
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight opens a new window for N-heterocyclic carbene catalysis
Jing
Liu
a,
Xiao-Ning
Xing
b,
Jin-Hai
Huang
a,
Liang-Qiu
Lu
 *a and
Wen-Jing
Xiao
*a
*a and
Wen-Jing
Xiao
*a
aKey Laboratory of Pesticide & Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, 152 Luoyu Road, Wuhan, Hubei 430079, China. E-mail: luliangqiu@mail.ccnu.edu.cn; wxiao@mail.ccnu.edu.cn; Web: http://chem-xiao.ccnu.edu.cn
bAnyang Academy of Agricultural Sciences, 833 Wenming Road, Anyang, Henan, China
First published on 11th August 2020
Abstract
N-Heterocyclic carbenes (NHCs) are efficient Lewis basic catalysts for the umpolung of various polarized unsaturated compounds usually including aldehydes, imines, acyl chlorides and activated esters. NHC catalysis involving electron pair transfer steps has been extensively studied; however, NHC catalysis through single-electron transfer (SET) processes, despite having the potential to achieve chemical transformations of inert chemical bonds and using green reagents, has long been a challenging task in organic synthesis. In parallel, visible-light-induced photocatalysis and photoexcitation have been established as powerful tools to facilitate sustainable organic synthesis, as they enable the generation of various reactive radical intermediates under extremely mild conditions. Recently, a number of elegant visible-light-induced, NHC-catalyzed transformations were developed for accessing valuable organic compounds. As a result, this minireview will highlight the recent advances in this field.
1. Introduction
N-Heterocyclic carbenes (NHCs) are a class of intriguing nitrogen-containing heterocyclic compounds possessing a bivalent carbon atom with six valence electrons.1 According to their general properties and applications in modern chemistry, NHCs are approximately categorized into three sections: (i) ligands for transition metals; (ii) coordination to p-block elements and (iii) organocatalysts. Since the early 2000s, NHCs have been employed extensively as powerful and efficient nucleophilic or Brønsted base catalysts2 for chemical bond formations, including cross-benzoin reactions, Stetter reactions, cycloadditions and so on. Several comprehensive reviews have summarized these important advances.3 However, most of these investigations presumably involved electron pair transfer steps. With nucleophilic NHC catalysis as an example, the majority of these processes are initiated by nucleophilic attack of the carbene catalyst on the carbonyl group. Subsequently, the generated Breslow intermediate, homoenolate or dienolate, usually reacts with strong electrophiles with sp2 carbon, such as active ketones and imines. Weak electrophiles with sp3 carbon, such as alkyl halides, are inert in NHC-catalyzed reactions. In contrast to electron pair transfer pathways, the development of NHC-catalyzed reactions through single-electron transfer (SET) processes is still in its infancy.4 Although Fukuzumi and co-workers carefully studied the redox or electron transfer characteristics of Breslow intermediates in the late 1990s,5 it wasn’t until 2008 that Studer and co-workers reported pioneering work on the biomimetic, carbene-catalyzed oxidation reaction of aldehydes, which involves two successive single-electron oxidations of the Breslow intermediate by 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO).6 After that, Rovis,7 Chi8 and Ye9 demonstrated that single-electron oxidations of NHC-bound intermediates are indeed useful for developing new chemical transformations. However, these studies are primarily limited to specific oxygen or functionalized carbon-centred radicals. In 2019, Nagao and Ohmiya developed an NHC-catalyzed decarboxylative alkylation of aldehydes using redox-active esters.10a Soon after, the same catalysis strategy was applied to two three-component coupling reactions via a radical relay process.10b,c Despite these substantial contributions, there are still only limited radical reactions that take place through single NHC catalysis.Over the past decade, a wide range of visible-light-induced organic photochemical reactions have been successfully explored to meet the requirement of green synthetic chemistry.11 Key to these successes is the use of the strategy of visible light photocatalysis to generate highly reactive radical species under environment- and user-friendly conditions. In addition, nature always inspires synthetic chemists to mimic its multienzymatic systems for constructing molecular complexity.12 In this context, the combination of photocatalysis with other catalytic modes,13 including NHC catalysis, has been established as an important platform for modern green synthesis. This distinct catalysis mode could generate products with remarkable selectivity, which are inaccessible through a single NHC catalyst system. This minireview focuses on recent advances in visible-light-induced, NHC-catalyzed organic reactions, in which light is used to excite the additional photosensitizer (photoredox catalysis mechanism) or the NHC-bound intermediate itself (direct photoactivation mechanism).
2. Synergistic NHC/photoredox catalysis
In recent years, synergistic NHC/photoredox catalysis has been used to realize several significant organic transformations under oxidative and neutral conditions. In these dual catalysis systems, an independent NHC catalyst is used for umpolung of carbonyls via the formation of Breslow intermediates or transient acyl azoliums. In parallel, a photocatalyst (PC) is employed to absorb visible light and the excited photocatalyst can promote the generation of highly reactive radical species via SET events with substrates (Fig. 1a) or NHC-intermediates (Fig. 1b). Compatibility between these two catalysts is crucial to make sure these radical reactions proceed well with remarkable efficiency and selectivity. | ||
| Fig. 1 Two representative activation modes of synergistic NHC/photoredox catalysis. | ||
2.1 Visible-light-driven oxidative reactions
In 2012, Rovis and co-workers first reported that photoredox catalysis can be combined with chiral NHC catalysis, realizing the catalytic asymmetric acylation of N-aryl tetrahydroisoquinolines with aldehydes by using an aminoindanol-derived NHC catalyst and a Ru(bpy)3Cl2 photocatalyst (Scheme 1).14 In this process, using a sub-stoichiometric quantity of the weak organic oxidant meta-dinitrobenzene (m-DNB) is essential for obtaining high reaction efficiency. Stronger oxidants such as BrCCl3 may result in oxidative decomposition of the NHC catalyst. A series of aliphatic aldehydes and tertiary amines participate smoothly in this transformation, affording the corresponding α-amino ketones in good yields and excellent stereoselectivities. Notably, sensitive functionalities such as olefins, thioethers and esters were well tolerated. However, electron-donating substituents on the N-aryl group are essential for this reaction in order to reduce the competing radical dimerization.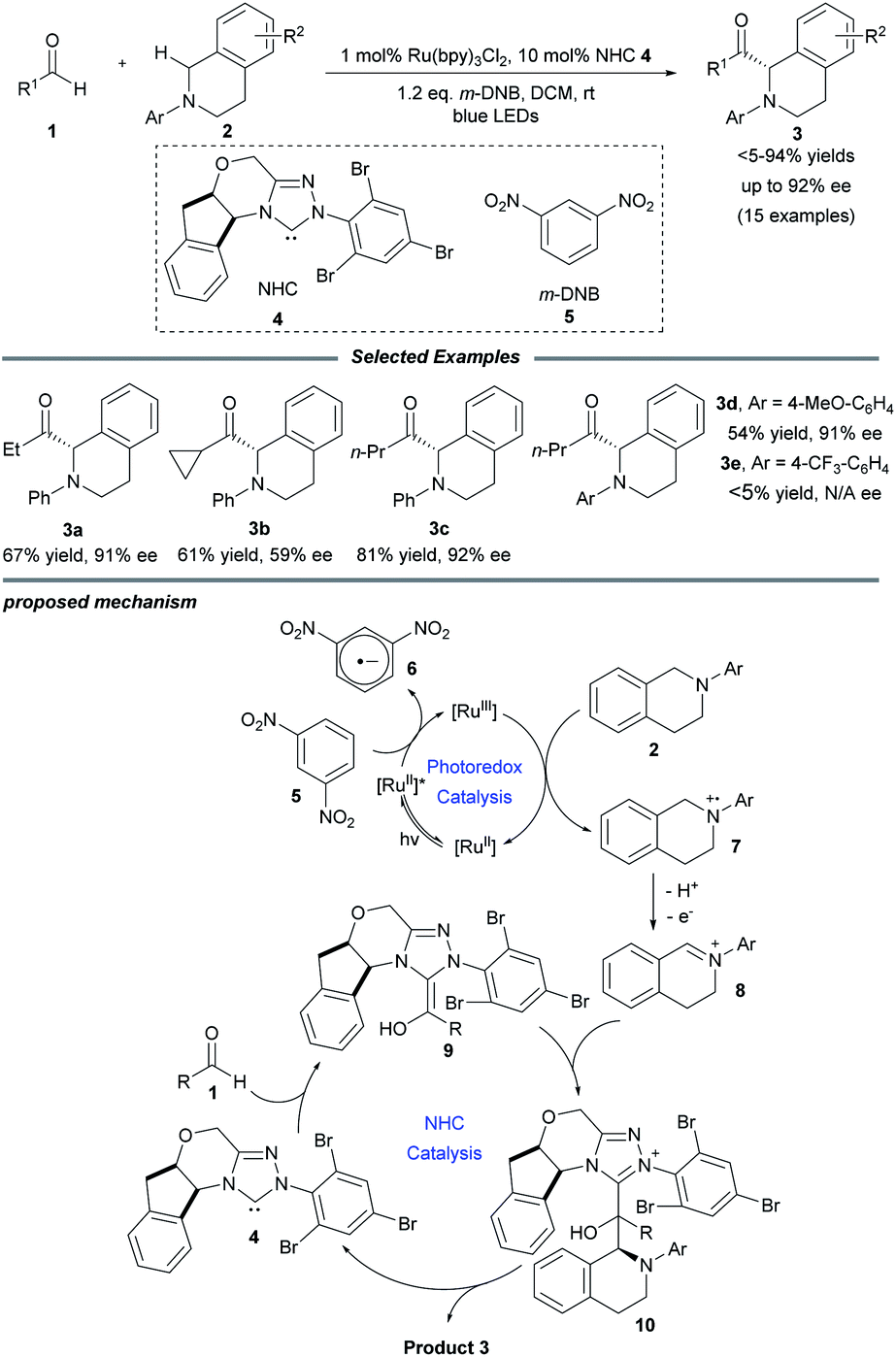 | ||
| Scheme 1 Photo/NHC-cocatalyzed enantioselective acylation of N-aryl tetrahydroisoquinolines with aldehydes. bpy: 2,2′-bipyridine. | ||
Mechanistically, Ru(bpy)33+ is generated upon the oxidative quenching of excited *Ru(bpy)32+ by m-DNB accompanied by the release of arene radical anion 6. Then tetrahydroisoquinoline 2 is oxidized by the Ru(bpy)33+ to form amine radical cation 7, followed by release of a proton and an electron to deliver the iminium ion species 8. Meanwhile, NHC catalyst 4 reacts with aliphatic aldehyde 1 to generate the activated Breslow intermediate 9. Stereoselective addition of this nucleophilic intermediate to iminium ion 8 leads to the formation of a C–C bond to yield aminoalcohol derivative 10. Finally, the enantio-enriched α-amino ketone product 3 is obtained and the NHC catalyst is regenerated by the following elimination of adduct 10, and the NHC catalytic cycle is then completed.
In 2018, Miyabe and co-workers disclosed a facile protocol for oxidative esterification of cinnamaldehydes by using the strategy of NHC/photoredox dual catalysis (Scheme 2).15 A series of methyl cinnamates bearing electron-donating or -withdrawing groups were obtained under mild conditions. In addition, 2-naphthaldehyde could also undergo this transformation smoothly and be converted into the corresponding ester 12d. A stoichiometric amount of the external oxidant BrCCl3 is necessary to prevent reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. The mechanism initiates with the interaction of the NHC with the aldehyde to form the Breslow intermediate 15. In path A, subsequent protonation of 15 by MeOH leads to the enolate ion pair 17 through a stable azolium intermediate 16. Then, a SET oxidation of enolate 17 by the excited-state organophotocatalyst rhodamine 6G delivers a radical cation 18, which reacts with BrCCl3 to generate the brominated cation intermediate 19. Then a resonance-stabilized cation 22 is obtained via the elimination of HBr. Ultimately, cation 22 reacts with MeOH to provide the ester product 12 and regenerate the NHC catalyst 14. In another possible pathway (path B), acylium ion equivalent 22 can be delivered through two consecutive photocatalytic SET oxidations of Breslow intermediate 15.
C bond. The mechanism initiates with the interaction of the NHC with the aldehyde to form the Breslow intermediate 15. In path A, subsequent protonation of 15 by MeOH leads to the enolate ion pair 17 through a stable azolium intermediate 16. Then, a SET oxidation of enolate 17 by the excited-state organophotocatalyst rhodamine 6G delivers a radical cation 18, which reacts with BrCCl3 to generate the brominated cation intermediate 19. Then a resonance-stabilized cation 22 is obtained via the elimination of HBr. Ultimately, cation 22 reacts with MeOH to provide the ester product 12 and regenerate the NHC catalyst 14. In another possible pathway (path B), acylium ion equivalent 22 can be delivered through two consecutive photocatalytic SET oxidations of Breslow intermediate 15.
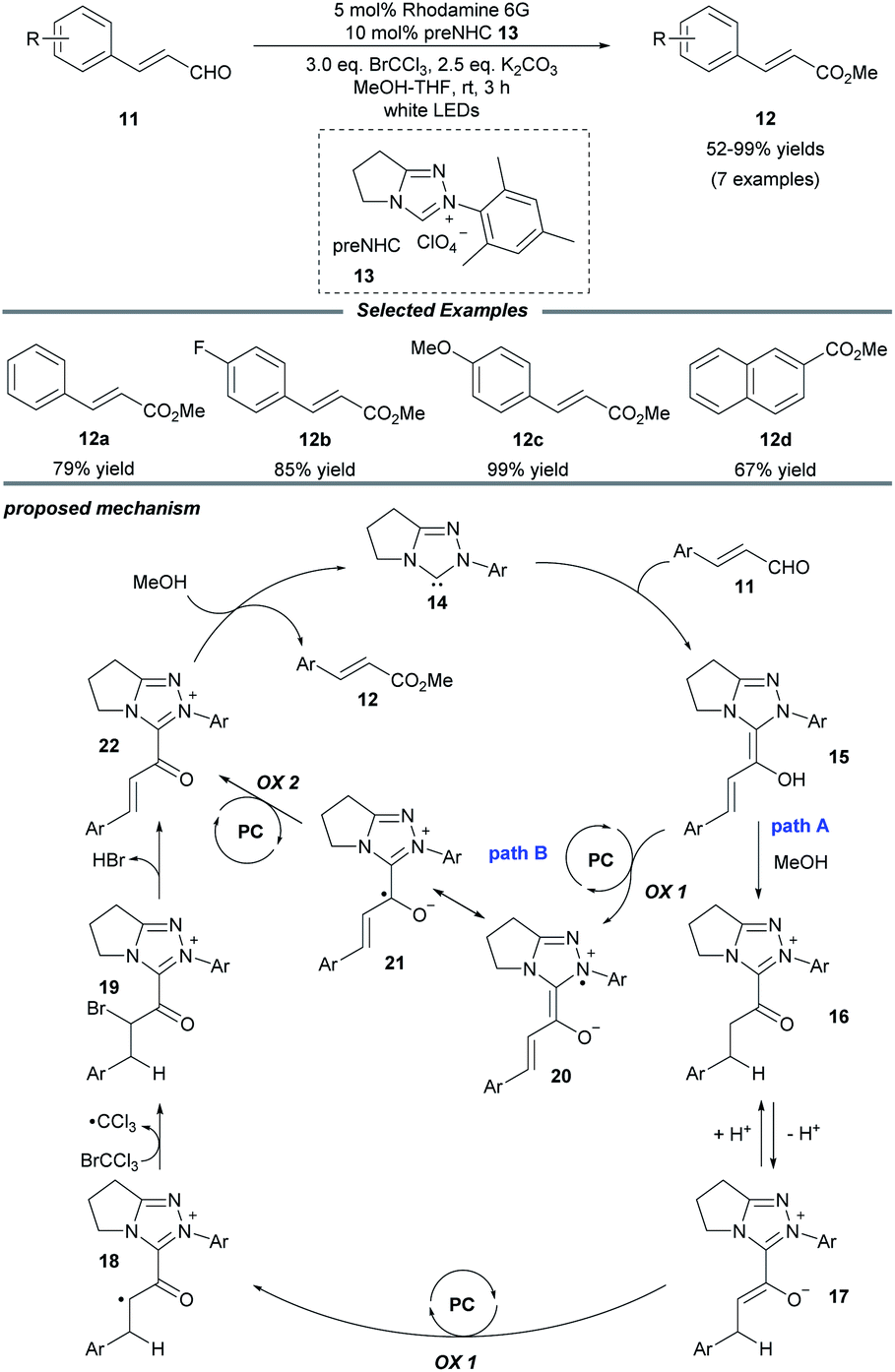 | ||
| Scheme 2 Photo/NHC-cocatalyzed oxidative esterification of cinnamaldehyde derivatives. | ||
Molecular oxygen is a beneficial and green oxidant in chemical oxidation reactions. Quite recently, Ye and colleagues documented an NHC/photo-cocatalyzed, aerobic oxidative Smiles rearrangement of o-aryl salicylaldehydes for the preparation of aryl salicylates (Scheme 3).16 With the use of an achiral triazolium preNHC catalyst and the Fukuzumi photocatalyst, aryl groups with different electronic properties migrate well in the rearrangement process. NaI is a prerequisite additive to accelerate the electron transfer. A series of control experiments show that the Breslow intermediate is oxidized by molecular oxygen in the presence of NaI and acridium salt. A plausible catalytic cycle is proposed in Scheme 3. The attack of NHC 26 on salicylaldehyde generates the key Breslow intermediate 27, which can be oxidized by O2 through a SET process. Subsequent deprotonation of the generated intermediate 28 by a superoxide anion affords a zwitterionic radical species 29 and a hydroperoxide radical, which can further oxidize 29 to deliver acyl azolium 30. Then the acyl azolium undergoes a hydrolysis process to furnish the salicylic acid 31, completing the oxidative NHC catalytic cycle. Meanwhile, photoexcitation of the acridium salt by visible light affords the excited photocatalyst, [Mes–Acr+]*, which can undergo reductive quenching by the carboxylic anion 32 to give carboxyl radical 33 and an Acr–Mes radical. Following this, the intramolecular Smiles rearrangement generates the oxygen radical 35 through a spirocyclic radical intermediate 34. Then the oxygen radical oxidizes the Acr–Mes radical to regenerate the photocatalyst and form the oxygen anion species 36, which undergoes protonation to yield the desired product 24.
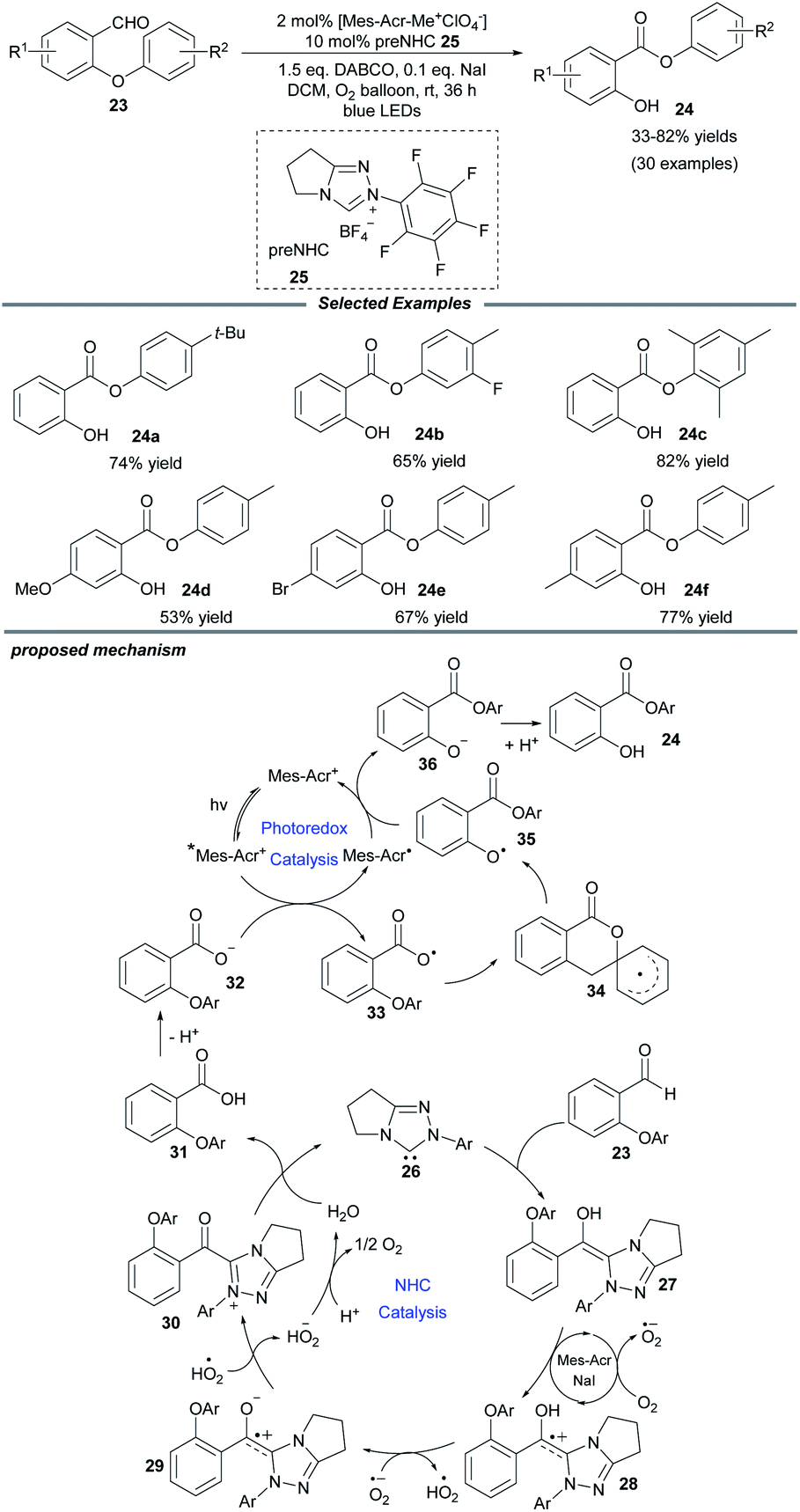 | ||
| Scheme 3 Photo/NHC-cocatalyzed oxidative Smiles rearrangement. DABCO: 1,4-diazabicyclo[2.2.2]octane. | ||
2.2 Visible-light-driven redox neutral reactions
The dihalomethylene group widely occurs in natural products and biologically active molecules.17 In 2016, the Sun group described one example of the γ-dichloromethylenation method by employing [Ir(dtbbpy)(ppy)2]PF6 as the photoredox catalyst and triazolium salt 40 as the NHC precatalyst (Scheme 4).18 Under the irradiation of a blue LED light, the unsaturated δ,δ-dihalo ester product 39 was generated from aldehyde 37 and CCl4 with a perfect regioselectivity in 42% yield. Presumably, the initial trichloromethylation product 41 is the key intermediate, which could be formed during the cocatalyzed process and undergo elimination of HCl to give the final product. The authors observed that the reaction yield can be further improved by using the more labile radical precursors CBr4 and CCl3Br even without additional light irradiation.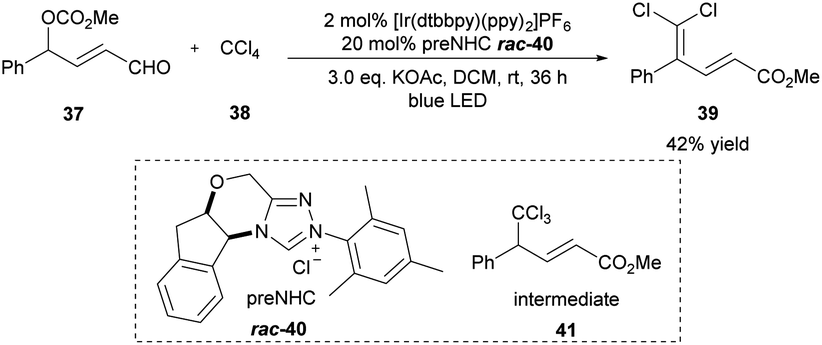 | ||
| Scheme 4 Photo/NHC-cocatalyzed γ-dichloromethylenation of enal. ppy: 2-phenylpyridine, dtbbpy: 4,4-di-tert-butyl-2,2′-bipyridine. | ||
Later in 2019, Ye and co-workers realized an NHC/photoredox dual catalytic protocol for γ- and ε-alkylations of enals, affording multisubstituted unsaturated esters with good efficiency and exclusive regioselectivity (Scheme 5).19 Under mild conditions, this method showed a good substrate scope and functional group compatibility for both constituents. Comfortingly, γ-dialkylated enals could also be tolerated and the desired products containing all-carbon quaternary centers, which are a challenging topic in organic synthesis, could be obtained in moderate to good yields. Notably, a series of primary, secondary and tertiary radical precursors all worked well for this transformation. Through a trienolate intermediate, the ε-functionalization of enals 43 that bear alkenyl substituents at the γ-position was also established. Beyond that, a catalytic enantioselective alkylation was examined and a promising enantioselectivity with 29% ee was afforded when the chiral NHC precursor 48 was employed. It was observed that the alkylation process was inhibited in the absence of light, photocatalyst and NHC catalyst. Quenching experiments with the radical scavenger TEMPO confirmed that the transformation should proceed through an alkyl radical pathway.
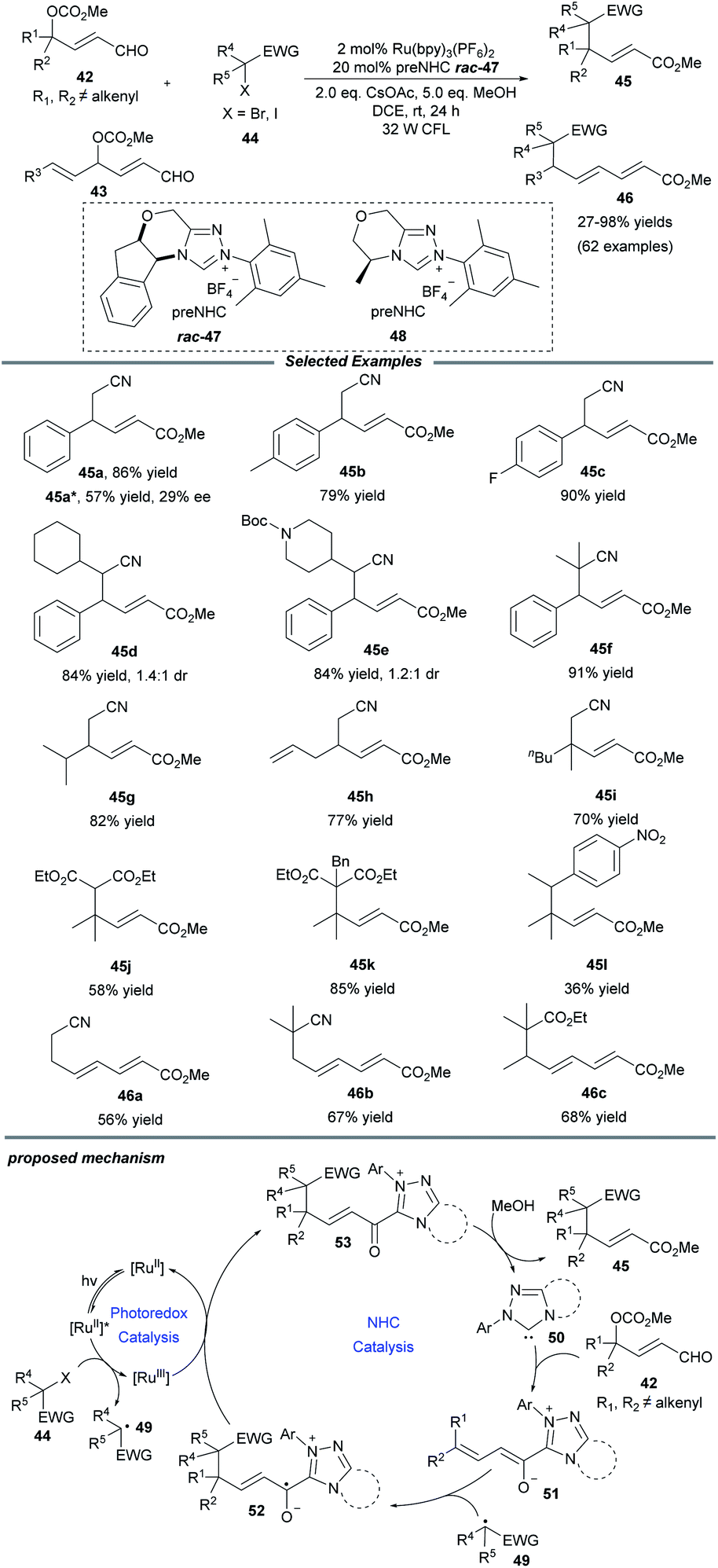 | ||
| Scheme 5 Photo/NHC-cocatalyzed γ- and ε-alkylations of enals with alkyl radicals. | ||
For the mechanism, Ye proposed that the excited state *Ru(bpy)32+ is probably quenched by halide to give alkyl radical 49 and the Ru(bpy)33+ photocatalyst. At the same time, NHC 50 reacts with peroxidized enal 42 to deliver the dienolate intermediate 51, which can be captured by alkyl radical 49 to furnish the homoenolate radical 52. The oxidation of radical 52 by Ru(bpy)33+ reforms Ru(bpy)32+ and generate the acyl azolium intermediate 53, followed by attack by MeOH to obtain the final product 45 and close the NHC catalytic cycle.
Shortly afterwards, the same group extended this photo/NHC dual catalysis system to the ring opening/γ-alkylation reaction of cyclopropane enal 54 (Scheme 6).20 A radical-trapping experiment with TEMPO suggests that an alkyl radical relay process is involved. Under mild conditions, a variety of alcohols and halogenated alkylation reagents participate smoothly in this reaction, furnishing the corresponding γ-alkylated α,β-unsaturated esters with moderate to excellent yields. The authors also pointed out that epoxy enals were not suitable for this transformation. Notably, a preliminary asymmetric variant was also studied with enal 54 and α-benzyl-α-bromomalonate as the substrates by using the chiral NHC precursor 57, affording the product 56e in a promising enantioselectivity with 22% ee. Mechanistically, NHC 50 reacts with γ-cyclopropane enal 54 to give the dienolate intermediate 59, which can be trapped by radical 58 which is generated from the alkyl halide via photoredox catalysis. The afforded homoenolate radical 60 undergoes a subsequent SET oxidation and alcoholysis to regenerate the NHC catalyst and provide the final product 56.
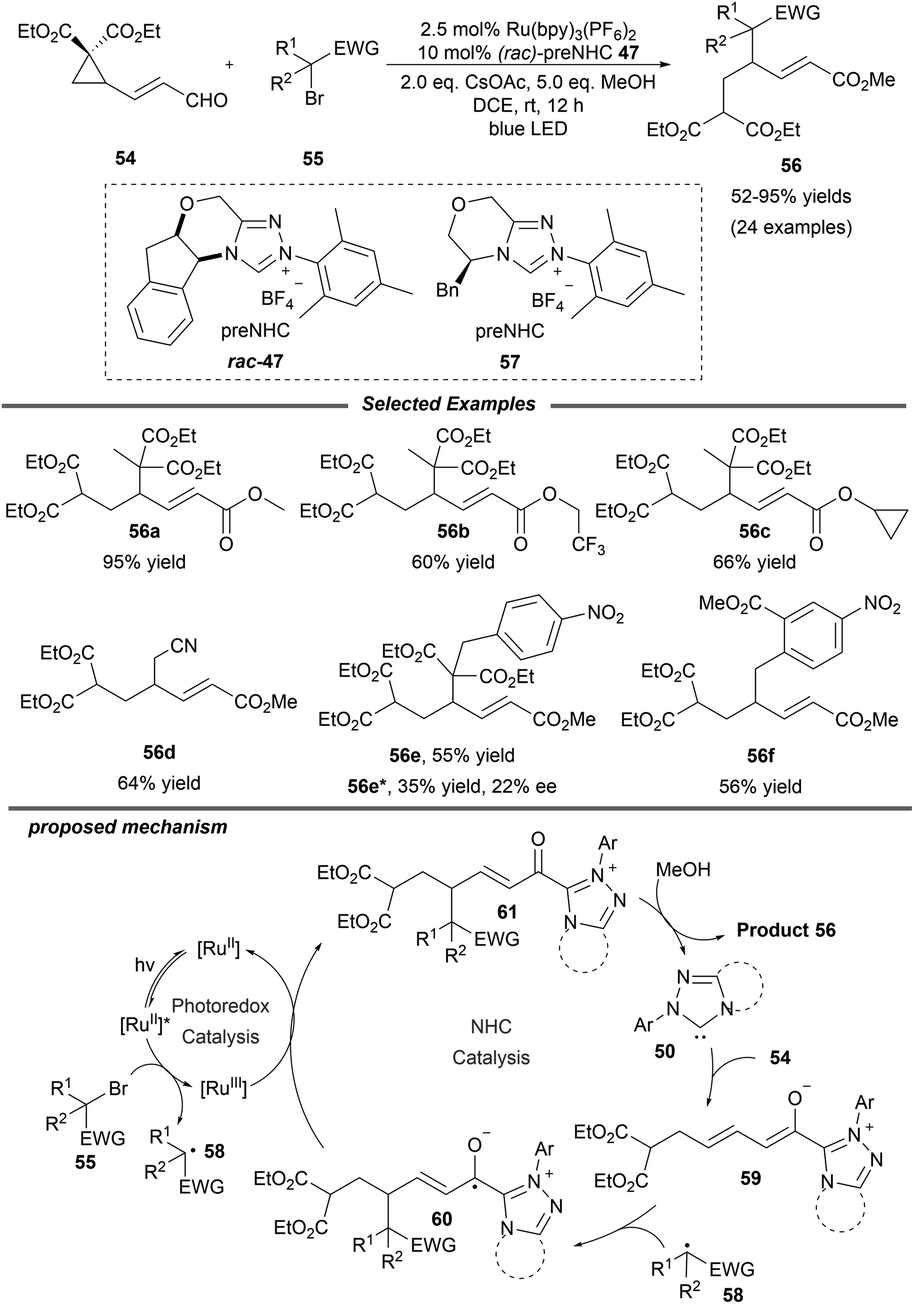 | ||
| Scheme 6 Ring opening and γ-alkylation of cyclopropane enal. | ||
Very recently, Scheidt and coworkers realized an elegant single-electron alkylation reaction of acyl azoliums for the synthesis of ketones from carboxylic acids (Scheme 7).21 Semi-high-throughput experimentation (HTE) facilely showed that a Hantzsch ester served as a suitable alkyl radical precursor and acyl imidazole was a suitable azolium radical precursor. This reaction employing 2 mol% iridium photocatalyst and 15 mol% azolium precatalyst exhibits a broad scope for both coupling partners. A series of functional groups, e.g., vinyl, alkynyl, ester, and heteroaryl motifs, were well tolerated. Additionally, to illustrate the ease and practicality of this NHC/photoredox cocatalyzed methodology, the authors also achieved the one-step late-stage functionalization of three carboxylic acid-containing pharmaceutical compounds, producing the desired ketone compounds in moderate to good yields. Notably, when a chiral NHC precursor 66 was used instead of 65, a promising enantioselectivity of the ketone product 64g was observed. It is a novel asymmetric acyl-like radical–radical cross-coupling reaction to construct ketones bearing an α-stereogenic center. A radical trapping experiment with TEMPO proved that radical species are involved in this process. The proposed reaction pathway starts from the reductive quenching of excited state *[IrIII] by the Hantzsch ester to afford photocatalyst [IrII], together with radical cation species 67 which can undergo fragmentation to form pyridine by-product 68 and alkyl radical 69. Then acyl triazolium 70, generated from the acyl imidazole, the NHC precursor and base, undergoes a single-electron reduction to yield the radical intermediate 71 and reform the ground-state photocatalyst [IrIII]. Finally, radical–radical coupling of intermediates 72 and 69 provides the desired product 64 and releases the NHC catalyst 73.
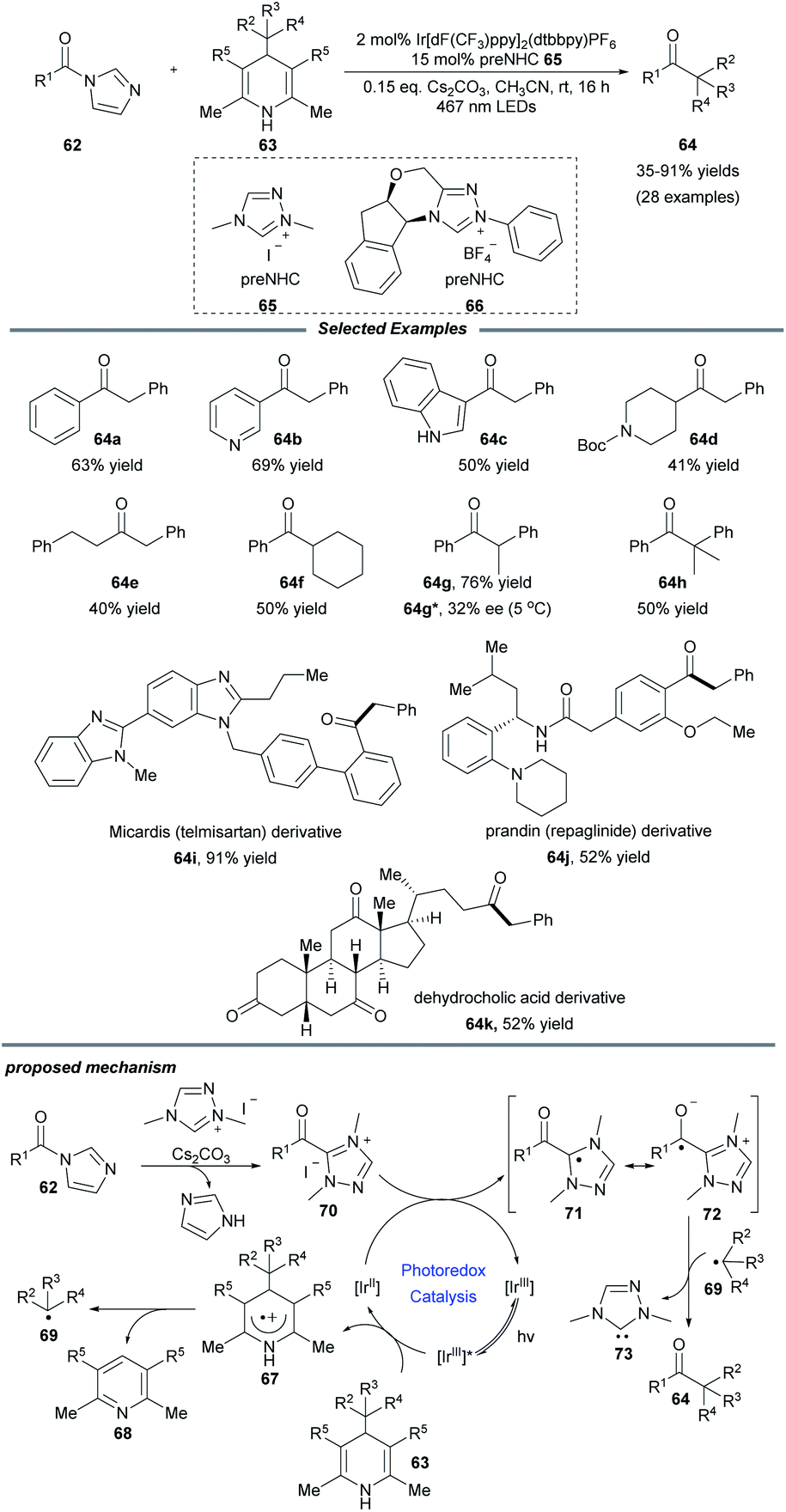 | ||
| Scheme 7 Alkylation of acyl azoliums for the synthesis of ketones. dF(CF3)ppy: 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine. | ||
3. Photoexcited NHC-catalyzed transformations
In addition to the aforementioned dual catalysis, single-NHC-catalyzed radical reactions under UVA or visible light irradiation were also developed. In these transformations, addition of the NHC catalyst to a carbonyl substrate generates an acyl azolium or Breslow intermediate, which can reach an excited state upon irradiation. Following this, these excited species can undergo further transformations through different pathways leading to the final products (Fig. 2a). Also, some α-diazo carbonyl substrates can absorb light directly and then generate active ketene intermediates, which can be captured by the NHC catalyst to afford enolate species (Fig. 2b).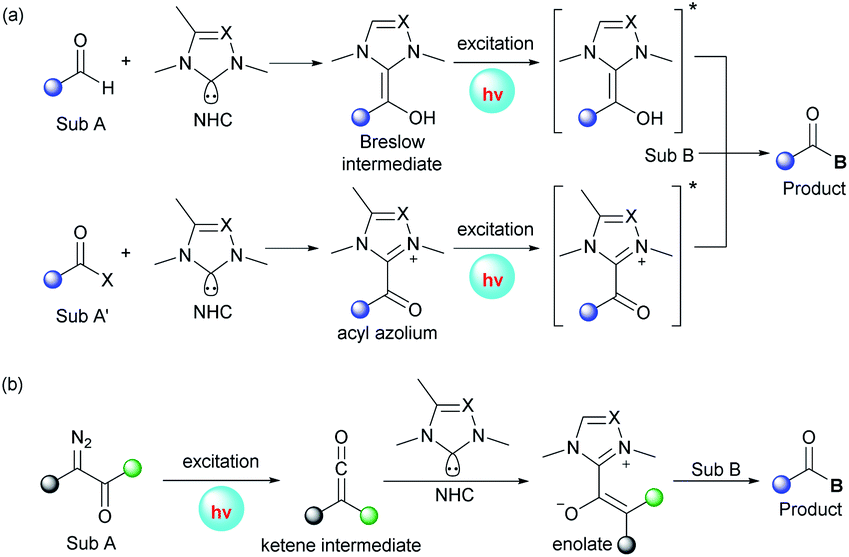 | ||
| Fig. 2 Activation modes of photoexcited NHC-catalyzed transformations. | ||
UVA-light-induced photochemical transformations of aromatic aldehydes and ketones have been well established;22 however, carboxylic acid substrates are always limited due to their shorter absorbing wavelength than that of the corresponding ketones. To address this issue, in 2019, the Hopkinson group utilized a combination of photoactivation with NHC organocatalysis to accomplish a sequential photoenolization/Diels–Alder (PEDA) reaction of acyl fluorides 74, resulting in the formation of isochroman-1-one derivatives 76 in moderate to high yields (Scheme 8).23 It is particularly noteworthy that o-ethyl and o-benzyl-substituted acid fluorides, which could not be employed to construct isochroman-1-ones by previous NHC-catalyzed methods, successfully participated in this reaction, too. Regrettably, some dienophiles such as isatins or 2-ketoesters failed to furnish the desired product, possibly owing to their competitive absorption of intense UVA light.
 | ||
| Scheme 8 NHC-catalyzed photoenolization/Diels–Alder reaction. | ||
The authors proposed a possible mechanism to illustrate this PEDA process. The imidazolylidene IMe 78, generated in situ by deprotonation of the NHC precursor 77 with base, selectively attacks the acyl fluoride, releasing a fluoride anion and giving o-toluoyl azolium 79. This benzophenone-like intermediate can be photoexcited via intersystem crossing to generate a triplet excited state 80, which undergoes intramolecular 1,5-hydrogen atom transfer to afford triplet dienol biradical species 81. Following rotation, 81 gives rise to a hydroxyl o-quinodimethane intermediate 82, which can react with the ketone to form the cycloadduct 84via the transient species 83. Finally, the catalytic cycle is completed with the elimination of NHC catalyst 78 and generation of the desired PEDA product 76.
Soon after, a particularly NHC-catalyzed photooxidation reaction of tetrahydroisoquinoline (THIQ)-tethered aldehydes 85 was described by Ye and coworkers (Scheme 9).24 In this intramolecular cross dehydrogenative coupling process, molecular oxygen was chosen as the optimal oxidant and a catalytic amount of NaI was necessary to improve the efficiency. Substrates with different substituents on the benzaldehyde and THIQ skeletons were compatible with this protocol and furnished various oxidative cyclization products 86 in moderate to excellent yields. This transformation could be scaled up without apparent loss of yield. Control experiments suggested a radical process and the fluorescence spectra showed that an excited Breslow intermediate was generated during the photooxidation process.
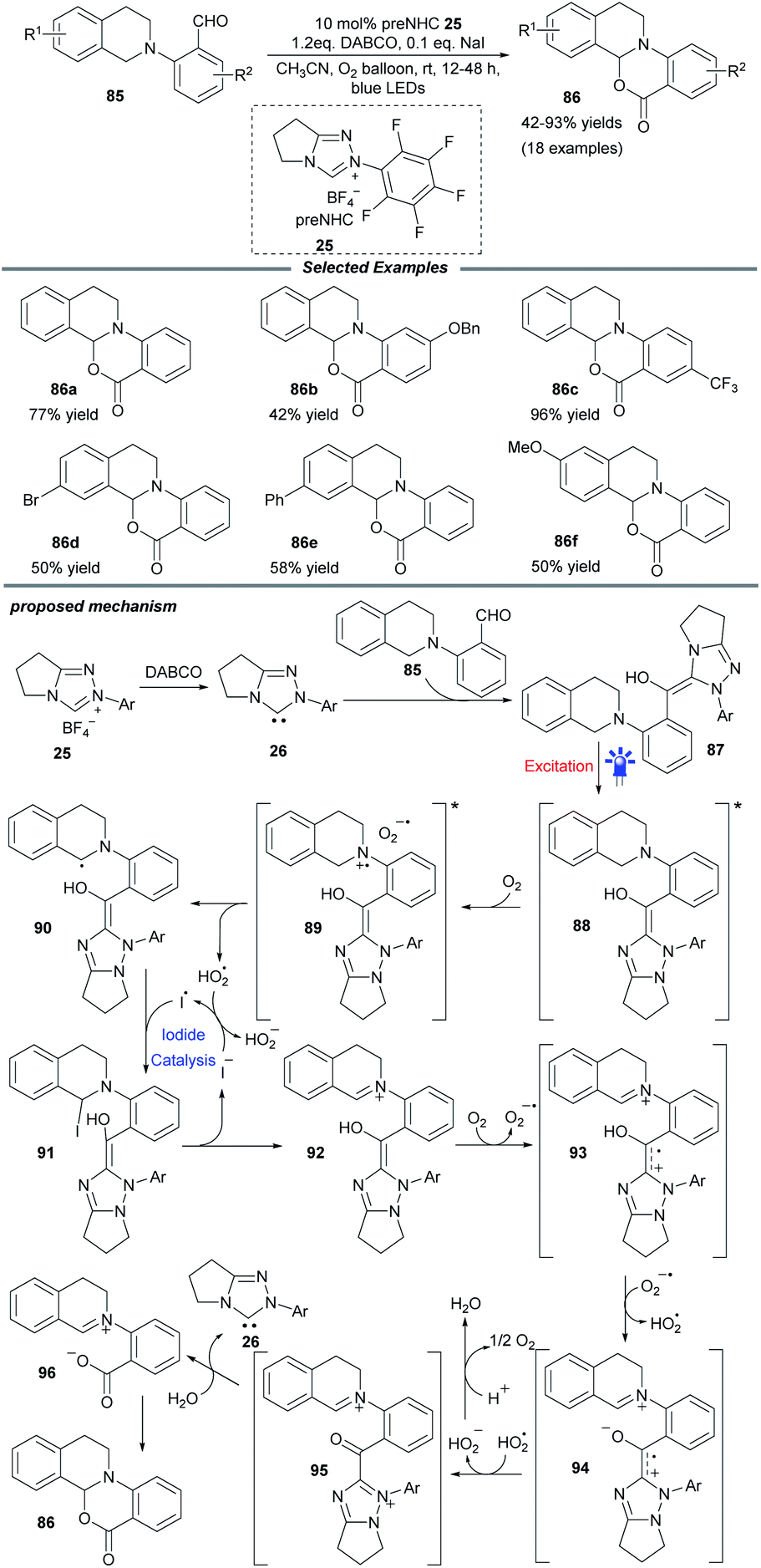 | ||
| Scheme 9 NHC-catalyzed intramolecular cross dehydrogenative coupling. | ||
Mechanistically, the interaction of NHC 26 with aldehyde 85 generates the Breslow intermediate 87, which can be promoted to excited state 88 under blue light. The following SET process between 88 and molecular oxygen furnishes radical pair species 89. Subsequently, α-amino radical 90 is formed by sequential 1,2-H shift and proton abstraction of 89. Under iodide catalysis, this α-amino radical can be further transformed into iminium species 92via α-iodoamine intermediate 91. Then the oxidation of iminium by oxygen delivers radical cation 93, which undergoes deprotonation to give zwitterionic radical 94. After further oxidation by the hydroperoxide radical, radical 94 is converted into acyl azolium intermediate 95, which undergoes consecutive hydrolysis and deprotonation to reform NHC catalyst 26 and afford the iminium/carboxylate species 96. A final intramolecular nucleophilic addition in 96 yields the cyclization product 86.
The visible-light-driven Wolff rearrangement is an effective method for the conversion of readily available α-diazoketones into reactive ketenes.25 Very recently, Hui and coworkers combined this photolytic process with NHC catalysis to realize an asymmetric [4 + 2] cycloaddition, affording a variety of chiral tetrahydropyrano[2,3-b]indoles in 57–96% yields with high diastereo- and enantioselectivity (Scheme 10).26 The catalytic cycle begins with the photoactivation of α-diazoketones, which can promote the Wolff rearrangement to generate ketene 101. This reactive intermediate is then attacked by NHC catalyst 102 to form enolate species 103. The following stereoselective [4 + 2] cycloaddition of 103 with 3-alkylenyloxindole gives the ion pair 104, which delivers the final cyclization product with elimination of the active NHC catalyst 102. Notably, a similar photochemical transformation was also realized by Song and coworkers with the use of a chiral isothiourea catalyst.25c
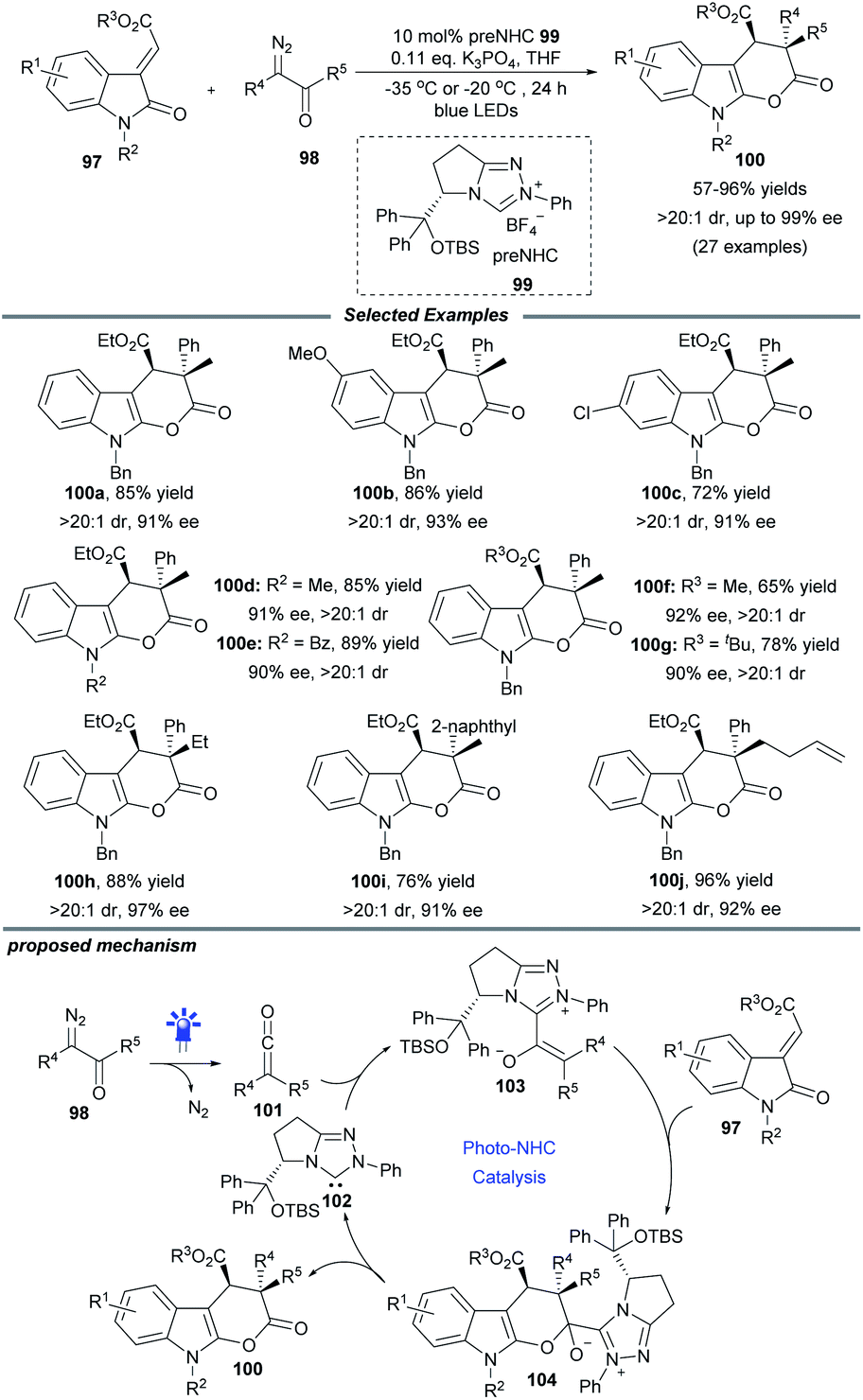 | ||
| Scheme 10 NHC-catalyzed stereoselective [4 + 2] cycloaddition. | ||
4. Conclusion and perspectives
In summary, a variety of successful transformations have undoubtedly demonstrated that the combination of visible-light photocatalysis and photoactivation with NHC catalysis provides a unique and efficient strategy for accessing valuable structural motifs from simple starting materials. This cooperative strategy can usually provide unique reaction pathways compared with classic two-electron NHC catalysis, thus expanding the application of NHC catalysis in modern organic synthesis. Moreover, high reaction selectivity, substrate diversity and functional group tolerance are enabled because of the mild reaction conditions.Despite this impressive progress, the development of photochemical NHC catalysis is still in its infancy. There is still some significant potential to be exploited. First, there are only two reports regarding asymmetric catalysis systems. Broadly applicable, enantioselective transformations with high levels of regio- and enantio-control are awaited. Second, the transformations discussed above are mainly focused on the construction of carbon–carbon and carbon–oxygen bonds, and other reactions to forge carbon–heteroatom bonds (C–N, C–P, C–S, C–Si and so on) are still unexplored. Third, in-depth experimental and theoretical studies are highly desirable to help chemists design and discover novel chemical transformations, as well as new catalysts or catalyst systems. Ultimately, we deeply believe that the collaborative photo/NHC catalysis strategy will evolve into a practical synthetic tool in the construction of natural products and pharmaceuticals. We hope that this review will inspire more research efforts and further investigations in this intriguing and emerging area.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Science Foundation of China (No. 21822103, 21820102003, 21772052, 21772053 and 91956201), the Program of Introducing Talents of Discipline to Universities of China (111 Program, B17019), the Natural Science Foundation of Hubei Province (2017AHB047) and the International Joint Research Center for Intelligent Biosensing Technology and Health for support of this research.Notes and references
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- J. Chen and Y. Huang, N-Heterocyclic Carbenes as Brønsted Base Catalysts, Wiley-VCH, 2018, ch. 9, pp. 261–286 Search PubMed.
- For selected reviews, see: (a) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC; (b) X.-Y. Chen and S. Ye, Org. Biomol. Chem., 2013, 11, 7991–7998 RSC; (c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (d) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed; (e) M.-H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS PubMed; (f) X.-Y. Chen, Z.-H. Gao and S. Ye, Acc. Chem. Res., 2020, 53, 690–702 CrossRef CAS PubMed; (g) Q. Zhao, G.-R. Meng, M. Szostak and S. P. Nolan, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed.
- For a recent review, see: T. Ishii, K. Nagao and H. Ohmiya, Chem. Sci., 2020, 11, 5630–5636 RSC.
- (a) I. Nakanishi, S. Itoh, T. Suenobu, H. Inoue and S. Fukuzumi, Chem. Lett., 1997, 26, 707–708 CrossRef; (b) I. Nakanishi, S. Itoh, T. Suenobu and S. Fukuzumi, Chem. Commun., 1997, 1927–1928 RSC; (c) I. Nakanishi, S. Itoh, T. Suenobu and S. Fukuzumi, Angew. Chem., Int. Ed., 1998, 37, 992–994 CrossRef CAS; (d) I. Nakanishi, S. Itoh and S. Fukuzumi, Chem.–Eur. J., 1999, 5, 2810–2818 CrossRef CAS.
- J. Guin, S. D. Sarkar, S. Grimme and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 8727–8730 CrossRef CAS PubMed.
- (a) N. A. White and T. Rovis, J. Am. Chem. Soc., 2014, 136, 14674–14677 CrossRef CAS PubMed; (b) N. A. White and T. Rovis, J. Am. Chem. Soc., 2015, 137, 10112–10115 CrossRef CAS PubMed.
- (a) Y. Du, Y.-H. Wang, X. Li, Y.-L. Shao, G.-H. Li, R. D. Webster and Y. R. Chi, Org. Lett., 2014, 16, 5678–5681 CrossRef CAS PubMed; (b) Y.-X. Zhang, Y. Du, Z.-J. Huang, J.-F. Xu, X.-X. Wu, Y.-H. Wang, M. Wang, S. Yang, R. D. Webster and Y. R. Chi, J. Am. Chem. Soc., 2015, 137, 2416–2419 CrossRef CAS PubMed; (c) B.-S. Li, Y.-H. Wang, R. S. J. Proctor, Y.-X. Zhang, R. D. Webster, S. Yang, B.-A. Song and Y. R. Chi, Nat. Commun., 2016, 7, 12933–12940 CrossRef PubMed; (d) X.-X. Wu, Y.-X. Zhang, Y.-H. Wang, J. Ke, M. Jeret, R. N. Reddi, B.-A. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2017, 56, 2942–2946 CrossRef CAS PubMed.
- X.-Y. Chen, K.-Q. Chen, D.-Q. Sun and S. Ye, Chem. Sci., 2017, 8, 1936–1941 RSC.
- (a) T. Ishii, Y. Kakeno, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 3854–3858 CrossRef CAS PubMed; (b) T. Ishii, K. Ota, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 14073–14077 CrossRef CAS PubMed; (c) K. Ota, K. Nagao and H. Ohmiya, Org. Lett., 2020, 22, 3922–3925 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5362 CrossRef CAS PubMed; (d) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (e) M. Peña-López, A. Rosas-Hernández and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 5006–5008 CrossRef PubMed; (f) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (g) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed; (h) Y. Wei, Q.-Q. Zhou, F. Tan, L.-Q. Lu and W.-J. Xiao, Synthesis, 2019, 51, 3021–3054 CrossRef CAS.
- A. Galván, F. J. Fañanás and F. Rodríguez, Eur. J. Inorg. Chem., 2016, 1306–1313 CrossRef.
- For selected reviews, see: (a) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874–3886 CrossRef CAS PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (c) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052–0070 CrossRef CAS.
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094–8097 CrossRef CAS PubMed.
- E. Yoshioka, M. Inoue, Y. Nagoshi, A. Kobayashi, R. Mizobuchi, A. Kawashima, S. Kohtani and H. Miyabe, J. Org. Chem., 2018, 83, 8962–8970 CrossRef CAS PubMed.
- Z.-H. Xia, L. Dai, Z.-H. Gao and S. Ye, Chem. Commun., 2020, 56, 1525–1528 RSC.
- (a) A. Arefolov and J. S. Panek, J. Am. Chem. Soc., 2005, 127, 5596–5603 CrossRef CAS PubMed; (b) P. Cheruku, J. L. Keffer, C. Dogo-Isonagie and C. A. Bewley, Bioorg. Med. Chem. Lett., 2010, 20, 4108–4111 CrossRef CAS PubMed; (c) W. Schmidt, T. M. Schulze, G. Brasse, E. Nagrodzka, M. Maczka, J. Zettel, P. G. Jones, J. Grunenberg, M. Hilker, U. Trauer-Kizilelma, U. Braun and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 7698–7702 CrossRef CAS PubMed.
- W. Yang, W.-M. Hu, X.-Q. Dong, X. Li and J.-W. Sun, Angew. Chem., Int. Ed., 2016, 55, 15783–15786 CrossRef CAS PubMed.
- L. Dai, Z.-H. Xia, Y.-Y. Gao, Z.-H. Gao and S. Ye, Angew. Chem., Int. Ed., 2019, 58, 18124–18130 CrossRef CAS PubMed.
- L. Dai and S. Ye, Org. Lett., 2020, 22, 986–990 CrossRef CAS PubMed.
- A. V. Davies, K. P. Fitzpatrick, R. C. Betori and K. A. Scheidt, Angew. Chem., Int. Ed., 2020, 59, 9143–9148 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed; (b) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (c) J. A. Dantas, J. T. M. Correia, M. W. Paixão and A. G. Corrêa, ChemPhotoChem, 2019, 3, 506–520 CrossRef CAS.
- A. Mavroskoufis, K. Rajes, P. Golz, A. Agrawal, V. Ruß, J. P. Götze and M. N. Hopkinson, Angew. Chem., Int. Ed., 2020, 59, 3190–3194 CrossRef CAS PubMed.
- Z.-H. Gao, Z.-H. Xia, L. Dai and S. Ye, Adv. Synth. Catal., 2020, 362, 1819–1824 CrossRef CAS.
- For selected examples, see: (a) M.-M. Li, Y. Wei, J. Liu, H.-W. Chen, L.-Q. Lu and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 14707–14713 CrossRef CAS PubMed; (b) Y. Wei, S. Liu, M.-M. Li, Y. Li, Y. Lan, L.-Q. Lu and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 133–137 CrossRef CAS PubMed; (c) T. Fan, Z.-J. Zhang, Y.-C. Zhang and J. Song, Org. Lett., 2019, 21, 7897–7901 CrossRef CAS PubMed; (d) J. Meng, W.-W. Ding and Z.-Y. Han, Org. Lett., 2019, 21, 9801–9805 CrossRef CAS PubMed; (e) Q.-L. Zhang, Q. Xiong, M.-M. Li, W. Xiong, B. Shi, Y. Lan, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 14096–14100 CrossRef CAS PubMed; (f) M.-M. Li, Q. Xiong, B.-L. Qu, Y.-Q. Xiao, Y. Lan, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006366.
- C.-Y. Wang, Z.-Y. Wang, J. Yang, S.-H. Shi and X.-P. Hui, Org. Lett., 2020, 22, 4440–4443 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |