
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding medicinal chemistry into 3D space: metallofragments as 3D scaffolds for fragment-based drug discovery†
Christine N.
Morrison‡§
 a,
Kathleen E.
Prosser‡
a,
Ryjul W.
Stokes‡
a,
Anna
Cordes
b,
Nils
Metzler-Nolte
b and
Seth M.
Cohen
*a
a,
Kathleen E.
Prosser‡
a,
Ryjul W.
Stokes‡
a,
Anna
Cordes
b,
Nils
Metzler-Nolte
b and
Seth M.
Cohen
*a
aDepartment of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093, USA. E-mail: scohen@ucsd.edu
bLehrstuhl für Anorganische Chemie 1, Bioanorganische Chemie, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany
First published on 12th December 2019
Abstract
Fragment-based drug discovery (FBDD) is a powerful strategy for the identification of new bioactive molecules. FBDD relies on fragment libraries, generally of modest size, but of high chemical diversity. Although good chemical diversity in FBDD libraries has been achieved in many respects, achieving shape diversity – particularly fragments with three-dimensional (3D) structures – has remained challenging. A recent analysis revealed that >75% of all conventional, organic fragments are predominantly 1D or 2D in shape. However, 3D fragments are desired because molecular shape is one of the most important factors in molecular recognition by a biomolecule. To address this challenge, the use of inert metal complexes, so-called ‘metallofragments’ (mFs), to construct a 3D fragment library is introduced. A modest library of 71 compounds has been prepared with rich shape diversity as gauged by normalized principle moment of inertia (PMI) analysis. PMI analysis shows that these metallofragments occupy an area of fragment space that is unique and highly underrepresented when compared to conventional organic fragment libraries that are comprised of orders of magnitude more molecules. The potential value of this metallofragment library is demonstrated by screening against several different types of proteins, including an antiviral, an antibacterial, and an anticancer target. The suitability of the metallofragments for future hit-to-lead development was validated through the determination of IC50 and thermal shift values for select fragments against several proteins. These findings demonstrate the utility of metallofragment libraries as a means of accessing underutilized 3D fragment space for FBDD against a variety of protein targets.
Introduction
Fragment-based drug discovery (FBDD) is an increasingly successful strategy for the discovery of small molecule therapeutics.1–3 The FBDD pipeline begins with the development of a library of small ‘fragment’ molecules. Fragments are generally designed to be ‘rule-of-three’ compliant: molecular weight (MW) ≤300 Da, calculated partition coefficient (clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) ≤ 3, number of hydrogen bond donors/acceptors ≤3, and ≤3 rotatable bonds.4 In FBDD, the fragment library is screened against a protein target associated with a disease phenotype, and fragments that inhibit protein activity beyond a defined threshold are designated as ‘hits’.5–7 Once hits are identified, strategies of fragment growth, linking, and/or merging are employed to develop lead-like inhibitors. Fragment libraries have been touted as more effectively covering chemical space/diversity compared to high-throughput screening (HTS) libraries, which consist of larger, more drug-like molecules.8–10 FBDD realizes greater chemical diversity even while employing libraries that are a fraction of the size (100–1000 fragments for FBDD) of those used for traditional HTS campaigns (100000–1000000 compounds for HTS).9 Recently, several therapeutics discovered by FBDD have gained FDA approval, thereby validating the FBDD approach for new drug discovery.6,7
P) ≤ 3, number of hydrogen bond donors/acceptors ≤3, and ≤3 rotatable bonds.4 In FBDD, the fragment library is screened against a protein target associated with a disease phenotype, and fragments that inhibit protein activity beyond a defined threshold are designated as ‘hits’.5–7 Once hits are identified, strategies of fragment growth, linking, and/or merging are employed to develop lead-like inhibitors. Fragment libraries have been touted as more effectively covering chemical space/diversity compared to high-throughput screening (HTS) libraries, which consist of larger, more drug-like molecules.8–10 FBDD realizes greater chemical diversity even while employing libraries that are a fraction of the size (100–1000 fragments for FBDD) of those used for traditional HTS campaigns (100000–1000000 compounds for HTS).9 Recently, several therapeutics discovered by FBDD have gained FDA approval, thereby validating the FBDD approach for new drug discovery.6,7
Although FBDD has proven to be a successful method of drug discovery that achieves great chemical diversity, it remains a challenge to create the same degree of structural diversity in fragment libraries.8–12 Structural diversity is highly desired because molecular shape is among the most important factors dictating biological effects of molecules.8,9,13,14 Also, increased 3D shape can lead to greater aqueous solubility due to greater solvation and poorer solid-state crystal lattice packing, as well as improved ADMET properties (absorption, distribution, metabolism, excretion, and toxicity).13 As a result, increasing the 3D shape of molecules has been correlated to broader biological activity.8,14 It has been shown that molecular shape is more strongly dictated by the core compound scaffold rather than the shape or positioning of substituents decorating the core scaffold.8,14 Thus, fragment libraries consisting of a variety of 3D scaffolds are expected to display a wider range of biological activities compared to single scaffold libraries.8,9,12–14
Conventional organic fragments tend to be linear or flat molecules.15 For example, a previously reported analysis of 18534 organic fragments from the ZINC database (a collection of commercially available chemicals used for virtual screening) showed that the majority (∼75%) of conventional fragments have a linear (1D) or planar (2D) shape (Fig. 1).8 The ZINC database was analyzed using the method of Sauer and Schwartz,9 which employs the normalized principal moments of inertia (PMI) for each fragment and benchmarks them against three molecular standards: 2-butyne (intrinsically 1D), benzene (intrinsically 2D), and adamantane (intrinsically 3D). PMI is a measure of a molecule's resistance to angular acceleration around the principal axes (I1, I2, and I3); conventionally, I1 ≤ I2 ≤ I3. These values allow the comparison of molecular shapes by normalizing the PMI values and plotting the ratios (I1/I3, I2/I3) for each compound on a graph in which points occupy a triangular region (Fig. 1). In the resulting plots the PMIs of ZINC fragments overwhelmingly fall along the edge between 1D (2-butyne, top left corner) and 2D (benzene, bottom corner) shapes, with relatively few populating the 3D region of space.8,9
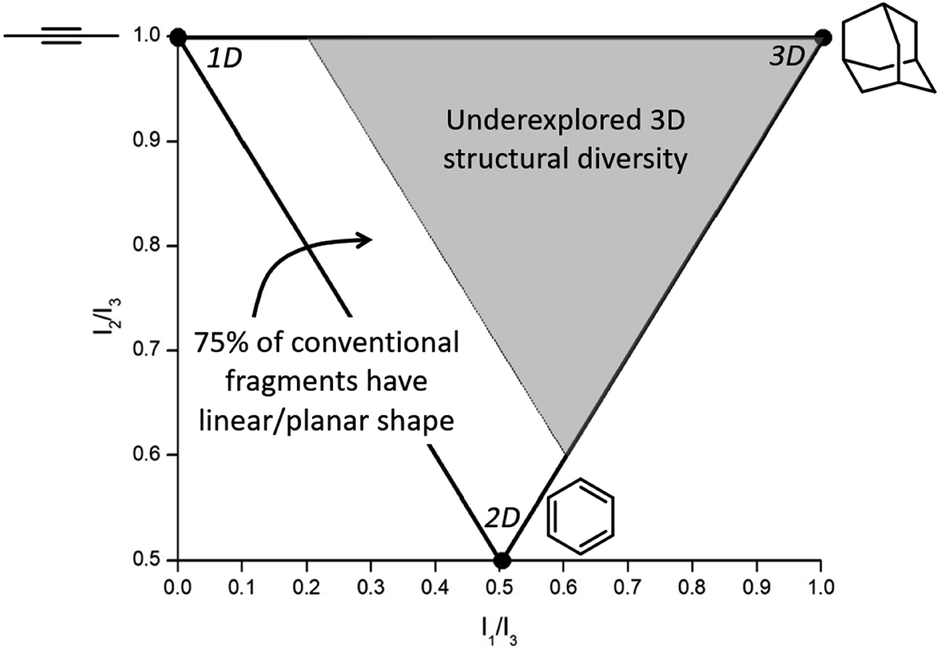 | ||
| Fig. 1 Normalized PMI values of a molecule are plotted to assess molecular topology. Analysis of the ZINC database shows that ∼75% of conventional fragments have a linear/planar shape (fall in the white region of the plot),8 indicating that fragments with 3D topology (gray region of the plot) are vastly underexplored in FBDD. | ||
The lack of structural diversity in fragment libraries is in large part due to the challenge of producing small, organic molecules with inherent 3D shape.8 Efforts to create 3D organic fragments have included diversity-oriented synthesis,8,11,14 combinatorial libraries,9 incorporation of cubanes,16 and incorporation of chiral carbon atoms,10 all of which pose significant synthetic challenges. Herein, this issue is addressed by introducing the first metallofragment (mF) library composed entirely of small, inorganic complexes with inherent 3D topologies. This proof-of-concept library consists of 71 compounds divided into 13 different classes based on metal center and structural homology. The prospective value of our mF library is demonstrated by screening the library against three therapeutic targets, including an antiviral target, an antibacterial target, and an anticancer target. The specific proteins screened in this study are the PA N-terminal (PAN) endonuclease domain of the RNA-dependent RNA polymerase complex of the influenza A virus, New Delhi metallo-β-lactamase-1 (NDM-1), and the N-terminal domain of heat shock protein 90-α (Hsp90), respectively. As a demonstration, select fragments from one class were further characterized with thermal shift assays (TSA) as an orthogonal screening method, as well as dose response assays to determine IC50 values. Taken together, the findings presented here show that 3D mFs are an innovative and potentially useful new tool for FBDD that are capable of targeting topological space not readily accessible by conventional organic fragment libraries.
Metallofragment library design
The bioinorganic community has explored the use of coordination and organometallic compounds as inhibitors or as auxiliary groups to augment existing organic moieties,17–22 and some of these metal-containing inhibitors having entered clinical trials, such as ferroquine.23,24 A few uses of organometallic groups to augment existing organic inhibitors have produced spectacular results, including highly selective and active kinase inhibitors.18,25 However, none of these efforts have approached the use of coordination compounds as fragments for FBDD. This is an important distinction from prior studies: rather than using the metal complex alone or to augment an existing molecule, the approach presented here starts with the coordination compound as a core structural scaffold. Additionally, Dyson and coworker have recently presented a new type of fragment-based approach using metal complexes.26 Their approach involves linking known, bioactive metal compounds to create bimetallic compounds with the potential to form short- or long-range crosslinks in DNA and protein targets.26 This is substantially different from our approach, which focuses on 3D mononuclear complexes as structural scaffolds that can be elaborated into more drug-like molecules through modification/elaboration of the ligand components of the fragments.The heart of this work is a novel mF library, which consists of 13 classes of various sandwich, half-sandwich, and octahedral metal complexes (Fig. 2). Members within each class share the same metal and core geometry, but feature ligands with different functional groups and/or heterocycles. Approximately 15% of the library was purchased from commercial sources and used without further modification, while the remaining majority of complexes were prepared according to literature procedures (see ESI† for details). Of the prepared metal compounds, ∼30% (19/71) represent previously unreported chemical entities. The majority of these novel complexes are ruthenium arene derivatives and rhenium tricarbonyl complexes (Classes J and K). In addition to their described application in this work as mFs, these new compounds have potential applications in the development of new anticancer agents (Ru(II) arenes)27–30 and model agents for imaging applications (Re(I) tricarbonyls).31 The majority of ligands in Classes J and K are derived from previously reported metal-binding pharmacophores.32 The other synthesized compounds include metallocene and piano-stool derivatives with substituent modifications carried out on their respective aromatic rings (Classes A, B, D, E),33 and a variety of carbene and diimine Re(I) complexes that have been reported extensively elsewhere.34–37 One particular advantageous aspect of this library is that most complexes, both reported and novel, were prepared in one or two steps from commercially available starting materials. This presents the opportunity in this and future studies to rapidly expand the contents of the mF library, in part due to the intrinsically modular nature of ligand and metal complex syntheses. In addition to library expansion, the modularity of ligand synthesis will allow for fragment growth or linking, as is the practice in traditional FBDD campaigns. Indeed, the mFs could potentially be screened in tandem with organic fragment libraries, providing insight to the appropriate ligand substituents needed to achieve potent target binding. Every class of mFs contain positions for modification, via ligand modification, that will facilitate future hit-to-lead or fragment building efforts.
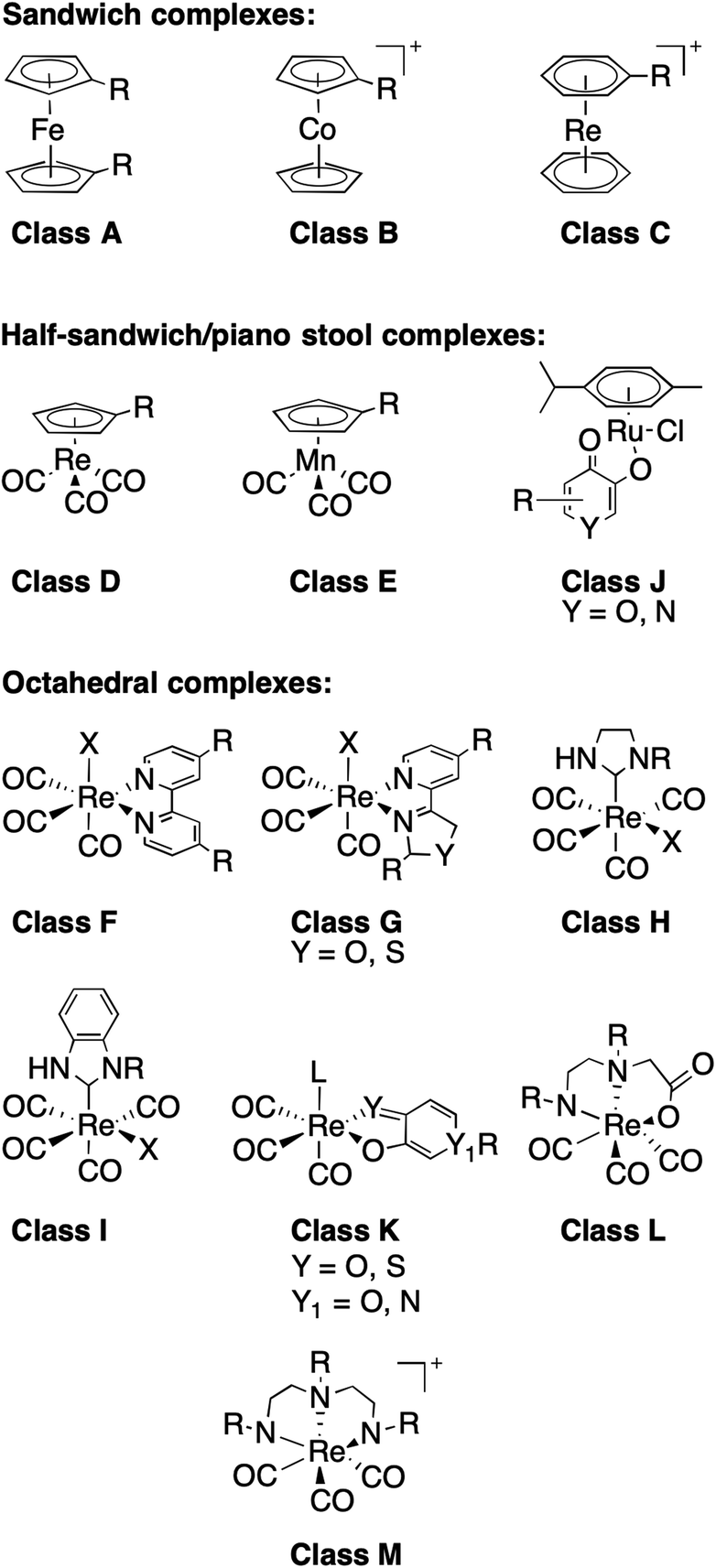 | ||
| Fig. 2 Classes of compounds in the metallofragment library, separated into sub-groups defined by their overall geometry. | ||
To explore extensive chemical space, features such as charge (e.g., neutral Class Aversus cationic Class B), hydrophobicity, synthetic accessibility, structural diversity, aqueous stability, and rule-of-three compliance (see below) were used to guide this initial library design.4 Fragments were selected with the aim of having a kinetically and thermodynamically stable core, ideally posing no significant pharmacokinetic challenge beyond that found for conventional organic fragments. Each compound in the library presented herein consists of only one metal ion. Other studies have examined the utility of metal cluster compounds in drug discovery;26,38 however, these tend to be much larger compounds, which makes them less suitable for FBDD.
Within the metallofragment library there are three sub-groups described by their general structure (Fig. 2): sandwich or metallocene complexes (Classes A–C), half-sandwich or piano-stool complexes (Classes D, E and J), and six-coordinate octahedral complexes (Classes F–I and K–M). Class A contains ferrocene derivatives, which are one of the most common metal-containing scaffolds explored in medicinal bioinorganic chemistry due to their ease of functionalization, stability, and low cost.19,39 Ferrocene was first introduced as a bioisostere for aryl/heteroaryl rings, and ferrocene has been utilized to improve anticancer, antimalarial, and antibacterial properties of organic therapies.19,29,39 Classes B and C are comprised of cobaltocenes and bis(arene)rhenium scaffolds, which are structurally similar to Class A but possess a positive charge.
The half-sandwich compounds included in our library are Re(I) compounds (Class D) that have been used for biomedical imaging applications,40 Mn(I) complexes (Class E) that have been used as CO releasing agents, and Ru(II) agents (Class J) that have been extensively studied as potential therapeutics in their own right.19,24,28,29 Despite the large number of reports on the biological activity of the Ru(II) arene complexes, this work represents, to the best of our knowledge, the first attempt to use Ru(II) arene complexes as core structural scaffolds.
All the octahedral complexes presented in our mF library contain Re(I) with 3–4 carbonyl ligands (Classes F–I and K–M, Fig. 2). Complexes similar to those in Classes F and G with bidentate N,N donors have been investigated for their anticancer properties.22,36 Classes H and I are carbene complexes; most reported biologically active metal–carbene complexes are prepared with Ag(I) and Au(I) and have shown anticancer and antimicrobial properties.19,29 Class K molecules consist of O,O and S,O heterocyclic bidentate ligands, while Classes L and M are prepared with N,N,O (Class L) and N,N,N (Class M) tridentate donor ligands. Again, to the best of our knowledge, this work is the first time that the Re(I)-based compounds in Classes H, I, K, L, and M are being utilized in FBDD. It is worth noting that octahedral complexes in the library containing asymmetric bidentate ligands (Classes G and K) form enantiomeric mixtures due to different binding orientations of the bidentate ligand. Small molecule crystal structures of some Class K metallofragments (Fig. S14†) show that both enantiomers are present in the product. No attempt to separate the enantiomers was made as it is beyond the scope of this work. Use of enantiomeric mixtures in early stage drug discovery is commonplace and as such does not represent a significant shortcoming of the mF library. Future work dedicated to hit-to-lead development of Class G and K molecules will address purification of the enantiomers.
Redefining the ‘rule of three’ for mFs
The concept of ‘drug-like properties’ is constantly evolving. For example, it was recently shown that the average molecular weight of drug molecules has increased substantially in the last 20 years, and its validity as an indicator of drug-likeness has been called into question.41 As stated earlier, fragments for FBDD are generally designed to be ‘rule-of-three’ compliant, which includes MW ≤ 300 Da, clogP ≤ 3, number of hydrogen bond donors ≤3, number of hydrogen bond acceptors ≤3, and ≤3 rotatable bonds.4 mFs generally satisfy all of these rules except MW ≤ 300 Da. This rule – much like the Lipinski rule stating a 500 Da cutoff for drug-like molecules42 – can be considered a proxy to account for molecular size, which can impact permeability and uptake, rather than a strict restriction on MW. Although transition metal ions have a much higher atomic weight than a carbon atom, the actual molecular volume (MV, Å3) of transition metal ions is not proportionally larger than a carbon atom.
With this in mind, an analysis of representative mFs was performed to redefine the rule-of-three parameter for MW in terms of molecular size. This redefinition was validated by comparing the heavy atom count (HAC; the number of non-hydrogen atoms) and ‘apparent MW’ of mFs (where the atomic weight of the metal ion is substituted for a carbon atom) to that of conventional organic fragments. The MV of representative mFs was evaluated against their apparent MW and their HAC. The result of this analysis (Fig. S15†) shows that the MV of mFs varies in a manner that is indistinguishable from the MV of conventional organic fragments based on HAC and apparent MW. Thus, although the mFs have a greater MW compared to organic fragments, they are not proportionally greater in size, and hence should operate as suitable scaffolds for FBDD. Based on this analysis, in lieu of MW ≤ 300 Da, we propose a new rule-of-three for mFs: MV ≤ 300 Å3. As a representative example, the chemical structure, X-ray structure, and molecular surface of mF K5 is shown in Fig. 3; the molecular volume of K5 is 292 Å3 (calculated by Molecular Operating Environment, v. 2019.0101).43
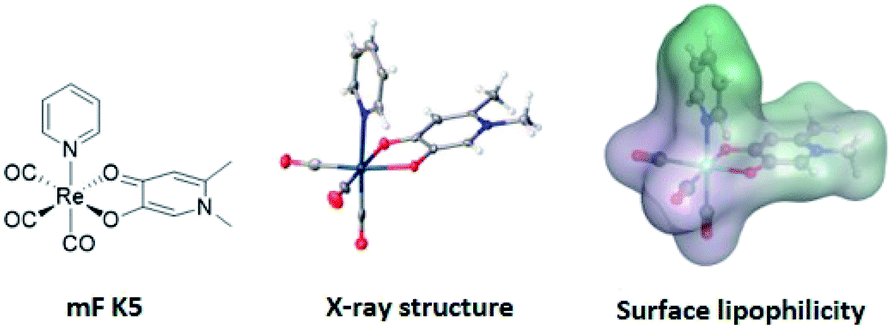 | ||
| Fig. 3 Representation of mF K5 (from left-to-right): chemical structure, X-ray structure, and molecular surface colored by lipophilicity. Hydrophilic and lipophilic regions are represented by pink and green surfaces, respectively. The molecular volume of K5 was determined to be 292 Å3 and only one enantiomer is shown from the X-ray structure (see ESI† for details). | ||
3-Dimensional analysis of metallofragments
To confirm that the initial fragment selection encompassed the desired 3D molecular space, a normalized PMI analysis of each mF was performed as described using the Molecular Operating Environment program (version 2019.0101, see ESI† for details).9 The normalized PMI ratios were benchmarked using the same standards as previously reported (Fig. 4): 2-butyne (1D), benzene (2D), and adamantane (3D). As shown in Fig. 4, the mF library broadly covers the 3D section of the normalized PMI plot. When compared to existing fragment libraries, such as the ZINC library (Fig. 1), the metallofragment library covers a much broader 3D topological space using far fewer compounds. This was quantified by determining the percentage of mFs above the left-hand boundary given by the equation x + y = 1.2 or (I1/I3) + (I2/I3) = 1.2. In our analysis, which is based on the ZINC library analysis performed by Hung and coworkers,8 fragments that satisfy the equation (I1/I3) + (I2/I3) > 1.2 are considered to have a 3D shape. Of the 71 mFs in the library, 55 (77%) satisfy (I1/I3) + (I2/I3) > 1.2 and can be considered to have a 3D shape. Comparatively, Hung's analysis of the ZINC library showed that only ∼25% of conventional fragments have 3D shape.8 Thus, by the normalized PMI metric to analyze molecular shape, the mF library clearly achieves the goal of providing greater access to 3D scaffolds.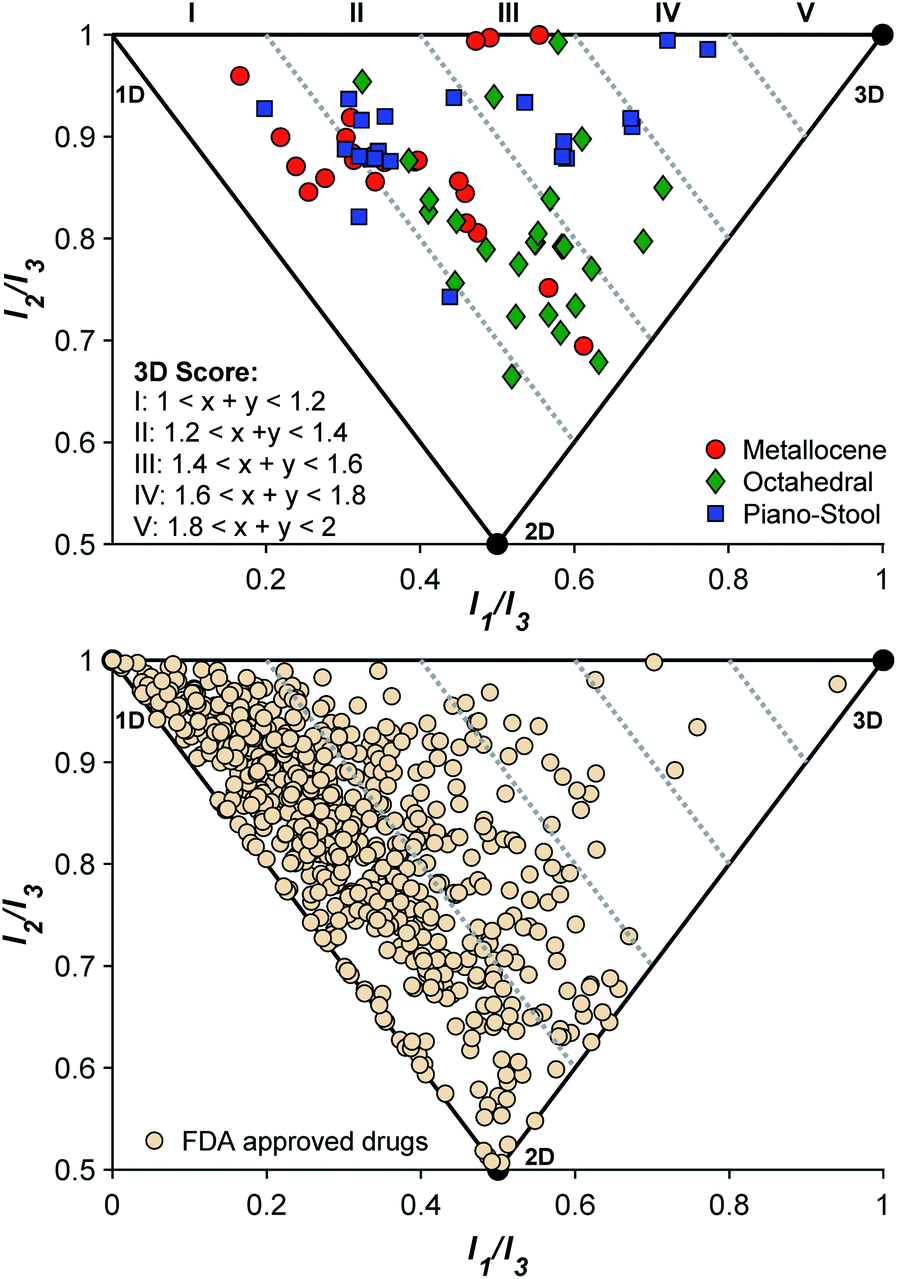 | ||
| Fig. 4 (Top) Normalized PMI analysis of the entire mF library shows that the mF library broadly populates 3D topological space. mFs of each complex type tend to have a related topology. Within the 3D region, metallocene complexes (red circles) are more linear/planar, piano-stool complexes (blue squares) are more linear/spherical, and octahedral complexes (green diamonds) are more planar/spherical. (Bottom) Normalized PMI analysis of approved drug molecules in DrugBank (v. 5.1.3). In both plots, the degree of increasing 3D topologies, or the 3D scores, is delineated by lines with x + y ≤ 1.2 (I linear/flat), 1.4 (II), 1.6 (III), 1.8 (IV), and x + y > 1.8 (V). | ||
The topology of the mFs was also analyzed by complex type (sandwich, half-sandwich, and octahedral complexes). The normalized PMI plots (Fig. 4) show that mFs belonging to the same complex type tend to have similar topology. The sandwich mFs cluster near the linear region of the plot, half-sandwich mFs occupy the top of the plot in the linear to spherical region, and octahedral mFs are concentrated between the planar and spherical region. This analysis validates the notion that the core scaffold of a molecule contributes more to its overall shape than does its substituents.8,14 Knowledge of the general topology of various scaffolds may be useful in targeted FBDD.
For comparison to the mF library, the molecular topology of FDA-approved drugs was assessed (Fig. 4), using structures in the DrugBank (version 5.1.3 from 4 April 2019). Structures were downloaded as ‘3D’, meaning that the downloaded structure represents the lowest energy conformer of the free drug molecule. Normalized PMI calculations of the lowest energy conformer were performed on all structures, and the results are shown in Fig. 4. To the best of our knowledge, this 3D analysis of approved drugs has not previously been performed and it serves as an interesting insight to the current scope of therapeutic structural diversity. Of the approved drugs with structures in the DrugBank database, 23% (161/712) have 3D scores falling above 1.2 ((I1/I3) + (I2/I3) > 1.2). The largely 2D character of drug molecules has been previously described,13 but it is important to note that these energy-minimized structures may not reflect the protein-bound or solution confirmations of the drugs.13,44 These energy-minimized structures may also demonstrate some bias towards the left side of the PMI plot due to the steric interactions of side-chains, producing some artificial linearity/planarity. Even concerted efforts in FBDD campaigns to drive towards 3D diversity fail to achieve high 3-dimensionality (3D score > III).8 Of the 712 approved therapies examined here, only 5 (0.7%) compounds are considered highly 3D (3D score > III), while with only 71 entries, the mF platform places 2 complexes, ∼3% of the library, in this space. Given the 3D nature of these core fragment scaffolds, the mF library offers more direct access to molecules that occupy the previously underexplored 3D space at both the fragment and drug level.
Metallofragment library evaluation and screening
As described above, many of the sandwich, half-sandwich, and octahedral complexes that comprise the mF library have been broadly examined for their biological activity. In this work the mFs are intended to serve as inert scaffolds upon which fragment growth can be carried out. To determine the general stability of each fragment class, 1H NMR analysis was carried out in deuterated DMSO prior to screening against potential protein targets. Spectra were collected of the first entry in each Class and for each complex A-I1 and L-M1 only a single species was observed (Fig. S19†). Complexes in Class K undergo partial solvation through the loss of the monodentate heterocycle to produce a second species with a coordinated DMSO (Fig. S18†). Such complexes have been reported,45 and it is anticipated that all components of these solutions will be aquated once dissolved in aqueous media. Similarly, the Ru(arene) scaffolds in Class J exhibit some ligand exchange in DMSO; the speciation of such complexes has been studied extensively in both organic and aqueous media.46 While these particular scaffolds may not be an ideal mF motif, the relevance of these compounds to the bioinorganic literature and their 3D topological diversity prompted the inclusion of Class J as a starting point for these studies.To assess the utility of the mF library for FBDD, the library was screened against three therapeutically relevant targets (Fig. 5). The selected targets were: the polymerase acidic N-terminal (PAN) endonuclease domain from the H1N1 influenza A virus (antiviral target), New Delhi metallo-β-lactamase-1 (NDM-1; antibacterial target), and the N-terminal domain of heat shock protein 90-α (Hsp90; anticancer target).47,48 The role of these enzymes in their respective diseases and inhibitor development for each are presented elsewhere.47–49 Briefly, PAN endonuclease is one of three proteins in the RNA-dependent RNA polymerase complex of the influenza A virus, along with the polymerase basic protein 1 (PB1) and the polymerase basic protein 2 (PB2).50 PAN endonuclease contains a dinuclear metal active site, with two Mn2+ or Mg2+ cations that promote endonuclease activity.51 A functional RNA polymerase complex is essential to viral replication,50 and the first therapeutic targeting this protein, Baloxavir marboxil, has now gained FDA approval.52 Baloxavir, along with other leading drug discovery efforts, have targeted the metal centers in the large active site of PAN as a means of enzyme inhibition. NDM-1 is a protein found in both Gram-negative and Gram-positive bacteria that has been shown to hydrolyze clinically relevant β-lactam antibiotics.53 The active site of NDM-1 is largely hydrophobic, with fluctional conformations and two Zn2+ ions that participate in antibiotic hydrolysis.54 There has been substantial effort to develop NDM-1 inhibitors and the target is still considered promising,47 although NDM-1 inhibitors have yet to gain FDA approval. The final target, Hsp90, is a ubiquitous molecular chaperone with many diverse functions including the folding, stability, and activity of many proteins (‘clients’).55,56 Several Hsp90 clients have been identified as oncoproteins that are associated with cancer hallmarks.48,56 The binding site of the reported inhibitors is a 15 Å deep pocket capable of binding polypeptide chains.57 More than a dozen Hsp90 inhibitors have entered clinical trials, but none have received FDA approval.48,56
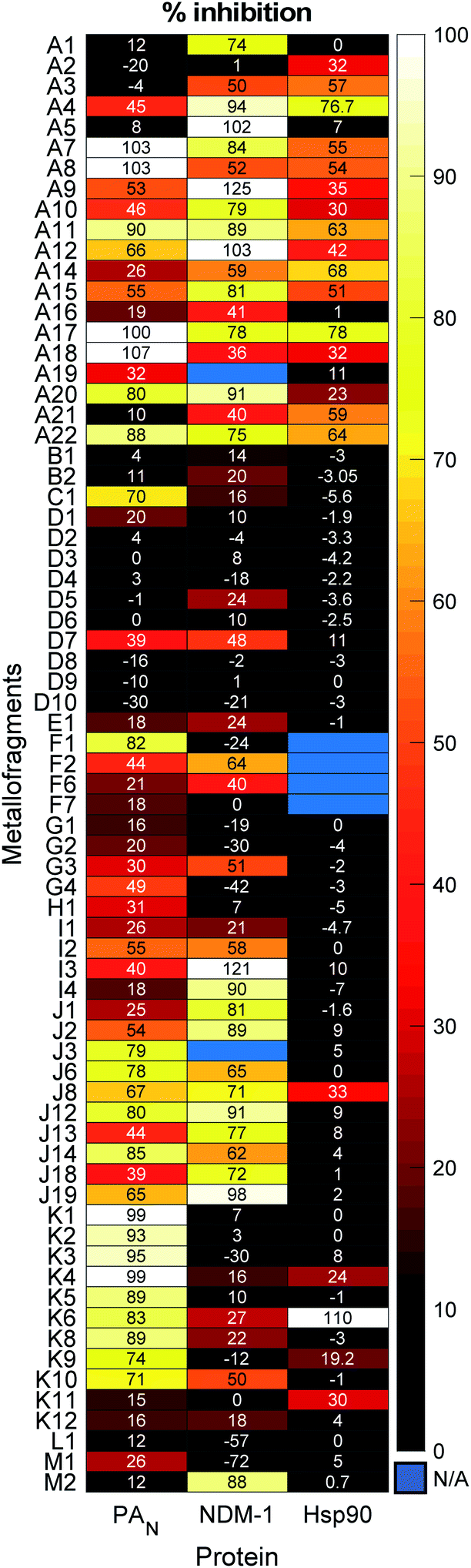 | ||
| Fig. 5 Screening results, presented as percent inhibition, for the mF library tested at 200 μM mF concentration against the viral target PAN, the bacterial target NDM-1, and the human cancer target Hsp90. | ||
The mF library was screened at a fragment concentration of 200 μM against all three targets using established screening assays for each protein (Fig. 5 and S17†).51,58 Considering the library as an entirety, a hit rate (percent inhibition > 50%) of approximately ∼40% was achieved against PAN and NDM-1, while Hsp90 had a hit rate of ∼15% (Fig. S16†). Classic high-throughput screening (HTS) of drug-like molecules have reported hit rates between 0.001% and 0.2%, while organic fragment libraries are reported to have hit rates ranging from 3% to 30%.59,60 Thus, the mF library performs as well as or better than both traditional screening libraries, speaking to the promise of this screening platform.
The metallocene subgroup, particularly the Class A ferrocene derivatives, performed well against each of the targets (Fig. 5), achieving hits rates between 41–68% (Fig. S16†). While these compounds may present promising scaffolds for future work on fragment growth and elaboration, it is important to recognize their general lack of specificity. Excessive lipophilicity, along with properties such as redox activity and metal chelation are considered to be hallmarks of potential ligand promiscuity.61 The lipophilicity of ferrocene and ferrocene derivatives, discussed in terms of the partition coefficient logP, are typically between 2–5.62 These values fall on the higher end of what is generally considered acceptable for drug-like molecules (logP < 5), and it is likely that their lipophilicity would only increase with fragment growth. Given this, should a Class A mF be selected as a hit compound in future efforts, precautions are advised to ensure that the complex binds to the desired binding pocket rather than to non-specific hydrophobic patches on a protein target. While the class performed broadly well, suggesting promiscuity and non-specific interactions, some differences in their activity against the three targets was observed. For example, mF A5, ferrocenemethylamine (Fig. S1†), completely inhibited NDM-1 in the 200 μM screen but failed to inhibit PAN endonuclease and Hsp90. Such results demonstrate the promise of the metallocene subgroup to serve as fragment scaffolds in future FBDD campaigns.
In addition to validating the suitability of the mF platform for future FBDD campaigns, these screening results allow for the examination of the relationship between mF 3D topologies and their biological activity. By calculating the 3D score of each fragment ((I1/I3) + (I2/I3), Fig. 4), the 3D topologies of each mF can be plotted against the percent inhibition as determined through each assay (Fig. 6). Based on the PMI analysis presented above, the majority of mFs have 3D scores of II (1.2 ≤ (I1/I3) + (I2/I3) < 1.4) compared to the ZINC fragment library where ∼75% of fragments have (I1/I3) + (I2/I3) < 1.2, a 3D score of I.8 Within II there are mFs from each subgroup, and they all exhibit a broad range of inhibitory effects against the three targets. Of the most 3D fragments, those that fall into IV, none of them achieved percent inhibition values above 50%. There are only 2 mFs with this score, representing ∼3% of the library, and both are from the half-sandwich complex subgroup. It is challenging to draw any significant conclusions regarding their moderate biological activity given the limited scope. Nonetheless, both of these highly 3D fragments achieved percent inhibition values >20% at 200 μM against at least one of the targets, and as such could potentially be pursued for fragment growth and lead development. In future studies it will be important to populate these higher 3D scores so that more informative analyses of the relationship between 3-dimensionality and biological activity can be undertaken.
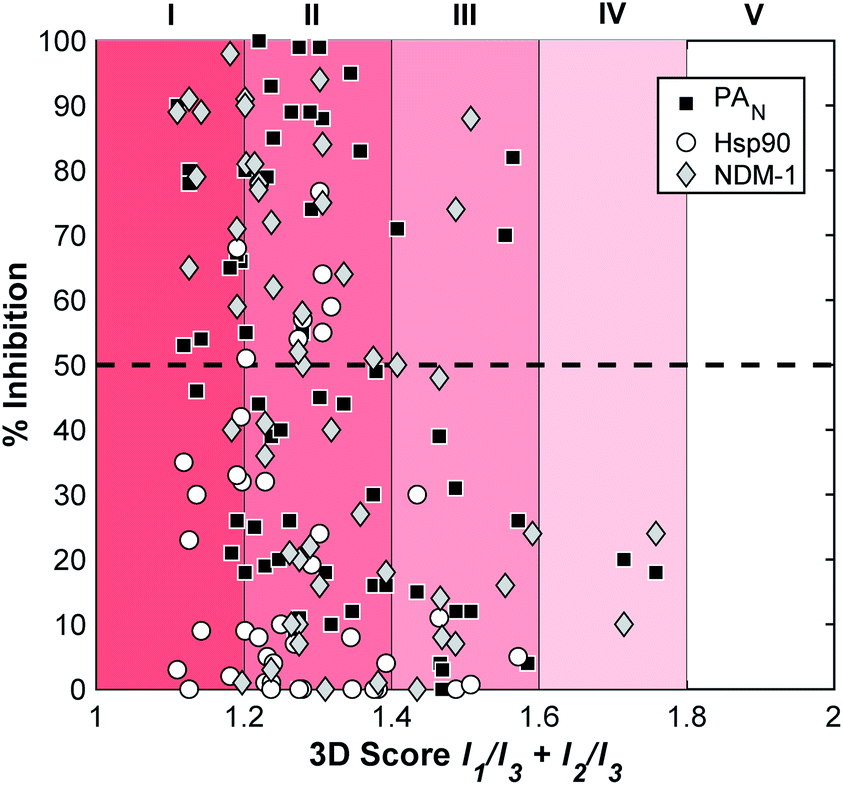 | ||
| Fig. 6 Analysis of the inhibitory data and the 3D topologies of the mF library tested against PAN, NDM-1, and Hsp90 at fragment concentrations of 200 μM. | ||
Structure–activity relationship of mFs
To explore the suitability of the mFs for further analysis and development, the activity of four compounds from Class A (A4, A7, A11, and A12) was validated using dose response assays and an orthogonal screening technique, the thermal shift assay (TSA). The results of these studies are summarized in Table 1, and demonstrate the range of inhibitory responses that were generated by this small collection of mFs. The dose responses of these fragments against PAN endonuclease, NDM-1, and Hsp90 demonstrated that the IC50 values of each fragment was under 100 μM against each protein in all but two cases, indicating that most of these mFs could serve as viable candidates to initiate a hit-to-lead campaign against these three targets. Among this small subset of mFs, an inhibitor with an IC50 value under 25 μM was identified against each protein. Additionally, these fragments all demonstrated good ligand efficiency (LE), which is determined using the IC50 values and heavy atom count (HAC).1 The LE values of these mF hits against the three protein targets are all near or above the optimal LE value of ≥0.3 (kcal mol−1)/HAC for fragments.63| mF | HAC | PAN endonuclease | NDM-1 | Hsp90 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50a (μM) | LEb | ΔTMc | IC50 (μM) | LE | ΔTM | IC50 (μM) | LE | ΔTM | ||
| a IC50 values reported in μM with the 95% CI indicated. b Ligand efficiency (LE; kcal per mol per HAC). c Reported in °C. | ||||||||||
| A4 | 14 | >500 | <0.33 | −1.1 ± 0.3 | 33 ± 17 | 0.45 | 0.7 ± 0.1 | 33 ± 2 | 0.45 | 0 ± 1 |
| A7 | 14 | 80 ± 20 | 0.41 | −1.5 ± 0.1 | 33 ± 5 | 0.45 | 0.10 ± 0.04 | 24 ± 12 | 0.46 | 2 ± 0.6 |
| A11 | 20 | 18 ± 8 | 0.33 | 0.8 ± 0.5 | 12 ± 5 | 0.34 | 0.12 ± 0.03 | 24 ± 4 | 0.32 | 2 ± 1 |
| A12 | 19 | 55 ± 15 | 0.31 | −3.7 ± 0.4 | 50 ± 20 | 0.32 | 1.0 ± 0.1 | >500 | <0.24 | −2 ± 0.6 |
TSA experiments, which measure the change of the melting (unfolding) temperature (TM) of a protein, were carried out on the select Class A mFs. Inhibitors that stabilize a protein increase the melting temperature of the protein, while inhibitors that destabilize the protein decrease the melting temperature. The TSA data is provided in Table 1 and is reported as ΔTM (in °C), which refers to the difference in melting temperature of the inhibitor-bound protein compared to the native protein. While large ΔTM values for any protein with inhibitor were not observed, moderate ΔTM values suggest a range of different stabilizing and destabilizing interactions between the protein and the mF. Within the small subset of fragments examined here there are no immediate correlations between the ΔTM values and their determined IC50 values. However, the ability to measure and compare these different markers of the inhibitor–protein interaction will allow future FBDD campaigns to take full advantage of the topological diversity afforded by the mF library.
In an effort to elucidate the possible mechanism of inhibition, preliminary docking exercises were carried out on a representative mF, K6. Fragment K6 showed good inhibitory activity against PAN endonuclease, and also stood out as a potent inhibitor of Hsp90, providing a model mF for this docking study. The coordinates of an aquated version of K6 were determined from the X-ray crystal structure of K5 (see ESI Fig. S14 and Table S1†) and the complex was docked against a reported inhibitor-bound PAN endonuclease structure (6E3M) and an inhibitor-bound crystal structure of Hsp90 (1YET) using the Molecular Operating Environment program (version 2019.0101).64 The best scoring pose against each protein is shown in Fig. 7. While these poses may not indicate the true binding mode of K6 against these two targets, they serve as an initial starting point in the rational development of new inhibitors. In both docking studies, the proteins and mF are mapped from pink to green based on their lipophilicities (Fig. 7). Interestingly, many of the mF library entries do not have hydrogen bond donating or accepting entities, as is the case for K6. As such, an analysis of the molecular interactions shows that the docking of the mF in the pockets of PAN endonuclease and Hsp90 is driven primarily by the steric interactions, directly related to the fragment 3-dimensionality.
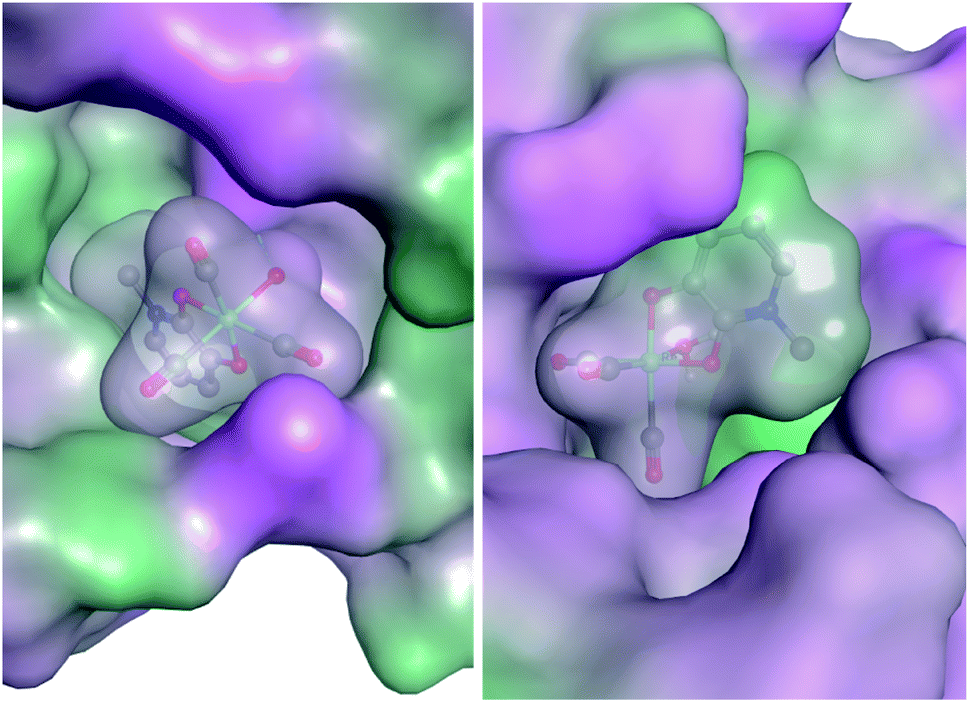 | ||
| Fig. 7 Aquated fragment K6 docked against PAN endonuclease (left) and Hsp90 (right). Fragments and protein are shown with molecular surface maps colored to indicate lipophilicity. Hydrophilic and lipophilic regions are represented by pink and green surfaces, respectively. | ||
Conclusions
The use of metal complexes to complement organic fragment libraries for drug discovery applications has not been considered elsewhere. The benefit this approach aims to impart on the FBDD methodology is the ability to access underexplored 3D chemical topologies. To evaluate the feasibility and the validity of such an approach, we designed, synthesized and characterized a modest library of coordination and organometallic complexes with diverse 3D topologies. A comparative shape analysis by the PMI method impressively demonstrates the validity of our approach, with 77% populating the 3D region, as opposed to only 25% from the much larger ZINC library of purely organic compounds. As a proof-of-concept, the mF library was then screened against three different, relevant biological enzyme targets, i.e. PAN endonuclease, NDM-1, and Hsp90 at 200 μM fragment concentration. These assays generated a range of inhibitory responses, in which some mF classes performed well, while others achieved only moderate to poor inhibition. The fragment-like behavior of selected ferrocene-derivatives was examined through dose–response and thermal shift assays, demonstrating that mFs can be suitably studied using traditional medicinal chemistry approaches. Through the combination of fragment screening, dose response assays, TSA, and molecular docking, we have shown that these mFs are capable of the same types of analyses undergone by traditional organic fragments in medicinal chemistry campaigns.An analysis of the 3D topologies of ∼700 approved therapeutics demonstrates that a large majority fall under a linear/flat regime. Because of the limitations in the synthesis of 3D rich drug/fragment libraries, it is difficult to know if these flat/linear structures represent ideal geometries, or if they are simply a consequence of the tools used to prepare drug discovery libraries. The mFs presented here have comparatively high 3D topologies, but this space has been sufficiently challenging to access such that the potential of truly 3D scaffolds has yet to be determined. Overall, this work showcases the utility of a novel mF library of modest size with 3D diversity that exhibits a broad range of biological responses. Future efforts will build on these exciting proof-of-concept studies by expanding the mF library to include more highly 3D fragments with careful consideration of their kinetic and thermodynamic stability. With a second-generation library in development, these mFs can be further developed into lead-like molecules for addressing previously inaccessible or challenging targets.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Dr Yongxuan Su (UC San Diego, Molecular Mass Spectrometry Facility) for aid with mass spectrometry analysis. We thank Dr Curtis Moore and Dr Milan Gembicky for assistance with X-ray crystallography, and Dr Cy V. Credille, Allie Y. Chen, Rebecca N. Adamek, and Benjamin L. Dick for helpful discussions. We acknowledge Dr Elbek Kurbanov for assembling and characterizing some Class A fragments. We gratefully acknowledge the laboratory of Dr Michael W. Crowder for providing the NDM-1 protein used in these experiments and Allie Y. Chen for assisting with the NDM-1 activity and thermal shift assays. This work was supported by grants from the National Institutes of Health (R21 AI138934; F32 GM125233 to C. N. M.), by the University of California President's Postdoctoral Fellowship (to C. N. M.), a Natural Sciences and Engineering and Research Council of Canada Postdoctoral Fellowship (to K. E. P.), and the National Science Foundation Graduate Research Fellowship Program (DGE-1650112 to R. W. S.). This project was also enabled by the Ruhr University Research School PLUS, funded by Germany's Excellence Initiative [DFG GSC 98/3], through a VIP grant (to S. M. C) and an IRB grant (to A. C.).References
- R. A. Carr, M. Congreve, C. W. Murray and D. C. Rees, Drug Discovery Today, 2005, 10, 987–992 CrossRef CAS PubMed.
- M. Congreve, G. Chessari, D. Tisi and A. J. Woodhead, J. Med. Chem., 2008, 51, 3661–3680 CrossRef CAS PubMed.
- D. E. Scott, A. G. Coyne, S. A. Hudson and C. Abell, Biochemistry, 2012, 51, 4990–5003 CrossRef CAS PubMed.
- M. Congreve, R. A. E. Carr, C. W. Murray and H. Jhoti, Drug Discovery Today, 2003, 8, 876–877 CrossRef PubMed.
- A. G. Coyne, D. E. Scott and C. Abell, Curr. Opin. Chem. Biol., 2010, 14, 299–307 CrossRef CAS PubMed.
- G. Bollag, P. Hirth, J. Tsai, J. Zhang, P. N. Ibrahim, H. Cho, W. Spevak, C. Zhang, Y. Zhang, G. Habets, E. A. Burton, B. Wong, G. Tsang, B. L. West, B. Powell, R. Shellooe, A. Marimuthu, H. Nguyen, K. Y. Zhang, D. R. Artis, J. Schlessinger, F. Su, B. Higgins, R. Iyer, K. D'Andrea, A. Koehler, M. Stumm, P. S. Lin, R. J. Lee, J. Grippo, I. Puzanov, K. B. Kim, A. Ribas, G. A. McArthur, J. A. Sosman, P. B. Chapman, K. T. Flaherty, X. Xu, K. L. Nathanson and K. Nolop, Nature, 2010, 467, 596–599 CrossRef CAS PubMed.
- J. Tsai, J. T. Lee, W. Wang, J. Zhang, H. Cho, S. Mamo, R. Bremer, S. Gillette, J. Kong, N. K. Haass, K. Sproesser, L. Li, K. S. Smalley, D. Fong, Y. L. Zhu, A. Marimuthu, H. Nguyen, B. Lam, J. Liu, I. Cheung, J. Rice, Y. Suzuki, C. Luu, C. Settachatgul, R. Shellooe, J. Cantwell, S. H. Kim, J. Schlessinger, K. Y. Zhang, B. L. West, B. Powell, G. Habets, C. Zhang, P. N. Ibrahim, P. Hirth, D. R. Artis, M. Herlyn and G. Bollag, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3041–3046 CrossRef CAS PubMed.
- A. Hung, A. Ramek, Y. Wang, T. Kaya, J. Wilson, P. Clemons and D. Young, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6799–6804 CrossRef CAS PubMed.
- W. H. B. Sauer and M. K. Schwarz, J. Chem. Inf. Comput. Sci., 2003, 43, 987–1003 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- S. L. Kidd, T. J. Osberger, N. Mateu, H. F. Sore and D. R. Spring, Front. Chem., 2018, 6, 460 CrossRef PubMed.
- M. Aldeghi, S. Malhotra, D. L. Selwood and A. W. Chan, Chem. Biol. Drug Des., 2014, 83, 450–461 CrossRef CAS PubMed.
- N. C. Firth, N. Brown and J. Blagg, J. Chem. Inf. Model., 2012, 52, 2516–2525 CrossRef CAS PubMed.
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 CrossRef PubMed.
- J. Meyers, M. Carter, N. Y. Mok and N. Brown, Future Med. Chem., 2016, 8, 1753–1767 CrossRef PubMed.
- T. A. Reekie, C. M. Williams, L. M. Rendina and M. Kassiou, J. Med. Chem., 2019, 62, 1078–1095 CrossRef CAS PubMed.
- D. Can, B. Spingler, P. Schmutz, F. Mendes, P. Raposinho, C. Fernandes, F. Carta, A. Innocenti, I. Santos, C. T. Supuran and R. Alberto, Angew. Chem., Int. Ed., 2012, 57, 3354–3357 CrossRef PubMed.
- M. Dorr and E. Meggers, Curr. Opin. Chem. Biol., 2014, 19, 76–81 CrossRef PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- M. Huisman, J. P. Kodanko, K. Arora, M. Herroon, M. Alnaed, J. Endicott, I. Podgorski and J. J. Kodanko, Inorg. Chem., 2018, 57, 7881–7891 CrossRef CAS PubMed.
- R. M. Vaden, K. P. Guillen, J. M. Salvant, C. B. Santiago, J. B. Gibbons, S. S. Pathi, S. Arunachalam, M. S. Sigman, R. E. Looper and B. E. Welm, ACS Chem. Biol., 2019, 14, 106–117 CrossRef CAS PubMed.
- C. C. Konkankit, B. A. Vaughn, S. N. MacMillan, E. Boros and J. J. Wilson, Inorg. Chem., 2019, 58, 3895–3909 CrossRef CAS PubMed.
- V. B. A. M. Galanski, M. A. Jakupec and B. K. Keppler, Curr. Pharm. Des., 2003, 9, 2078–2089 CrossRef PubMed.
- S. Parveen, F. Arjmand and S. Tabassum, Eur. J. Med. Chem., 2019, 175, 269–286 CrossRef CAS PubMed.
- S. Blanck, Y. Geisselbrecht, K. Kräling, S. Middel, T. Mietke, K. Harms, L.-O. Essen and E. Meggers, Dalton Trans., 2012, 41, 9337–9348 RSC.
- L. K. Batchelor and P. J. Dyson, Trends in Chemistry, 2019, 1, 644–655 CrossRef.
- G. Meola, H. Braband, P. Schmutz, M. Benz, B. Spingler and R. Alberto, Inorg. Chem., 2016, 55, 11131–11139 CrossRef CAS PubMed.
- A. F. A. Peacock, A. Habtemariam, R. Fernández, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739–1748 CrossRef CAS PubMed.
- P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5–14 CrossRef CAS.
- E. Trifonova, D. Perekalin, K. Lyssenko and A. R. Kudinov, J. Organomet. Chem., 2013, 727, 60–63 CrossRef CAS.
- J. Klenc, M. Lipowska, P. L. Abhayawardhana, A. T. Taylor and L. G. Marzilli, Inorg. Chem., 2015, 54, 6281–6290 CrossRef CAS PubMed.
- S. M. Cohen, Acc. Chem. Res., 2017, 50, 2007–2016 CrossRef CAS PubMed.
- A. Baramee, A. Coppin, M. Mortuaire, L. Pelinski, S. Tomavo and J. Brocard, Bioorg. Med. Chem., 2006, 14, 1294–1302 CrossRef CAS PubMed.
- D. Siegmund, PhD, Ruhr University Bochum, 2018.
- J.-M. Heldt, N. Fischer-Durand, M. Salmain, A. Vessières and G. Jaouen, J. Organomet. Chem., 2004, 689, 4775–4782 CrossRef CAS.
- K. M. Knopf, B. L. Murphy, S. N. MacMillan, J. M. Baskin, M. P. Barr, E. Boros and J. J. Wilson, J. Am. Chem. Soc., 2017, 139, 14302–14314 CrossRef CAS PubMed.
- N. E. Kolobova, Z. P. Valueva and M. Y. Solodova, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1980, 29, 1701–1705 CrossRef.
- E. L.-M. Wong, R. W.-Y. Sun, N. P. Y. Chung, C.-L. S. Lin, N. Zhu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 4938–4939 CrossRef CAS PubMed.
- A. Singh, I. Lumb, V. Mehra and V. Kumar, Dalton Trans., 2019, 48, 2840–2860 RSC.
- C. Policar, J. B. Waern, M.-A. Plamont, S. Clède, C. Mayet, R. Prazeres, J.-M. Ortega, A. Vessières and A. Dazzi, Angew. Chem., Int. Ed., 2011, 50, 860–864 CrossRef CAS PubMed.
- M. D. Shultz, J. Med. Chem., 2019, 62, 1701–1714 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), 2019.0101, Chemical Computing Group ULC, Montreal, QC, Canada, 2019 Search PubMed.
- A. D. Morley, A. Pugliese, K. Birchall, J. Bower, P. Brennan, N. Brown, T. Chapman, M. Drysdale, I. H. Gilbert, S. Hoelder, A. Jordan, S. V. Ley, A. Merritt, D. Miller, M. E. Swarbrick and P. G. Wyatt, Drug Discovery Today, 2013, 18, 1221–1227 CrossRef PubMed.
- M. Grzegorczyk, A. Kapturkiewicz, J. Nowacki and A. Trojanowska, Inorg. Chem. Commun., 2011, 14, 1773–1776 CrossRef CAS.
- M. Patra, T. Joshi, V. Pierroz, K. Ingram, M. Kaiser, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, Chem.–Eur. J., 2013, 19, 14768–14772 CrossRef CAS PubMed.
- A. Y. Chen, R. N. Adamek, B. L. Dick, C. V. Credille, C. N. Morrison and S. M. Cohen, Chem. Rev., 2019, 119, 1323–1455 CrossRef CAS PubMed.
- K. Sidera and E. Patsavoudi, Recent Pat. Anti-Cancer Drug Discovery, 2014, 9, 1–20 CAS.
- C. V. Credille, C. N. Morrison, R. W. Stokes, B. L. Dick, Y. Feng, J. Sun, Y. Chen and S. M. Cohen, J. Med. Chem., 2019, 62, 9438–9449 CrossRef CAS PubMed.
- A. Dias, D. Bouvier, T. Crépin, A. A. McCarthy, D. J. Hart, F. Baudin, S. Cusack and R. W. H. Ruigrok, Nature, 2009, 458, 914–918 CrossRef CAS PubMed.
- C. V. Credille, B. L. Dick, C. N. Morrison, R. W. Stokes, R. N. Adamek, N. C. Wu, I. A. Wilson and S. M. Cohen, J. Med. Chem., 2018, 61, 10206–10217 CrossRef CAS PubMed.
- Y.-A. Heo, Drugs, 2018, 78, 693–697 CrossRef CAS PubMed.
- P. Nordmann, L. Poirel, T. R. Walsh and D. M. Livermore, Trends Microbiol., 2011, 19, 588–595 CrossRef CAS PubMed.
- Z. Sun, L. Hu, B. Sankaran, B. V. V. Prasad and T. Palzkill, Nat. Commun., 2018, 9, 4524 CrossRef PubMed.
- H. Wegele, L. Muller and J. Buchner, Rev. Physiol., Biochem. Pharmacol., 2004, 151, 1–44 CAS.
- L. M. Butler, R. Ferraldeschi, H. K. Armstrong, M. M. Centenera and P. Workman, Mol. Cancer Res., 2015, 13, 1445–1451 CrossRef CAS PubMed.
- C. E. Stebbins, A. A. Russo, C. Schneider, N. Rosen, F. U. Hartl and N. P. Pavletich, Cell, 1997, 89, 239–250 CrossRef CAS PubMed.
- A. Y. Chen, P. W. Thomas, Z. Cheng, N. Y. Xu, D. L. Tierney, M. W. Crowder, W. Fast and S. M. Cohen, ChemMedChem, 2019, 14, 1271–1282 CrossRef CAS PubMed.
- P. J. Hajduk, J. R. Huth and S. W. Fesik, J. Med. Chem., 2005, 48, 2518–2525 CrossRef CAS PubMed.
- S. Ansgar, R. Simon, M. Andreas, J. Wolfgang, S. Paul and J. Edgar, Curr. Top. Med. Chem., 2005, 5, 751–762 CrossRef PubMed.
- J. Blagg and P. Workman, Cancer Cells, 2017, 32, 9–25 CrossRef CAS PubMed.
- M. Maschke, H. Alborzinia, M. Lieb, S. Wölfl and N. Metzler-Nolte, ChemMedChem, 2014, 9, 1188–1194 CrossRef CAS PubMed.
- S. Schultes, C. de Graaf, E. E. J. Haaksma, I. J. P. de Esch, R. Leurs and O. Krämer, Drug Discovery Today: Technol., 2010, 7, e157–e162 CrossRef CAS PubMed.
- S. Vilar, G. Cozza and S. Moro, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, characterization, and detailed assay procedures. CCDC 1962326–1962330. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05586j |
| ‡ These authors contributed equally to this work. |
| § Current address: Colorado School of Mines, 1500 Illinois Street, Golden, CO 80401, USA. |
| This journal is © The Royal Society of Chemistry 2020 |