
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetrosynthetic strategies and their impact on synthesis of arcutane natural products
Shelby V.
McCowen
 ,
Nicolle A.
Doering
and
Richmond
Sarpong
*
,
Nicolle A.
Doering
and
Richmond
Sarpong
*
Department of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: rsarpong@berkeley.edu
First published on 21st April 2020
Abstract
Retrosynthetic analysis is a cornerstone of modern natural product synthesis, providing an array of tools for disconnecting structures. However, discussion of retrosynthesis is often limited to the reactions used to form selected bonds in the forward synthesis. This review details three strategies for retrosynthesis, focusing on how they can be combined to plan the synthesis of polycyclic natural products, such as atropurpuran and the related arcutane alkaloids. Recent syntheses of natural products containing the arcutane framework showcase how these strategies for retrosynthesis can be combined to plan the total synthesis of highly caged scaffolds. Comparison of multiple syntheses of the same target provides a unique opportunity for detailed analysis of the impact of retrosynthetic disconnections on synthesis outcomes.
 Shelby V. McCowen | Shelby V. McCowen received her B.S. and M.S. in Chemistry at the University of Texas at San Antonio. During her Master's she worked under Professor Stanton McHardy on developing small molecules that inhibit triple negative breast cancer. Currently, she is a PhD candidate in the Chemistry Department at the University of California–Berkeley under the supervision of Professor Richmond Sarpong. Her research focuses on the synthesis of complex natural products, including arcutinidine and marine meroterpenoids. |
 Nicolle A. Doering | Nicolle A. Doering received her B.S. in Chemistry from the University of California–Santa Barbara in 2015. During that time, she carried out research with Professor Armen Zakarian on enantioselective α-functionalization of carboxylic acid derivatives, as well as on enantioselective total synthesis. She is currently a PhD student in the Chemistry program at the University of California–Berkeley, where she conducts her research under the supervision of Professor Richmond Sarpong. Her current research is focused on strategies to enable the total synthesis of diterpenoid alkaloid and terpene natural products. |
 Richmond Sarpong | Richmond Sarpong was born in Ghana, West Africa in 1974. He received his B.A. from Macalester College in 1995. During that time he carried out research with Professor Rebecca Hoye. He conducted his PhD research with Professor Martin F. Semmelhack at Princeton University and completed his degree in 2000. After three and a half years as a postdoctoral fellow with Professor Brian Stoltz at Caltech he began his independent career at the University of California–Berkeley where he is Professor of Chemistry. His current research interests include development of new strategies for synthesis of complex natural products including diterpenoid alkaloids. |
1. Introduction
Diterpenoid alkaloids are an exceptionally large class of natural products with highly caged scaffolds, which have long been of interest to synthetic chemists.1,2 The arcutane-type diterpenoid alkaloids represent a small, recently discovered subset of the family that contain an unusual [5.3.3.04,9.04,12]pentacyclic ring system (ABCDE ring system, Fig. 1). The first reported isolation of an arcutane alkaloid was of arcutine (1) from Aconitum arcuatam Maxim by Tashkhodzhaev and coworkers in 2000.3 A year later, the same group reported the isolation of arcutinine (2), also from A. arcuatam, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture with arcutine.4 In basic methanol, the arcutane alkaloid mixture converged to produce arcutinidine (3) via methanolysis of the ester side chain. An unusual iminium ion-bearing arcutane-type alkaloid, aconicarmicharcutinium (4), was also isolated from the roots of Aconitum carmichaelii Debx.5 In 2009, Wang and coworkers reported the isolation of a diterpenoid with the arcutane skeleton, atropurpuran (5), from the roots of Aconitum hemsleyanum var. atropurpureum, expanding the known set of arcutane-type natural products beyond alkaloids.6
:2 mixture with arcutine.4 In basic methanol, the arcutane alkaloid mixture converged to produce arcutinidine (3) via methanolysis of the ester side chain. An unusual iminium ion-bearing arcutane-type alkaloid, aconicarmicharcutinium (4), was also isolated from the roots of Aconitum carmichaelii Debx.5 In 2009, Wang and coworkers reported the isolation of a diterpenoid with the arcutane skeleton, atropurpuran (5), from the roots of Aconitum hemsleyanum var. atropurpureum, expanding the known set of arcutane-type natural products beyond alkaloids.6
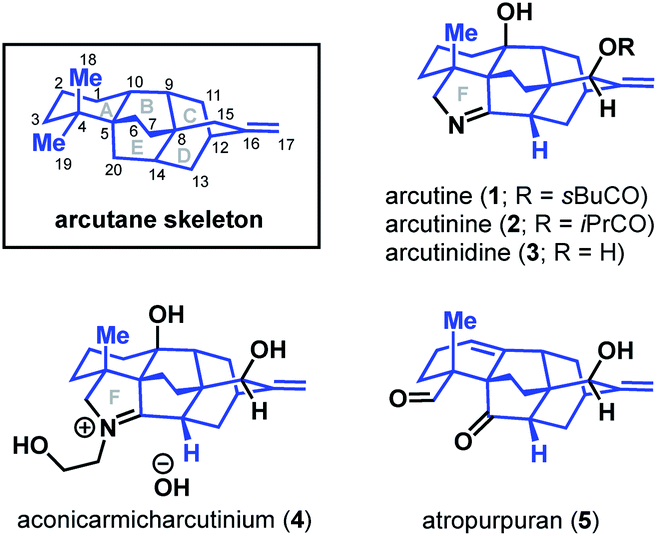 | ||
| Fig. 1 The arcutane framework and arcutane-containing natural products. | ||
The broader class of diterpenoid alkaloids are known to possess a range of biological activities, largely due to their interactions with ion channels.1,2,7 The biological activity of the arcutane alkaloids, however, has not been evaluated. This is also true of atropurpuran. However, there have been a number of patents filed regarding the use of atropurpuran derivatives to treat myriad conditions, including—but not limited to—pancreatic fibrosis,8 hypoxia,9 and inflammation.10 Synthetic access to a larger quantity of arcutane-type natural products could allow for detailed study into their biological activity as well as evaluate their potential as therapeutics.
The remainder of this review is divided into four sections. The first is focused on retrosynthetic strategies, including discussion of the use of biosynthetic pathways as inspiration, application of bond-network analysis, as well as of complementary functional group- and transform- oriented analyses. The second compares total syntheses of atropurpuran, while the third discusses total syntheses of the arcutane alkaloids. The final section includes other studies of bridged polycycles en route to these alkaloids.
2. Retrosynthetic strategies
In the process of evaluating a pathway for the synthesis of a natural product, synthetic chemists often develop a strategy of taking the target molecule back to simpler building blocks, a process known as retrosynthesis. Several plans may be evaluated, and the analysis of the various pathways is referred to as retrosynthetic analysis.2.1 Proposed biosynthetic pathways as inspiration
Nature, and especially the biosynthetic pathways that lead to secondary metabolites, have been a source of inspiration for retrosynthesis for over a century.11 Such bioinspired or biomimetic approaches can enable highly efficient syntheses and challenge chemists to explore new transformations. A strong understanding of biosynthesis is not necessarily required; in fact, Hoffmann argues that consideration of potentially biosynthetic pathways itself advances synthesis strategy, underscoring the utility of this retrosynthetic perspective.11 While biosyntheses of some diterpenoid alkaloids have been well-studied,1,12,13 the biosynthetic pathway to the arcutane natural products has yet to be elucidated.Currently, there are two proposed biosynthetic pathways to the arcutane natural products. These pathways provide many opportunities from which to draw inspiration. In their report detailing the isolation of atropurpuran,6 Wang and coworkers proposed that an oxygenated hetidine-type scaffold, such as 6, could fragment along the C13–C14 bond to produce dialdehyde 7 (Scheme 1A).6 Further fragmentation would release ethylene to generate hemi-quinone 8, into which hydroxide could add in a conjugate fashion to give enolate 9. The dienyl moiety in 9 would then recombine with ethylene to forge the central BE bicycle (10). Dehydration to form 11, followed by an aldol cyclization, would then close the D ring, completing the arcutane framework. Dicarbonyl compound 12 would then undergo deoxygenation at C13 followed by allylic oxidation to afford atropurpuran (5). The Wang group did not discuss the biosynthesis of the arcutane alkaloids.
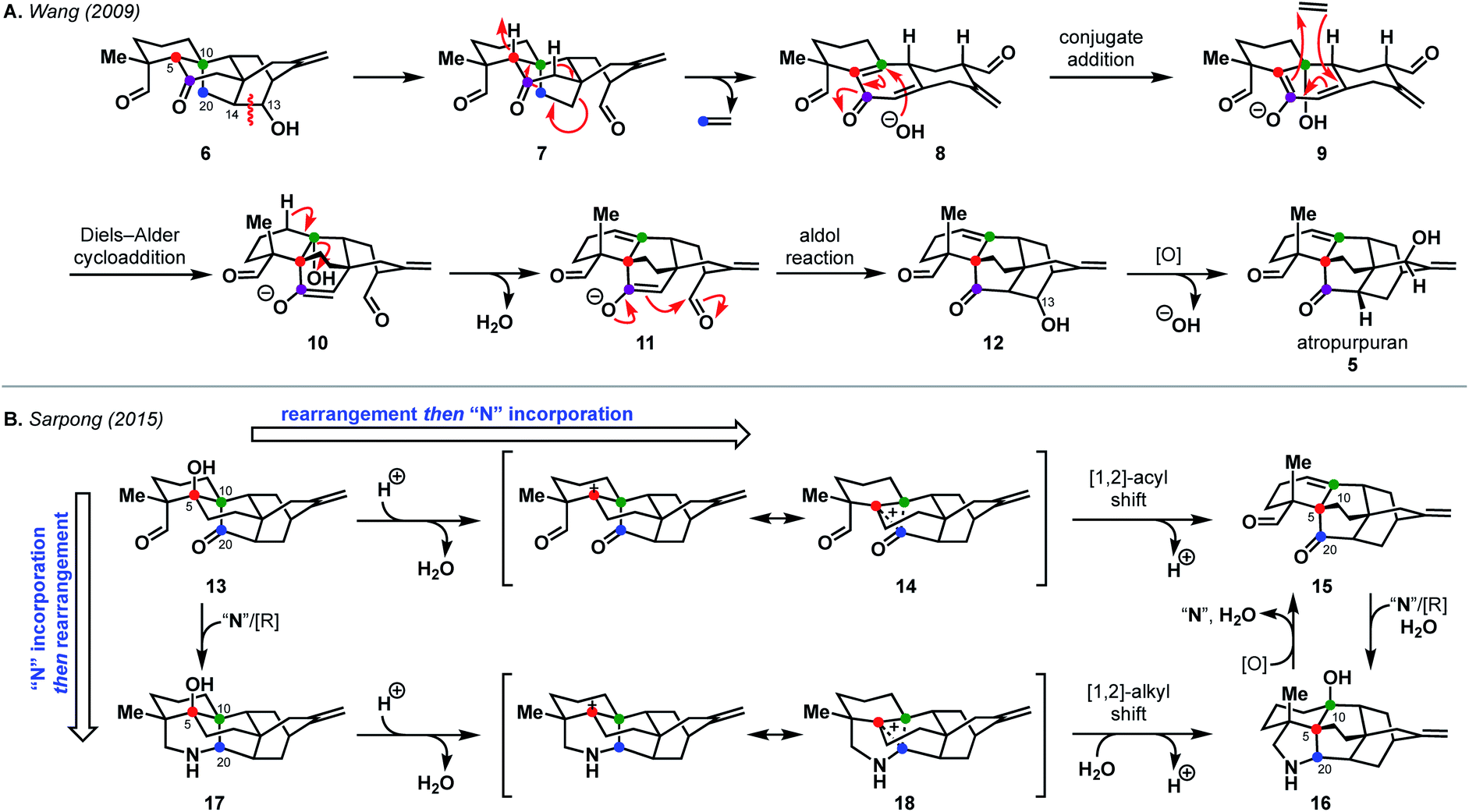 | ||
| Scheme 1 Postulated biosyntheses of atropurpuran by Wang (A) and of arcutane natural products by Sarpong (B). | ||
An alternative biosynthesis was proposed by Sarpong and coworkers,14 who believed aspects of the Wang proposal were unlikely (see 7 → 8, 9 → 10). While the authors agreed that the arcutane framework likely arises from an oxygenated hetidine scaffold, they postulated that conversion from the hetidine to arcutane framework occurs through a deep-seated rearrangement (Scheme 1B). Beginning with alcohol 13, protonation and loss of water could produce non-classical carbocation 14. A [1,2]-acyl shift to forge the C20–C5 bond with concomitant cleavage of the C20–C10 bond followed by elimination, would give dioxygenated arcutane 15. Hydration of the double bond and introduction of a nitrogen atom “N” through a reductive process would then give the arcutane alkaloids, represented by 16. In a complementary fashion, nitrogen could be introduced directly to diketone 13 to produce 17. In an analogous manner as before, loss of water could produce cation 18 that could, in turn, undergo a related [1,2]-alkyl shift (Wagner–Meerwein rearrangement) followed by trapping with water to give 16. The Sarpong group supported their hypothesis with DFT calculations, which suggested that the transition state energies for the diterpenoid and diterpenoid alkaloid [1,2] shifts are +4.8 and +7.7 kcal mol−1 in the gas phase, starting from 14 and 18, respectively.14 This is consistent with previous studies, which showed greater migratory aptitude for acyl groups than for alkyl in Wagner–Meerwein rearrangements.15
In the case of other diterpenoid alkaloids, the nitrogen atom is believed to come from either serine or ethanolamine.1,16 As such, aconicarmicharcutinium (4) could be an intermediate in the biosynthetic pathway from terpenoid 15 to alkaloid 16, where ethanolamine has been incorporated but the ethanol moiety is yet to be cleaved. Feeding studies, similar to those used to determine the source of the nitrogen atom in other alkaloids,16 could be used to interrogate arcutane alkaloid biosynthesis. In addition, isotopic labeling of the C20 carbon16 (marked with a blue dot in Scheme 1) on either proposed precursor 6 or 13 could provide insight into the veracity of both proposed biosynthetic pathways.
Both proposed biosyntheses illustrated in Scheme 1 could provide inspiration to synthetic chemists targeting arcutane natural products to develop non-obvious and efficient routes for their syntheses. Given that both biosynthetic proposals for the arcutane natural products begin with an oxygenated hetidine scaffold, it may also be possible to leverage existing hetidine syntheses17–21 to examine the feasibility of the proposed biosynthetic pathways in a laboratory setting.
2.2 Bond-network analysis of arcutane natural products
In contrast to bioinspired synthesis strategies, which tend to lend themselves more to both skeletal and functional group-based analysis, another approach to developing synthesis strategies focuses primarily on the disconnection of the framework of a target natural product. In 1975, Corey provided guidelines for identifying disconnections in a topology-oriented retrosynthesis, specifically in the context of bridged polycycles.22,23 This network analysis begins with the identification of the maximally bridged ring within the framework of interest, defined as “those rings which are bridged at the greatest number of sites”.22 Once the maximally bridged ring has been identified, the bonds within that ring are analyzed to determine which are considered strategic. These are bonds that, when broken in the retrosynthetic sense, directly lead back to a less bridged, simplified precursor without introducing new synthetic challenges, such as large (>7-membered) rings or acyclic fragments containing chiral centers. If multiple bonds in a ring are strategic, one is selected for disconnection on the basis of complementary strategies for retrosynthesis, such as those based on functional groups or known transforms. The precursor identified in this way can then be analyzed iteratively until a precursor lacking any bridged ring systems is obtained. More common functional group- and transform-based strategies then prove complementary to network analysis when determining how to forge the selected bonds in the synthetic direction.Such an approach to retrosynthesis focuses primarily on the topology of the target, rendering consideration of functional group manipulation secondary. In a recent perspective,24 Sarpong and Hoffmann analyzed the strategic disconnections utilized in other recent diterpenoid alkaloid syntheses and argued that the rules Corey laid out for network analysis, while well-suited to their original goal of automating retrosynthesis, should be applied more flexibly when analyzing such highly bridged natural products. For example, consideration should be given not to only single-bond disconnections, but also to two-bond disconnections as well (e.g., bicyclization transforms). The following discussion will include consideration of multi-bond disconnections and will refer to this modified version of Corey's protocol as ‘bond-network analysis’.
This bond-network analysis can aid significantly in the development of a retrosynthetic strategy for the caged, architecturally complex, arcutane scaffold. In addition to containing a caged, highly bridged framework, the functional groups in the arcutane natural products are mostly peripheral, allowing for an analysis that almost completely disregards functional groups, making it an ideal candidate for bond-network analysis. The maximally bridged ring of the arcutane framework (see emphasis in Fig. 2A) was identified as a central six-membered primary ring containing 4 bridging atoms in the diterpenoid scaffold and 6 bridging atoms in the arcutane alkaloids, which contain an additional pyrrolidine ring. Analysis of the strategic bonds reveals three potential options for the first retrosynthetic disconnection. First is a two-bond disconnection across either the C (purple bonds), D (red bonds), or the maximally bridged E (in the case of atropurpuran) rings, which would lead directly back to an all fused precursor containing no bridging elements (option 1, Fig. 2B). A second option is disconnection of a strategic single-bond (highlighted in green, option 2, Fig. 2C),† which would lead back to a bridged precursor. Alternatively, two-bond disconnection involving ring A (red bonds) and/or F (purple bonds, option 3, Fig. 2C) would also lead back to a bridged precursor. In either of the latter two cases, the remaining CD bicycle could then be subjected to bond-network analysis, which furnishes options for single-bond (highlighted in green, option 2.2/3.2, Fig. 2D) or two-bond (highlighted in blue and red, option 2.1/3.1) disconnection, either of which would produce a fused precursor. Considering the options outlined in this thought exercise, there are nearly 50 sets of possible bond-network analysis derived disconnections, however only 3 disconnections (those delineated in option 1) lead directly back to a fused precursor.
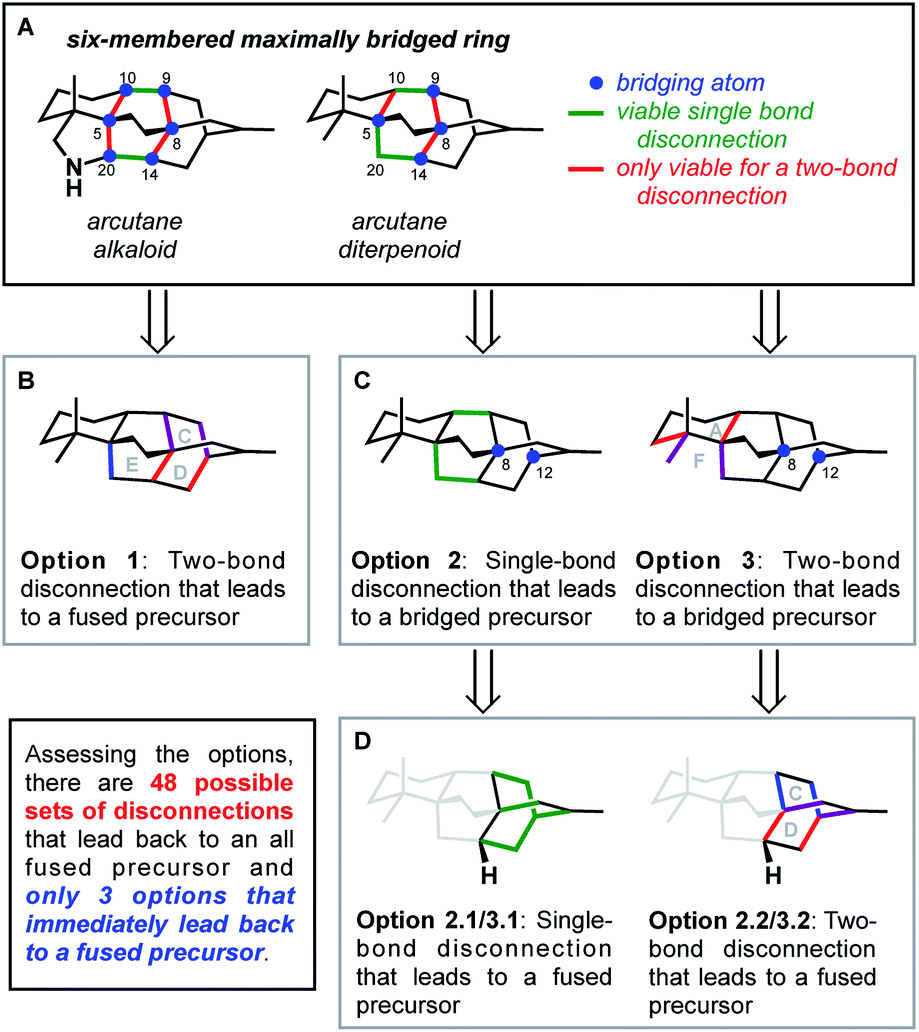 | ||
| Fig. 2 Bond-network analysis of the arcutane skeleton. (A) Maximally bridged ring in arcutane natural products with possible (B) two-bond and (C) single-bond disconnections. (D) Possible disconnections of a bridging precursor. | ||
2.3 Functional group- and transform-oriented analysis of arcutane natural products
Complementary to topology-oriented retrosynthesis, which gives primary consideration to the skeleton of a natural product, functional group-oriented retrosynthesis aims to take advantage of existing and potential functional groups to make disconnections.23,25 In many ways, this is a subjective approach due to the large variety of synthetic choices that are available. One can also recognize retrons, such as a six-membered ring, which can trigger transform-based disconnections.In the case of the arcutane scaffold, the heteroatom-based functional groups are mainly peripheral. However, the framework still provides ample possibilities for functional group- and transform-based disconnections (Fig. 3). For example, one could imagine that a [2.2.2]bicycle (highlighted in blue in Fig. 3) could arise from a Diels–Alder reaction between a dienophile and a cyclic diene; however, this transform requires a double bond within the bicyclic structure (highlighted in red), which would necessitate an extra step to adjust the oxidation state of the ring system.
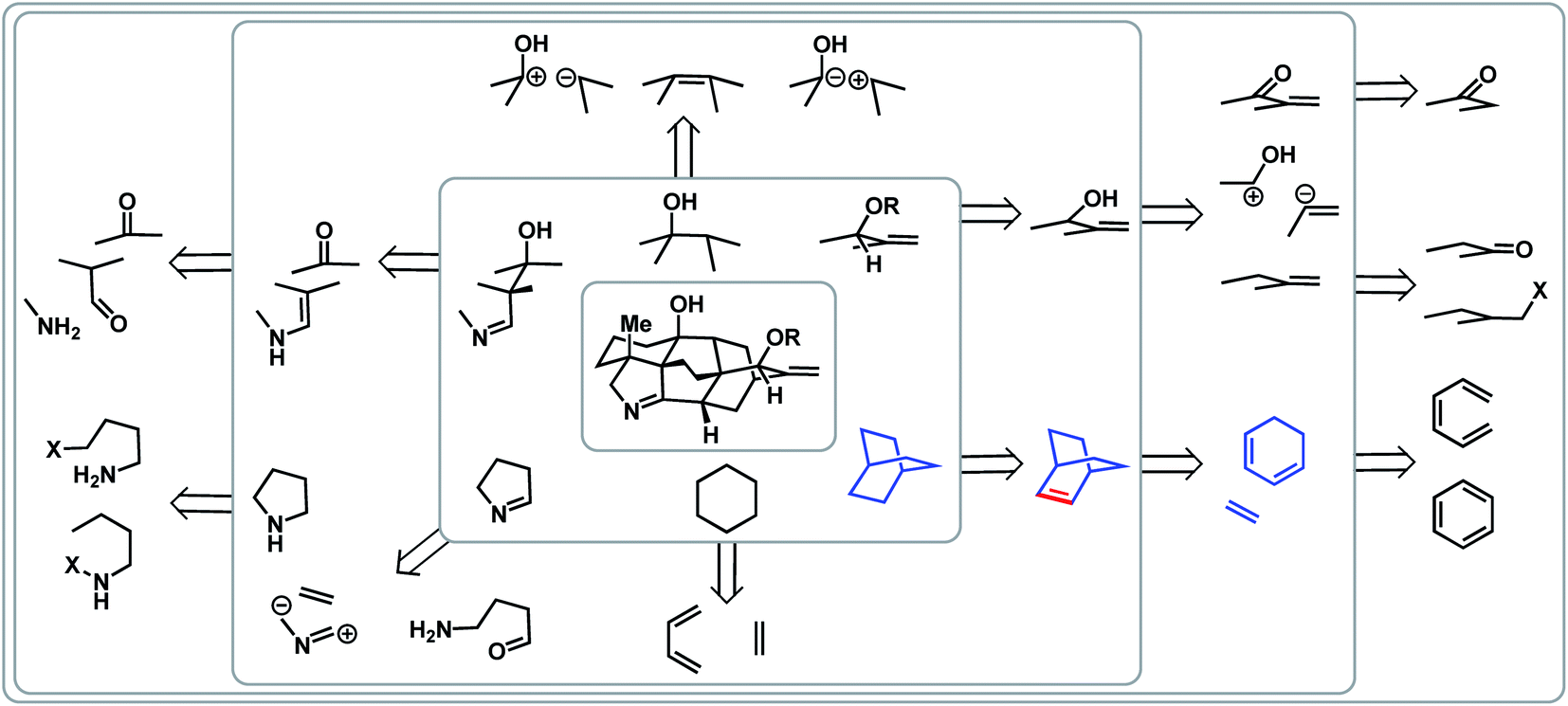 | ||
| Fig. 3 Functional group- and transform-based disconnection scenarios within the arcutane alkaloids. | ||
If one were to map all the potential functional group-oriented transforms for a given molecule, a large array of retrosynthetic plans would emerge, many of which would be unproductive. However, when considered together with bond-network analysis, these strategies can provide the means for comprehensive retrosynthetic planning, as well as provide flexibility for the development of novel transforms.
The ideas of bond-network analysis, functional group-oriented retrosynthesis, and transform-oriented retrosynthesis will guide the discussion throughout this review as we examine how closely syntheses of arcutane natural products and related structures follow principles from each type of retrosynthesis and how the choice of strategy contributes to efficiency‡ in the forward synthesis. This review aims to not only familiarize the reader with the current literature on the arcutane framework, but to also provide insight from each synthesis, specifically through the lens of retrosynthetic analysis.
3. Syntheses of atropurpuran
In the ten years since atropurpuran was first isolated, there have been two reported total syntheses, one by the Qin group and the other by the Xu group. Both syntheses utilize disconnections in line with those suggested by bond-network analysis; however, the two syntheses are distinguished by the specific disconnections chosen and the functional groups introduced to facilitate the construction of strategic bonds in the forward sense.3.1 Qin (2016)
Qin and coworkers reported the first total synthesis of atropurpuran, which also represented the first reported synthesis of the complete arcutane scaffold (Scheme 2).26 The Qin retrosynthesis echoes disconnections outlined previously as option set 2.2 (see Fig. 2), bringing atropurpuran back to monocycle 20via bridged intermediate 19. In the forward synthesis, guaiacol 20 was obtained in three steps from known aldehyde 21, which was itself made in one step from commercial starting materials.27 At this point, the CD bicycle was forged through an oxidative dearomatization/Diels–Alder sequence. Following four functional group manipulation steps, aldehyde 22 underwent a Knoevenagel condensation with 1,3-cyclohexadione to install the A ring, giving thioester 23. Reduction of thioester 23 set the stage for a tetrabutylammonium fluoride (TBAF)-induced Mukaiyama aldol reaction, which completed the E ring to give alcohol 24. Following protection of the free hydroxy group as a TBS ether, a SmI2-mediated ketyl–olefin coupling furnished alcohol 25, completing the arcutane core. Qin and coworkers found that the introduction of the bulky TBS group was necessary to bring the ketyl group and double bond in close enough proximity for cyclization. Dehydration, followed by acetal cleavage, gave diketone 26. The final two carbon atoms of the arcutane skeleton were then installed via Wittig olefination at C16 and Corey–Chaykovsky epoxidation at C4. The epoxide was then opened and rearranged to desired aldehyde 27, which was then converted into atropurpuran in six additional steps. The Qin group was unable to elaborate atropurpuran to arcutinidine, citing the C4 quaternary center and the C1–C10 alkene as possible reasons for the lack of desired reactivity.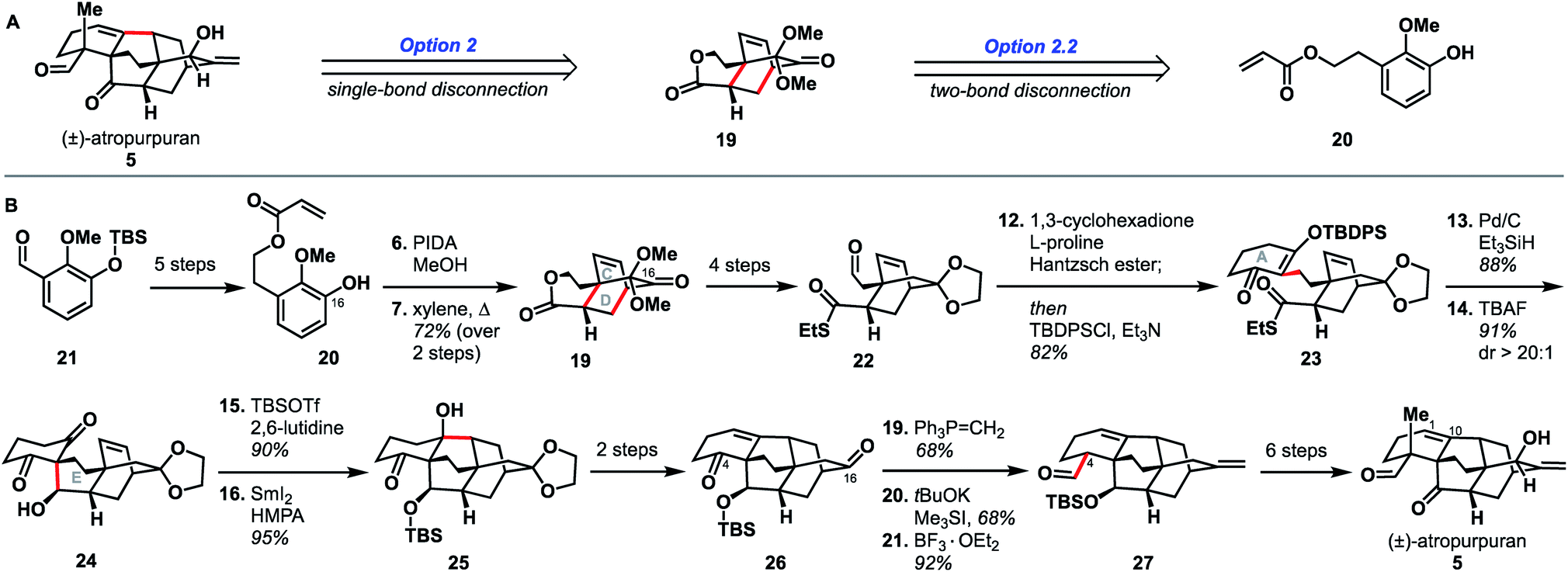 | ||
| Scheme 2 Key bond forming steps in Qin's synthesis of (±)-atropurpuran. | ||
Overall, Qin's racemic synthesis of atropurpuran was completed in 27 steps from known materials (28 from commercial). While the oxidative dearomatization/Diels–Alder sequence is well-precedented for construction of the CD bicycle of other diterpenoid alkaloids, the decision to do so early in this synthesis is unique and proved effective.17,20,21 The Qin group also took advantage of the symmetry of functionality required in the A ring precursor to introduce a relatively simple A-ring structural moiety, exemplifying another principle of efficient synthesis: exploitation of hidden symmetry in synthetic intermediates.28,29 This strategic decision provided an advantage over their initial approach,30 which had involved the synthesis of a more elaborate A ring. As the first completed synthesis of the arcutane skeleton, the Qin atropurpuran synthesis serves as a benchmark against which subsequent syntheses may be measured.
3.2 Xu (2019)
The second reported synthesis of atropurpuran was completed by the Xu group in 2019.31 The Xu group chose an ideal disconnection based on a bond-network analysis: a bicyclization transform that leads directly back to a fused precursor (see option 1, Fig. 2B). In the forward sense, this translated into a 14-step total synthesis of atropurpuran from commercially available 5-methoxytetralone (29, Scheme 3). Tetralone 29 was sequentially acylated and alkynylated to give alkyne 30. In an inspired series of transformations, alkyne 30 was subjected to enyne metathesis to close the A ring and introduce a pendant vinyl group, followed by cleavage of the phenolic methyl ether and global reduction of the carbonyl groups in a single step to give 28. Regioselective double oxidative dearomatization, followed by intramolecular Diels–Alder cycloaddition, yielded 31, which contains the complete arcutane core. Diol 31 was then advanced to triflate 32 in two steps. From there, the C19 carbon was installed through a reductive carbonylative coupling to afford alcohol 33 after DIBAL reduction of the resulting aldehyde. Redox manipulation of 33 gave tricarbonyl compound 34. Alkylation to install the C18 methyl group gave compound 35, at which point treatment with SmI2 led to a reductive cleavage of the dimethoxy acetal group, setting the stage for methylenation to install C17 and afford enone 36. Reduction of this penultimate intermediate with NaBH(OMe)3 gave atropurpuran (5).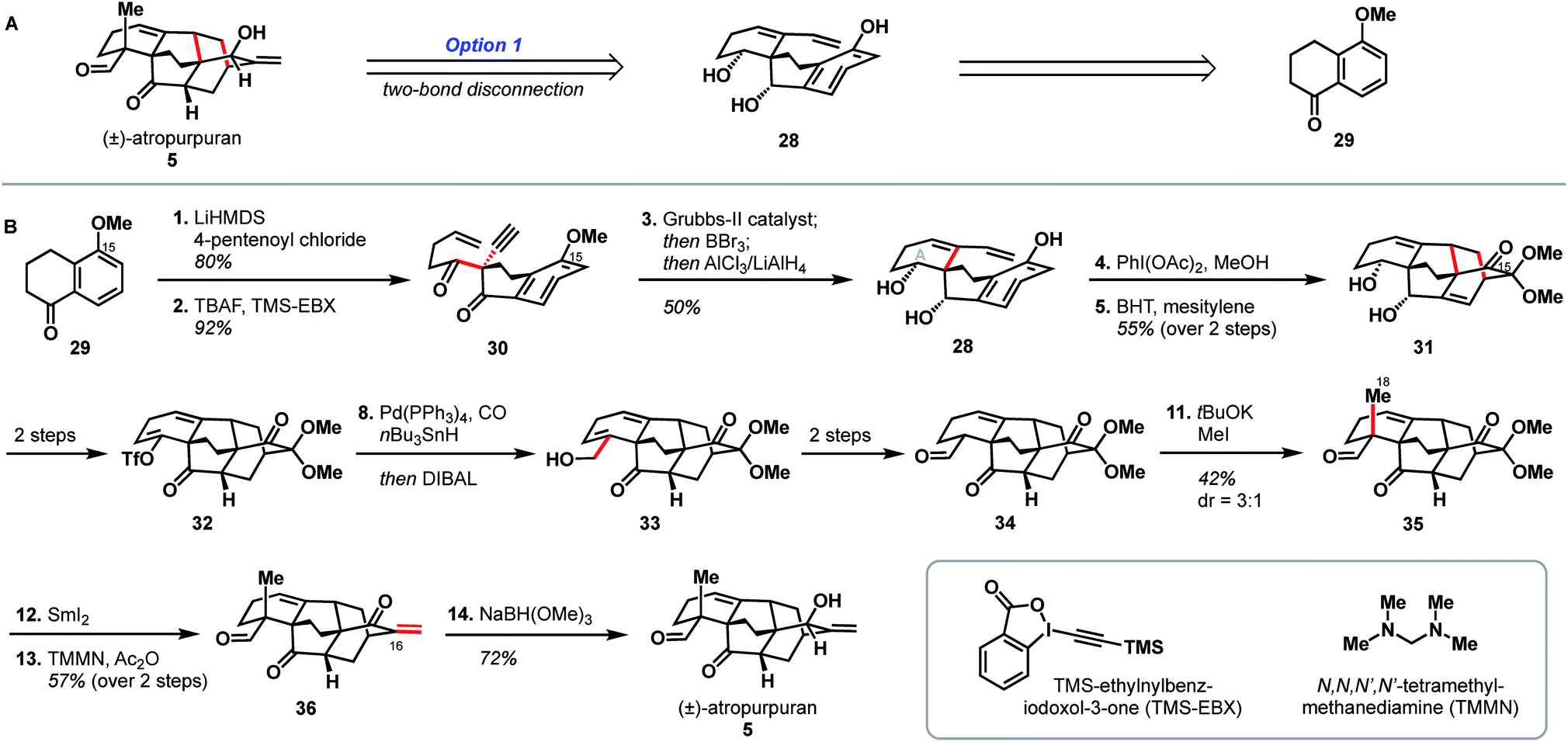 | ||
| Scheme 3 Key bond forming steps in Xu's synthesis of (±)-atropurpuran. | ||
Xu and coworkers' synthesis of atropurpuran benefited greatly from leveraging the most efficient disconnections, as identified by bond-network analysis, to access the tetracyclo[5.3.3.04,9.04,12]tridecane arcutane core in just four steps from commercial starting materials, showcasing the power of bond-network analysis as a tool for retrosynthesis of highly caged polycycles. Still, elements from the earlier synthesis of atropurpuran by Qin are also evident in Xu's strategy. For example, Xu's introduction of C18 and reduction of the penultimate intermediate (36), both proceeded analogously to transformations in the Qin synthesis. In addition, both groups utilized an oxidative dearomatization/intramolecular Diels–Alder sequence, a powerful, well-established way to generate [2.2.2]bicycles in diterpenoid alkaloid synthesis.17–21
Perhaps more intriguing than the similarities between the Qin and Xu syntheses are the differences—arising from small variations in the functional groups employed—that underpin the two routes to atropurpuran. The means by which the C19 aldehyde was installed, for example, differed significantly despite both routes utilizing C4 ketones as precursors with the same carbon backbone (see 37 and 39, Scheme 4A). In the case of the Xu synthesis, the β-ketone moiety at C20 in 39 (highlighted in purple) complicated the introduction of a carbon nucleophile into C4 since direct addition led to retro-aldol ring opening or non-specific decomposition. Xu therefore introduced the C19 aldehyde in a roundabout manner (see 31 → 35, Scheme 3). In contrast, these issues were circumvented in the Qin synthesis through the use of 37, which contained a silyl ether at C20. The other major difference is seen in the site of hydroxylation on the arene used in the shared oxidative dearomatization/Diels–Alder sequence. In the Qin synthesis, the hydroxy-bearing carbon in the arene maps onto the C16 carbonyl (see abbreviated structure 41, Scheme 4B). After olefination at this site, an allylic oxidation was required to introduce the C15 hydroxy group (41 → 42). Unfortunately, allylic oxidation favored the undesired diastereomer, necessitating two additional redox manipulation steps. Xu was able to avoid this issue by mapping the arene hydroxy group onto C15, facilitating a more direct conversion of the Diels–Alder adduct to the desired allylic alcohol (43 → 44), highlighting the dependence of synthetic efficiency not only on the sites of bond formation, but also on the functional handles chosen to build those bonds.
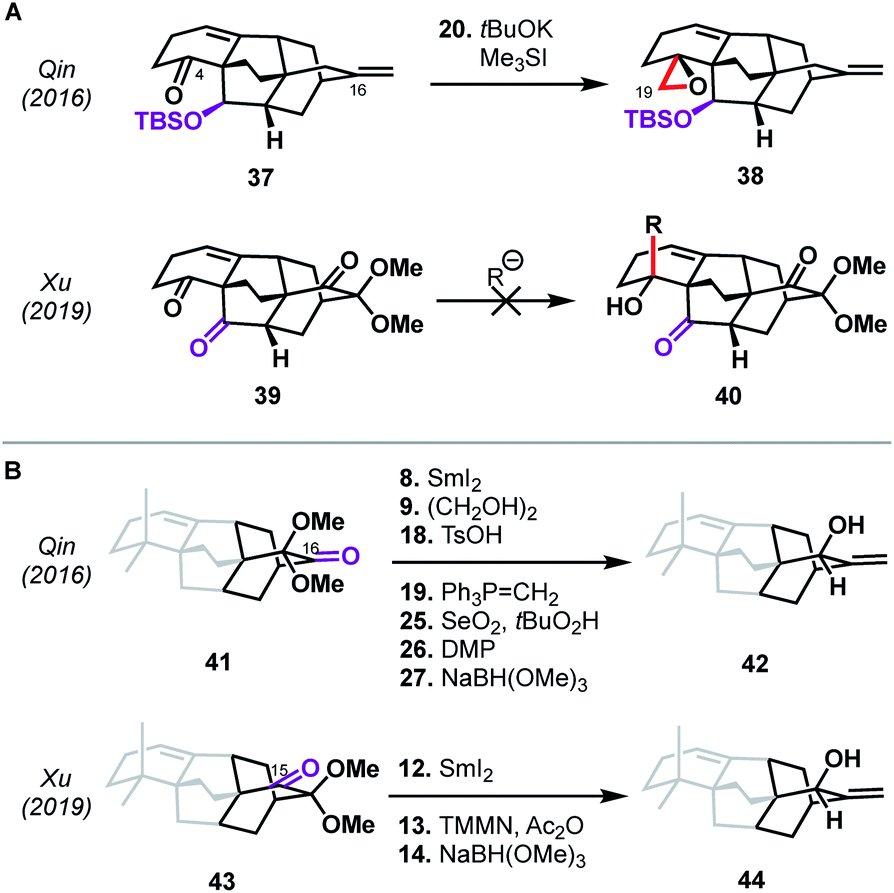 | ||
| Scheme 4 Comparison of the Qin and Xu syntheses of atropurpuran. (A) Installation of the C19 carbon. (B) Functional group manipulations at C15 and C16. The ABE ring system has been omitted for clarity. | ||
4. Syntheses of arcutane alkaloids
There are three reported syntheses of arcutane alkaloids to date—by the Qin,32 Sarpong,33 and Li34 groups—all of which were published in close succession in 2019. Qin and Sarpong identified similar bond-network disconnections, but with very different outcomes. In contrast, Li followed an intriguing bioinspired strategy.4.1 Qin (2019)
In addition to being the first group to report the synthesis of atropurpuran, Qin and coworkers were the first to report the syntheses of arcutinidine and arcutinine (Scheme 5A).32 As mentioned previously, the Qin group was unable to elaborate atropurpuran to arcutinidine, prompting the group to devise a new approach to the synthesis of the alkaloid congeners of the family. Notably, they utilized the same bond-network derived disconnection set 2.2 (Scheme 5A) but altered the timing of ring connections in the forward sense. The Qin synthesis began with the reductive Knoevenagal condensation of 1,3-cyclohexadione (47) and aldehyde 48 (ref. 35) to produce the coupled product (49). A Stork–Danheiser protocol involving methyl lithium addition into vinylogous ester 49, followed by hydrolysis to unveil an enone and conjugate addition of cyanide, generated nitrile 50. Elaboration of the silyl enol ether product in three steps gave ketone 51. Reduction of the nitrile moiety, followed by protection of the resulting primary amine with a tosyl group, formed alcohol 52, with all the desired functionality in place to begin stitching together the bridged elements of the arcutane framework. Upon protection of the C10 hydroxy group as a TMS ether, an aza-Wacker reaction forged the C5–C20 bond to produce pyrrolidine 53. TMS protection of the C10 hydroxy group proved essential to achieve the desired diastereoselectivity at C20. Pyrrolidine 53 was then progressed to phenol 46 in another three steps, at which point an oxidative dearomatization/intramolecular Diels–Alder sequence forged the CD bicycle, generating alkene 45. Much like in their atropurpuran synthesis, the final skeletal bond (C10–C9) was forged using a SmI2-mediated ketyl–olefin cyclization. The resultant alcohol (54) was then subjected to five additional steps to complete arcutinine (2), which was then converted to arcutinidine (3) through methanolysis.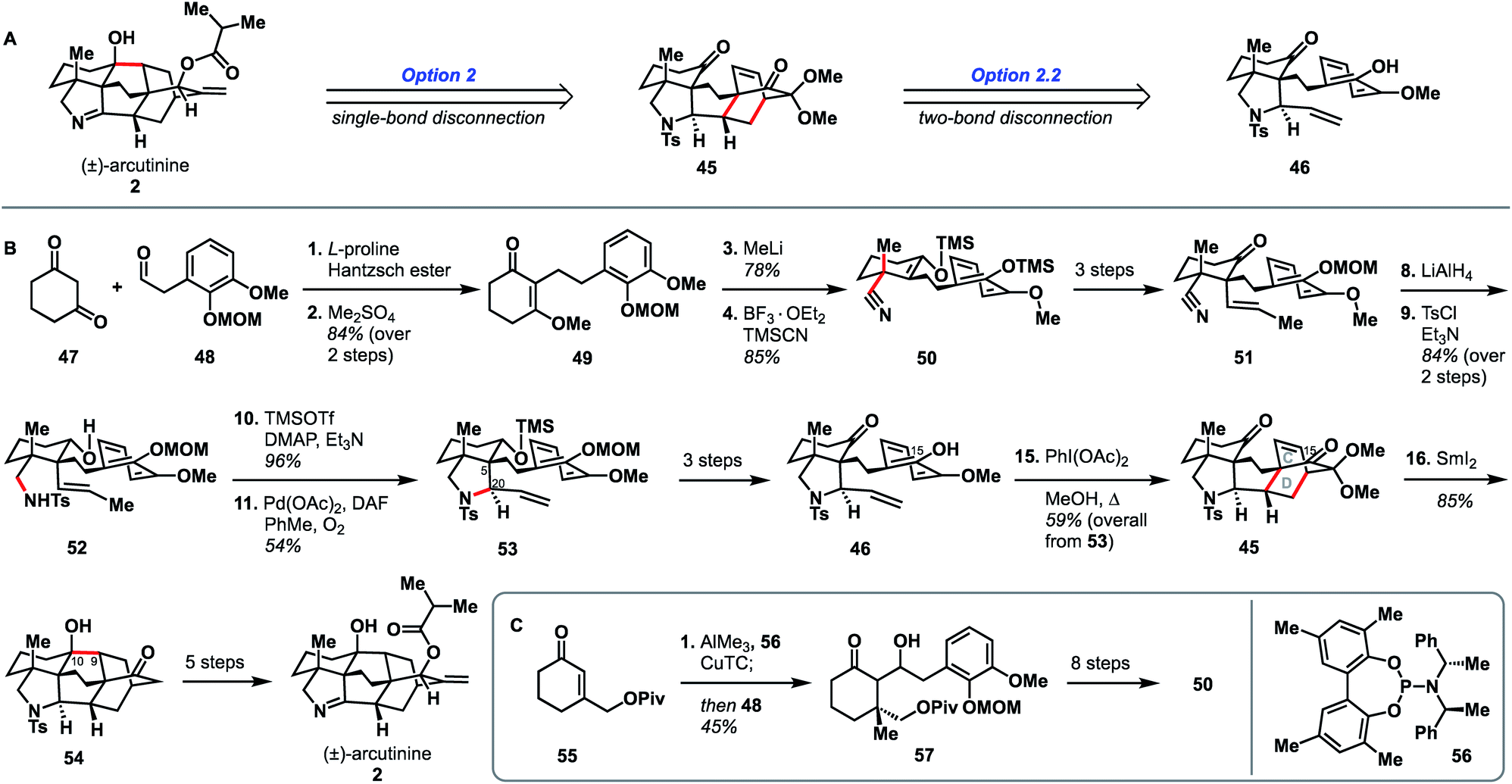 | ||
| Scheme 5 Bond-network analysis (A) and key bond forming steps (B) in Qin's synthesis of (±)-arcutinine. (C) Synthesis of enantioenriched 50. | ||
Although the initial 21-step (25 from commercial materials) synthesis of 2 by the Qin group was racemic, they rendered it enantioselective through an alternative route to access enantioenriched 50 (Scheme 5B). The modified synthesis began with an enantioselective conjugate addition of a methyl group into cyclohexenone 55,36,37 mediated by a copper complex generated from phosphoramidite 56, CuTC, and AlMe3. The resultant enol ether was then treated with aldehyde 48 to give aldol product 57 in 92% ee. Alcohol 57 was then converted to enantioenriched 50 in 8 steps. The enantioselective synthesis of (−)-arcutinine was achieved in 26 steps from known starting materials (30 from commercial).
It is valuable to compare the two Qin syntheses (of atropurpuran and arcutinine) and examine how they differ in their strategies despite both deriving from the same bond-network analysis derived option set (2.2, Fig. 2) and using the same key ring forming steps (oxidative dearomatization/intramolecular Diels–Alder and SmI2-mediated ketyl–alkene cyclization). One of the larger differences is the point at which the CD bicycle is built. In the atropurpuran synthesis, the CD bicycle was constructed relatively early (step 7) whereas in the arcutinine synthesis, it was built toward the end of the synthesis (step 15). By constructing the [2.2.2]bicycle early in the atropurpuran synthesis, the synthetic intermediates benefited from the greater conformational rigidity, facilitating high levels of diastereoselectivity. In contrast, amine 52 had larger conformational freedom, requiring the addition of a bulky TMS group to achieve the desired diastereoselectivity of the aza-Wacker cyclization (2.5:1 d.r., see Scheme 5, 52 → 53). Once the cis-fused AF ring system was installed with the desired stereochemistry at C20, the remaining synthesis proceeded smoothly and with diastereocontrol. In addition, by having the pyrrolidine ring in place, the ketyl–alkene cyclization occurred without the necessity of introducing a bulky group to force C9 and C10 close together, unlike in their atropurpuran synthesis (see Scheme 2, 24 → 25). In addition, the phenolic isomer that Qin used to build the arcutinine CD ring system is different from that used to build atropurpuran. Pursued contemporaneously with Xu's atropurpuran synthesis, Qin's arcutinine synthesis again highlights the improved efficiency imparted by mapping the phenolic hydroxy group onto C15 instead of C16 (see Scheme 5, 46 → 45), as had been done in the original atropurpuran synthesis (see Scheme 2, 20 → 19).
4.2 Sarpong (2019)
Shortly after the publication of Qin and coworkers' synthesis of arcutinine, related syntheses were reported by the Sarpong and Li groups. Here, we discuss the Sarpong group's synthesis first.33 Similar to Qin's retrosynthetic strategy, the Sarpong synthesis took advantage of the bond-network derived disconnection set 2.2. The synthesis began by bringing together known starting materials 60 and 61 through a Wittig reaction. DBU-mediated isomerization of the resulting double bond gave citraconamide 62 (Scheme 6B). Treatment of 62 with triflic acid effected a Friedel–Crafts cyclization to furnish hemiaminal 63. The hemiaminal (63) was then ionized with AlCl3 to produce an oxopyrollium dienophile, which participated in a Diels–Alder cycloaddition to generate tetracycle 64, which was itself carried forward in five steps to phenol 59. As in the previous syntheses, phenol 59 was subjected to an oxidative dearomatization/intramolecular Diels–Alder sequence to forge the CD bicycle. However, in contrast to previous syntheses, the dienophile was appended onto the phenol through a Wessely-type oxidation,38 rather than through tethering to the carbon skeleton. The Diels–Alder adduct (65) was then converted to diketone 58 in three steps. The last C9–C10 bond was forged using a SmI2-mediated cyclization to give pinacol 66, which was advanced to arcutinidine in another nine steps. Overall, Sarpong's racemic synthesis of the natural product derivative, arcutinidine, was completed in 25 steps from commercially available material.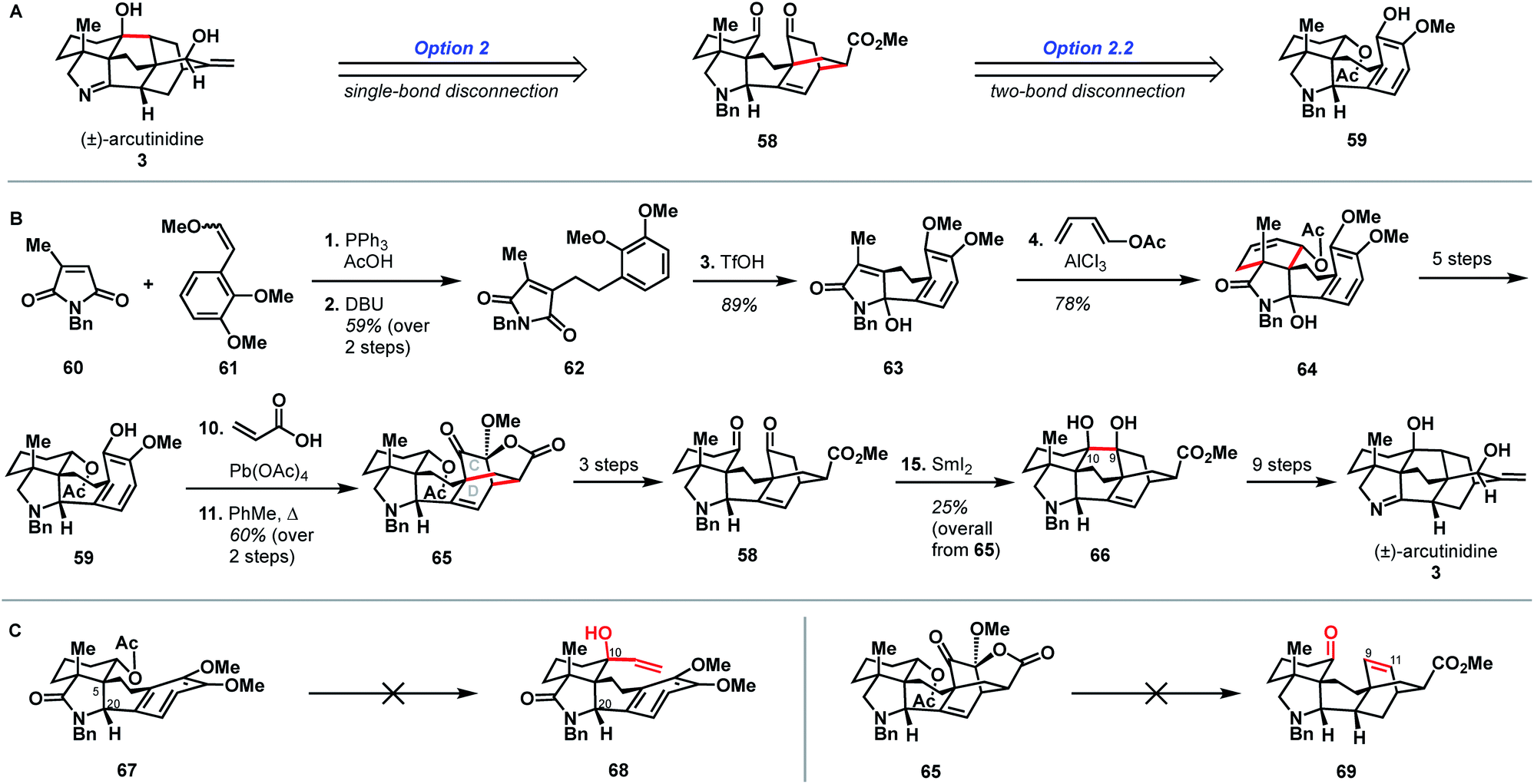 | ||
| Scheme 6 Bond-network analysis (A) and key bond forming steps (B) in Sarpong's synthesis of (±)-arcutinidine. (C) Unsuccessful transformations in Sarpong's synthesis. | ||
It is informative to compare the Qin and Sarpong syntheses as both take advantage of the bond-network derived disconnection option 2.2. Both leverage the same strategic disconnections: the C9–C10 bond of the maximally bridged ring, followed by two-bond disconnection of the CD ring system to give a fused precursor. Qin's two-bond disconnection cleaved the D ring, whereas Sarpong's removed the C15–C16 bridge common to the C and D rings. This unusual disconnection led to the use of a Wessely-type oxidative dearomatization to append a dienophile that facilitated the formation of the CD bicycle. However, Diels–Alder adduct 65 contained superfluous oxygenation at C9, which could not be converted into a suitable substrate for ketyl–olefin coupling (5) and required deoxygenation in two subsequent concession steps (see Scheme 5B, 45 → 54 and Scheme 6C, 65 → 69).
While the choice to disconnect the CD C15–C16 bridge in the retrosynthetic sense is what enabled the completion of arcutinidine, it was not the original plan. The Sarpong group originally sought to use option 1 from the bond-network analysis, as realized by Xu in the context of atropurpuran (Scheme 6C). There, the maximally bridged ring would have been forged through cycloaddition of a fused precursor, ideally allylic alcohol 68. However, all attempts to introduce a vinyl group at C10 from intermediate 67 were unsuccessful, likely due to steric encumbrance from the C5 quaternary center, as well as the concavity of the molecule created by the cis-fused AF ring system and the C20 stereocenter. The lack of success in this context showcases how bicyclization transforms, while rapidly increasing structural complexity, also rapidly increase steric congestion, which can introduce new challenges in the forward synthesis. Turning this impasse into an opportunity, the concavity of the molecule was leveraged to effect a diastereoselective Wessely oxidation. Changing the disconnection option ultimately enabled the completion of a synthesis of arcutinidine, however at the cost of a significantly longer synthesis.
4.3 Li (2019)
Li's synthesis of arcutane alkaloids34 takes an elegant bioinspired approach that is significantly different from the Qin and Sarpong approaches. Specifically, the Li group leverages a Prins/Wagner–Meerwein cascade to convert a hetidine-type scaffold to an arcutane skeleton; a rearrangement that had been described in one of the two biosynthetic proposals (Scheme 1B). Li's synthesis began from enantioenriched alcohol 72,39,40 which was elaborated to aldehyde 73 in two steps. A Diels–Alder reaction between dienophile 73 and diene 74 afforded adduct 75. Alkene 75 was advanced over two steps to 71, which upon deprotonation with LiHMDS underwent a second, intramolecular, anionic Diels–Alder reaction to forge the CD bicycle. The resultant alcohol (76) was then dehydrated to give alkene 70. Ionization of the MOM group gave oxocarbenium ion 77 that then underwent the key Prins reaction to produce cation 78 followed by a [1,2]-alkyl shift to produce 79 of the arcutane framework. The newly formed arcutane framework was then brought forward in another seven steps to alcohol 80. Reductive amination of 80 with hydroxylamine, followed by cyclization to the pyrrolidine and diastereoselective reduction of the remaining carbonyl, produced arcutinidine (3). With arcutinidine in hand, esterfication with iPrCO2H or (S)-sBuCO2H gave arcutinine (2) or arcutine (1), respectively. In addition to synthesizing almost all of the arcutane alkaloids, the Li group was able to correct the stereochemistry of the s-butyl ester side chain, which had been misassigned in the original isolation report.3 Overall, the Li synthesis, with its key bioinspired rearrangement, is the shortest reported synthesis of an arcutane alkaloid to date (18 steps from known alcohol 72, 21 steps from commercial).There is a lot to be learned from Li and coworkers' bioinspired synthesis. First, it provides experimental evidence for the plausibility of Sarpong's proposed biosynthesis (Scheme 1B). In addition to proving that the rearrangement between the hetidine and arcutane skeletons is feasible, Li also showed that it is possible to introduce nitrogen and complete the pyrrolidine ring from the corresponding keto-aldehyde (Scheme 7, 80 → 3). The Qin group had been unable to effect the same transformation from atropurpuran, citing the C4 quaternary center and the C1–C10 alkene as likely impediments. On the contrary, Li showed that the C4 quaternary center does not prevent N incorporation (see 80 → 3); it is possible that the C1–C10 alkene impedes cyclization. That said, the Li group did not advance intermediate 80 to atropurpuran. This was not commented upon in their report.
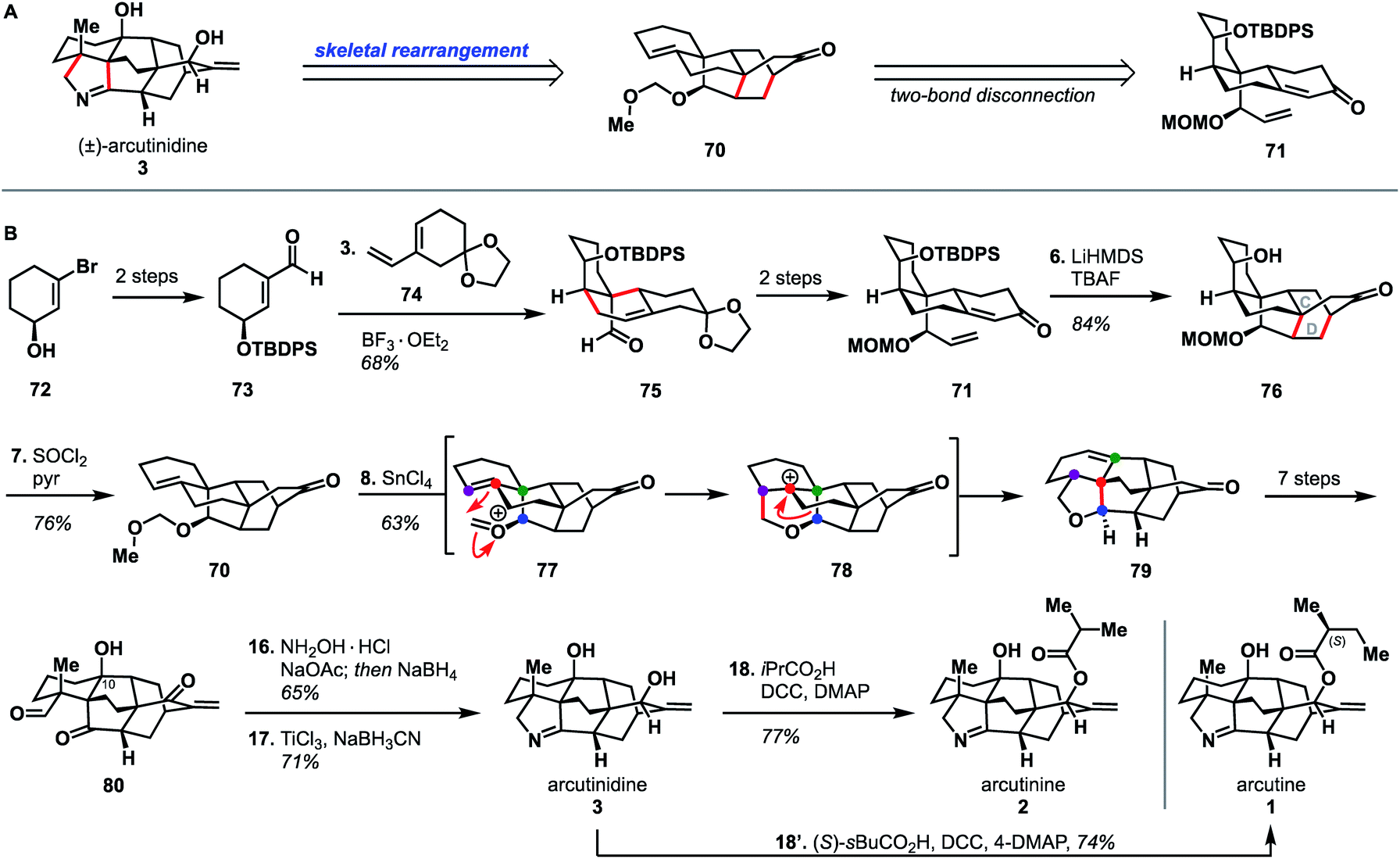 | ||
| Scheme 7 Bond-network analysis (A) and key bond forming steps (B) in Li's synthesis of the arcutane alkaloids. | ||
In addition to lending credence to the proposed arcutane biogenesis, the Li synthesis also exposes a potential limitation of bond-network analysis. In network analysis, emphasis is placed on disconnection at the maximally bridged ring, which is believed to represent structural complexity. As skeletal rearrangements typically involve both building and breaking sigma bonds, network analysis does not align itself well with identifying potential rearrangements. The value of such rearrangements is difficult to define; however, they can provide the ability to trace back a complex bridged framework to a more readily accessible one and have been used in other diterpenoid alkaloid syntheses.18,41–45 In the case of the Li synthesis of the arcutane-type alkaloids, the bioinspired rearrangement is central to what is the shortest reported syntheses to date. That said, bioinspired syntheses and retrosyntheses borne out of bond-network analysis are not necessarily mutually exclusive; Li's rearrangement forges the maximally bridged ring at a late stage and all other disconnections follow the tenets of bond-network analysis.
The Li synthesis is also noteworthy in the use of a convergent46 Diels–Alder reaction to bring together fragments 73 and 74 (see Scheme 7), allowing for a reduced linear step count. By using this atom economical28 Diels–Alder cycloaddition, the Li group avoided the introduction of additional heteroatom-containing functional groups. This strategy was also utilized in the construction of the CD bicycle (see 71 → 76), further contributing to the overall efficiency of the synthesis. Li's Prins/Wagner–Meerwein cascade also takes advantage of functionality and unsaturation inherent within the arcutanes. The Xu group similarly relied on C–C π bonds to effect the desired bond forming transforms (see Scheme 3, 30 → 28 → 31). While the Li and Xu syntheses took very different approaches to arrive at the arcutane framework, both groups were able to minimize the introduction of heteroatom-containing functional groups that would later need to be removed, which allowed them to achieve the shortest syntheses of arcutinidine and atropurpuran, respectively.
5. Studies of bridged polycycles en route to the arcutane skeleton
In addition to the completed syntheses of the arcutane natural products, there have been a number of synthesis studies toward the arcutane skeleton. While not all these studies led to completed syntheses of the full arcutane framework, they each provided key insights.5.1 Kobayashi (2011)
The earliest report of synthetic progress toward the arcutane natural products was by Kobayashi and coworkers, who constructed the arcutane ABCDE ring system (81).47 The Kobayashi studies toward atropurpuran began from phenol 83 (Scheme 8), which was advanced in 6 steps to ketone 84 in an approach featuring retrosynthetic disconnections that correspond to option 1 from the bond-network analysis (see Scheme 8A). Double allylation of 84, followed by debenzylation, gave guaiacol 85. Grignard addition of allyl magnesium bromide into the carbonyl moiety furnished triene 86. Selective ring-closing metathesis closed the A ring to give diene 82. In keeping with the theme of the previously discussed atropurpuran syntheses, fused tricycle 86 underwent an oxidative dearomatization/intramolecular Diels–Alder sequence to build the CD ring system (87), completing the arcutane ring system. Bicyclization of 86 and subsequent ring-closing metathesis was also achieved. However, 85 was not a suitable substrate for bicyclization as it spontaneously dimerized upon oxidative dearomatization. Diene 87 was then pushed forward through two additional steps to alkene 81, which lacks substitution at C4 and C20. These missing substituents make 81 an unlikely candidate for further advancement to atropurpuran; such an endeavor would necessitate significant changes to the earlier stage of the synthesis. Building on this proof of concept, Kobayashi also published a separate synthesis of the two fused bicyclo[2.2.2]octane units (termed dibarrelane, which corresponds to the BCDE rings of the arcutane skeleton).48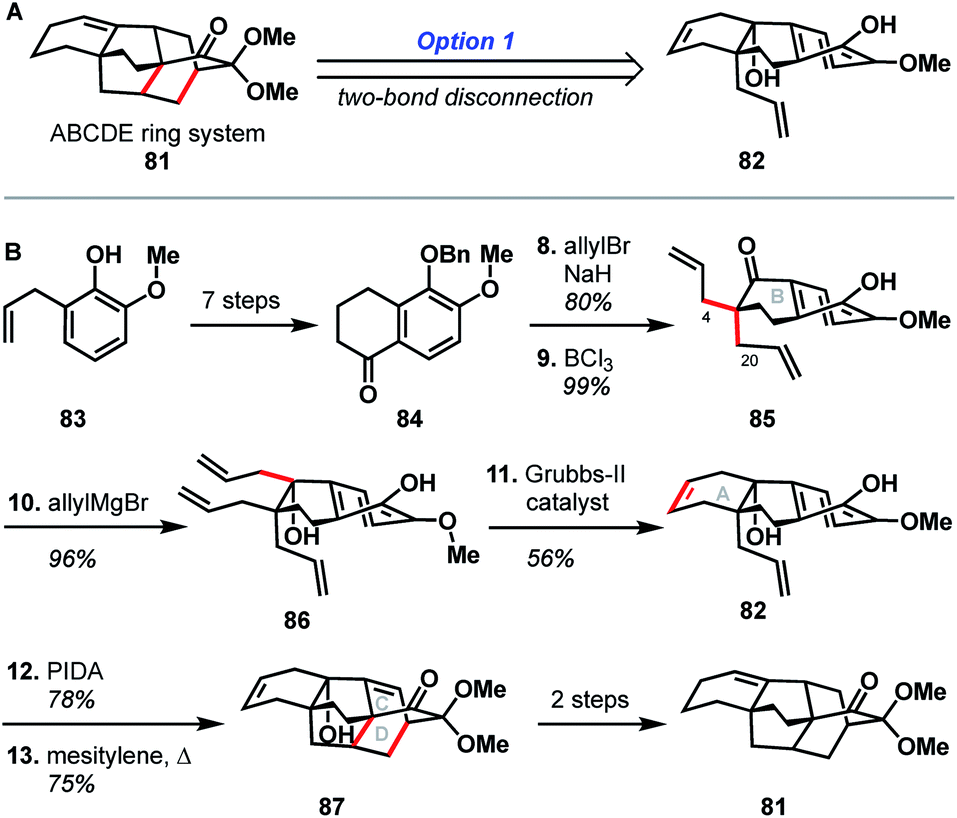 | ||
| Scheme 8 Bond-network analysis (A) and key bond forming steps (B) in Kobayashi's synthesis of the arcutane framework. | ||
While Kobayashi and coworkers did not ultimately complete a total synthesis of atropurpuran, this work appears to have laid part of the groundwork for Xu and coworkers' synthesis of atropurpuran (see Section 3.2). In both syntheses, an oxidative dearomatization/intramolecular Diels–Alder cycloaddition is employed to simultaneously construct all the bridged ring systems in the arcutane scaffold (see option 1, Fig. 2). Comparison of the fused precursors also provides further insight into the similarities of the two plans. Xu's ketone 29 resembles Kobayashi's 84. Ketone 29 is, however, commercially available as opposed to ketone 84, which was synthesized in 7 steps from ortho-eugenol (83). Both groups then append unsaturated groups alpha to the carbonyl to be utilized in a Grubbs ring-closing metathesis to forge the A ring. For this purpose, Kobayashi uses two equivalent allyl groups whereas Xu employs more highly functionalized substituents. Kobayashi's inspiring report of the synthesis of the arcutane ABCDE ring system thus represented a major advancement toward the total synthesis of atropurpuran, the completion of which would not be reported for another five years by others.
5.2 Hsung (2012)
In 2012, Hsung and coworkers reported their progress toward atropurpuran, using a strategy that relies heavily on pericyclic reactions (see Scheme 9).49 Their synthesis began with the pairing of allenamide 89 (ref. 50) and allyl bromide 88 to produce coupled product 90. Treatment of 90 with CSA led to a formal [1,3]-hydride shift to give tetraene 91. A 6π electrocyclic ring closure of tetraene 91 yielded diene 92, which then underwent an intramolecular Diels–Alder reaction in the same pot to furnish 93. Tricycle 93 was then elaborated to allylic alcohol 94 in three additional steps. Hsung and coworkers believed the enantiomer of 94 (95) would map onto the BCD ring system of the arcutane framework. Alternatively, due to the symmetry inherent in the BCDE ring system, allylic alcohol 94 could also map onto the CDE ring system (Scheme 9). In either case, however, the authors concede that the stereochemistry at C9 on 95 (C14 for 94) would need to be inverted in order to advance to a successful total synthesis of the arcutane natural products.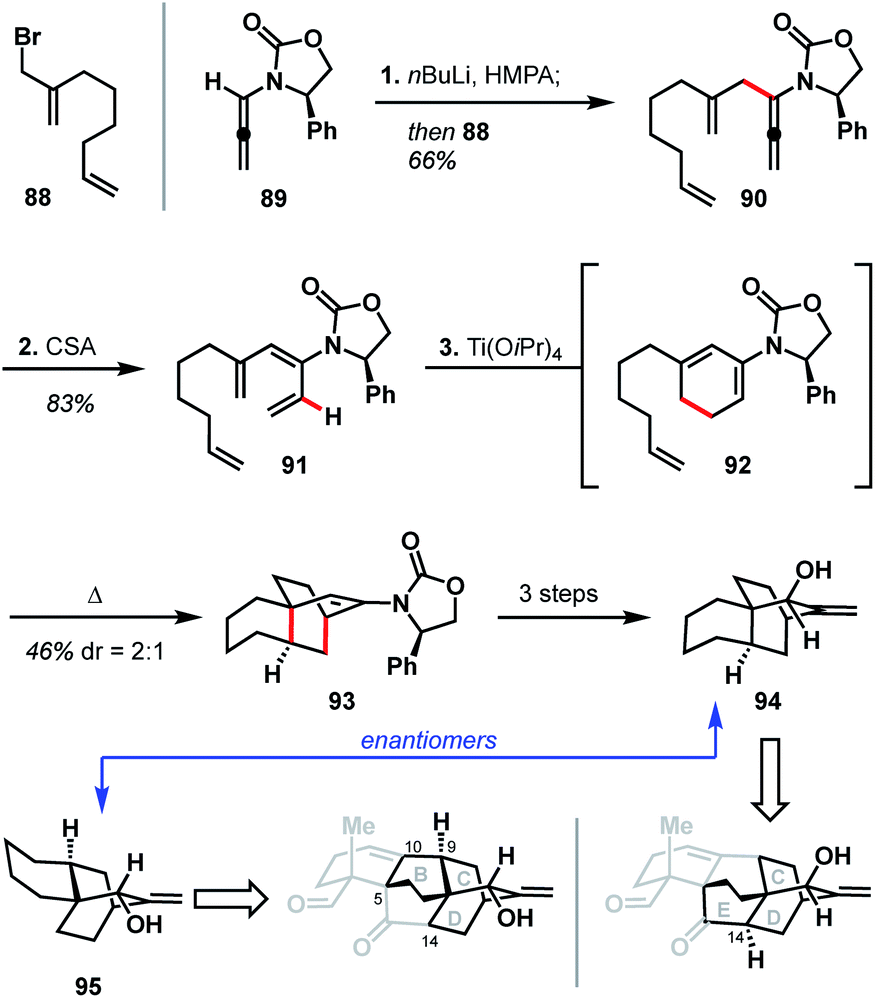 | ||
| Scheme 9 Key bond forming steps in Hsung's synthesis of the arcutane BCD ring system. | ||
The Hsung retrosynthesis of atropurpuran highlights the cyclization methodology developed: a unique use of a triene as a masked diene for an intramolecular Diels–Alder to produce the BCD ring system. While this method provides an expedient route to three of the five rings of atropurpuran, it did not prove fruitful for the complete synthesis of atropurpuran. Furthermore, the transformations reported by Hsung did not feature functional groups at the key bridging sites (C5, C10, and C14) of 95 nor did it provide ways to introduce functionality, which would be required for a successful total synthesis. However, the Hsung approach does highlight the potential of reactions that forge multiple C–C bonds in a single step, as well as the utility of pericyclic reactions in constructing heteroatom-poor natural products. In addition, the Hsung approach would minimize functional group manipulation, allowing production of the BCD carbon scaffold in only three steps from known materials (seven steps from commercial in the longest linear sequence).
5.3 Singh (2016)
The Singh group was drawn to the [5.3.3.04,9.04,12]tetracycle (BCDE ring system) within the arcutane framework and reported a unique synthesis of this scaffold.51 Their synthesis began with the oxidative dearomatization of diol 96, followed by Diels–Alder cycloaddition with ethyl acrylate to produce epoxide 97. The epoxide was then carried forward six steps to aldehyde 98, excising the epoxide group rather than retaining what could have become C17. Wittig olefination effected a two-carbon homologation to produce unsaturated ester 99, which was then advanced to alkene 100 in three steps. Formation of the C9–C10 bond through the intermediacy of an acyl radical gave diketone 101. Cleavage of the methyl ether with BBr3 gave alcohol 102, which then underwent a domino oxidation/aldol sequence to furnish the complete BCDE ring system (103).Examination of the Singh synthesis through the lens of bond-network analysis is impeded by the fact that the A ring was never installed. However, by analogy to a full arcutane scaffold and the other reported syntheses, some insights can be gleaned. Similar to Qin's synthesis of atropurpuran, the Singh approach began with an oxidative dearomatization/Diels–Alder cycloaddition sequence to build the CD bicycle. From there, the Singh group made an interesting choice of excising the C17 carbon (see Scheme 10, 97 → 98) which would be required to complete a synthesis of atropurpuran. The maximally bridged ring was then forged before the last bridging element was secured, in stark contrast to the other syntheses where closure of the maximally bridged ring completed the installation of bridging elements.
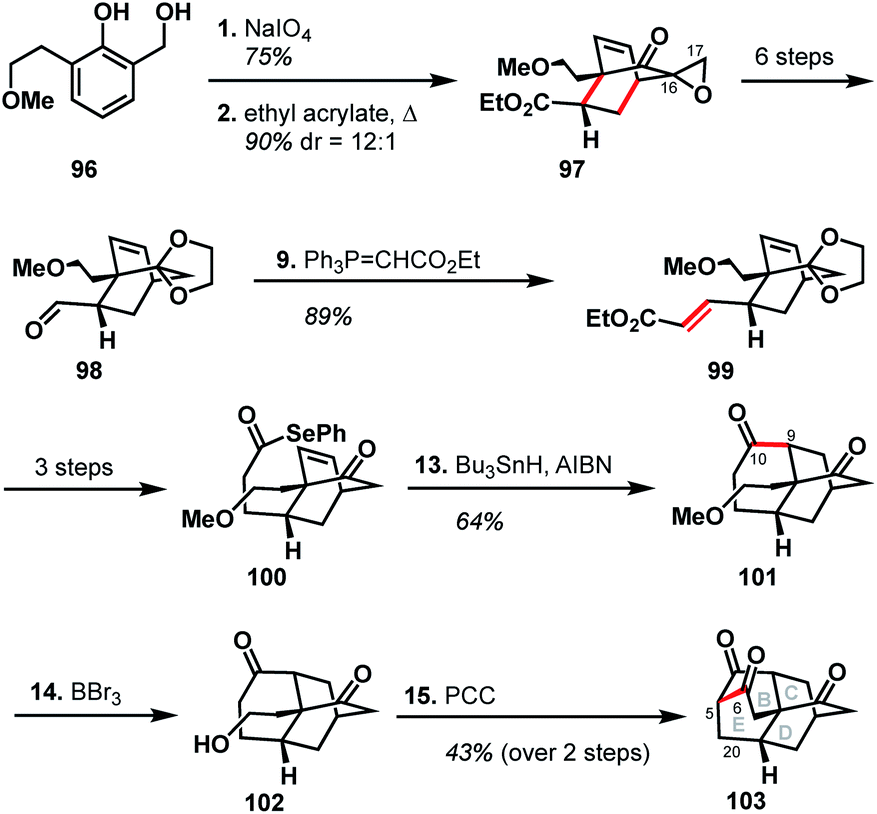 | ||
| Scheme 10 Key bond forming steps in Singh's synthesis of the arcutane BCDE ring system. | ||
There are several clues as to why this synthesis was not advanced to the full arcutane framework. First, dibarrelane-like 103 lacked the required functionality at C20, as well as at the C5 bridgehead, which would be challenging to install later in the synthesis. While such elements could be installed earlier in the synthesis when the intermediates were more highly functionalized, the Singh group instead removed functional handles that could have been leveraged for this purpose (see the loss of C17 in 97 → 98 and loss of unsaturation in 99 → 100). Additionally, the three functional groups resident in 103 are all ketone moieties that are not readily distinguishable. The synthesis also relied on different heteroatom-based functional groups for each stage of the synthesis, which would have led to a significantly longer synthesis than many of those discussed previously.
5.4 Qin (2016)
In the same year that they published their successful synthesis of atropurpuran, Qin and coworkers also reported a distinctly different route to the arcutane ABC ring system.52 The Qin approach began with ketoester 106, which was advanced to aldehyde 107 in five steps (Scheme 11). Prolinol derivative 108 was utilized to effect enantioselective closure of the C ring and produce bicycle 109, in a manner similar to their organocatalytic synthesis of a potential A ring fragment.30 After selective protection of the aldehyde carbonyl in 109, intramolecular Michael addition gave lactone 110, which was converted to aldehyde 111 in six additional steps. Addition of an organolithium species generated from stannane 112 into aldehyde 111 yielded alcohol 113. Conversion of 113 to diketone 114 over two steps set the stage for a second conjugate addition to close the A ring. Tricycle AY was intended to map onto the ABC ring system of the arcutane scaffold. However, 115 was the C8 epimer of the desired product, resulting in C15 and C16 mapping onto the incorrect carbons of atropurpuran.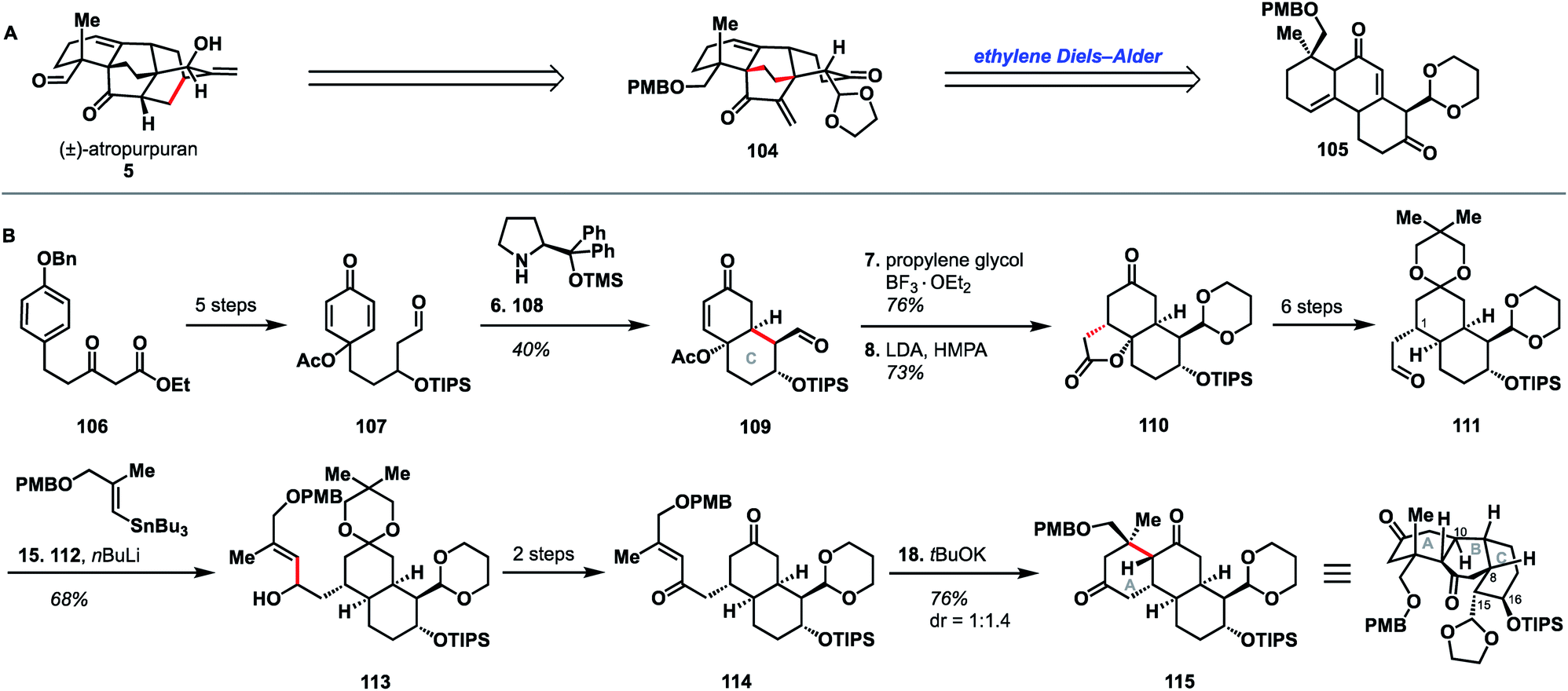 | ||
| Scheme 11 (A) Retrosynthetic analysis of atropurpuran. (B) Key bond forming steps (B) in Qin's synthesis of the arcutane ABC ring system. | ||
The Qin retrosynthesis, which did not align with the bond-network analysis for this scaffold, was designed to probe the validity of the ethylene Diels–Alder reaction that Wang had proposed for the biosynthesis of atropurpuran.6 Unfortunately, given the undesired stereochemistry at C8 in tricycle 115, Wang's hypothesis remained untested. It is worth noting that this study from Qin is the only reported attempt to synthesize the arcutane scaffold in which the authors began with one of the central rings (E or B) and worked outward, annealing the other rings sequentially. This appears to have reduced overall synthetic efficiency; synthesis of tricycle 115 took 18 steps (21 from commercial), as opposed to the 27 total steps (28 from commercial) needed for their total synthesis of pentacyclic atropurpuran. That said, the disparity in step count was exacerbated by the reliance of the route outlined in Scheme 11 on carbonyl chemistry, which necessitated repeated manipulation of protecting groups. Still, the unviability of a synthesis of atropurpuran from 115, regardless of the number of steps involved, further validates the efficacy of bond network analysis as a tool in retrosynthesis.
6. Conclusions
These syntheses of atropurpuran and the arcutane alkaloids, both complete and partial, showcase the synergy between the described strategies for retrosynthesis. The successful syntheses of the arcutane natural products additionally open doors for investigation of the biological activity and biosynthesis of these fascinating diterpenoids. Studies that resulted in the synthesis of the full arcutane core are summarized in Fig. 4, which highlights key retrosynthetic disconnections, the number of steps required to reach each intermediate, as well as the overall yield for each synthesis. Trends in disconnections and associated strategies utilized in the forward syntheses are discussed in the context of the arcutane-type natural products, including both completed and partial syntheses.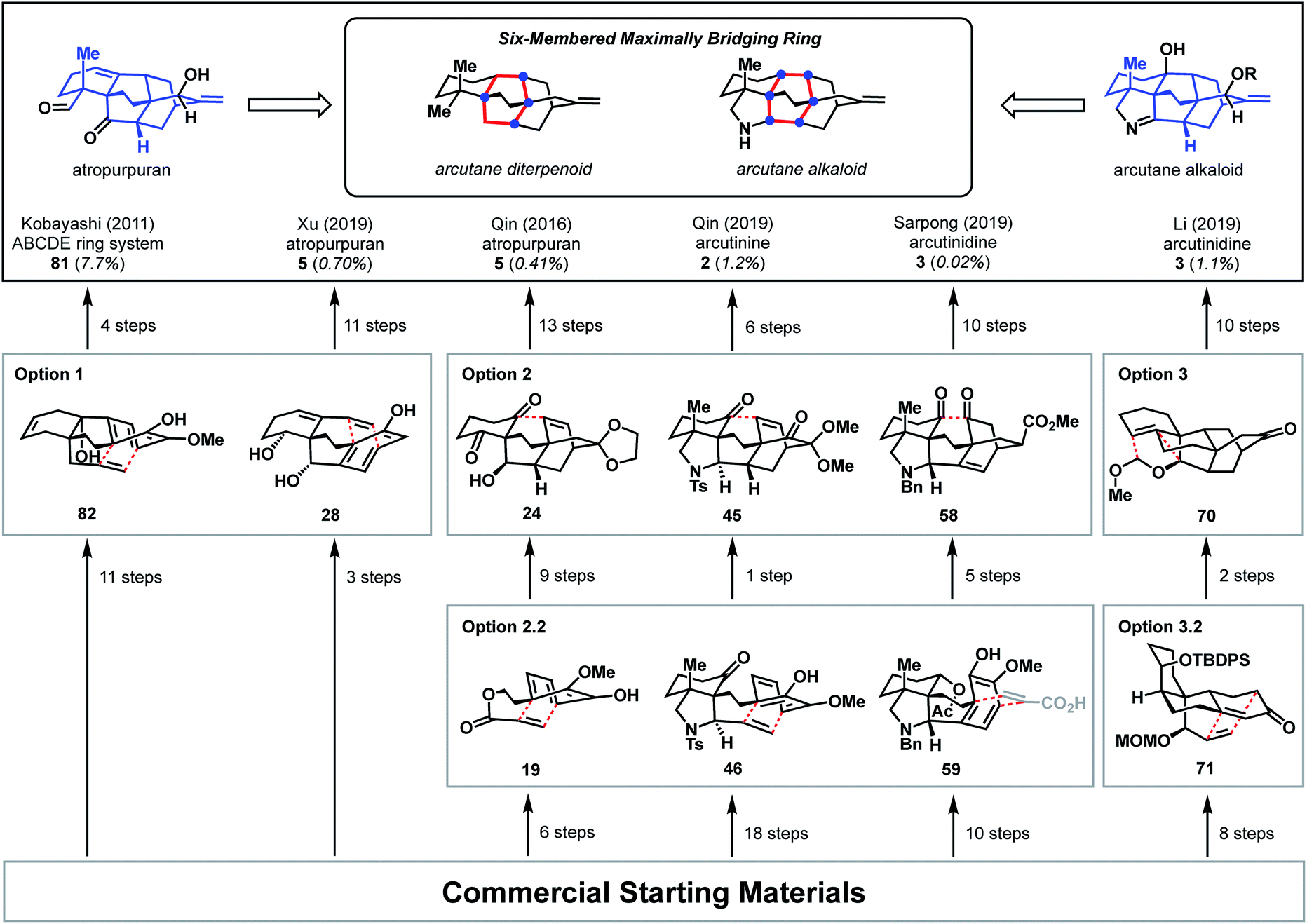 | ||
| Fig. 4 Comparison of bond-network disconnections utilized to synthesize the arcutane framework. | ||
Taken together, these syntheses show that bond-network analysis can provide an effective starting point in retrosynthesis, guiding the synthetic chemist in the development of an efficient synthesis. The tenets of bond-network analysis were by-and-large followed in all of the completed arcutane syntheses, whether intentionally or not. While both one- and two-bond disconnections can be considered strategic, two-bond disconnections proved particularly effective. All successful syntheses included at least one bicyclization step. This fact highlights not only the reliability and the ubiquity of the Diels–Alder transform, the most common method used herein for two-bond disconnection of [2.2.2]bicycles, but also the inherent preference for the application of bicyclization transforms to achieve a rapid decrease in complexity in the retrosynthetic direction. Not only was this transform utilized in the total synthesis of the arcutane-type natural products, but also in many of the efforts toward the arcutane scaffold. However, a remaining challenge in the synthesis of such highly bridged polycycles is to leverage these disconnections for convergent synthesis. Of the syntheses surveyed here, only Li's arcutane alkaloid synthesis involves coupling of two cyclic components.
Additionally, although two-bond disconnections tended to correlate positively to step efficiency, the functional handles utilized to forge skeletal bonds in the forward sense were also shown to have a large impact. Functional group-oriented analysis is thus an important secondary consideration in retrosynthesis. The challenge is to minimize use of functional handles that one would need to remove later, leveraging functionality present in the natural product wherever possible. In the context of arcutane natural products, which contain relatively few heteroatoms, this presents an additional challenge: how to forge skeletal C–C bonds with minimal reliance on heteroatom-based functional groups. While alkenes are often utilized throughout the syntheses discussed herein—enabling various Diels–Alder cycloadditions, Xu's enyne metathesis, and more—there remains significant room for advancement in this area as the majority of these transformations still involved at least one heteroatom that did not appear in the target structure. The exception to this was Hsung's series of C–C bond-forming pericyclic reactions en route to the arcutane scaffold.
As bond-network analysis is based on bond disconnection, it necessarily focuses on structural—rather than synthetic—complexity and, thus, emphasizes the closure of new rings over rearrangement processes. This is an area where transform-oriented and bioinspired retrosynthesis can augment the topology-oriented bond-network analysis. The potential benefits of such a hybrid approach become apparent when one considers the Li synthesis, which features a bioinspired Prins/Wagner–Meerwein cascade and is the shortest synthesis of the arcutane alkaloids to date.
Bond-network analysis, unbiased by known reactions, can identify strategic disconnections for which no precedent exists and can thus challenge chemists to find creative solutions and drive development of new reactions. This critical examination of the arcutane syntheses underscores this idea, as well as the inventiveness of the synthetic groups towards solving problems. While there were not many examples of new reactivity—such as Sarpong's use of the oxopyrollium Diels–Alder reaction or Hsung's pericyclic cyclization sequence—these syntheses contribute to the paradigm shift in the use of “protecting groups”. A number of the syntheses utilized traditional protecting groups to enforce the desired diastereoselectivity in reactions (e.g., the use of a TMS group in Qin's arcutinine syntheses) or as carbon sources (e.g., Li's use of a MOM group in their arcutinidine synthesis). The creativity displayed is also highlighted by the fact that multiple groups chose similar disconnection types but arrived at the natural products through very different means.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided to R. S. by the National Institutes of Health (NIGMS R35 GM130345). N. A. D. thanks the NSF for a graduate research fellowship (DGE 1106400).Notes and references
- F. P. Wang and X. T. Liang, Alkaloids, 2002, 59, 1–280 Search PubMed.
- F. P. Wang, Q. H. Chen and X. Y. Liu, Nat. Prod. Rep., 2010, 27, 529–570 RSC.
- B. Tashkhodzhaev, Sh. A. Saidkhodzhaeva, I. A. Bessonova and M. Y. Antipin, Chem. Nat. Compd., 2000, 36, 79–83 CrossRef CAS.
- Sh. A. Saidkhodzhaeva, I. A. Bessonova and N. D. Abdullaev, Chem. Nat. Compd., 2001, 37, 466–469 CrossRef.
- X.-H. Meng, Z.-B. Jiang, Q.-L. Guo and J.-G. Shi, Chin. Chem. Lett., 2017, 28, 588–592 CrossRef CAS.
- P. Tang, Q.-H. Chen and F. P. Wang, Tetrahedron Lett., 2009, 50, 460–462 CrossRef CAS.
- A. Ameri, Prog. Neurobiol., 1998, 56, 211–235 CrossRef CAS PubMed.
- W. Zhuoting, Chinese Pat., 201611205012, 2017.
- W. Zhuoting, Chinese Pat., 201611206414, 2017.
- W. Zhuoting, Chinese Pat., 201611205627, 2017.
- Biomimicry in Organic Synthesis, in Bioinspiration and Biomimicry in Chemistry: Reverse-Engineering Nature, ed. R. W. Hoffmann and G. F. Swiegers, Wiley, Hoboken, NJ, 2012, pp. 415–453. also see references therein Search PubMed.
- Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2010, 132, 5375–5386 CrossRef CAS PubMed.
- E. C. Cherney and P. S. Baran, Isr. J. Chem., 2011, 51, 391–405 CrossRef CAS PubMed.
- M. Weber, K. Owens and R. Sarpong, Tetrahedron Lett., 2015, 56, 3600–3603 CrossRef CAS PubMed.
- C. Le Drain and P. Vogel, Helv. Chim. Acta, 1987, 70, 1703–1720 CrossRef.
- P.-J. Zhao, S. Gao, L.-M. Fan, J.-L. Nie, H.-P. He, Y. Zeng, Y.-M. Shen and X.-J. Hao, J. Nat. Prod., 2009, 72, 645–649 CrossRef CAS PubMed.
- S. Zhou, R. Guo, P. Yang and A. Li, J. Am. Chem. Soc., 2018, 140, 9025–9029 CrossRef CAS PubMed.
- E. C. Cherney, J. M. Lopchuk, J. C. Green and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 12592–12595 CrossRef CAS PubMed.
- A. M. Hamlin, D. Lapointe, K. Owens and R. Sarpong, J. Org. Chem., 2014, 79, 6783–6800 CrossRef CAS PubMed.
- J. Liu and D. Ma, Angew. Chem., Int. Ed., 2018, 57, 6676–6680 CrossRef CAS.
- X.-H. Li, M. Zhu, Z.-X. Wang, X.-Y. Liu, H. Song, D. Zhang, F. P. Wang and Y. Qin, Angew. Chem., Int. Ed., 2016, 55, 15667–15671 CrossRef CAS PubMed.
- E. J. Corey, W. J. Howe, H. W. Orf, D. A. Pensak and G. Petersson, J. Am. Chem. Soc., 1975, 97, 6116–6124 CrossRef CAS.
- E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, New York, 1995 Search PubMed.
- N. A. Doering, R. Sarpong and R. W. Hoffmann, Angew. Chem., Int. Ed., 2020 DOI:10.1002/ange.201909656.
- R. W. Hoffmann, Elements of Synthesis Planning, Springer-Verlag, Berlin Heidelberg, 2009 Search PubMed.
- J. Gong, H. Chen, X.-Y. Liu, Z.-X. Wang, W. Nie and Y. Qin, Nat. Commun., 2016, 7, 12183 CrossRef CAS PubMed.
- T. Harayama, T. Sato, Y. Nakano, H. Abe and Y. Takeuchi, Heterocycles, 2003, 59, 293–301 CrossRef CAS.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- C. S. Poss and S. L. Schreiber, Acc. Chem. Res., 1994, 27, 9–17 CrossRef CAS.
- H. Chen, D. Zhang, F. Xue and Y. Qin, Tetrahedron, 2013, 69, 3141–3148 CrossRef CAS.
- S. Xie, G. Chen, H. Yan, J. Hou, Y. He, T. Zhao and J. Xu, J. Am. Chem. Soc., 2019, 141, 3435–3439 CrossRef CAS PubMed.
- W. Nie, J. Gong, Z. Chen, J. Liu, D. Tian, H. Song, X.-Y. Liu and Y. Qin, J. Am. Chem. Soc., 2019, 141, 9712–9718 CrossRef CAS PubMed.
- K. Owens, K. S. V. McCowen, K. A. Blackford, S. Ueno, Y. Hirooka, M. Weber and R. Sarpong, J. Am. Chem. Soc., 2019, 141, 13713–13717 CrossRef CAS PubMed.
- S. Zhou, K. Xia, X. Leng and A. Li, J. Am. Chem. Soc., 2019, 141, 13718–13723 CrossRef CAS PubMed.
- K. Y. Tsang and M. A. Brimble, Tetrahedron, 2007, 63, 6015–6034 CrossRef CAS.
- V. Heguaburu, V. Schapiro and E. Pandolfi, Tetrahedron Lett., 2010, 51, 6921–6923 CrossRef CAS.
- H. Lechuga-Eduardo, M. Romero-Ortega and H. F. Olivo, Eur. J. Org. Chem., 2016, 2016, 51–54 CrossRef CAS.
- P. Yates and H. Auksi, Can. J. Chem., 1979, 57, 2853–2863 CrossRef CAS.
- M. Attolini, F. Bouguir, G. Iacazio, G. Peiffer and M. Maffei, Tetrahedron, 2001, 57, 537–543 CrossRef CAS.
- G. Carrea, B. Danieli, G. Palmisano, S. Riva and M. Santagostino, Tetrahedron: Asymmetry, 1992, 3, 775–784 CrossRef CAS.
- Y. Nishiyama, S. Yokoshima and T. Fukuyama, Org. Lett., 2017, 19, 5833–5835 CrossRef CAS.
- H. Cheng, L. Xu, D.-L. Chen, Q.-H. Chen and F. P. Wang, Tetrahedron, 2012, 68, 1171–1176 CrossRef CAS.
- K. Kou, S. Kulyk, C. J. Marth, C. J. Lee, N. A. Doering, B. X. Li, G. M. Gallego, T. P. Lebold and R. Sarpong, J. Am. Chem. Soc., 2017, 139, 13882–13896 CrossRef CAS PubMed.
- K. Wiesner, T. Y. R. Tsai, K. Huber, S. E. Bolton and R. Vlahov, J. Am. Chem. Soc., 1974, 96, 4990–4992 CrossRef CAS.
- K. Weisner, T. Y. R. Tsai and K. P. Nambiar, Can. J. Chem., 1978, 56, 1451–1454 CrossRef.
- For a recent review of convergent strategies in natural product synthesis: D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207–9231 CrossRef CAS PubMed.
- T. Suzuki, A. Sasaki, N. Egashira and S. Kobayashi, Angew. Chem., Int. Ed., 2011, 50, 9177–9179 CrossRef CAS PubMed.
- T. Suzuki, H. Okuyama, A. Takano, S. Suzuki, I. Shimizu and S. Kobayashi, J. Org. Chem., 2014, 79, 2803–2808 CrossRef CAS PubMed.
- R. Hayashi, Z.-X. Ma and R. P. Hsung, Org. Lett., 2012, 14, 252–255 CrossRef CAS PubMed.
- L. L. Wei, H. Xiong, C. J. Douglas and R. P. Hsung, Tetrahedron Lett., 1999, 40, 6903–6907 CrossRef CAS.
- B. D. Jarhad and V. Singh, J. Org. Chem., 2016, 81, 4304–4309 CrossRef PubMed.
- H. Chen, X.-H. Li, J. Gong, H. Song, X.-Y. Liu and Y. Qin, Tetrahedron, 2016, 72, 347–353 CrossRef CAS.
Footnotes |
| † In the case of the arcutane alkaloids, a single bond disconnection across the C5–C20 bond would not be considered strategic due to the macrocyclic intermediate it would form. |
| ‡ The term efficiency, for the purpose of this review, will only refer to step count. A “one-pot” reaction will count as a single step only if none of the components of the reaction mixture are removed (including removal of solvent). The step counts for the following syntheses are adjusted from their original reports to follow this criterion. |
| This journal is © The Royal Society of Chemistry 2020 |