-5 provides sufficient active area for uptaking and releasing of Na+, thus enhancing the Na-ion storage capacity. Second, the Na storage behavior is more surface controlled in ultrafine BiVO4 particles, BVO@rGO-5, which is similar to that of supercapacitor materials with pseudocapacitive mechanisms,48,49 and thus is believed to deliver improved rate capability. Third, the microstructural damage induced by strains during (de)sodiation process was depressed in ultrafine BiVO4 particles, thus eliminating the main structural instabilities of BiVO4 and giving rise to the steadily enhanced cycle stability. This can be collaborated by the SEM observation of BiVO4 with different sizes, such as BVO@rGO-50 and BVO@rGO-bulk electrodes after 200 cycles (Fig. S19†). The BVO@rGO-50 electrode still remained structurally integrated and well distributed, while the BVO@rGO-bulk electrode has produced visible cracks and agglomerated seriously.
Conclusions
In summary, we propose an innovative laser irradiation technique for constructing multinary metal oxide based inorganics (e.g., BiVO4) colloids with well-defined particle size. The easy regulation of the irradiation parameters results in BiVO4 colloids with rich oxygen vacancies that demonstrates visually black colour, which is as far as we know a new type of artificial black material. When evaluated as an anode material for SIBs, the BVO@rGO electrodes deliver outstanding rate capability and high reversible capacity, which is much superior to the as-reported BiVO4-based electrodes for SIBs, benefitting from the oxygen vacancies as well as the dramatically reduced dimensions of BiVO4 colloids. Therefore, our study may break new ground in designing multinary metal oxide-based inorganics and pave the way for high-performance electrode materials for energy storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the project of the National Natural Science Foundation of China (No. 51911530212, 51872240, 51702262, 51972270, 51672225), the China Postdoctoral Science Foundation (2018T111094, 2018M643734, 2018BSHYDZZ57), the Fundamental Research Funds for the Central Universities (3102019JC005, 3102019ghxm004), the Research Fund of the State Key Laboratory of Solidification Processing (NPU), China (Grant No. 2019-QZ-03), the Key Research and Development Program of Shaanxi Province (2019TSLGY07-03), the Top International University Visiting Program for Outstanding Young scholars of Northwestern Polytechnical University, the DFG (KA 1698/27-1) and BMBF Excellence Center BaMoSa. H. Wang acknowledges support from the 1000 Youth Talent Program of China. We would like to thank the Analytical & Testing Center of Northwestern Polytechnical University for SEM and TEM characterizations.References
- S. B. Sant, Mater. Manuf. Processes, 2012, 27, 1462–1463 CrossRef CAS.
- K. J. Klabunde and R. M. Richards, Nanoscale Materials in Chemistry, John Wiley and Sons, 2009 Search PubMed.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS.
- Y. Barak, I. Meir, A. Shapiro, Y. Jang and E. Lifshitz, Adv. Mater., 2018, 30, 1801442 CrossRef.
- A. L. Rogach, D. V. Talapin, E. V. Shevchenko, A. Kornowski, M. Haase and H. Weller, Adv. Funct. Mater., 2002, 12, 653–664 CrossRef CAS.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- D. V. Talapin, J. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- P. Guo, X. Yang, Q. Ye, J. Zhang, H. Wang, H. Yu, W. Zhao, C. Liu, H. Yang and H. Wang, Adv. Energy Mater., 2019, 9, 1901341 CrossRef.
- P. Guo, Q. Ye, X. Yang, J. Zhang, F. Xu, D. Shchukin, B. Wei and H. Wang, J. Mater. Chem. A, 2019, 7, 2497–2506 RSC.
- N. T. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS.
- l. T. Lu, N. T. Dung, L. D. Tung, C. T. Thanh, O. K Quy, N. V. Chuc, S. Maenosono and N. T. K. Thanh, Nanoscale, 2015, 7, 19596–19610 RSC.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- K. J. Griffith, K. M. Wiaderek, G. Cibin, L. E. Marbella and C. P. Grey, Nature, 2018, 559, 556–563 CrossRef CAS.
- M. Rui, X. Li, L. Gan, T. Zhai and H. Zeng, Adv. Funct. Mater., 2016, 26, 5051–5060 CrossRef CAS.
- J. Jian, G. Jiang, R. van de Krol, B. Wei and H. Wang, Nano Energy, 2018, 51, 457–480 CrossRef CAS.
- J. Jian, Y. Xu, X. Yang, W. Liu, M. Fu, H. Yu, F. Xu, F. Feng, L. Jia, D. Friedrich, R. van de Krol and H. Wang, Nat. Commun., 2019, 10, 2609 CrossRef PubMed.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- F. Chen, Q. Yang, Y. Wang, J. Zhao, D. Wang, X. Li, Z. Guo, H. Wang, Y. Deng, C. Niu and G. Zeng, Appl. Catal., B, 2017, 205, 133–147 CrossRef CAS.
- T. Sivakumar and J. Gopalakrishnan, Chem. Mater., 2002, 14, 3984–3989 CrossRef CAS.
- Y. Sun, Y. Xie, C. Wu, S. Zhang and S. Jiang, Nano Res., 2010, 3, 620–631 CrossRef CAS.
- S. S. Shin, E. J. Yeom, W. S. Yang, S. Hur, M. G. Kim, J. Im, J. Seo, J. H. Noh and S. Seok, Science, 2017, 356, 167–171 CrossRef CAS.
- J. Yu and A. Kudo, Adv. Funct. Mater., 2006, 16, 2163–2169 CrossRef CAS.
- A. Pyatenko, H. Wang and N. Koshizaki, J. Phys. Chem. C, 2014, 118, 4495–4500 CrossRef CAS.
- H. Wang, K. Kawaguchi, A. Pyatenko, X. Li, Z. S. Swiatkowska-Warkocka, Y. Katou and N. Koshizaki, Chem.–Eur. J., 2012, 18, 163–169 CrossRef CAS PubMed.
- H. Wang, A. Pyatenko, K. Kawaguchi, X. Li, Z. Swiatkowska-Warkocka and N. Koshizaki, Angew. Chem., Int. Ed., 2010, 49, 6361–6364 CrossRef CAS PubMed.
- J. Xu, Z. Bian, X. Xin, A. Chen and H. Wang, Chem. Eng. J., 2018, 337, 684–696 CrossRef CAS.
- S. Wang, P. Chen, J. H. Yun, Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2017, 56, 8500–8504 CrossRef CAS PubMed.
- G. Wang, Y. Ling, X. Lu, F. Qian, Y. Tong, J. Z. Zhang, V. Lordi, C. R. Leao and Y. Li, J. Phys. Chem. C, 2013, 117, 10957–10964 CrossRef CAS.
- Z. Chen, H. Jiang, W. Jin and C. Shi, Appl. Catal., B, 2016, 180, 698–706 CrossRef CAS.
- W. Li, Y. Zhang, G. Tian, S. Xie, Q. Xu, L. Wang, J. Tian and Y. Bu, J. Mol. Catal. A: Chem., 2016, 423, 356–364 CrossRef CAS.
- H. Wang, N. Koshizaki, L. Li, L. Jia, K. Kawaguchi, X. Li, A. Pyatenko, Z. Swiatkowska-Warkocka, Y. Bando and D. Golberg, Adv. Mater., 2011, 23, 1865–1870 CrossRef CAS PubMed.
- X. Chen, D. Zhao, K. Liu, C. Wang, L. Liu, B. Li, Z. Zhang and D. Shen, ACS Appl. Mater. Interfaces, 2015, 7, 16070–16077 CrossRef CAS.
- M. Kawasaki, J. Phys. Chem. C, 2011, 115, 5165–5173 CrossRef CAS.
- T. W. Kim, Y. Ping, G. A. Galli and K. S. Choi, Nat. Commun., 2015, 6, 8769 CrossRef CAS.
- D. D. Qin, T. Wang, Y. M. Song and C. L. Tao, Dalton Trans., 2014, 43, 7691–7694 RSC.
- S. Yang, Y. Liu, Y. Hao, X. Yang, W. A. Goddard III, X. Zhang and B. Cao, Adv. Sci., 2018, 5, 1700659 CrossRef.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS.
- T. Jin, Q. Han, Y. Wang and L. Jiao, Small, 2018, 14, 1703086 CrossRef PubMed.
- D. Ma, Y. Li, H. Mi, S. Luo, P. Zhang, Z. Lin, J. Li and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8901–8905 CrossRef CAS.
- Y. Xu, M. Zhou, X. Wang, C. Wang, L. Liang, F. Grote, M. Wu, Y. Mi and Y. Lei, Angew. Chem., Int. Ed., 2015, 127, 8892–8895 CrossRef.
- J. Sottmann, M. Herrmann, P. Vajeeston, A. Ruud, C. Drathen, H. Emerich, D. S. Wragg and H. Fjellvåg, Chem. Mater., 2017, 29, 2803–2810 CrossRef CAS.
- R. Muruganantham and W. Liu, ChemistrySelect, 2017, 2, 8187–8195 CrossRef CAS.
- Y. Fang, X. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2018, 57, 1–6 CrossRef.
- H. Si, L. Seidl, E. M. L. Chu, S. Martens, J. Ma, X. Qiu, U. Stimming and O. Schneider, J. Electrochem. Soc., 2018, 165, A2709–A2717 CrossRef CAS.
- Y. Xiao, D. Su, X. Wang, S. Wu, L. Zhou, Y. Shi, S. Fang, H. Cheng and F. Li, Adv. Energy Mater., 2018, 8, 1800930 CrossRef.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS PubMed.
- Y. Tang, Y. Zhang, X. Rui, D. Qi, Y. Luo, W. R. Leow, S. Chen, J. Guo, J. Wen, W. Li, J. Deng, Y. Lai, B. Ma and X. Chen, Adv. Mater., 2016, 28, 1567–1576 CrossRef CAS PubMed.
- F. Xu, Z. Tang, S. Huang, L. Chen, Y. Liang, W. Mai, H. Zhong, R. Fu and D. Wu, Nat. Commun., 2015, 6, 7221 CrossRef PubMed.
- F. Xu, J. Xu, H. Xu, Y. Lu, H. Yang, Z. Tang, Z. Lu, R. Fu and D. Wu, Energy Storage Materials, 2017, 7, 8–16 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta13021g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |