
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe endeavor of vibration-induced emission (VIE) for dynamic emissions
Zhiyun
Zhang
 ,
Guangchen
Sun
,
Wei
Chen
,
Jianhua
Su
and
He
Tian
*
,
Guangchen
Sun
,
Wei
Chen
,
Jianhua
Su
and
He
Tian
*
Key Laboratory for Advanced Materials, Feringa Nobel Prize Scientist Joint Research Center, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science & Technology, 130 Meilong Road, Shanghai, 200237, China. E-mail: tianhe@ecust.edu.cn
First published on 7th May 2020
Abstract
Organic chromophores with large Stokes shifts and dual emissions are fascinating because of their fundamental and applied interest. Vibration-induced emission (VIE) refers to a tunable multiple fluorescence exhibited by saddle-shaped N,N′-disubstituted-dihydribenzo[a,c]phenazines (DHPs), which involves photo-induced configuration vibrations from bent to planar form along the N–N axis. VIE-active molecules show intrinsic long-wavelength emissions in the unconstrained state (planar state) but bright short-wavelength emissions in the constrained state (bent state). The emission response for VIE-active luminogens is highly sensitive to steric hindrance encountered during the planarization process such that a tiny structural variation can induce an evident change in fluorescence. This can often be achieved by tuning the intensity ratio of short- and long-wavelength bands. In some special cases, the alterations in the emission wavelength of VIE fluorophores can be achieved step by step by harnessing the degree of bending angle motion in the excited state. In this perspective, we summarize the latest progress in the field of VIE research. New bent heterocyclic structures, as novel types of VIE molecules, are being developed, and the general features of the chemical structures are also being proposed. Technologically, novel emission color-tuning approaches and VIE-based probes for visualizing biological activity are presented to demonstrate how the dynamic VIE effect can be exploited for cutting-edge applications.
1. Introduction
Shedding light on the fluorescent mechanisms of chromophores with large Stokes-shifted and dual emissions is of great significance, as it could provide reliable guidance for the development of high-performance multi-colour emission materials.1–10 As shown in the Jablonski diagram in Fig. 1a, the dual bands of an individual fluorophore can generally be divided into small (normal) and large (abnormal) Stokes-shifted emissions.11–13 The overlap of the normal short-wavelength emission with the lowest-lying absorption band originates from the initial emissive state near the Franck–Condon excited state, while the red-shifted emission far from the absorption band arises from the structurally transformed emissive state formed by additional excited-state transformations, such as enhanced charge transfer,14–19 energy transfer,20–25 complex formation,26–31 and structural and/or conformational changes.32–55 Due to the significant configurational and/or structural changes in the excited state, the ratio of short- and long-wavelength bands can be manipulated in a controllable and dynamic manner, activating unprecedented multi-colour luminescence.9,56–61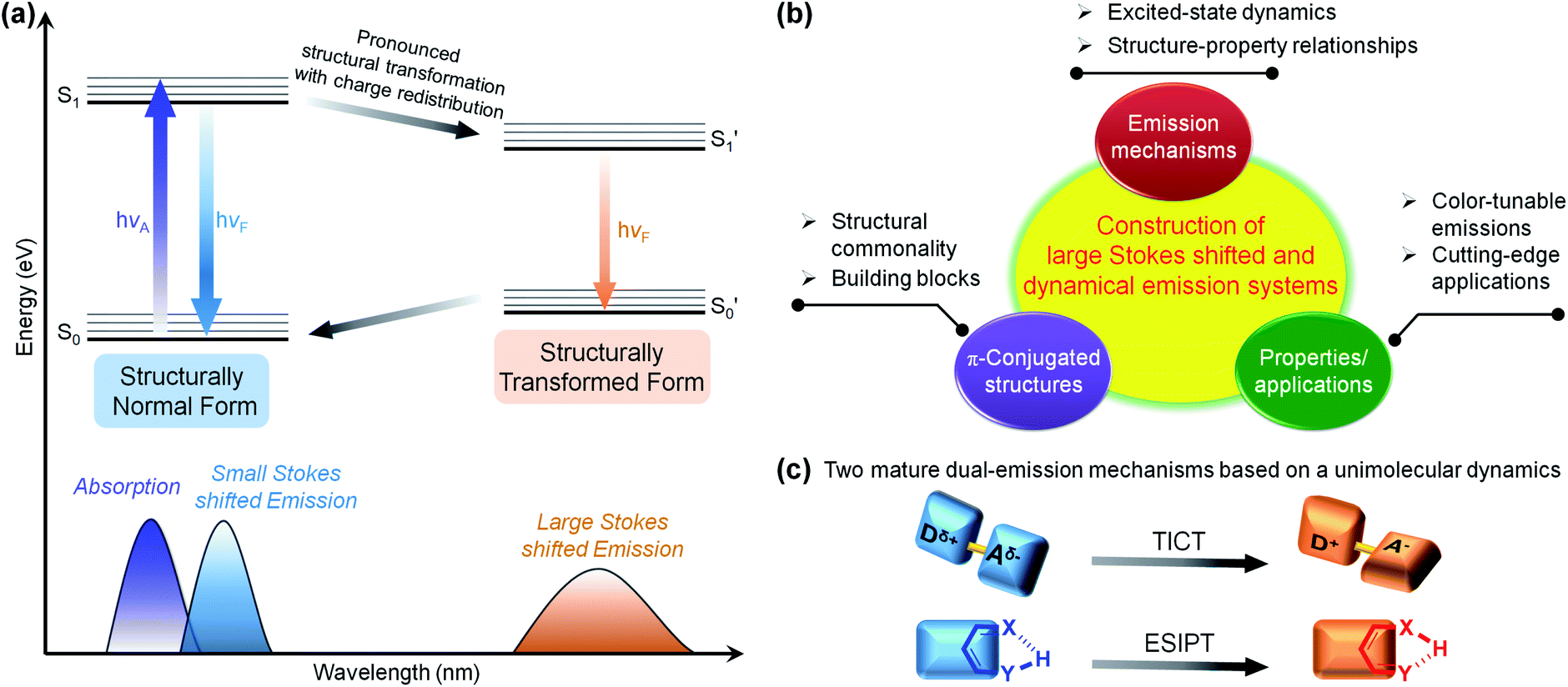 | ||
| Fig. 1 (a) Jablonski diagram for the normal and transformed two-state model resulting in a large Stokes shift and dual fluorescence. (b) Route for the construction of a specific emission system and concept. (c) Illustrative schemes of two mature dual-emission mechanisms based on unimolecular dynamics: TICT and ESIPT. | ||
Regarding the specific π-conjugated structures and luminescent mechanisms, scientists have made notable achievements with intriguing systems and concepts, such as the excimer/exciplex,26–28,30,31 fluorescence resonance energy transfer (FRET),20–25 twisted intramolecular charge transfer (TICT),48–55 and excited-state intramolecular proton transfer (ESIPT).36–42 Most of the mechanisms originate from the accidental discovery of a specific fluorescence phenomenon,62–73 after which the excited-state dynamics and the structure–property relationships were extensively studied and well established. These discoveries have been regarded as guidelines for exploring a range of applications. A self-contained luminescence system/concept can be constructed (Fig. 1b)50,65 that includes: (1) basic building blocks with common characteristics, (2) unique excited-state processes, and (3) amazing properties and functions that have not been achieved by other luminescence systems. A well-known example of a dual-emission system based on a unimolecular reaction is the TICT concept,50 for which Grabowski and co-workers formulated a structural hypothesis based on the molecule N,N-dimethylaminobenzonitrile (DMABN) in 1973.74 Donor and acceptor moieties linked by single bonds build typical TICT molecules. Upon photoexcitation, the TICT molecules are involved in rapid electron transfer from the donor to acceptor units, accompanied by mutually perpendicular donor and acceptor round the single bond. The TICT process is highly dependent on the polarity and viscosity of the solvent, so TICT fluorophores hold great promise in polarity, viscosity, and temperature sensing.54 Another well-known and important unimolecular type is the ESIPT process,75,76 and salicylic acid was the first reported ESIPT dye by Weller in 1955.77 Typical ESIPT molecules possess an intramolecular hydrogen bond (H-bond) between an O–H or N–H proton donor and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or N– proton acceptor. Upon photoexcitation, the ESIPT process involves fast proton transfer from the proton donor to the acceptor. Generally, the ESIPT process can be affected by polar and H-bond donating solvents or other molecules, so the ESIPT fluorophores are wonderful options for fluorescence sensors and imaging agents.40
O or N– proton acceptor. Upon photoexcitation, the ESIPT process involves fast proton transfer from the proton donor to the acceptor. Generally, the ESIPT process can be affected by polar and H-bond donating solvents or other molecules, so the ESIPT fluorophores are wonderful options for fluorescence sensors and imaging agents.40
Adding a new chapter to the subject of large Stokes shifts and dual fluorescence, we demonstrated a novel type of structure–photophysics relationship for the first time by examination of a set of N,N′-disubstituted-dihydrophenazines (DHPs).78,79 The preliminary structure N,N′-diphenyl-dihydrodibenzo[a,c]phenazine (DPAC) and the proposed structural evolution upon photoexcitation are shown in Fig. 2a and b, respectively. A benzene ring and a phenanthrene ring are fused by a flexible cyclic 8π-electron ring dihydropyrazine as the junction, resulting in a V-shaped polycyclic skeleton in DPAC, i.e., dihydrodibenzo[a,c]phenazine. Furthermore, the considerable steric hindrance, between the phenanthrene moiety and N,N′-diphenyl groups, results in a dramatically puckered dihydrodibenzo[a,c] phenazine in DPAC. As a result, the two nitrogen lone pair orbitals may partially participate in the π-conjugation in the bent ground state. Therefore, the π-conjugation, aromaticity (Baird's rule80–82) and steric hindrance effects contribute to enhancing the geometry-bending or geometry-flattening for the ground and excited states, respectively. Upon photoexcitation, DPAC molecules encounter steric hindrance, producing an initially bent form (Fig. 2b), which subsequently overcomes the internal energy barrier and relaxes to the final planar form with fully extended π-conjugation.79 According to Baird's rule,80–82 the bent-to-planar vibration of DPAC is driven by the excited-state aromaticity reversal in the lowest excited state (S1). We termed this unconventional abnormal large Stokes shifted emission of DPAC vibration-induced emission (VIE),78,83,84 in which the photoinduced bent-to-planar transformation involves out-of-plane bending of a V-shaped polycyclic conjugated structure.
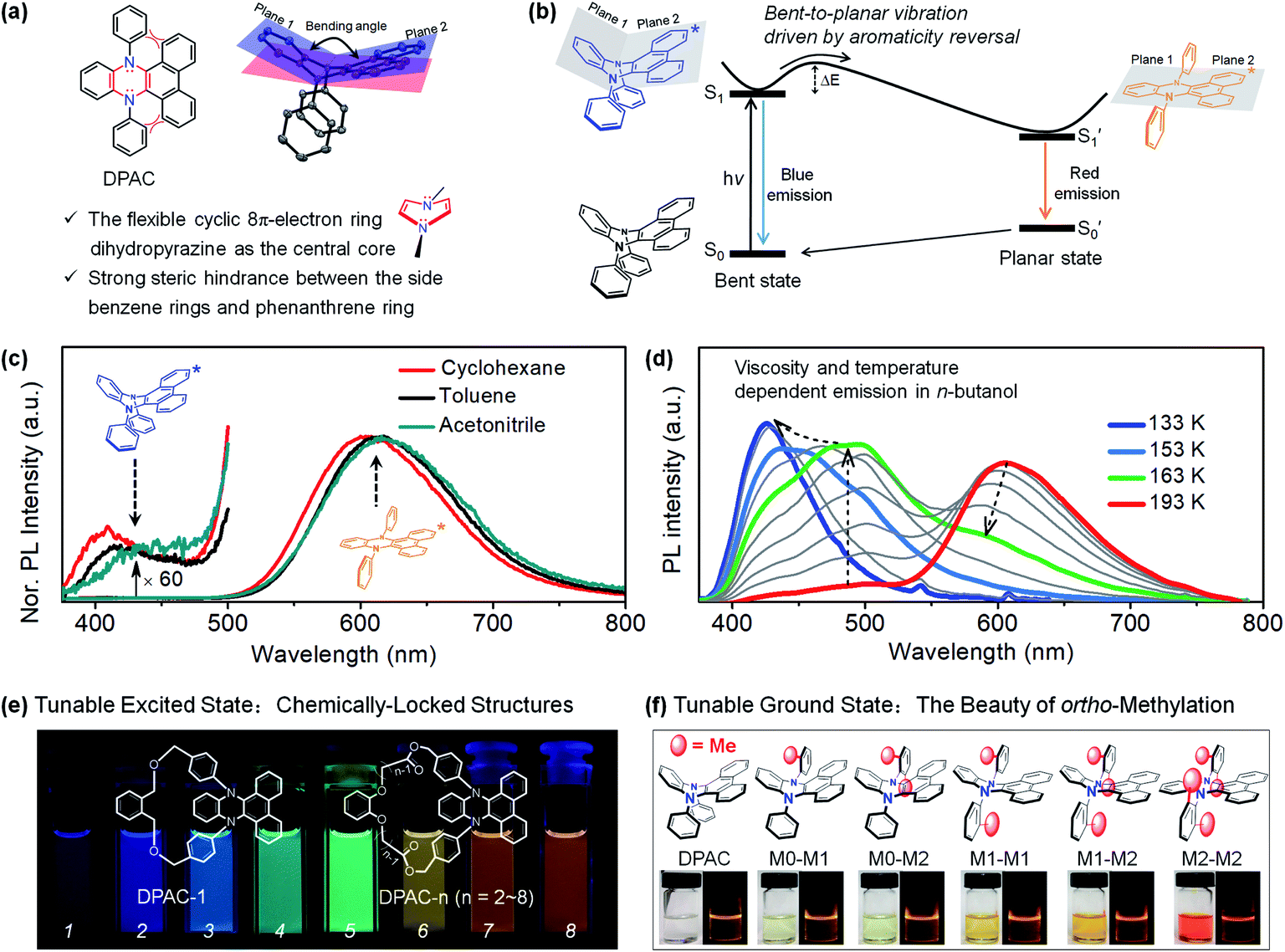 | ||
| Fig. 2 (a) Chemical and crystal structure of N,N′-diphenyl-dihydrodibenzo[a,c]phenazines (DPAC), together with their structural characteristics. (b) Diagrammatic sketch illustrating the vibration-induced emission (VIE) involving a photo-induced configuration vibration from bent to planar form along the flexible N–N axis, driven by excited-state aromaticity reversal. (c) Steady-state emission spectra at room temperature in solvents with different polarities. (d) Temperature/viscosity dependent emission spectra of DPAC in n-butanol from 233 K to 133 K. (e) Chemically-locked structures with tunable excited states. (f) ortho-Methyl effect induced tunable ground states. Reproduced from ref. 79 (c and d) and ref. 95 (e) with permission from the American Chemical Society, Copyright 2015 and 2017. Reproduced from ref. 97 (f) with permission from the Wiley-VCH, Copyright 2018. | ||
The VIE mechanism has been utilized as a persuasive rationalization for large Stokes shifts and full-colour-tunable fluorescence of DPAC and its analogues under various conditions.84 In regular solutions (Fig. 2c), an excited molecule of DPAC transforms the configuration from a bent state to a planar state in S1, releasing a very weak blue emission (bent state) and prominent orange-red fluorescence (planar state). However, the photoinduced bent-to-planar transformation can be somewhat hampered in highly viscous media, and the bright blue fluorescence is emitted from the retained saddle-shaped conformation. Therefore, a vivid fluorescence colour-tuning process of VIE-active molecules, from orange-red to blue via white, could be realized by regulating the local properties (Fig. 2d). Along these lines, the very distinctive feature of VIE-active molecules is that a single fluorophore can exhibit a very large Stokes shift and a dynamic change in full-colour emission in the visible region. As an emerging class of opto-functional materials, VIE-active molecules have been utilized in thermometers,85,86 viscometers,87 chemosensors,88–90 biosensors87,91,92 and white light emitters.93,94 To date, cogent spectroscopic and theoretical proofs of the excited state processes of VIE molecules have been presented and the structure–photophysics relationship has been firmly established.84 To systematically reveal the correlation between the conformational and electronic variables of the VIE process, the covalent approach has been utilized to fine-tune the excited-state planarization of DPAC (Fig. 2e),95,96 producing highly colour-tunable fluorescent images. Furthermore, to mimic the excited-state structural evolution, the strategy of using ortho-methyl effects has been applied to regulate the ground-state geometry of DPAC from a bent to a planar configuration step by step (Fig. 2f).97
This accidental discovery is turning into a sustainable achievement. In this perspective, we aim to highlight the noticeable progress in VIE-active luminogens over the past year, including (1) novel bending heterocyclic cores for the molecular library of the VIE system, (2) new strategies for dynamic emission colour changes and real-time functions, (3) applications as biological probes, and (4) excimer formation of a VIE-active luminogen in a crystal. We also discuss the present pros and cons of the VIE system and look forward to its further development.
2. Designing new bending heterocyclic cores for the VIE molecular family
DPAC, acting as the most typical DHP core, has been widely utilized in VIE research (Fig. 3a).84 However, such a monotonous structure greatly restricts the development of VIE research both fundamentally and in applications. Therefore, there is an urgent need to exploit new VIE cores. To meet this requirement, replacing the N-phenyl groups by other heteroatoms with lone electron pairs has been the primary consideration, for which the aim has been to retain the central core as a flexible 8π-electron ring (Fig. 3a). Additionally, according to our previous research on DHPs, the steric hindrance between the phenanthrene moiety and N-substituted aryl groups plays a key role in the VIE process (Fig. 3a). Based on this perspective, a series of DHP derivatives were designed and synthesized, in which one of the N-aryl groups was replaced by an O (PNO), S (PNS), or Se (PNSe) atom and dimethyl–methanediyl (PNC) (Fig. 3b).98 As a result, these three compounds exhibited ultraviolet lowest-lying absorption bands located at 330–400 nm and strongly bathochromic shifted emissions at 465, 555, and 570 nm, with corresponding large Stokes shifts of 7906, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480, and 11955 cm−1 (PNO < PNS < PNSe, see Fig. 3c), respectively. The order of the Stokes shifts resulted mainly from the better polarization of the S and Se atoms than that of the O atom, which could extend the π-conjugation of the global minimum state (i.e. planarization state) and thus narrow the energy gap. Notably, there was a negligible emission band at 429 nm in PNSe, which was very similar to that in DPAC, indicating a pronounced VIE characteristic. In contrast, PNC only exhibited a normal emission band at 417 nm with a Stokes shift of 5313 cm−1 since the elongated π-conjugation of PNC was interrupted by the non-conjugated bridge CMe2. The steady-state results combined with further time-resolved and computational data proved the ubiquity of VIE in DHP derivatives and provided a sufficient theoretical basis for the development of new VIE cores.
480, and 11955 cm−1 (PNO < PNS < PNSe, see Fig. 3c), respectively. The order of the Stokes shifts resulted mainly from the better polarization of the S and Se atoms than that of the O atom, which could extend the π-conjugation of the global minimum state (i.e. planarization state) and thus narrow the energy gap. Notably, there was a negligible emission band at 429 nm in PNSe, which was very similar to that in DPAC, indicating a pronounced VIE characteristic. In contrast, PNC only exhibited a normal emission band at 417 nm with a Stokes shift of 5313 cm−1 since the elongated π-conjugation of PNC was interrupted by the non-conjugated bridge CMe2. The steady-state results combined with further time-resolved and computational data proved the ubiquity of VIE in DHP derivatives and provided a sufficient theoretical basis for the development of new VIE cores.
 | ||
| Fig. 3 (a) General characteristics of VIE-active molecules. (b) Chemical structures of PNO, PNS and PNSe as novel kinds of VIE-active molecules and reference compound PNC. (c) Absorption and emission spectra in various solvents at room temperature. Reproduced from ref. 98 with permission from Wiley-VCH, Copyright 2019. | ||
By further exploring the relationship between the photoinduced structural planarization and steric hindrance, phenothiazine derivatives were chosen for further study due to their similar structures to those of DHPs and potential applications in bioimaging. Therefore, we developed a series of new compounds NAS, NAS-1, and NAS-2 (Fig. 4a).99 According to X-ray analysis of the three compounds, the order of the bending angles (Θa) in the phenothiazine moiety was NAS (152.08°) > NAS-1 (147.89°) > NAS-2 (131.12°). Notably, the N-phenyl showed a perpendicular orientation to the phenothiazine core in NAS and NAS-1 but a parallel orientation in NAS-2. These results could safely demonstrate that the bent angle Θa in NAS-2 was derived from the hindrance between the naphthalene ring and N-phenyl. Further proof could also be obtained from the steady-state spectra (Fig. 4b). NAS and NAS-1 showed coincident onset (purple circles), which was consistent with the characteristics of normal emission. However, NAS-2 showed a distinct separated onset (blue and red circles), which was the same as the absorption and emission of DPAC. This separation indicated that NAS-2 possessed different configurations from the initial ground state to the final S1 state. Accordingly, it was concluded that there was indeed a VIE phenomenon, similar to that of DHPs, in NAS-2, which was further demonstrated by the time-resolved data and computational approaches. This work established a clearer relationship between the VIE and steric hindrance and facilitated the design of VIE cores, which greatly extended the application of the VIE mechanism.
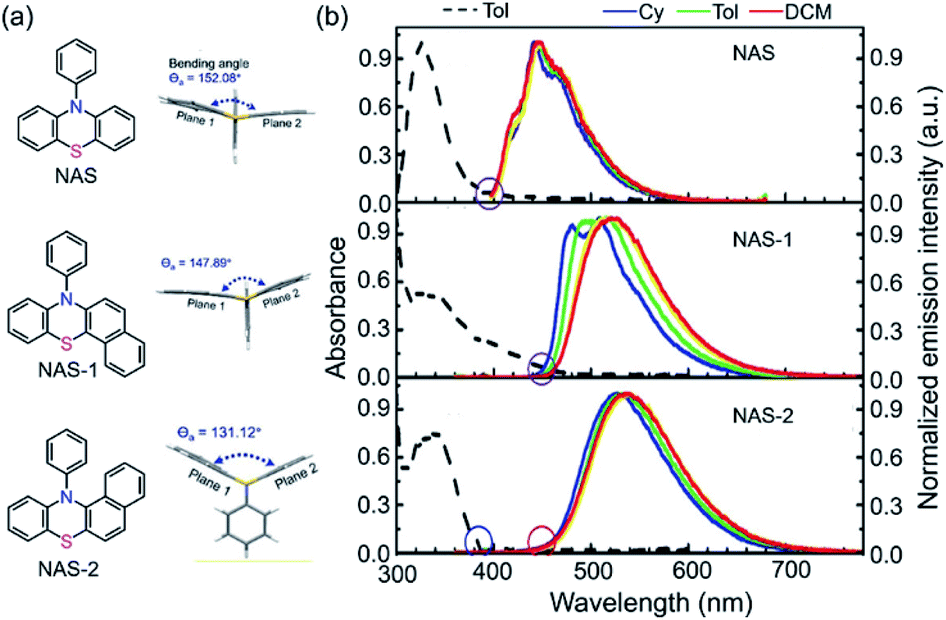 | ||
| Fig. 4 (a) Chemical and single-crystal structures for NAS, NAS-1 and NAS-2. (b) Absorption and photoluminescence spectra in various solvents at room temperature. Reproduced from ref. 99 with permission from the Wiley-VCH, Copyright 2019. | ||
The two studies mentioned above indicated that steric hindrance plays an important role in the dual emission in the VIE cores. However, what is the real driving force behind the photoinduced structural planarization process?82 Such a structural relaxation was reported for the conjugated 8π system dibenz[b,f]oxepin in 1992 (Fig. 3a).33 Its preferential bent configuration in the ground state can be explained by Hückel's rule,100 in which the planar structure possessed unfavourable antiaromatic properties due to the 8π electron array in the middle ring. Upon excitation, however, the equilibrium configuration tended to be planar, driven by excited-state aromaticity reversal, which can be explained by Baird's aromaticity rule.80–82 Such a structural planarization allowed for the global conjugation of the whole molecule and led to a large Stokes shift. Therefore, it can be speculated that the 8π electron array is another important factor in the photoinduced structural planarization process. Recently, Naka et al. reported a class of 8π system dibenzo[b,f]arsperins,101 which also exhibited severely bent boat-like configurations in the ground state (Fig. 5a). Both 3-Me and 3-Ph exhibited intrinsic emission bands (∼370 nm) and longer-wavelength emission broad bands (500–700 nm), with large Stokes shifts of 7249 and 6830 cm−1 (Fig. 3b), respectively. The longer-wavelength emission intensity ratio was solvent dependent. It decreased with increasing solvent polarity due to the lower polarity of the planar structure in the excited state (Fig. 3b and c). Moreover, a high viscosity and PMMA matrix environment could also weaken the longer-wavelength emission intensity ratio, very similar to the behaviour of DHPs, indicating that the structural planarization process was impeded in such environments. Therefore, there should be an activation energy (ΔEa) in the structural planarization process from the Franck–Condon state to the relaxed state, which was estimated to be 3.2 kJ mol−1. According to these results, a typical photoinduced structural planarization process can be observed in such conjugated 8π systems, which provides a clear direction for molecule design with the VIE property.
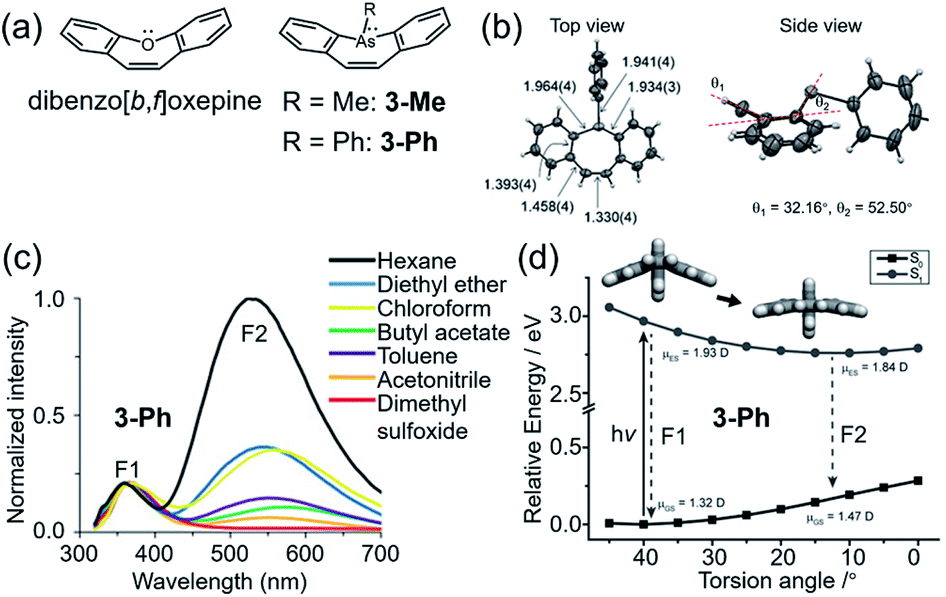 | ||
| Fig. 5 (a) Chemical structures of dibenzo[b,f]oxepine and dibenzo[b,f]arsepin derivatives 3-Me and 3-Ph. (b) Single-crystal structure of 3-Ph. (c) Photoluminescence spectra of 3-Ph in various solvents. (d) Calculated potential energy surface of 3-Ph with the variation in the torsion angle in the S0 and S1 states. Reproduced from ref. 101 with permission from the Wiley-VCH, Copyright 2019. | ||
Combined with our previous work, we can determine the general characteristics of VIE molecules (Fig. 3a). On the one hand, annular conjugated 8π systems provide the internal driving forces behind the bent-to-planar process due to their aromaticity in different states and configurations (i.e., bent/planar configuration in the ground/excited state) based on Hückel's rule100 and Baird's rule.80–82 On the other hand, the steric hindrance between the X(Y)-substituted aryls (X(Y) is the heteroatom in the internal core) and the extended conjugated parts of the core further enlarges the torsion angle and finely tunes the planarization process. By suitable utilization of the steric hindrance in annular conjugated 8π systems, larger Stokes shifts and colour-tunable multiple emission of a single fluorophore can be achieved.
3. Dynamic emission colour change
The most distinctive feature of VIE-active molecules is that they can achieve very large Stokes shifts and dynamic emission changes in color.84 A preferential strategy to achieve dynamic emission colour changes is to modulate the intramolecular vibrations of DHPs and thus control their excited configurations.95,96 The supramolecular approach is an ideal choice and has been widely employed to design smart materials due to its facile molecular design and synthesis.102,103 Moreover, the reversible non-covalent interaction in supramolecular assemblies generates a controllable system, and thus, it might achieve unexpected properties. As a general rule, there are two main supramolecular approaches: (i) directly modulating the excited configuration of the DHPs from a molecular level and (ii) taking advantage of the supramolecular assembly induced aggregation to restrict the vibrations of DHPs.In view of this, we developed a novel fluorescent chemosensor DPAC-bisC4P consisting of DPAC and two appended calix[4]pyrrole moieties (Fig. 6a).104 Unlike conventional sensors, the DPAC-bisC4P chemosensor could not only quantitatively determine specific dicarboxylate dianions (DC2−) but also differentiate certain linear saturated DC2− and three phthalate DC2− (Fig. 6c). In this work, DPAC-bisC4P could form pseudomacrocyclic noncovalent linked host–guest complexes with various DC2− due to calix[4]pyrrole-mediated dianion recognition, thus imposing various degrees of constraint on the planarization of DPAC (Fig. 6b and d). In acetonitrile, an obvious red-shifted emission colour change was evident as the length of the linear saturated DC2− chains increased. Various emission colour changes could be also observed between the three phthalate DC2− due to different structural effects (Fig. 6e and f). According to the distinct chromaticity readouts, these DC2− can be easily differentiated by DPAC-bisC4P, which is vividly compared to a molecular cursor calliper. This work highlights a novel recognition-based approach for dianion sensing and even provides an effective way to achieve the facile sensing of other analytes via a rational smart design.
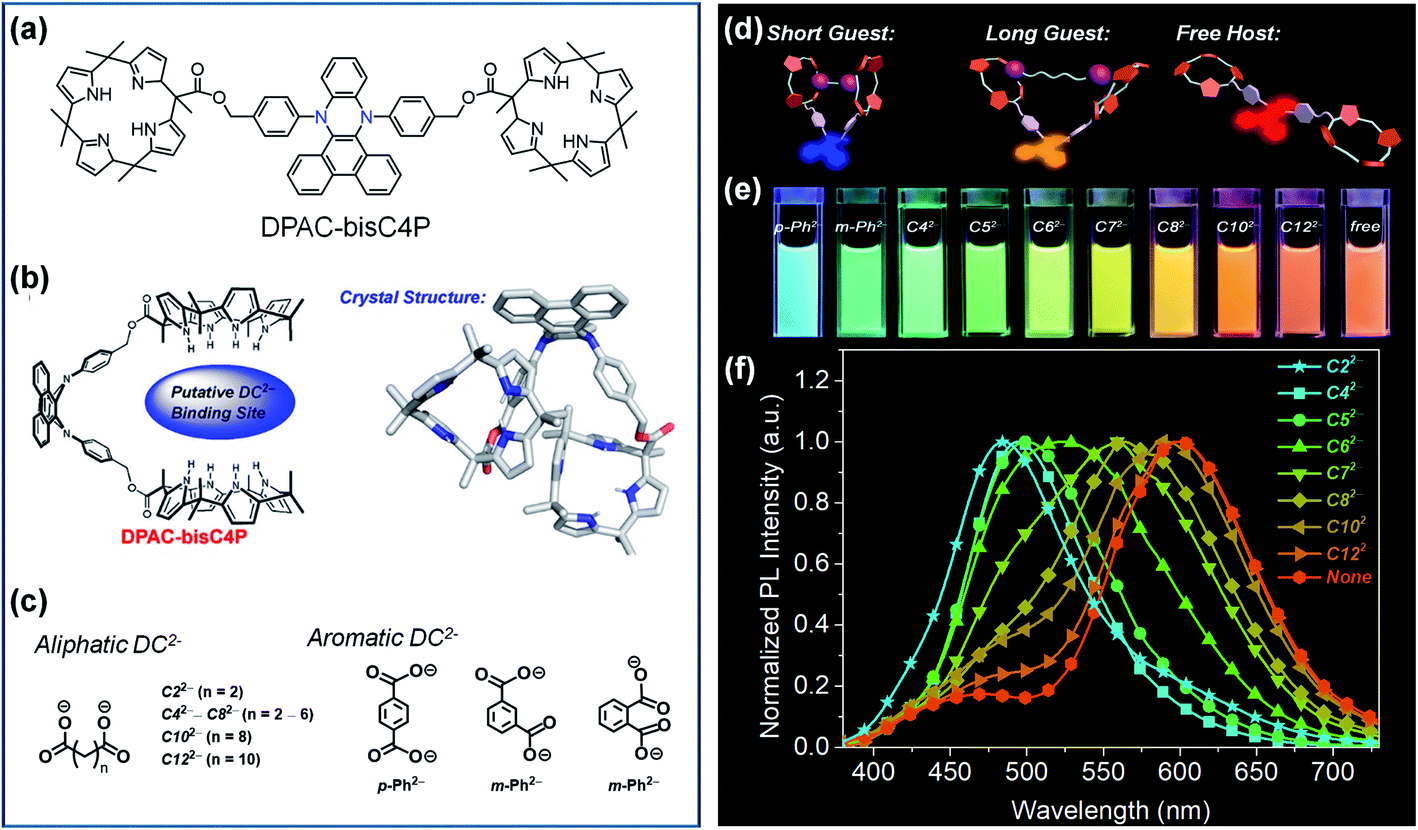 | ||
| Fig. 6 (a) Chemical and (b) crystal structure of DPAC-bisC4P, as well as (c) chemical structures for various dicarboxylate anionic guests (DC2−). (d) Working principle of the fluorescence tuning of DPAC-bisC4P⊃DC2−. (e) Photographs of DPAC-bisC4P⊃DC2− and DPAC-bisC4P in acetonitrile obtained under 365 nm UV light illumination. (f) Normalized photoluminescence (PL) spectra of DPAC-bisC4P in the presence of various linear saturated dicarboxylates at the end of titrations. Reproduced from ref. 104 with permission from the American Chemical Society, Copyright 2019. | ||
To explore the effect of supramolecular-assembly-induced aggregation, two benzo-15-crown-5 parts were appended on the DPAC moiety at the para site of 9,14-phenyl through ester groups to form a potassium ion (K+) responsive molecule DPAC-[B15C5]2 (Fig. 7),105 in which DPAC was the fluorescent signaller and benzo-15-crown-5 was the K+ recognition site. 15-crown-5 could form a 2:1 sandwich-like supramolecular assembly with K+. Meanwhile, the diameter of K+ (2.66 Å) was so much smaller than the distance of the centres of the two 15-crown-5 moieties in DPAC-[B15C5]2 (23.18 Å) that only an intermolecular supramolecular assembly could be formed, thus causing aggregation of the system and constraining the planarization of the DPAC moiety. By fine tuning the degree of aggregation, the emission colour changed from orange-red via white to blue with the gradual titration of K+. Conversely, a reversible process could clearly be seen in which the emission colour recovered from blue via white and orange-red with the titration of 18-crown-6, because 18-crown-6 possessed a higher complexing ability with K+ and thus caused disaggregation of the system. Moreover, water was found to have a similar effect on 18-crown-6. Even trace amounts could generate evident emission colour changes in the system. In the utilization of this supramolecular assembly, the detection limit of water in toluene was calculated to be as low as 0.035 wt‰. This work provided sufficient proof for and a deep understanding of the relationship among supramolecular interactions, aggregation, and the VIE process.
 | ||
| Fig. 7 Schematic representation of the supramolecular assembly and disassembly processes of DPAC-[B15C5]2. Reproduced from ref. 105 with permission from the American Chemical Society, Copyright 2020. | ||
Temperature is another important factor that influences the molecular vibrations, which makes DHPs an ideal candidate for the construction of thermometers.85,86 To be specific, on the one hand, a lower temperature provides less energy for the thermal motion of DHPs, and may keep them in bent excited states even if they are in the form of a single molecular state. On the other hand, a low temperature tends to reduce the solubility of DHPs in solution and cause aggregation, thus restricting their excited state planarization process. However, only ultra-low temperatures (below ∼233 K) can suppress the vibration of DHPs in normal solutions, which severely limits their application in the field of daily temperature detection. Therefore, the dilemma between daily temperature requirements and the vibration restriction of DHPs must be overcome. In this regard, we developed an ultra-sensitive and range-tunable ratiometric thermometer DPC (Fig. 8a).106 Its excited bent-to-planar process could be tuned via temperature-induced aggregation and disaggregation. Based on the different solubilities of DPC in ethanol/glycerol mixed solvents at different temperatures and glycerol fractions, the degree of aggregation could be controlled. An increasing temperature would dissociate the aggregates and enhance the orange-red emission, while a decreasing temperature would re-aggregate the dissociated DPC molecules and enhance the blue emission, thereby achieving a significant ratiometric response to temperature (Fig. 3b). Moreover, the range-tunable property could be realized by simply tuning the fraction of each component of the mixed solvent, since ethanol is a good solvent for DPC while glycerol is a poor one. As a result, it was realized for the first time that this fluorescent ratiometric thermometer possessed an ultra-high sensitivity at ∼2000% °C−1 and a broad overall linear response region from −11.4 °C to 37.7 °C. More interestingly, the fluorescent signal at various temperatures exhibited a good linear relationship on the CIE chromaticity diagram (Fig. 8c and d). In this manner, temperature information could be invaluably read with only a smartphone instead of a professional spectrometer. This work perfectly resolved the conflict between high sensitivity and broad response range and provided a novel perspective for developing thermometry schemes.
 | ||
| Fig. 8 (a) Chemical structure of DPC and an illustration of the VIE process. (b) Mechanism of ratiometric thermometers established by the aggregation and disaggregation of VIE-active molecules. (c) CIE chromaticity diagram, where the symbols are the measured values and the line segments are the calculated analogous values. (d) The fitted curves of the CIE coordinates corresponding to the FL spectra of DPC in an ethanol/glycerol mixture with fg = 40% at different temperatures. Reproduced from ref. 106 with permission from the Royal Society of Chemistry, Copyright 2020. | ||
In addition to using solvents as the media, DHPs incorporated into polyurethanes (PUs) can be applied not only for temperature detection but also for exploring the microphase separation of PUs.107 PUs possess particular properties due to the thermodynamic incompatibility between the soft and hard segments, thus making it possible to tune the excited intramolecular vibrations of the DHPs. An alternative strategy is to introduce DHPs into PUs through physical blending or chemical bonding. Therefore, we used DPAC doped in PUs (PU-4701-1) and its derivatives grafted to PUs in different forms (PU-4701-2, 3, 4). As shown in Fig. 9a, the DPAC moieties were external to the PU-4701-1 chain and subjected to little restriction in the planarization process. Consequently, they exhibited pink emission colours at room temperature. In the case of PU-4701-3, however, DPAC was grafted to PUs via bilateral chemical bonds, which greatly suppressed the excited bent-to-planar process and emitted strong blue fluorescence. As for PU-4701-2 and PU-4701-4, the unilateral chemical bonds between DPAC and PUs imposed weaker restrictions than PU-4701-3, but stronger than PU-4701-1, thus emitting pink-purple fluorescence. Temperature is an important factor in the change in the microphase structures of PUs. For example, PU-4701-2 exhibited a cross-regional emission colour change from blue to orange-red in the region of −30 °C to 90 °C (Fig. 8b). The high consistency between the continuous temperature cycles and the small-angle X-ray scattering experiments provided an alternative scheme for measuring the degree of microphase separation in the PUs as a “fluorescent ruler” (Fig. 8c). This work opens up a new path for polymer investigation through fluorescence techniques.
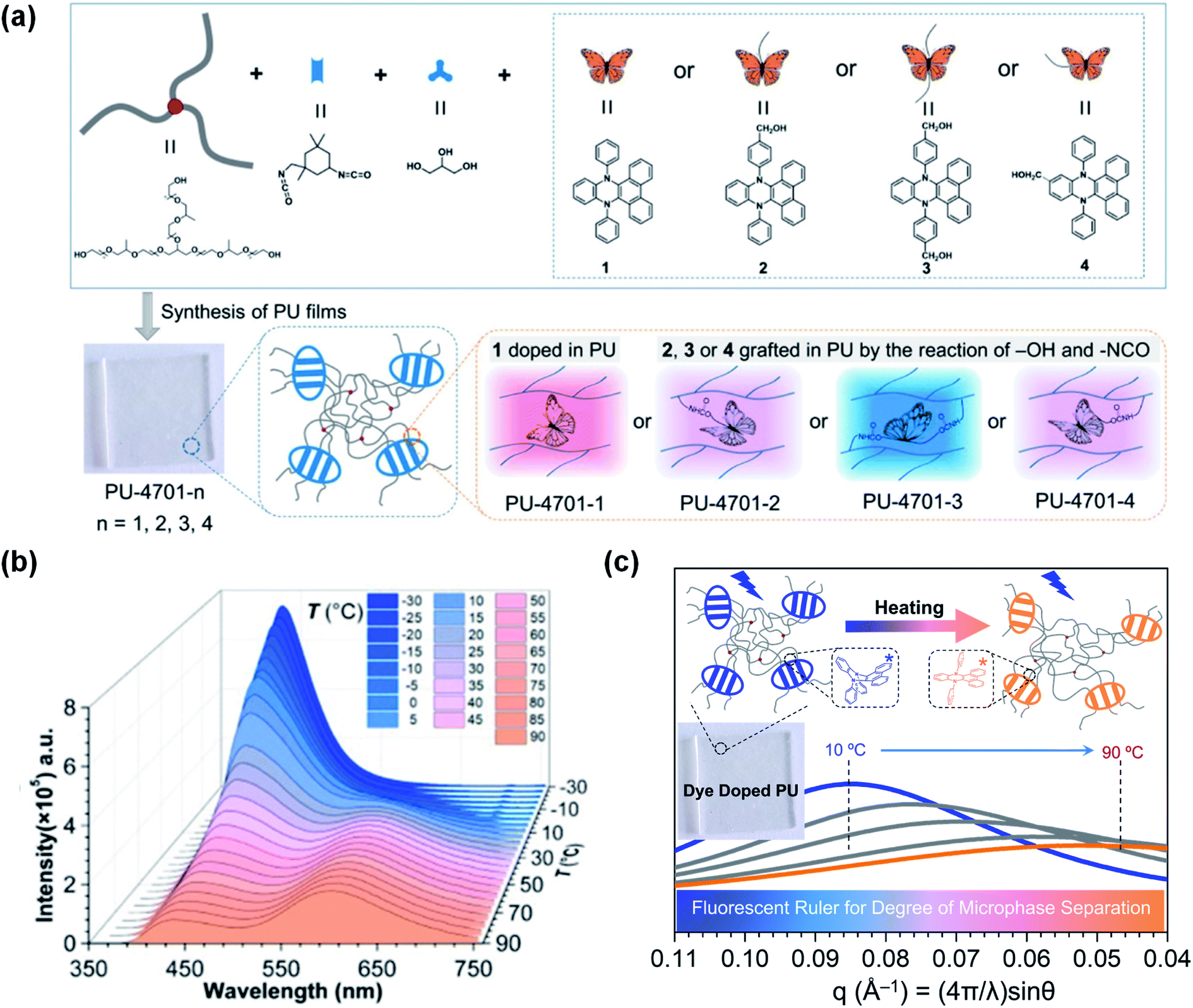 | ||
| Fig. 9 (a) Diagrammatic sketch illustrating the DPAC-doped polyurethanes PU-4701-1–PU-4701-4. (b) PL spectra of PU-4701-2 at various temperatures (−30 to 90 °C). (c) Scattering curves of PU-4701-1 achieved by small-angle X-ray scattering at various temperatures (10–90 °C). Reproduced from ref. 107 with permission from the American Chemical Society, Copyright 2019. | ||
4. Biological probes
VIE-active compounds can act as ideal signal moieties because of their unique dual emission properties, which is beneficial for achieving ratiometric detection.108–111 Hence, developing VIE-based probes for visualizing biological activity is extraordinarily fascinating.87,91,92 The enzymatic fluorescent probe Q1 based on the VIE mechanism was designed and synthesized for detecting and imaging the protein tyrosine phosphatase 1B (PTP1B) in living cells (Fig. 10a).112Q1 was composed of the DPAC moiety and a phosphate radical and dissolved well in solvent, thereby exhibiting a free emissive state. When Q1 reacted with PTP1B, hydroxyl-substituted DPAC was acquired and showed poor solubility, thereby exhibiting a fixed emissive state and enhanced blue emission (Fig. 10b). Unlike the conventional VIE phenomenon, this probe showed almost constant orange-red emissions at ∼600 nm during the enzymatic reaction process with PTP1B, which could act as an “internal standard”. However, the increasing blue emission intensity could be applied for detecting PTP1B as the reaction proceeded. Through this mechanism, Q1 was determined to have a detection limit of 5.7 ng μL−1 for PTP1B, and it showed a rapid response, and good selectivity and stability. Furthermore, Q1 could be used to detect PTP1B in living cells (Fig. 10c–e). Under the conditions of constant emissions in the red channels, Q1 emitted brighter emissions in the green channel in HeLa cells, indicating that HeLa cells contained more PTP1B. This work provided an idea for the construction of VIE-based biological probes.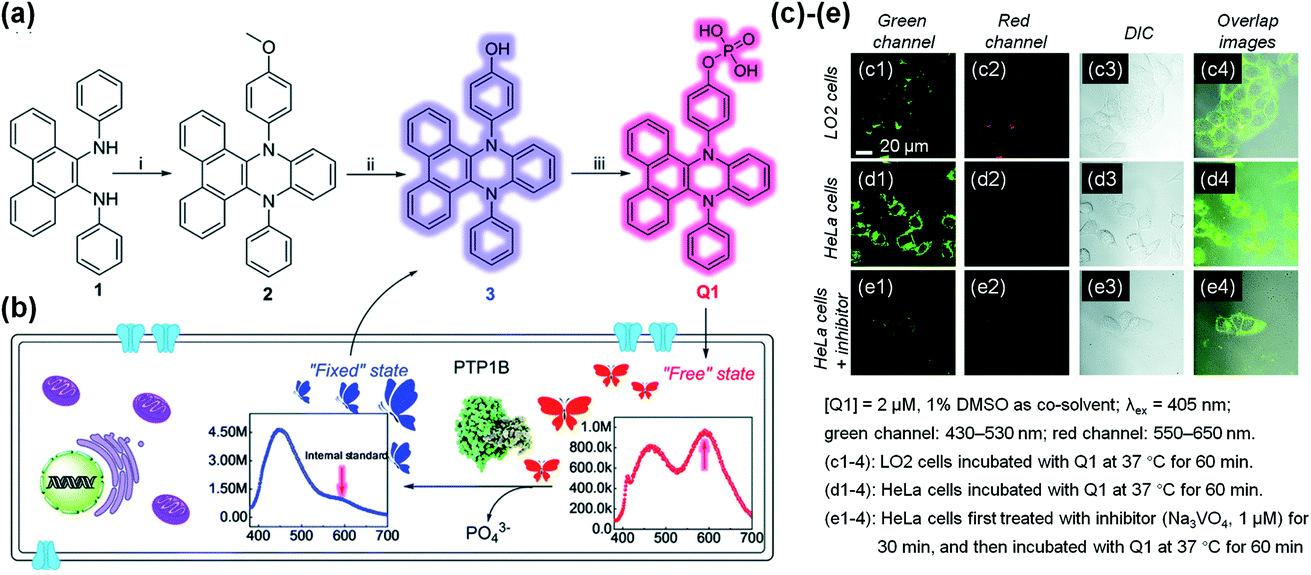 | ||
| Fig. 10 (a) Synthesis of VIE-based sensor Q1. (b) Diagrammatic sketch illustrating the working principle of Q1, showing VIE properties before and after reacting with phosphatase in living cells. (c)–(e) Fluorescence images of LO2 and HeLa cells. Reproduced from ref. 112 with permission of the Royal Society of Chemistry, Copyright 2020. | ||
Hydrophobic DHPs are a series of ideal molecules for developing core–shell structures in the field of biosensors. Based on this viewpoint, He and co-workers developed a sialyllactosyl probe for influenza virus (IV) detection and blocking.113DPAC acted as the core structure and the aggregation-enhanced blue fluorophore due to its poor solubility and restricted configuration in aqueous solutions. DCM-based sialyllactosides were applied to assemble with DPAC to form a sialyllactosyl probe, emitting a red fluorescence via Förster resonance energy transfer (FRET) from DPAC to DCM (Fig. 11a and b). Since the specific binding blocked the FRET process, a strong DPAC channel (blue) could be observed while the DCM channel (red) was nearly absent when the probe was treated with H3N2 or H7N9. However, the non-specific collisions between H10N8 and the probe particles had little influence on the dual emissions, thus exhibiting only a strong DCM channel (Fig. 10d). Apart from the sensing ability, the probe could also suppress the IVs entering human cells by antagonizing the interactions between the viruses' surface-bound hemagglutinin and cell-surface sialylglycans (Fig. 11c). This study provided further understanding of the construction of biological probes through the smart design of DHPs by utilizing their aggregation-enhanced fluorescence property.
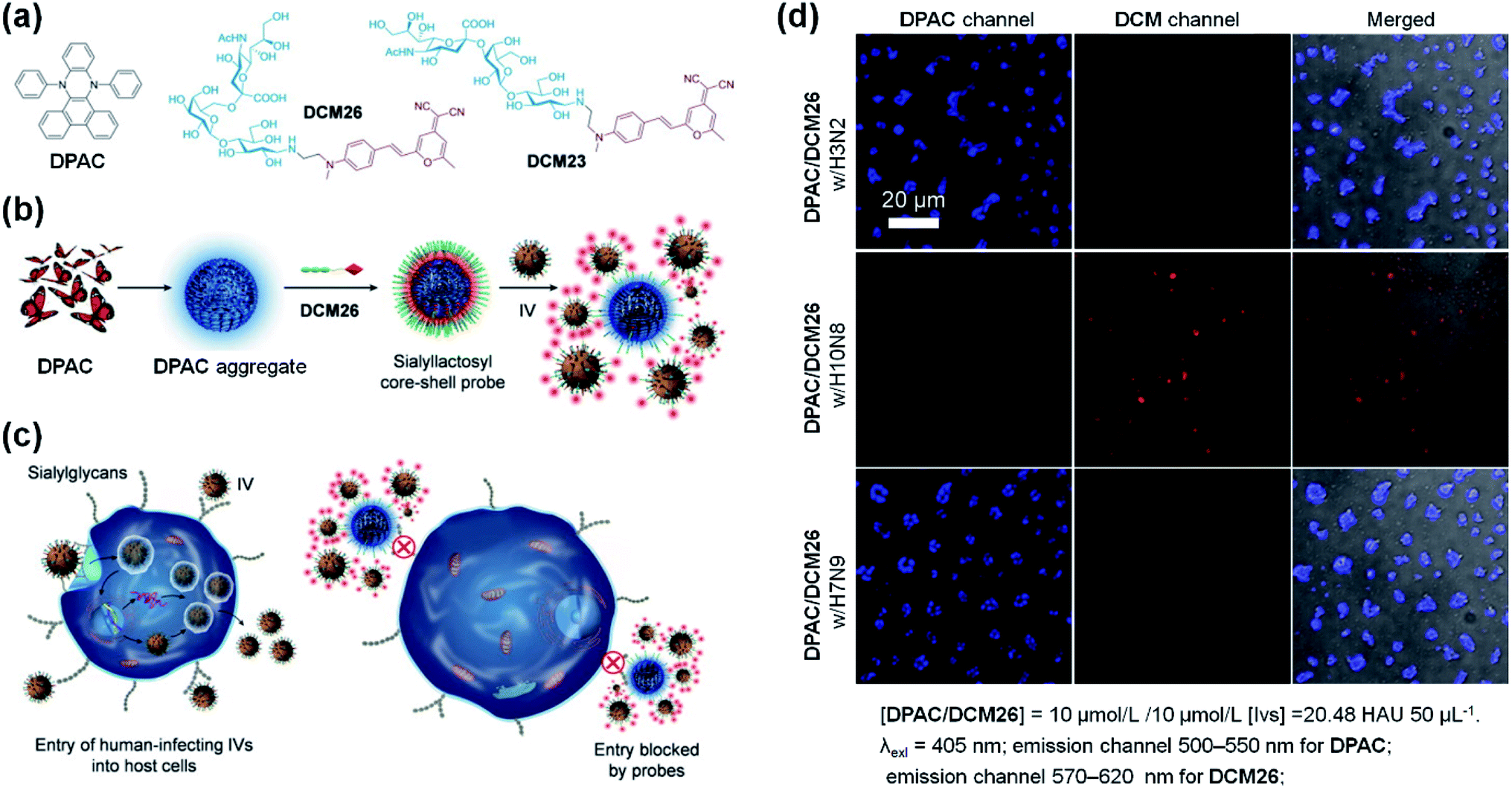 | ||
| Fig. 11 (a) Molecular structures of DPAC, DCM26 and DCM23. (b) Scheme for the stepwise self-assembly to prepare a core–shell sialyllactosyl sensor for the ratiometric fluorescence sensing of influenza viruses. (c) Illustration of the sensor utilized for blocking the entry of human-infecting influenza viruses (IVs) into host cells. (d) Fluorescence images of DPAC/DCM26 sensor in the presence of IV with different HA specificities. Reproduced from ref. 113 with the permission of Elsevier, Copyright 2019. | ||
5. Excimer formation of VIE-active luminogen in a crystal
Excimer emission26–28,30,31 and emission mechanochromism114–117 have been found in the crystalline solids of the VIE-active compound 12-phenyl-12H-benzo[a]phenothiazine (NAS-2, see Fig. 4a and 12).99 Normally, NAS-2 crystals are expected to emit normal Stokes-shifted emissions due to the lattice energy, which could prohibit the planarization process. However, the NAS-2 crystal exhibited dual emissions in the crystal state, located at 390 and 520 nm (Fig. 12a). Compared to the emission of the DPAC crystal located at ∼386 nm, it is believed that the short-wavelength emission originated from the monomer bent configuration of NAS-2. Since the planarization process resulting in the long-wavelength emission was excluded due to the lattice energy, intermolecular interactions can be considered to be the main factor, most likely as an excimer. This speculation was first proven by molecular packing in the crystal (Fig. 12b and c). The different bending angles of NAS-2 and NAS-1 lead to different orientations of N-phenyl due to the steric hindrance. As a result, NAS-2 is more suitable for forming proximal π–π interactions for the naphthalene unit, while NAS-1 avoids any possible π–π stacking. By investigating the emission behaviour of the NAS-2 crystal based on the depth from its surface, there was mainly a 390 nm emission (purple-blue) at the surface, and dual emission (pale blue) at the kernel of the crystal (Fig. 12a and d). Hence, it was concluded that excimer formation was preferred as the depth increased, while π–π stacking ruptured at the surface due to the dangling molecules. Furthermore, the existence of the excimer was also proven by the transient photoluminescence (Fig. 12e). The decay component of 20.8 ps monitored at the normal emission of 400 nm was well matched with the rising component of 21.3 ps monitored at the excimer emission of 500 nm. The discovery of the excimer in NAS-2 is significant, opening up an avenue for the study of the π–π interactions of VIE-active luminogens.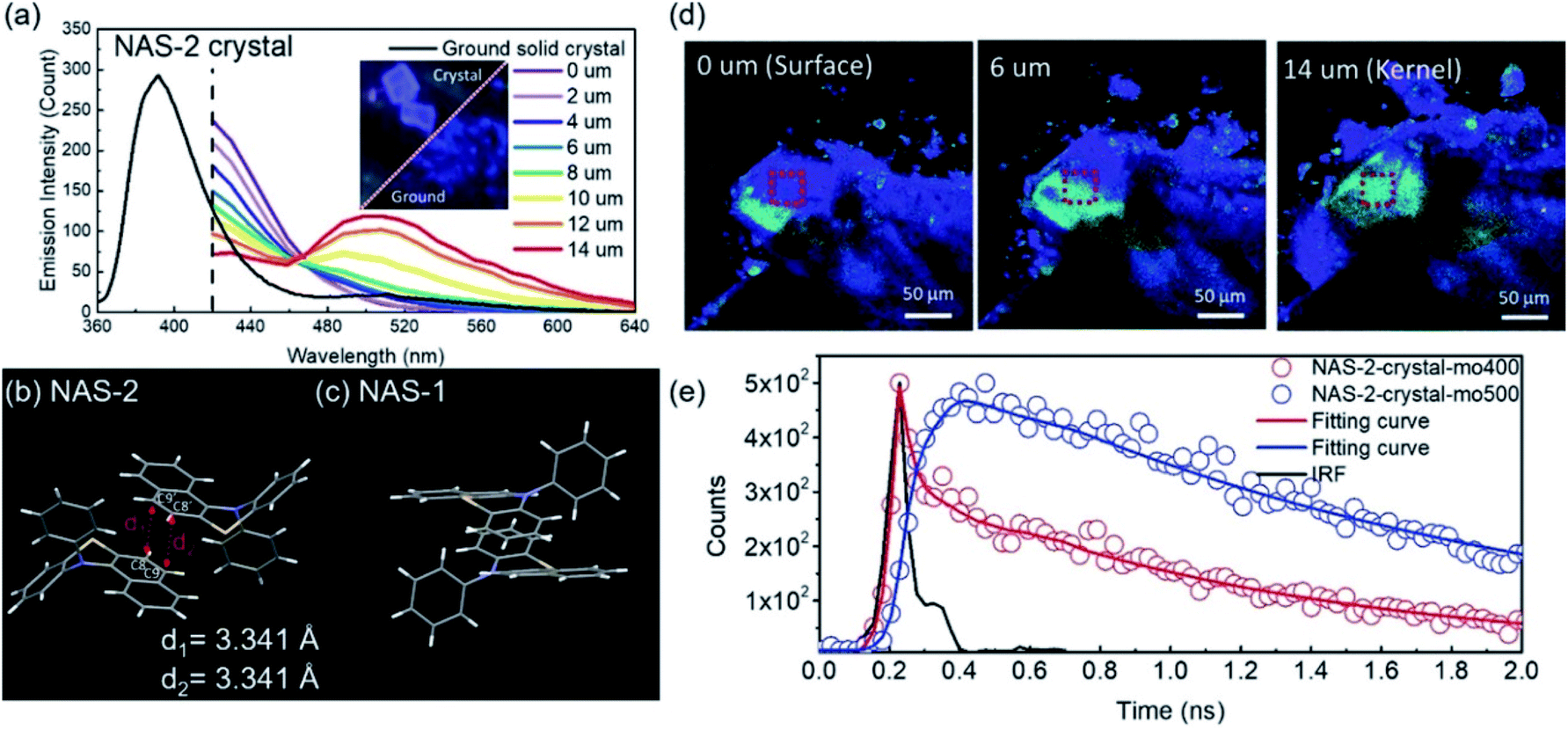 | ||
| Fig. 12 (a) Photoluminescent spectra of NAS-2: black solid line for the ground powder, and coloured solid lines for the red square in (d) of the crystal with different depths from the surface to the kernel. Packing of (b) NAS-2 and (c) NAS-1 in the crystal lattice. (e) Transient photoluminescence characteristics of NAS-2 in the crystal as monitored at various wavelengths. Reproduced from ref. 99 with permission of the Wiley-VCH, Copyright 2019. | ||
Broadly speaking, the role of excimer or dimer is one of the most important issues for organic luminogens in the solid state, which has been explored in the field of room-temperature phosphorescence (RTP).118–120 Utilizing VIE design principles to achieve tunable RTP may be feasible. To achieve this, it is very important to understand the actual role of the π–π interactions. In this regard, Tang, Li, and co-workers provided a clear direction by exploring the effect of the dimer in persistent RTP in detail.121 By adopting the phenothiazine 5,5-dioxide group as the donor and dimethyl isophthalate as the acceptor, a new compound, CS-2COOCH3, revealed the origin of all the photoluminescence species, including the persistent RTP, singlet excimer and thermally activated delayed fluorescence (TADF) emission (Fig. 13). Upon excitation, both the singlet excitons and excimers showed delayed fluorescence emissions with different wavelengths and lifetimes. However, only one molecule was involved in the natural transition orbitals of the triplet state for the dimer, despite strong π–π interactions (Fig. 13). Therefore, the persistent RTP was a monomer-dominated emission species, with the π–π interactions weakening the phosphorescence radiative and non-radiative decay. As for the triplet excimer, there was too low a radiative rate to exhibit phosphorescence. This study was significant for achieving a deep understanding of the RTP process. Without being limited to π–π interactions, a clear insight into the excited state electron transition in various photophysical processes greatly contributes to extending the applications. Our research along this line based on VIE cores is also in progress, and it is expected to further our understanding of the relationship between VIE and other photophysical processes.
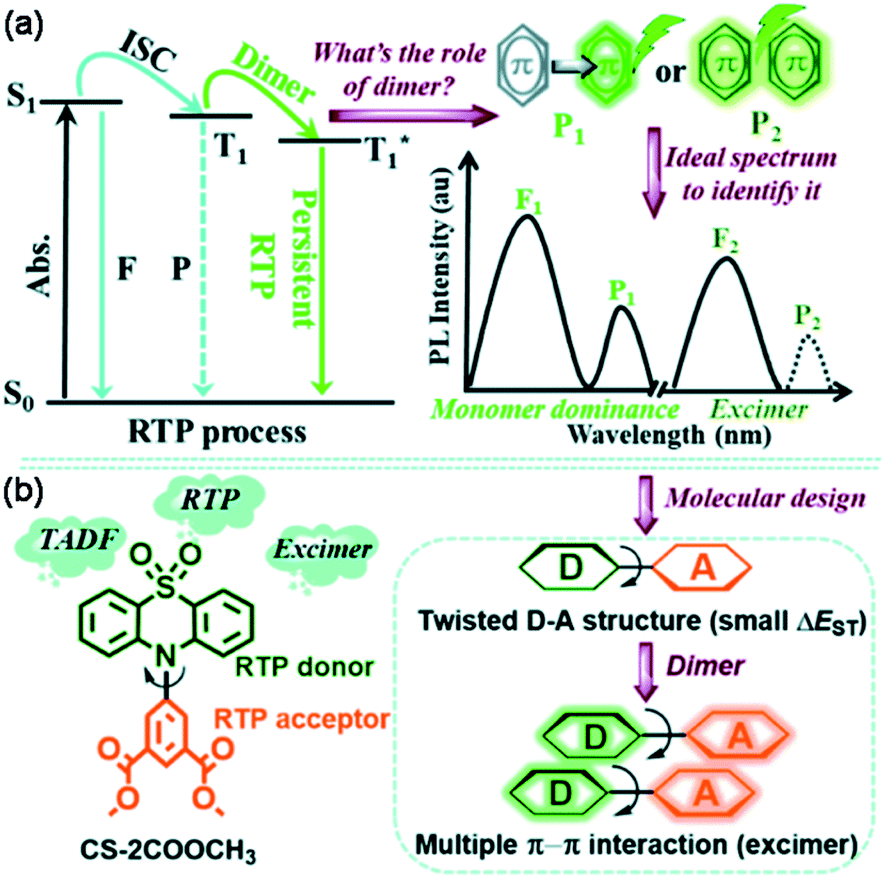 | ||
| Fig. 13 (a) Issue for the function of the dimer in the persistent room-temperature phosphorescence (RTP) effect and the proposed hypothesis, in which F1 denotes fluorescence from the monomer (singlet state, S1), P1 denotes RTP from the monomer (triplet state, T1), F2 denotes fluorescence from the excimer (S1), and P2 denotes RTP from the excimer (T1). (b) The chemical structure of CS-2COOCH3 together with the design strategy. Reproduced from ref. 121 with permission of the Royal Society of Chemistry, Copyright 2020. | ||
6. Summary and perspectives
In this perspective, we have reviewed the recent achievements in the design of significant VIE-active luminogens. Deep insight into the photoinduced conformation planarization has been gained by tailoring the new bending heterocyclic cores for the VIE molecular family. Such brilliant molecular engineering of the VIE system clearly reveals the ubiquity of the VIE process in DHP derivatives. It also proved that the annular conjugated 8π units provided the internal driving forces behind the photoinduced bent-to-planar process, while the steric hindrance enlarged the torsion angle and finely tuned the planarization process. Thus, stimuli-responsive multiple emission systems have been achieved by functionalized VIE-active molecules, which endowed them with precise ratiometric fluorescence detection for supramolecular polymer thermometers, as well as bio-probes.Despite the great progress in VIE studies over the past five years, there are abundant challenges to broadening the fundamentals and applications of VIE territory. A major task remaining is to develop various new VIE cores. The maximum emission wavelength of the current VIE-active luminogens based on phenazine, phenothiazine, phenoxazines or phenoselenazine cannot reach the near infrared (NIR) region, which severely restricts the biological applications of the VIE concept. A common strategy to solve this issue is to extend the π-conjugation of the core by utilizing larger polycyclic skeletons or appending other conjugated systems. Furthermore, it is known that such annular conjugated 8π systems are only a particular case that achieves the VIE process. According to Hückel's rule and Baird's aromaticity rule, the aromaticity of the internal ring is the driving force behind the photoinduced structural vibrations. Therefore, designing larger annular systems with increasing numbers of conjugated electrons should be an alternative method to achieve NIR emission or even unexpected results.
Likewise, water-soluble VIE-active molecules are in great demand, since most of the existing ones can only be well dissolved in organic solvents, which also greatly limits their biological applications. The majority of the reported VIE-based bio-probes do not exhibit an obvious emission colour change because of the poor orange-red emission in aqueous environments. It has been reported that appending polar functional groups, such as quaternary ammonium and carboxyl groups, on a VIE core can facilitate water solubility, providing a practicable method for molecular design. In addition, grafting VIE-active probes on water-soluble macromolecules, such as polyethylene glycol (PEG), polysaccharides and peptides, may not only improve the water solubility but also instil good biocompatibility.
Another important issue is to explore the relationship between VIE and other dynamic processes, such as electron and energy transfer, which are probably the most ubiquitous light-initiated pathways. Further in-depth insight into the competition between the VIE process and electron/energy transfer can greatly advance our knowledge of the time scale of such an excited state configuration transformation and reveal some unknown problems. Most importantly, these complicated dynamic processes could mimic VIE-based bio-probes in the membranes of organelles, such as mitochondria and chloroplasts, because both the viscosity and the redox/energy transfer reaction, corresponding to our situations of planarization, PET and FRET respectively, can be probed by the emission of VIE molecules. That is to say, VIE-based bio-probes could measure two parameters simultaneously in biomembranes, providing a correlation between the membrane viscosity and the efficiency of cellular respiration or photosynthesis. It is inevitable that systematically studying the excited state process of complex systems will be of great significance both in terms of scientific value and in practical applications in the future. The final goal of ‘dynamic living molecular emitting probes for living cells’ would be realized based on this VIE-concept molecular design.
As an expanding field, VIE research simultaneously provides us with opportunities and challenges. We hope this perspective will provide guidance for the innovation of smart VIE-active light-emitting materials and facilitating applications in the field of bio-sensors and bio-imaging.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support by NSFC/China (21788102, 21905090, 21790361, 91859205), Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03), the Programme of Introducing Talents of Discipline to Universities (B16017), Shanghai Science and Technology Committee (17520750100). Z. Y. Zhang thanks Program for Eastern Scholar at Shanghai, and Natural Science Foundation of Shanghai (19ZR1412200).Notes and references
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- X. Jia, C. Shao, X. Bai, Q. Zhou, B. Wu, L. Wang, B. Yue, H. Zhu and L. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4816–4821 CrossRef CAS PubMed.
- M. Jiang, X. Gu, J. W. Y. Lam, Y. Zhang, R. T. K. Kwok, K. S. Wong and B. Z. Tang, Chem. Sci., 2017, 8, 5440–5446 RSC.
- S. Samanta, U. Manna and G. Das, New J. Chem., 2017, 41, 1064–1072 RSC.
- T. Seki, T. Ozaki, T. Okura, K. Asakura, A. Sakon, H. Uekusa and H. Ito, Chem. Sci., 2015, 6, 2187–2195 RSC.
- L. Shi, C. Yan, Z. Guo, W. Chi, J. Wei, W. Liu, X. Liu, H. Tian and W.-H. Zhu, Nat. Commun., 2020, 11, 793 CrossRef CAS PubMed.
- Q. Wang, Q. Zhang, Q.-W. Zhang, X. Li, C.-X. Zhao, T.-Y. Xu, D.-H. Qu and H. Tian, Nat. Commun., 2020, 11, 158 CrossRef PubMed.
- H. Wu, W. Chi, G. Baryshnikov, B. Wu, Y. Gong, D. Zheng, X. Li, Y. Zhao, X. Liu, H. Ågren and L. Zhu, Angew. Chem., Int. Ed., 2019, 58, 4328–4333 CrossRef CAS PubMed.
- Q.-W. Zhang, D. Li, X. Li, P. B. White, J. Mecinović, X. Ma, H. Ågren, R. J. M. Nolte and H. Tian, J. Am. Chem. Soc., 2016, 138, 13541–13550 CrossRef CAS.
- L. Zhu, M. T. Trinh, L. Yin and Z. Zhang, Chem. Sci., 2016, 7, 2058–2065 RSC.
- A. O. Doroshenko, Theor. Exp. Chem., 2002, 38, 135–155 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, 2013 Search PubMed.
- F. Vollmer, W. Rettig and E. Birckner, J. Fluoresc., 1994, 4, 65–69 CrossRef CAS.
- L. Ding, Z. Zhang, X. Li and J. Su, Chem. Commun., 2013, 49, 7319–7321 RSC.
- R. Greiner, T. Schlücker, D. Zgela and H. Langhals, J. Mater. Chem. C, 2016, 4, 11244–11252 RSC.
- F. Lu, N. Kitamura, T. Takaya, K. Iwata, T. Nakanishi and Y. Kurashige, Phys. Chem. Chem. Phys., 2018, 20, 3258–3264 RSC.
- J. Mei, J. Wang, J. Z. Sun, H. Zhao, W. Yuan, C. Deng, S. Chen, H. H. Y. Sung, P. Lu, A. Qin, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, Chem. Sci., 2012, 3, 549–558 RSC.
- J. Merz, A. Steffen, J. Nitsch, J. Fink, C. B. Schürger, A. Friedrich, I. Krummenacher, H. Braunschweig, M. Moos, D. Mims, C. Lambert and T. B. Marder, Chem. Sci., 2019, 10, 7516–7534 RSC.
- K. Wang, S. Huang, Y. Zhang, S. Zhao, H. Zhang and Y. Wang, Chem. Sci., 2013, 4, 3288–3293 RSC.
- K.-Y. Chen, C.-C. Hsieh, Y.-M. Cheng, C.-H. Lai, P.-T. Chou and T. J. Chow, J. Phys. Chem. A, 2006, 110, 12136–12144 CrossRef CAS PubMed.
- S. Farazi, F. Chen, H. Foster, R. Boquiren, S. R. McAlpine and R. Chapman, Polym. Chem., 2020, 11, 425–432 RSC.
- H.-G. Fu, H.-Y. Zhang, H.-Y. Zhang and Y. Liu, Chem. Commun., 2019, 55, 13462–13465 RSC.
- H. J. Lee, Y. G. Lee, J. Kang, S. H. Yang, J. H. Kim, A. B. T. Ghisaidoobe, H. J. Kang, S.-R. Lee, M. H. Lim and S. J. Chung, Chem. Sci., 2019, 10, 1000–1007 RSC.
- A. J. P. Teunissen, C. Pérez-Medina, A. Meijerink and W. J. M. Mulder, Chem. Soc. Rev., 2018, 47, 7027–7044 RSC.
- L. Yuan, F. Jin, Z. Zeng, C. Liu, S. Luo and J. Wu, Chem. Sci., 2015, 6, 2360–2365 RSC.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Chem. Sci., 2019, 10, 10629–10639 RSC.
- E. V. Bichenkova, A. R. Sardarian, A. N. Wilton, P. Bonnet, R. A. Bryce and K. T. Douglas, Org. Biomol. Chem., 2006, 4, 367–378 RSC.
- J. B. Birks, Rep. Prog. Phys., 1975, 38, 903–974 CrossRef CAS.
- N. Bunzmann, S. Weissenseel, L. Kudriashova, J. Gruene, B. Krugmann, J. V. Grazulevicius, A. Sperlich and V. Dyakonov, Mater. Horiz., 2020, 7, 1126–1137 RSC.
- N. Okamura, T. Maeda, H. Fujiwara, A. Soman, K. N. N. Unni, A. Ajayaghosh and S. Yagi, Phys. Chem. Chem. Phys., 2018, 20, 542–552 RSC.
- A. Yadav, S. Trivedi, V. Haridas, J. B. Essner, G. A. Baker and S. Pandey, Photochem. Photobiol. Sci., 2020, 19, 251–260 RSC.
- R. Kotani, H. Sotome, H. Okajima, S. Yokoyama, Y. Nakaike, A. Kashiwagi, C. Mori, Y. Nakada, S. Yamaguchi, A. Osuka, A. Sakamoto, H. Miyasaka and S. Saito, J. Mater. Chem. C, 2017, 5, 5248–5256 RSC.
- D. Shukla and P. Wan, J. Am. Chem. Soc., 1993, 115, 2990–2991 CrossRef CAS.
- C. Yuan, S. Saito, C. Camacho, S. Irle, I. Hisaki and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 8842–8845 CrossRef CAS PubMed.
- C. Yuan, S. Saito, C. Camacho, T. Kowalczyk, S. Irle and S. Yamaguchi, Chem.–Eur. J., 2014, 20, 2193–2200 CrossRef CAS PubMed.
- C.-L. Chen, Y.-T. Chen, A. P. Demchenko and P.-T. Chou, Nat. Rev. Chem., 2018, 2, 131–143 CrossRef CAS.
- A. P. Demchenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC.
- B. Li, L. Zhou, H. Cheng, Q. Huang, J. Lan, L. Zhou and J. You, Chem. Sci., 2018, 9, 1213–1220 RSC.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- N. Suzuki, K. Suda, D. Yokogawa, H. Kitoh-Nishioka, S. Irle, A. Ando, L. M. G. Abegão, K. Kamada, A. Fukazawa and S. Yamaguchi, Chem. Sci., 2018, 9, 2666–2673 RSC.
- Y.-C. Wei, Z. Zhang, Y.-A. Chen, C.-H. Wu, Z.-Y. Liu, S.-Y. Ho, J.-C. Liu, J.-A. Lin and P.-T. Chou, Commun. Chem., 2019, 2, 10 Search PubMed.
- J. Feng, K. Tian, D. Hu, S. Wang, S. Li, Y. Zeng, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2011, 50, 8072–8076 CrossRef CAS PubMed.
- M. Han, Y. Tian, Z. Yuan, L. Zhu and B. Ma, Angew. Chem., Int. Ed., 2014, 53, 10908–10912 CrossRef CAS PubMed.
- Y. Inoue, P. Jiang, E. Tsukada, T. Wada, H. Shimizu, A. Tai and M. Ishikawa, J. Am. Chem. Soc., 2002, 124, 6942–6949 CrossRef CAS PubMed.
- M. K. Kuimova, S. W. Botchway, A. W. Parker, M. Balaz, H. A. Collins, H. L. Anderson, K. Suhling and P. R. Ogilby, Nat. Chem., 2009, 1, 69–73 CrossRef CAS PubMed.
- P. Valat, V. Wintgens, J. Kossanyi, L. Biczok, A. Demeter and T. Berces, J. Am. Chem. Soc., 1992, 114, 946–953 CrossRef CAS.
- J. Catalán, C. Díaz, V. López, P. Pérez and R. M. Claramunt, J. Phys. Chem., 1996, 100, 18392–18398 CrossRef.
- W. Chi, Q. Qiao, R. Lee, W. Liu, Y. S. Teo, D. Gu, M. J. Lang, Y.-T. Chang, Z. Xu and X. Liu, Angew. Chem., Int. Ed., 2019, 58, 7073–7077 CrossRef CAS PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- K. Li, Y. Liu, Y. Li, Q. Feng, H. Hou and B. Z. Tang, Chem. Sci., 2017, 8, 7258–7267 RSC.
- L. M. Lifshits, D. S. Budkina, V. Singh, S. M. Matveev, A. N. Tarnovsky and J. K. Klosterman, Phys. Chem. Chem. Phys., 2016, 18, 27671–27683 RSC.
- X. Liu, Q. Qiao, W. Tian, W. Liu, J. Chen, M. J. Lang and Z. Xu, J. Am. Chem. Soc., 2016, 138, 6960–6963 CrossRef CAS PubMed.
- S. Sasaki, G. P. C. Drummen and G.-i. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- S. Wiedbrauk, B. Maerz, E. Samoylova, P. Mayer, W. Zinth and H. Dube, J. Phys. Chem. Lett., 2017, 8, 1585–1592 CrossRef CAS PubMed.
- J. P. Otto, L. Wang, I. Pochorovski, S. M. Blau, A. Aspuru-Guzik, Z. Bao, G. S. Engel and M. Chiu, Chem. Sci., 2018, 9, 3694–3703 RSC.
- Y. Sagara, M. Karman, A. Seki, M. Pannipara, N. Tamaoki and C. Weder, ACS Cent. Sci., 2019, 5, 874–881 CAS.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- Y.-g. Shi, J.-w. Wang, H. Li, G.-f. Hu, X. Li, S. K. Mellerup, N. Wang, T. Peng and S. Wang, Chem. Sci., 2018, 9, 1902–1911 RSC.
- Z. Wang, X. He, T. Yong, Y. Miao, C. Zhang and B. Zhong Tang, J. Am. Chem. Soc., 2020, 142, 512–519 CrossRef CAS PubMed.
- Z. Zhang, Y.-A. Chen, W.-Y. Hung, W.-F. Tang, Y.-H. Hsu, C.-L. Chen, F.-Y. Meng and P.-T. Chou, Chem. Mater., 2016, 28, 8815–8824 CrossRef CAS.
- Y. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- M. Khorloo, Y. Cheng, H. Zhang, M. Chen, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2020, 11, 997–1005 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- K. Matsuo and T. Yasuda, Chem. Sci., 2019, 10, 10687–10697 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Gao, S. Zhang, Y. Pan, L. Yao, H. Liu, Y. Guo, Q. Gu, B. Yang and Y. Ma, Phys. Chem. Chem. Phys., 2016, 18, 24176–24184 RSC.
- W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS.
- A. Abdurahman, Y. Chen, X. Ai, O. Ablikim, Y. Gao, S. Dong, B. Li, B. Yang, M. Zhang and F. Li, J. Mater. Chem. C, 2018, 6, 11248–11254 RSC.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
- K. Rotkiewicz, K. H. Grellmann and Z. R. Grabowski, Chem. Phys. Lett., 1973, 19, 315–318 CrossRef CAS.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- A. Weller, Sci. Nat., 1955, 42, 175–176 CrossRef CAS.
- W. Huang, L. Sun, Z. Zheng, J. Su and H. Tian, Chem. Commun., 2015, 51, 4462–4464 RSC.
- Z. Zhang, Y.-S. Wu, K.-C. Tang, C.-L. Chen, J.-W. Ho, J. Su, H. Tian and P.-T. Chou, J. Am. Chem. Soc., 2015, 137, 8509–8520 CrossRef CAS PubMed.
- T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383–6422 CrossRef CAS PubMed.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed.
- J. Toldo, O. El Bakouri, M. Solà, P.-O. Norrby and H. Ottosson, ChemPlusChem, 2019, 84, 712–721 CrossRef CAS PubMed.
- W. Chen, Z. Lin, J. Su and H. Tian, in Aggregation-Induced Emission: Materials and Applications Volume 1, American Chemical Society, 2016, ch. 3, vol. 1226, pp. 21–33 Search PubMed.
- Z. Zhang, W. Song, J. Su and H. Tian, Adv. Funct. Mater., 2020, 30, 1902803 CrossRef CAS.
- J. Chen, Y. Wu, X. Wang, Z. Yu, H. Tian, J. Yao and H. Fu, Phys. Chem. Chem. Phys., 2015, 17, 27658–27664 RSC.
- L. Shi, W. Song, C. Lian, W. Chen, J. Mei, J. Su, H. Liu and H. Tian, Adv. Opt. Mater., 2018, 6, 1800190 CrossRef.
- H. V. Humeniuk, A. Rosspeintner, G. Licari, V. Kilin, L. Bonacina, E. Vauthey, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 10559–10563 CrossRef CAS PubMed.
- Y. Li, Y. Liu, H. Zhou, W. Chen, J. Mei and J. Su, Chem.–Eur. J., 2017, 23, 9280–9287 CrossRef CAS PubMed.
- Y. Li, L. Shi, Y. Zhang, G. Sun, L. Sun and J. Su, Dyes Pigm., 2019, 160, 794–798 CrossRef CAS.
- H. Zhou, J. Mei, Y.-A. Chen, C.-L. Chen, W. Chen, Z. Zhang, J. Su, P.-T. Chou and H. Tian, Small, 2016, 12, 6542–6546 CrossRef CAS PubMed.
- W.-T. Dou, W. Chen, X.-P. He, J. Su and H. Tian, Faraday Discuss., 2017, 196, 395–402 RSC.
- N. Wang, C. Xin, Z. Li, G. Zhang, L. Bai, Q. Gong, C. Xu, X. Han, C. Yu, L. Li and W. Huang, Dyes Pigm., 2019, 163, 425–432 CrossRef CAS.
- H. Wang, Y. Li, Y. Zhang, J. Mei and J. Su, Chem. Commun., 2019, 55, 1879–1882 RSC.
- J. Wang, X. Yao, Y. Liu, H. Zhou, W. Chen, G. Sun, J. Su, X. Ma and H. Tian, Adv. Opt. Mater., 2018, 6, 1800074 CrossRef.
- W. Chen, C.-L. Chen, Z. Zhang, Y.-A. Chen, W.-C. Chao, J. Su, H. Tian and P.-T. Chou, J. Am. Chem. Soc., 2017, 139, 1636–1644 CrossRef CAS PubMed.
- Z. Zhou, D.-G. Chen, M. L. Saha, H. Wang, X. Li, P.-T. Chou and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 5535–5543 CrossRef CAS PubMed.
- Z. Zhang, C.-L. Chen, Y.-A. Chen, Y.-C. Wei, J. Su, H. Tian and P.-T. Chou, Angew. Chem., Int. Ed., 2018, 57, 9880–9884 CrossRef CAS PubMed.
- Y. Chen, D.-G. Chen, Y.-A. Chen, C.-H. Wu, K.-H. Chang, F.-Y. Meng, M.-C. Chen, J.-A. Lin, C.-Y. Huang, J. Su, H. Tian and P.-T. Chou, Chem.–Eur. J., 2019, 25, 16755–16764 CrossRef CAS PubMed.
- D.-G. Chen, Y. Chen, C.-H. Wu, Y.-A. Chen, M.-C. Chen, J.-A. Lin, C.-Y. Huang, J. Su, H. Tian and P.-T. Chou, Angew. Chem., Int. Ed., 2019, 58, 13297–13301 CrossRef CAS PubMed.
- K. P. C. Vollhardt and N. E. Schore, Organic Chemistry; Palgrave Version: Structure and Function, Macmillan International Higher Education, 2014 Search PubMed.
- I. Kawashima, H. Imoto, M. Ishida, H. Furuta, S. Yamamoto, M. Mitsuishi, S. Tanaka, T. Fujii and K. Naka, Angew. Chem., Int. Ed., 2019, 58, 11686–11690 CrossRef CAS PubMed.
- I. M. Atkinson and L. F. Lindoy, Self assembly in supramolecular systems, Royal Society of Chemistry, 2007 Search PubMed.
- W. Zhou, Y. Chen, Q. Yu, P. Li, X. Chen and Y. Liu, Chem. Sci., 2019, 10, 3346–3352 RSC.
- W. Chen, C. Guo, Q. He, X. Chi, V. M. Lynch, Z. Zhang, J. Su, H. Tian and J. L. Sessler, J. Am. Chem. Soc., 2019, 141, 14798–14806 CrossRef CAS PubMed.
- G. Sun, J. Pan, Y. Wu, Y. Liu, W. Chen, Z. Zhang and J. Su, ACS Appl. Mater. Interfaces, 2020, 12, 10875–10882 CrossRef PubMed.
- W. Song, W. Ye, L. Shi, J. Huang, Z. Zhang, J. Mei, J. Su and H. Tian, Mater. Horiz., 2020, 7, 615–623 RSC.
- Y. Zhang, Y. Li, H. Wang, Z. Zhang, Y. Feng, Q. Tian, N. Li, J. Mei, J. Su and H. Tian, ACS Appl. Mater. Interfaces, 2019, 11, 39351–39358 CrossRef CAS PubMed.
- A. P. Demchenko, Introduction to Fluorescence Sensing, Springer International Publishing, 2015 Search PubMed.
- S.-H. Park, N. Kwon, J.-H. Lee, J. Yoon and I. Shin, Chem. Soc. Rev., 2020, 49, 143–179 RSC.
- S. J. Park, V. Juvekar, J. H. Jo and H. M. Kim, Chem. Sci., 2020, 11, 596–601 RSC.
- X. Shi, N. Yan, G. Niu, S. H. P. Sung, Z. Liu, J. Liu, R. T. K. Kwok, J. W. Y. Lam, W.-X. Wang, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2020, 11, 3152–3163 RSC.
- Q. Gong, W. Qin, P. Xiao, X. Wu, L. Li, G. Zhang, R. Zhang, J. Sun, S. Q. Yao and W. Huang, Chem. Commun., 2020, 56, 58–61 RSC.
- W.-T. Dou, Z.-Y. Qin, J. Li, D.-M. Zhou and X.-P. He, Sci. Bull., 2019, 64, 1902–1909 CrossRef CAS.
- Y. Jiang, J. Wang, G. Huang, Z. Li, B. S. Li and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 11790–11796 RSC.
- Y. Xie and Z. Li, Mater. Chem. Front., 2020, 4, 317–331 RSC.
- H. Zhang, D. Zeng, Y. Pan, Y. Chen, Y. Ruan, Y. Xu, R. Boulatov, C. Creton and W. Weng, Chem. Sci., 2019, 10, 8367–8373 RSC.
- Z. Zhang, B. Xu, J. Su, L. Shen, Y. Xie and H. Tian, Angew. Chem., Int. Ed., 2011, 50, 11654–11657 CrossRef CAS PubMed.
- M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081–2093 RSC.
- J. Yuan, R. Chen, X. Tang, Y. Tao, S. Xu, L. Jin, C. Chen, X. Zhou, C. Zheng and W. Huang, Chem. Sci., 2019, 10, 5031–5038 RSC.
- J. Yuan, S. Wang, Y. Ji, R. Chen, Q. Zhu, Y. Wang, C. Zheng, Y. Tao, Q. Fan and W. Huang, Mater. Horiz., 2019, 6, 1259–1264 RSC.
- Y. Wang, J. Yang, Y. Tian, M. Fang, Q. Liao, L. Wang, W. Hu, B. Z. Tang and Z. Li, Chem. Sci., 2020, 11, 833–838 RSC.
| This journal is © The Royal Society of Chemistry 2020 |