
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFolic acid-sulfonamide conjugates as antibacterial agents: design, synthesis and molecular docking studies†
Shabnam Shahzada,
Muhammad Abdul Qadira,
Mahmood Ahmed *bd,
Saghir Ahmada,
Muhammad Jadoon Khanc,
Asad Gulzard and
Muhammad Muddassar*c
*bd,
Saghir Ahmada,
Muhammad Jadoon Khanc,
Asad Gulzard and
Muhammad Muddassar*c
aInstitute of Chemistry, University of the Punjab, Lahore-54590, Pakistan
bRenacon Pharma Limited, Lahore-54600, Pakistan. E-mail: mahmoodresearchscholar@gmail.com
cDepartment of Biosciences, COMSATS University Islamabad, Park Road, Islamabad, Pakistan. E-mail: mmuddassar@comsats.edu.pk
dDivision of Science and Technology, University of Education, Lahore, Pakistan
First published on 25th November 2020
Abstract
Dihydrofolate reductase (DHFR) inhibitors, as antibacterial agents, contain pyrimidine, pteridine, and azine moieties among many other scaffolds. Folic acid (FA), with a pteridine ring and amine group, was used as our focus scaffold, which was then conjugated with sulfonamides to develop new conjugates. The novel synthesized conjugates were characterized using infrared spectroscopy, and 1H and 13C nuclear magnetic resonance (NMR) spectral studies and consequently screened for antimicrobial activities against bacterial strains with ampicillin as a positive control. Compound DS2 has the highest zone of inhibition (36.6 mm) with a percentage activity index (%AI) value of 122.8% against S. aureus and a minimum inhibitory concentration (MIC) of 15.63 μg mL−1. DHFR enzyme inhibition was also evaluated using the synthesized conjugates through in vitro studies, and inhibition assays revealed that compound DS2 exhibited a 75.4 ± 0.12% (mean ± standard error of the mean (SEM)) inhibition, which is comparable with the standard DHFR inhibitor trimethoprim (74.6 ± 0.09%). The compounds attached to the unsubstituted aryl moiety of the sulfonamides revealed better inhibition against the bacterial strains as compared to the methyl substituted aryl sulfonamides. Molecular docking studies of the novel synthesized conjugates were also performed on the DHFR enzyme to identify the plausible binding modes to explore the binding mechanisms of these conjugates.
1. Introduction
The development of resistance by microorganisms to most of the existing antimicrobial drugs is a global health problem. To overcome this problem, there is an increasing need to synthesize novel antimicrobial drugs. Bacteria have the capability to adapt according to the environment of the antibacterial agent using mutations, genetic mechanisms, or selection. These multi drug resistant bacteria (MDR) cause a multitude of infections, which have aroused the need for novel antibiotics.1 Among the Gram negative class are Pseudomonas aeruginosa and Escherichia coli, whereas Gram positive bacteria including Proteus mirabilis and Staphylococcus aureus have evolved aggressively into nosocomial pathogens that can cause severe infections such as endocarditis, meningitis, bacteremia, and infections of the biliary system, urinary tracts and wounds. Different strategies are used to target the defense mechanisms of microorganisms and enzyme inhibition studies have evolved as a real solution to this problem. Dihydrofolate reductase (DHFR) enzyme inhibitors are potent against diverse bacteria and tumors. DHFR is significantly conserved in all domains, however, it is divergent in amino acid sequences, offering an opportunity for the selectivity of drugs to various organisms. Therefore, antifolate drugs showed success against parasitic and bacterial infections, as well as chemotherapy.2–4Sulfonamides are curative drugs used against infections caused by microbes. The significance of sulfa drugs can never be neglected in the pharmaceutical and agricultural fields. They have stimulated researchers to design novel drugs with a lower toxicity and cost and a significant activity profile. These are structurally similar to p-aminobenzoic acid (PABA), so as an analog of PABA, sulfonamides can compete with it efficiently to prevent the synthesis of proteins and nucleic acid which results in the inhibition of various microorganisms.5–8 Moreover, a sulfonamide is a versatile moiety owing to its diverse pharmacological activities from antimicrobial to anticancer activities9–16 and its enzyme inhibition, such as cyclooxygenase, carbonic anhydrase, urease, acetylcholinesterase, butyrylcholinesterase, dihydropteroate synthase and DHFR.17–25 Sulfonamides play a vital role as antibiotics, anti-malarial agents, protease inhibitors, antiretroviral agents in the treatment of HIV/AIDS, and for treating asthma, leukemia and epilepsy.26–32 To improve the activity of DHFR inhibitors, two strategies can play a vital role; improving the selectivity and potency of inhibitors through structure based design, and enhancing the permeability and solubility. Compounds containing pyrimidine, pteridine and azine moieties have already been reported as good DHFR inhibitors.33,34 The rate of folate metabolism accelerates during microbial infection owing to their particular DNA and for protein synthesis to maintain proper cell replication. Taking advantage of this accelerated rate of metabolism, folic acid antagonists have been used to treat various microbial infections.35 As folic acid (FA) has a pteridine ring and amine group, its conjugation with a sulfonyl group forms a scaffold containing both pteridine and sulfonamide, which confer better antibacterial activities to target the anti-folate pathway.36 The structural similarity of the conjugates under investigation with reported DHFR inhibitors can offer similar/better antibacterial activities and might suppress the resistance mechanism of microorganisms.37,38 These findings motivated us to develop novel conjugates by combining different pharmacophores into one structure with the aim of obtaining novel compounds that have significant biological activity.
2. Experimental
2.1. Chemistry
In this research, high purity chemicals were used that were acquired from Falcon Scientific, Lahore-Pakistan or Merck (Germany), and high purity water was produced in our own lab using a Milli-Q® water system (UK). Spectral studies including Fourier-transform infrared spectroscopy (FTIR) (FTIR spectrophotometer, Agilent Technologies, Carry 630), 1HNMR-500 MHz, 13CNMR-125 MHz (NMR spectrometer, Bruker, USA) and elemental analysis (C, H, N and S using a Flash elemental analyzer, HT+ Thermo Scientific, UK) were performed to elucidate the structure of the novel synthesized compounds. The melting points were determined using Gallenkamp apparatus and a PG-T80 UV-Vis spectrophotometer (PG, Instruments-UK) was used to calculate the λmax. Pre-coated silica plates (Merck, Germany) were spotted for thin layer chromatography (TLC) analysis and spots were detected under UV light to confirm the purity of the synthesized compounds.2.2. Protocol for sulfonamide synthesis
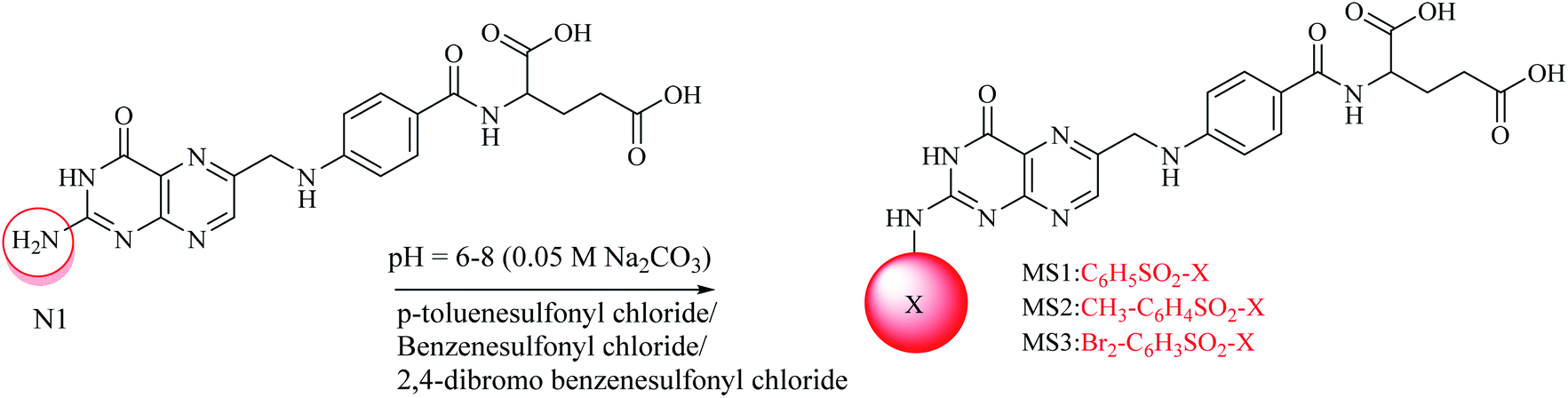
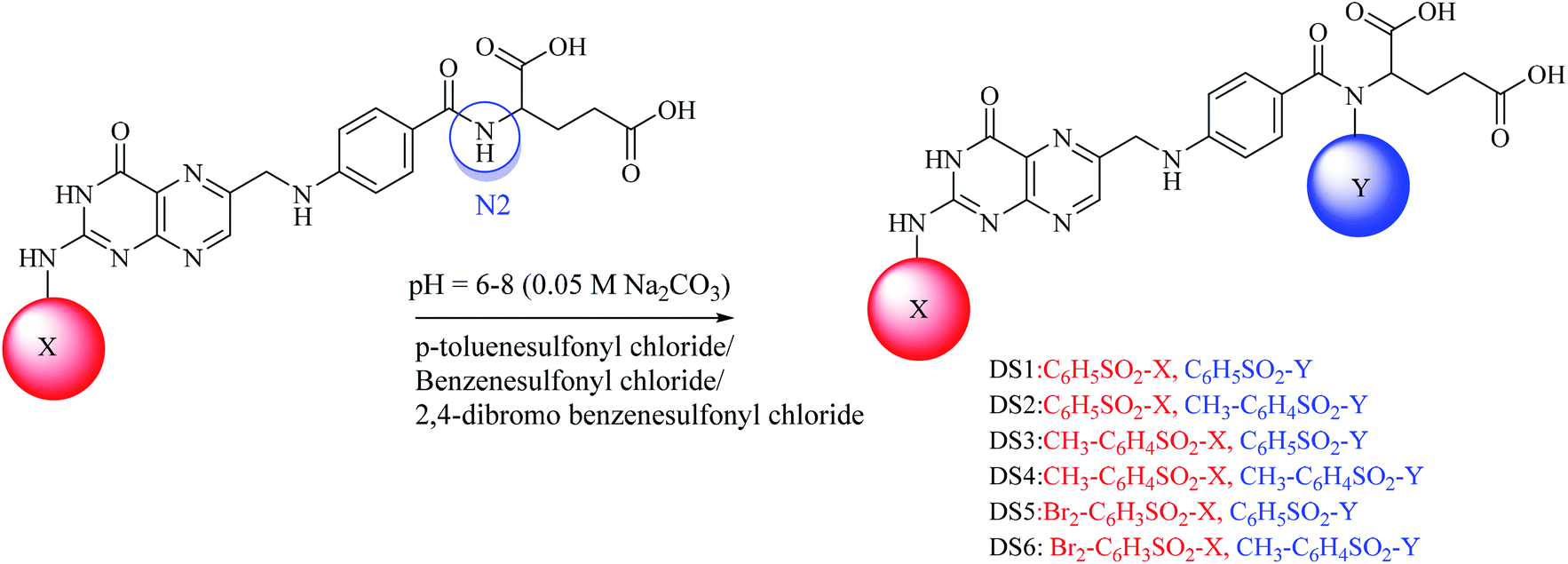
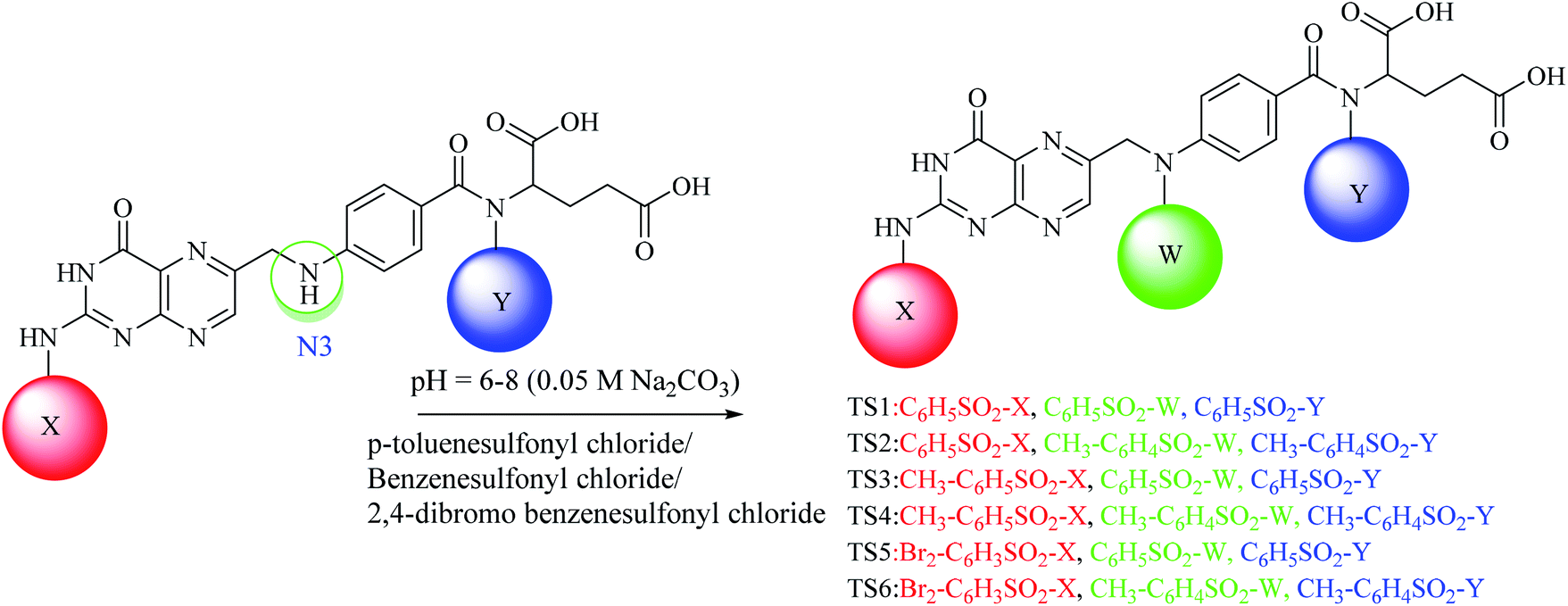
A convenient route under dynamic pH control in an aqueous medium was used for the synthesis of the sulfonamides.39,40 For mono substituted (MS1–MS3) folic acid sulfonamides (Scheme 1), in a 100 mL round-bottom flask, 1 mmol folic acid was dissolved in water/dimethylformamide (DMF). Then, 1 mmol each of p-toluenesulfonyl chloride, benzenesulfonyl chloride and 2,4-dibromo benzenesulfonyl chloride were weighed separately and poured into the above described mixture containing folic acid (1 mmol). The pH of the reaction was maintained at 6–8 by adding 0.05 M Na2CO3 drop-wise and the pH was monitored using a digital pH meter (WTW, Germany). The synthesis was performed in a round-bottom flask furnished with a magnetic stirrer at room temperature. The progress of the reaction was determined by the drop in pH owing to the HCl formation during the reaction. Hydrogen was easily removed owing to the aqueous basic medium. After the pH had been stabilized, the solvent was evaporated in a water bath to obtain the residues. Solvent (acetone) was used to dissolve the residues and filtration was performed using a filter paper (Whatman no. 42). The acetone in the filtrate was evaporated to obtain pure crystals of the products. Crystals were then washed several times and allowed to dry. Water soluble crystals were obtained. The formation of products was confirmed by TLC on pre-coated silica TLC plates. The mobile phase used was of dichloromethane, methanol and ammonia (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25:5) and spots were located in UV light. For di-substituted folic acid sulfonamides (Scheme 2), MS1–MS3 were reacted by following the procedure as explained above with p-toluenesulfonyl chloride, benzenesulfonyl chloride and 2,4-dibromo benzenesulfonyl chloride. For trisubstituted folic acid sulfonamides (Scheme 3), DS1–DS6 were reacted with p-toluenesulfonyl chloride, benzenesulfonyl chloride and 2,4-dibromo benzenesulfonyl chloride by following the above described procedure. The detailed synthesis of the folic acid linked sulfonamides with physiochemical and spectral data is explained below.
:25:5) and spots were located in UV light. For di-substituted folic acid sulfonamides (Scheme 2), MS1–MS3 were reacted by following the procedure as explained above with p-toluenesulfonyl chloride, benzenesulfonyl chloride and 2,4-dibromo benzenesulfonyl chloride. For trisubstituted folic acid sulfonamides (Scheme 3), DS1–DS6 were reacted with p-toluenesulfonyl chloride, benzenesulfonyl chloride and 2,4-dibromo benzenesulfonyl chloride by following the above described procedure. The detailed synthesis of the folic acid linked sulfonamides with physiochemical and spectral data is explained below.
 | ||
| Scheme 1 Synthesis of monosubstituted folic acid sulfonamides. | ||
 | ||
| Scheme 2 Synthesis of disubstituted folic acid sulfonamides. | ||
 | ||
| Scheme 3 Synthesis of trisubstituted folic acid sulfonamides. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1610 (imine –CHN–), 1484 (COOH), 1298 (–SO2–NH2-asym), 1186 (–SO2–NH2-sym), 1086 (–SO). UV λmax (nm): 285 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.95 (1H, s, –CHNH), 8.32 (1H, s, –NH–CO–), 8.02 (1H, s, –NH–CO–aryl), 7.56–7.84, (7H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (H, t, J = 7.8 Hz, –CH), 4.35 (2H, s, –CH2NH–), 4.29 (1H, s, –NH–CH), 2.35 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 174.7, 166.7, 164.2, 153.4, 151.3, 150.1, 133.7, 132.7, 130.7, 130.7, 128.9, 115.5, 115.2, 114.1, 55.2, 42.4, 34.9, 28.9. Anal. calc. for C25H23N7O8S (FW = 581.1 g mol−1): C, 51.63; H, 3.99; N, 16.86; S, 5.51%. Found: C, 51.54; H, 3.88; N, 16.74; S, 5.59%.O), 1648 (imine –CHN–), 1484 (COOH), 1298 (–SO2–NH2-asym), 1186 (–SO2–NH2-sym), 1086 (–SO). UV λmax (nm): 275 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.95 (1H, s, –CHN), 8.32 (1H, s, –NH–CO–), 8.02 (1H, s, –NH–CO–aryl), 7.67–7.84, (7H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.51 (H, t, J = 7.8 Hz, –CH), 4.35 (2H, s, –CH2NH–), 4.29 (1H, s, –NH–CH), 2.34 (3H, t, J = 7.1 Hz, –CH3), 2.07 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 174.7, 166.7, 164.2, 153.4, 151.3, 150.1, 133.7, 132.7, 130.7, 130.7, 128.9, 115.5, 115.2, 114.1, 55.2, 42.4, 34.9, 28.9, 21.1. Anal. calc. for C26H25N7O8S (FW = 595.1 g mol−1): C, 52.43; H, 4.23; N, 16.46; S, 5.38%. Found: C, 52.59; H, 4.38; N, 16.64; S, 5.49%.O), 1600 (imine –CHN–), 1451 (COOH), 1306 (–SO2–NH2-asym), 1173 (–SO2–NH2-sym), 1093 (–SO). UV λmax (nm): 255 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.95 (1H, s, –CHN), 8.43 (H, s, CH–CH–Br), 8.33 (1H, s, –NH–CO–), 8.03 (1H, s, –NH–CO–aryl), 7.56–7.84, (7H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.51 (H, t, J = 7.8 Hz, –CH), 4.35 (2H, s, –CH2NH–), 4.29 (1H, s, –NH–CH), 2.36 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 174.7, 166.7, 164.2, 153.4, 151.3, 150.1, 133.7, 132.7, 130.7, 130.7, 128.9, 115.5, 115.2, 114.1, 56.2, 42.4, 34.9, 28.9. Anal. calc. for C25H21Br2N7O8S (FW = 738.9 g mol−1): C, 40.61; H, 2.86; N, 13.26; S, 4.34%. Found: C, 40.41; H, 2.29; N, 13.37; S, 4.45%.O), 1601 (imine –CHN–), 1450 (COOH), 1298 (–SO2–NH2-asym), 1186 (–SO2–NH2-sym), 1060 (–SO). UV λmax (nm): 280 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.99 (1H, s, –CHN), 8.03 (1H, s, –NH–CO–aryl), 7.66–7.86, (12H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.51 (H, t, J = 7.8 Hz, –CH), 4.55 (2H, s, –CH2NH–), 4.32 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.8, 166.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 55.6, 49.4, 35.0, 34.7, 21.5. Anal. calc. for C31H27N7O10S2 (FW = 721.1 g mol−1): C, 51.59; H, 3.77; N, 13.59; S, 8.89%. Found: C, 51.41; H, 3.49; N, 13.47; S, 8.65%.O), 1601 (imine –CHN–), 1455 (COOH), 1298 (–SO2–NH2-asym), 1160 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 295 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.98 (1H, s, –CHN), 8.02 (1H, s, –NH–CO–aryl), 7.66–7.89, (12H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.51 (H, t, J = 7.8 Hz, –CH), 4.54 (2H, s, –CH2NH–), 4.33 (1H, t, J = 7.1 Hz, –N–CH), 2.34 (3H, t, J = 7.1 Hz, –CH3), 2.07 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.8, 166.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 55.6, 49.4, 35.0, 34.7, 23.4, 21.5. Anal. calc. for C31H27N7O10S2 (FW = 735.1 g mol−1): C, 52.24; H, 3.97; N, 13.33; S, 8.72%. Found: C, 52.41; H, 3.89; N, 13.49; S, 8.85%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1005 (–SO). UV λmax (nm): 290 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.98 (1H, s, –CHN), 8.02 (1H, s, –NH–CO–aryl), 7.66–7.89, (12H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (H, t, J = 7.8 Hz, –CH), 4.34 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (3H, t, J = 7.1 Hz, –CH3), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 166.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 55.6, 49.4, 35.0, 34.7, 23.4, 21.5. Anal. calc. for C31H27N7O10S2 (FW = 735.1 g mol−1): C, 52.24; H, 3.97; N, 13.33; S, 8.72%. Found: C, 52.41; H, 3.89; N, 13.49; S, 8.85%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1001 (–SO). UV λmax (nm): 275 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.98 (1H, s, –CHN), 8.33 (1H, s, –NH–CO–aryl), 7.73 (4H, d, J = 7.6 Hz, ArH), 7.54 (4H, d, J = 7.4 Hz, ArH), 7.34 (4H, d, J = 7.6 Hz, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (2H, s, –CH2NH–), 4.34 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (6H, t, J = 7.1 Hz, 2× CH3), 2.33 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 166.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 59.5, 49.4, 35.0, 34.7, 23.4, 21.5. C33H31N7O10S2 (FW = 749.1 g mol−1): C, 52.86; H, 4.17; N, 13.08; S, 8.55%. Found: C, 52.71; H, 4.09; N, 13.25; S, 8.65%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1176 (–SO2–NH2-sym), 1058 (–SO). UV λmax (nm): 240 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.96 (1H, s, –CHN), 8.43 (H, s, CH–CH–Br), 8.03 (1H, s, –NH–CO–aryl), 7.56–7.84, (11H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (H, t, J = 7.8 Hz, –CH), 4.33 (2H, s, –CH2NH–), 4.33 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.8, 165.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 59.5, 49.4, 35.0, 34.7, 23.4, 21.5. Anal. calc. for C31H25Br2N7O10S2 (FW = 878.9 g mol−1): C, 42.33; H, 2.87; N, 11.15; S, 7.29%. Found: C, 42.41; H, 2.79; N, 11.27; S, 7.45%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1176 (–SO2–NH2-sym), 1062 (–SO). UV λmax (nm): 245 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.96 (1H, s, –CHN), 8.43 (H, s, CH–CH–Br), 8.03 (1H, s, –NH–CO–aryl), 7.56–7.84, (10H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (H, t, J = 7.8 Hz, –CH), 4.33 (2H, s, –CH2NH–), 4.33 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (3H, t, J = 7.1 Hz, –CH3), 2.35 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.8, 165.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 59.5, 49.4, 30.4, 23.4, 21.5. Anal. calc. for C32H27Br2N7O10S2 (FW = 892.9 g mol−1): C, 43.01; H, 3.05; N, 10.97; S, 7.18%. Found: C, 42.91; H, 3.09; N, 11.17; S, 7.05%.O), 1606 (imine –CHN–), 1450 (COOH), 1284 (–SO2–NH2-asym), 1166 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 295 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.1 (2H, s, 2× OH), 8.99 (1H, s, –CHN), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.46, (17H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.7, 163.5, 161.5, 155.2, 138.9, 131.8, 130.1, 129.8, 128.6, 127.9, 124.9, 120.3, 117.7, 52.4, 49.6, 36.9, 29.9, 28.3, 26.6, 22.0. Anal. calc. for C37H31N7O12S3 (FW = 861.1 g mol−1): C, 51.56; H, 3.63; N, 11.38; S, 11.16%. Found: C, 51.41; H, 3.49; N, 11.47; S, 11.25%.O), 1606 (imine –CHN–), 1450 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 290 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.1 (2H, s, 2× OH), 8.99 (1H, s, –CHN), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.46, (17H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.9, 159.6, 157.2, 152.4, 143.6, 139.2, 135.4, 129.6, 129.3, 128.9, 128.1, 127.9, 52.6, 49.9, 37.2, 30.1, 28.5, 26.9, 22.2, 21.7. Anal. calc. for C39H35N7O12S3 (FW = 889.1 g mol−1): C, 52.64; H, 3.96; N, 11.02; S, 10.81%. Found: C, 52.41; H, 3.79; N, 11.16; S, 10.62%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 295 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.07 (2H, s, 2× OH), 8.87 (1H, s, –CHN), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.46, (16H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (3H, t, J = 7.1 Hz, CH3), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.9, 160.5, 157.2, 152.4, 143.6, 139.2, 135.4, 129.6, 129.3, 128.9, 128.1, 127.9, 52.6, 49.9, 37.2, 30.1, 28.5, 26.9, 22.2, 21.7. Anal. calc. for C38H33N7O12S3 (FW = 875.1 g mol−1): C, 52.11; H, 3.80; N, 11.19; S, 10.98%. Found: C, 52.32; H, 3.64; N, 11.25; S, 10.85%.O), 1606 (imine –CHN–), 1450 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 290 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.07 (2H, s, 2× OH), 8.87 (1H, s, –CHN), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.49, (16H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (3H, t, J = 7.1 Hz, CH3), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 179.7, 167.1, 163.1, 152.6, 149.5, 139.6, 139.5, 138.4, 130.8, 130.3, 129.5, 125.6, 125.2, 114.1, 55.8, 52.6, 51.5, 37.8, 30.2, 30.1, 29.9, 28.5, 26.8, 26.0 22.9, 22.5. Anal. calc. for C40H37N7O12S3 (FW = 903.1 g mol−1): C, 53.15; H, 4.13; N, 10.85; S, 10.64%. Found: C, 53.35; H, 4.21; N, 10.66; S, 10.71%.O), 1606 (imine –CHN–), 1450 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 280 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.07 (2H, s, 2× OH), 8.87 (1H, s, –CHN), 8.67 (1H, s, CH–CH–Br), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.48, (15H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 179.8, 167.1, 163.1, 152.7, 149.5, 139.7, 139.5, 138.4, 130.8, 130.3, 129.5, 125.6, 125.2, 114.0, 55.8, 52.6, 51.5, 37.8, 30.2, 30.1, 29.9, 28.5, 26.8, 26.0. Anal. calc. for C37H29Br2N7O12S3 (FW = 1018.9 g mol−1): C, 43.58; H, 2.87; N, 9.62; S, 9.43%. Found: C, 43.41; H, 2.79; N, 9.47; S, 9.25%.O), 1606 (imine –CHN–), 1450 (COOH), 1286 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 245 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.07 (2H, s, 2× OH), 8.87 (1H, s, –CHN), 8.67 (1H, s, CH–CH–Br), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.48, (15H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.24 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 179.6, 167.5, 163.5, 152.6, 149.5, 139.6, 139.5, 138.7, 130.9, 130.3, 129.5, 125.6, 125.2, 114.1, 55.8, 52.6, 51.5, 37.8, 30.2, 30.2, 29.8, 28.5, 26.8, 26.0 22.9, 22.5. Anal. calc. for C37H29Br2N7O12S3 (FW = 1046.9 g mol−1): C, 44.71; H, 3.17; N, 9.36; S, 9.18%. Found: C, 44.48; H, 3.29; N, 9.47; S, 9.25.
O), 1610 (imine –CHN–), 1484 (COOH), 1298 (–SO2–NH2-asym), 1186 (–SO2–NH2-sym), 1086 (–SO). UV λmax (nm): 285 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.95 (1H, s, –CHNH), 8.32 (1H, s, –NH–CO–), 8.02 (1H, s, –NH–CO–aryl), 7.56–7.84, (7H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (H, t, J = 7.8 Hz, –CH), 4.35 (2H, s, –CH2NH–), 4.29 (1H, s, –NH–CH), 2.35 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 174.7, 166.7, 164.2, 153.4, 151.3, 150.1, 133.7, 132.7, 130.7, 130.7, 128.9, 115.5, 115.2, 114.1, 55.2, 42.4, 34.9, 28.9. Anal. calc. for C25H23N7O8S (FW = 581.1 g mol−1): C, 51.63; H, 3.99; N, 16.86; S, 5.51%. Found: C, 51.54; H, 3.88; N, 16.74; S, 5.59%.O), 1648 (imine –CHN–), 1484 (COOH), 1298 (–SO2–NH2-asym), 1186 (–SO2–NH2-sym), 1086 (–SO). UV λmax (nm): 275 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.95 (1H, s, –CHN), 8.32 (1H, s, –NH–CO–), 8.02 (1H, s, –NH–CO–aryl), 7.67–7.84, (7H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.51 (H, t, J = 7.8 Hz, –CH), 4.35 (2H, s, –CH2NH–), 4.29 (1H, s, –NH–CH), 2.34 (3H, t, J = 7.1 Hz, –CH3), 2.07 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 174.7, 166.7, 164.2, 153.4, 151.3, 150.1, 133.7, 132.7, 130.7, 130.7, 128.9, 115.5, 115.2, 114.1, 55.2, 42.4, 34.9, 28.9, 21.1. Anal. calc. for C26H25N7O8S (FW = 595.1 g mol−1): C, 52.43; H, 4.23; N, 16.46; S, 5.38%. Found: C, 52.59; H, 4.38; N, 16.64; S, 5.49%.O), 1600 (imine –CHN–), 1451 (COOH), 1306 (–SO2–NH2-asym), 1173 (–SO2–NH2-sym), 1093 (–SO). UV λmax (nm): 255 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.95 (1H, s, –CHN), 8.43 (H, s, CH–CH–Br), 8.33 (1H, s, –NH–CO–), 8.03 (1H, s, –NH–CO–aryl), 7.56–7.84, (7H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.51 (H, t, J = 7.8 Hz, –CH), 4.35 (2H, s, –CH2NH–), 4.29 (1H, s, –NH–CH), 2.36 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 174.7, 166.7, 164.2, 153.4, 151.3, 150.1, 133.7, 132.7, 130.7, 130.7, 128.9, 115.5, 115.2, 114.1, 56.2, 42.4, 34.9, 28.9. Anal. calc. for C25H21Br2N7O8S (FW = 738.9 g mol−1): C, 40.61; H, 2.86; N, 13.26; S, 4.34%. Found: C, 40.41; H, 2.29; N, 13.37; S, 4.45%.O), 1601 (imine –CHN–), 1450 (COOH), 1298 (–SO2–NH2-asym), 1186 (–SO2–NH2-sym), 1060 (–SO). UV λmax (nm): 280 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.99 (1H, s, –CHN), 8.03 (1H, s, –NH–CO–aryl), 7.66–7.86, (12H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.51 (H, t, J = 7.8 Hz, –CH), 4.55 (2H, s, –CH2NH–), 4.32 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.8, 166.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 55.6, 49.4, 35.0, 34.7, 21.5. Anal. calc. for C31H27N7O10S2 (FW = 721.1 g mol−1): C, 51.59; H, 3.77; N, 13.59; S, 8.89%. Found: C, 51.41; H, 3.49; N, 13.47; S, 8.65%.O), 1601 (imine –CHN–), 1455 (COOH), 1298 (–SO2–NH2-asym), 1160 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 295 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.98 (1H, s, –CHN), 8.02 (1H, s, –NH–CO–aryl), 7.66–7.89, (12H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.51 (H, t, J = 7.8 Hz, –CH), 4.54 (2H, s, –CH2NH–), 4.33 (1H, t, J = 7.1 Hz, –N–CH), 2.34 (3H, t, J = 7.1 Hz, –CH3), 2.07 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.8, 166.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 55.6, 49.4, 35.0, 34.7, 23.4, 21.5. Anal. calc. for C31H27N7O10S2 (FW = 735.1 g mol−1): C, 52.24; H, 3.97; N, 13.33; S, 8.72%. Found: C, 52.41; H, 3.89; N, 13.49; S, 8.85%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1005 (–SO). UV λmax (nm): 290 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.98 (1H, s, –CHN), 8.02 (1H, s, –NH–CO–aryl), 7.66–7.89, (12H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (H, t, J = 7.8 Hz, –CH), 4.34 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (3H, t, J = 7.1 Hz, –CH3), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 166.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 55.6, 49.4, 35.0, 34.7, 23.4, 21.5. Anal. calc. for C31H27N7O10S2 (FW = 735.1 g mol−1): C, 52.24; H, 3.97; N, 13.33; S, 8.72%. Found: C, 52.41; H, 3.89; N, 13.49; S, 8.85%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1001 (–SO). UV λmax (nm): 275 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.98 (1H, s, –CHN), 8.33 (1H, s, –NH–CO–aryl), 7.73 (4H, d, J = 7.6 Hz, ArH), 7.54 (4H, d, J = 7.4 Hz, ArH), 7.34 (4H, d, J = 7.6 Hz, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (2H, s, –CH2NH–), 4.34 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (6H, t, J = 7.1 Hz, 2× CH3), 2.33 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 177.7, 166.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 59.5, 49.4, 35.0, 34.7, 23.4, 21.5. C33H31N7O10S2 (FW = 749.1 g mol−1): C, 52.86; H, 4.17; N, 13.08; S, 8.55%. Found: C, 52.71; H, 4.09; N, 13.25; S, 8.65%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1176 (–SO2–NH2-sym), 1058 (–SO). UV λmax (nm): 240 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.96 (1H, s, –CHN), 8.43 (H, s, CH–CH–Br), 8.03 (1H, s, –NH–CO–aryl), 7.56–7.84, (11H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (H, t, J = 7.8 Hz, –CH), 4.33 (2H, s, –CH2NH–), 4.33 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.8, 165.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 59.5, 49.4, 35.0, 34.7, 23.4, 21.5. Anal. calc. for C31H25Br2N7O10S2 (FW = 878.9 g mol−1): C, 42.33; H, 2.87; N, 11.15; S, 7.29%. Found: C, 42.41; H, 2.79; N, 11.27; S, 7.45%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1176 (–SO2–NH2-sym), 1062 (–SO). UV λmax (nm): 245 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.05 (2H, s, 2× OH), 8.96 (1H, s, –CHN), 8.43 (H, s, CH–CH–Br), 8.03 (1H, s, –NH–CO–aryl), 7.56–7.84, (10H, m, ArH), 6.68 (2H, d, J = 7.6 Hz, ArH), 4.55 (H, t, J = 7.8 Hz, –CH), 4.33 (2H, s, –CH2NH–), 4.33 (1H, t, J = 7.1 Hz, –N–CH), 2.35 (3H, t, J = 7.1 Hz, –CH3), 2.35 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.8, 165.8, 164.7, 162.9, 153.5, 151.3, 150.1, 133.3, 130.7, 130.6, 129.4, 128.9, 115.6, 115.7, 59.5, 49.4, 30.4, 23.4, 21.5. Anal. calc. for C32H27Br2N7O10S2 (FW = 892.9 g mol−1): C, 43.01; H, 3.05; N, 10.97; S, 7.18%. Found: C, 42.91; H, 3.09; N, 11.17; S, 7.05%.O), 1606 (imine –CHN–), 1450 (COOH), 1284 (–SO2–NH2-asym), 1166 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 295 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.1 (2H, s, 2× OH), 8.99 (1H, s, –CHN), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.46, (17H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.7, 163.5, 161.5, 155.2, 138.9, 131.8, 130.1, 129.8, 128.6, 127.9, 124.9, 120.3, 117.7, 52.4, 49.6, 36.9, 29.9, 28.3, 26.6, 22.0. Anal. calc. for C37H31N7O12S3 (FW = 861.1 g mol−1): C, 51.56; H, 3.63; N, 11.38; S, 11.16%. Found: C, 51.41; H, 3.49; N, 11.47; S, 11.25%.O), 1606 (imine –CHN–), 1450 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 290 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.1 (2H, s, 2× OH), 8.99 (1H, s, –CHN), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.46, (17H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.9, 159.6, 157.2, 152.4, 143.6, 139.2, 135.4, 129.6, 129.3, 128.9, 128.1, 127.9, 52.6, 49.9, 37.2, 30.1, 28.5, 26.9, 22.2, 21.7. Anal. calc. for C39H35N7O12S3 (FW = 889.1 g mol−1): C, 52.64; H, 3.96; N, 11.02; S, 10.81%. Found: C, 52.41; H, 3.79; N, 11.16; S, 10.62%.O), 1600 (imine –CHN–), 1451 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 295 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.07 (2H, s, 2× OH), 8.87 (1H, s, –CHN), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.46, (16H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (3H, t, J = 7.1 Hz, CH3), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 178.9, 160.5, 157.2, 152.4, 143.6, 139.2, 135.4, 129.6, 129.3, 128.9, 128.1, 127.9, 52.6, 49.9, 37.2, 30.1, 28.5, 26.9, 22.2, 21.7. Anal. calc. for C38H33N7O12S3 (FW = 875.1 g mol−1): C, 52.11; H, 3.80; N, 11.19; S, 10.98%. Found: C, 52.32; H, 3.64; N, 11.25; S, 10.85%.O), 1606 (imine –CHN–), 1450 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 290 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.07 (2H, s, 2× OH), 8.87 (1H, s, –CHN), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.49, (16H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (3H, t, J = 7.1 Hz, CH3), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 179.7, 167.1, 163.1, 152.6, 149.5, 139.6, 139.5, 138.4, 130.8, 130.3, 129.5, 125.6, 125.2, 114.1, 55.8, 52.6, 51.5, 37.8, 30.2, 30.1, 29.9, 28.5, 26.8, 26.0 22.9, 22.5. Anal. calc. for C40H37N7O12S3 (FW = 903.1 g mol−1): C, 53.15; H, 4.13; N, 10.85; S, 10.64%. Found: C, 53.35; H, 4.21; N, 10.66; S, 10.71%.O), 1606 (imine –CHN–), 1450 (COOH), 1288 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 280 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.07 (2H, s, 2× OH), 8.87 (1H, s, –CHN), 8.67 (1H, s, CH–CH–Br), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.48, (15H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.44 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 179.8, 167.1, 163.1, 152.7, 149.5, 139.7, 139.5, 138.4, 130.8, 130.3, 129.5, 125.6, 125.2, 114.0, 55.8, 52.6, 51.5, 37.8, 30.2, 30.1, 29.9, 28.5, 26.8, 26.0. Anal. calc. for C37H29Br2N7O12S3 (FW = 1018.9 g mol−1): C, 43.58; H, 2.87; N, 9.62; S, 9.43%. Found: C, 43.41; H, 2.79; N, 9.47; S, 9.25%.O), 1606 (imine –CHN–), 1450 (COOH), 1286 (–SO2–NH2-asym), 1158 (–SO2–NH2-sym), 1045 (–SO). UV λmax (nm): 245 nm. 1H NMR (500 MHz, DMSO-d6): δH 11.07 (2H, s, 2× OH), 8.87 (1H, s, –CHN), 8.67 (1H, s, CH–CH–Br), 8.42 (1H, s, –NH–CO–aryl), 6.88–7.48, (15H, m, ArH), 6.86 (2H, d, J = 7.6 Hz, ArH), 4.45 (H, t, J = 7.8 Hz, –CH), 4.45 (2H, s, –CH2N–), 4.28 (1H, t, J = 7.1 Hz, –N–CH), 2.24 (2H, t, J = 7.1 Hz, –CH2), 2.09 (2H, t, J = 7.6 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6): 179.6, 167.5, 163.5, 152.6, 149.5, 139.6, 139.5, 138.7, 130.9, 130.3, 129.5, 125.6, 125.2, 114.1, 55.8, 52.6, 51.5, 37.8, 30.2, 30.2, 29.8, 28.5, 26.8, 26.0 22.9, 22.5. Anal. calc. for C37H29Br2N7O12S3 (FW = 1046.9 g mol−1): C, 44.71; H, 3.17; N, 9.36; S, 9.18%. Found: C, 44.48; H, 3.29; N, 9.47; S, 9.25.2.3. Antibacterial activities
Disk diffusion and 96-well plate assay methods were employed to determine the zone of inhibition and the minimum inhibitory concentration (MIC) respectively, to investigate the antibacterial activities, where ampicillin was used as a reference drug as described in our previous studies,21,41 details of which are provided in the ESI.†
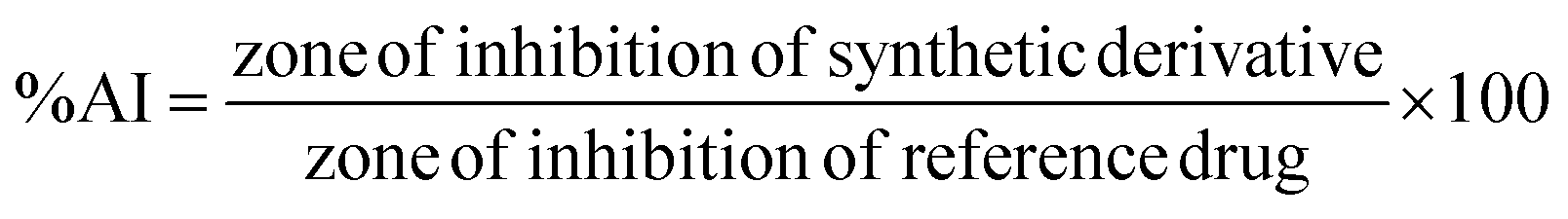 | (A) |
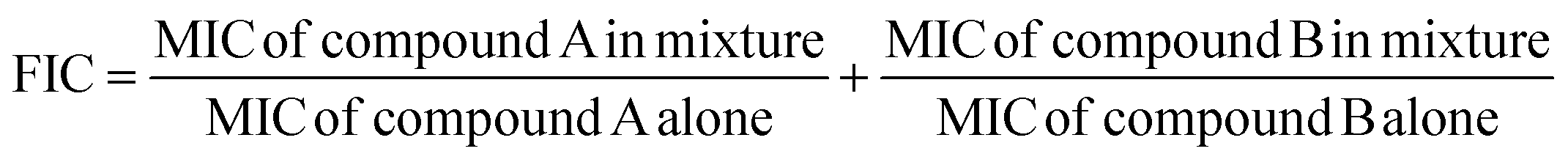 | (B) |
In which T is the absorbance of the inhibitory well and C is absorbance of 100% initial activity without inhibitor.

2.4. Molecular docking studies
3. Results and discussion
3.1. Chemistry
Conjugated sulfonamides were synthesized by the reaction of p-toluenesulfonyl chloride, benzenesulfonyl chloride and 2,4-dibromo benzenesulfonyl chloride with folic acid (details are explained in the Experimental section) and their biological activities were evaluated against bacterial strains such as Proteus mirabilis (ATCC 43071), Pseudomonas aeruginosa (ATCC-27853), Escherichia coli (ATCC-25922) and Staphylococcus aureus (ATCC-25923).The amine groups of FA can be numbered as N1, N2 and N3 according to their preference of availability for electrophile attack. The reaction was carried out at basic pH (0.05 M, Na2CO3) because folic acid is soluble in the basic environment, as well as assisting the electrophilic attack of sulfonyl chloride. HCl was formed as a by-product that raised the pH towards an acidic value which was neutralized by the base present in the reaction mixture. Mono-substituted FA-derivatives (MS1–MS3, Scheme 1) were obtained by the attack of the sulfur atom at N1, then monosubstituted derivatives were reacted with sulfonyl chlorides to obtain the disubstituted FA-derivatives (DS1–DS6, Scheme 2), consequently these were reacted again with the sulfonyl chlorides to obtain the trisubstituted FA-derivatives (TS1–TS6). The first sulfonyl chlorides attack was on N1 as it is a primary nitrogen (–NH2), that is, the most easily accessible, and then N2 and N3 as they are facing steric hindrance owing to the presence of aromatic rings in their neighborhoods. UV/Vis, FTIR, and 1H and 13C NMR spectroscopic techniques were used to characterize the novel synthesized compounds.
A strong absorption band in the 1148–1153 cm−1 (symmetrical) and 1315–1362 cm−1 (unsymmetrical) region for the MS1–MS3 compounds, the 1132–1170 cm−1 (symmetrical) and 1345–1375 cm−1 (unsymmetrical) region for the DS1–DS6 compounds and the 1125–1162 cm−1 (symmetrical) and 1344–1370 cm−1 (unsymmetrical) region confirm the presence of the –NH–SO group. Moreover, a characteristic sulfoxide absorption band appeared in the 1026–1032 cm−1 region for all the synthetic compounds. The peaks for the –NH proton of the –SO2NH– group appeared at δ 10.47–12.43 ppm and confirmed the formation of the sulfonamides. The compounds gave a singlet at δ 2.49–2.48 ppm assigned to a proton of –CH3 linked to the aryl moiety. In the 13C NMR spectra, the 143–163 ppm peaks were assigned to the presence of an imine carbon (–CN–) while the peaks at 165.2–179.8 ppm were assigned to the carbonyl carbon. The signals at 134.24–146.45 were assigned to the aryl carbons, as there are three types of nitrogen, and the highest value signals were at the N3 substitution of the sulfonyl chlorides, which is bound to two aromatic rings and offered a high delocalization. Therefore, the 13C NMR spectral analyses were consistent with the assigned structure of all the compounds.
3.2. Antibacterial activities
The novel synthesized derivatives were assessed as antibacterial agents. The disk diffusion method was utilized to measure the inhibition zone and MIC via the 96-well plate method by measuring the optical density (OD) at 600 nm using a two-fold serial dilution method. Four bacterial strains were used, including two from each group, Gram negative bacteria (P. aeruginosa, E. coli) and Gram positive (S. aureus, P. mirabilis). Ampicillin was used as a reference drug while trimethoprim was considered as a reference DHFR inhibitor. FA and respective sulfonyl chloride were also screened for antibacterial activity. The %AI of each synthetic compound was also measured as compared to the standard drug ampicillin. The zone of inhibition for each synthetic compound was measured in triplicate at three gradient concentrations (Tables S1 and S2, ESI†) such as 3, 1.5 and 0.75 mg mL−1.The compounds MS3, DS1, DS2, DS5, DS6 and TS5 have an appreciable antibacterial activity against all strains of bacteria (Tables 1 and 2). Compound DS2 has the highest zone of inhibition (36.6 mm) with a %AI value of 122.8% against S. aureus and a MIC of 15.63 μg mL−1. Sulfonyl chlorides did not show any response against the bacterial strains, while folic acid showed a small amount of activity for the highest concentration (3 mg mL−1). The FA derivatives showed a good activity that could be attributed to the presence of a pyrimidine ring in its scaffold and the resulting sulfonamides that can cause the inhibition of dihydrofolate reductase synthesis. Owing to the interaction of FA with bacterial cell membranes, it may offer enhanced permeability inside the microbial cells. Thus, the FA moiety can be an essential nutrient for DNA/RNA synthesis in bacteria, and may assist the transportation of sulfonamides via endocytosis into the cytoplasm, easily crossing the plasma membrane.45,46
| Compound | Gram (+) bacteria | Gram (−) bacteria | ||
|---|---|---|---|---|
| S. aureus | P. mirabilis | E. coli | P. aeruginosa | |
| a Control drug.b Reference DHFR inhibitor. The zone of inhibition was measured in mm ± SD, gradient concentrations of 3, 1.5, and 0.75 mg mL−1 were used. S. aureus = Staphylococcus aureus, E. coli = Escherichia coli, P. aeruginosa = Pseudomonas aeruginosa, and P. mirabilis = Proteus mirabilis. | ||||
| MS1 | 31.1 ± 0.06, 29.9 ± 0.06, 27.8 ± 0.06 | 29.3 ± 0.06, 27.9 ± 0.06, 22.0 ± 0.15 | 31.1 ± 0.12, 29.8 ± 0.06, 24.1 ± 0.12 | 30.9 ± 0.06, 28.2 ± 0.06, 17.8 ± 0.06 |
| MS2 | 29.8 ± 0.06, 28.8 ± 0.11, 26.8 ± 0.06 | 27.8 ± 0.06, 24.0 ± 0.12, 22.1 ± 0.06 | 29.8 ± 0.06, 28.9 ± 0.10, 23.7 ± 0.06 | 27.9 ± 0.06, 25.8 ± 0.06, 15.8 ± 0.06 |
| MS3 | 31.9 ± 0.06, 27.9 ± 0.06, 22.1 ± 0.06 | 28.1 ± 0.06, 24.3 ± 0.06, 22.5 ± 0.06 | 33.1 ± 0.12, 28.4 ± 0.06, 24.7 ± 0.06 | 29.2 ± 0.06, 27.8 ± 0.06, 20.3 ± 0.06 |
| DS1 | 32.8 ± 0.06, 30.9 ± 0.06, 28.7 ± 0.06 | 23.3 ± 0.12, 29.8 ± 0.06, 28.5 ± 0.06 | 34.9 ± 0.06, 32.3 ± 0.06, 29.3 ± 0.06 | 32.8 ± 0.06, 31.9 ± 0.06, 18.5 ± 0.06 |
| DS2 | 36.6 ± 0.06, 31.5 ± 0.06, 30.5 ± 0.06 | 35.8 ± 0.12, 30.1 ± 0.12, 29.6 ± 0.06 | 37.8 ± 0.10, 33.3 ± 0.06, 31.6 ± 0.06 | 34.8 ± 0.06, 32.1 ± 0.06, 30.9 ± 0.06 |
| DS3 | 30.8 ± 0.06, 27.8 ± 0.06, 26.8 ± 0.06 | 29.8 ± 0.12, 23.8 ± 0.06, 22.3 ± 0.06 | 31.6 ± 0.06, 29.3 ± 0.06, 23.7 ± 0.06 | 28.8 ± 0.06, 27.8 ± 0.06, 17.9 ± 0.06 |
| DS4 | 31.8 ± 0.06, 29.1 ± 0.06, 26.8 ± 0.06 | 30.8 ± 0.06, 28.9 ± 0.06, 26.4 ± 0.12 | 34.9 ± 0.06, 32.4 ± 0.10, 28.7 ± 0.12 | 30.9 ± 0.12, 29.4 ± 0.06, 27.4 ± 0.15 |
| DS5 | 34.8 ± 0.06, 30.8 ± 0.06, 28.6 ± 0.06 | 33.5 ± 0.06, 29.5 ± 0.06, 28.4 ± 0.12 | 36.5 ± 0.06, 32.3 ± 0.06, 28.9 ± 0.06 | 32.8 ± 0.06, 31.3 ± 0.06, 29.3 ± 0.12 |
| DS6 | 33.8 ± 0.06, 31.7 ± 0.06, 28.4 ± 0.11 | 33.2 ± 0.12, 29.2 ± 0.06, 28.7 ± 0.06 | 36.7 ± 0.06, 32.4 ± 0.06, 29.7 ± 0.12 | 34.0 ± 0.06, 31.3 ± 0.06, 29.9 ± 0.06 |
| TS1 | 32.5 ± 0.06, 26.5 ± 0.06, 23.8 ± 0.06 | 31.3 ± 0.06, 29.3 ± 0.06, 24.1 ± 0.06 | 34.7 ± 0.06, 30.8 ± 0.06, 26.0 ± 0.06 | 29.4 ± 0.06, 27.6 ± 0.10, 24.2 ± 0.06 |
| TS2 | 31.8 ± 0.06, 28.8 ± 0.06, 25.8 ± 0.06 | 30.7 ± 0.06, 27.7 ± 0.06, 26.2 ± 0.06 | 34.8 ± 0.05, 31.9 ± 0.05, 29.5 ± 0.09 | 31.2 ± 0.09, 29.3 ± 0.09, 27.3 ± 0.09 |
| TS3 | 32.5 ± 0.06, 26.8 ± 0.06, 23.4 ± 0.06 | 30.8 ± 0.06, 29.7 ± 0.06, 24.1 ± 0.06 | 34.6 ± 0.05, 30.1 ± 0.09, 24.4 ± 0.09 | 29.7 ± 0.09, 27.9 ± 0.05, 24.5 ± 0.19 |
| TS4 | 20.3 ± 0.06, 15.8 ± 0.06, 10.8 ± 0.06 | 18.3 ± 0.06, 14.4 ± 0.06, 10.8 ± 0.06 | 19.3 ± 0.09, 12.1 ± 0.09, 10.8 ± 0.09 | 20.1 ± 0.09, 17.7 ± 0.09, 14.3 ± 0.05 |
| TS5 | 34.8 ± 0.06, 31.7 ± 0.06, 28.4 ± 0.06 | 33.2 ± 0.06, 29.2 ± 0.06, 28.6 ± 0.06 | 36.7 ± 0.05, 32.4 ± 0.08, 29.7 ± 0.08 | 33.9 ± 0.05, 31.3 ± 0.09, 29.9 ± 0.09 |
| TS6 | 23.1 ± 0.06, 15.3 ± 0.06, 11.1 ± 0.06 | 19.0 ± 0.06, 14.4 ± 0.06, 10.7 ± 0.06 | 19.6 ± 0.05, 12.4 ± 0.09, 10.9 ± 0.05 | 20.9 ± 0.05, 17.8 ± 0.05, 14.6 ± 0.05 |
| Folic acid | —, —, — | —, —, — | —, —, — | —, —, — |
| p-Toluenesulfonyl chloride | —, —, — | —, —, — | —, —, — | —, —, — |
| Benzenesulfonyl chloride | —, —, — | —, —, — | —, —, — | —, —, — |
| 2,4-Dibromo benzenesulfonyl chloride | —, —, — | —, —, — | —, —, — | —, —, — |
| aAmpicillin | 29.8 ± 0.06, 28.1 ± 0.06, 26.7 ± 0.06 | 30.5 ± 0.06, 29.5 ± 0.06, 28.2 ± 0.06 | 33.6 ± 0.12, 30.1 ± 0.08, 29.1 ± 0.08 | 29.2 ± 0.08, 27.3 ± 0.05, 26.2 ± 0.08 |
| bTrimethoprim | 28.3 ± 0.05, 20.3 ± 0.05, 18.3 ± 0.09 | 25.0 ± 0.09, 20.3 ± 0.05, 14.4 ± 0.09 | 31.0 ± 0.17, 25.1 ± 0.09, 20.1 ± 0.08 | 14.2 ± 0.05, 9.3 ± 0.05, 4.1 ± 0.05 |
| Compound | Gram (+) bacteria | Gram (−) bacteria | ||||||
|---|---|---|---|---|---|---|---|---|
| S. aureus | P. mirabilis | E. coli | P. aeruginosa | |||||
| MIC | AI | MIC | AI | MIC | AI | MIC | AI | |
| a Control drug. %AI = percentage activity index, MIC = minimum inhibitory concentrations in μg mL−1. | ||||||||
| MS1 | 15.63 | 104.7 | 15.63 | 96.1 | 31.25 | 92.5 | 15.63 | 106.2 |
| MS2 | 31.25 | 100.3 | 31.25 | 91.5 | 31.25 | 89.3 | 15.63 | 95.5 |
| MS3 | 31.25 | 107.0 | 31.25 | 92.4 | 31.25 | 98.5 | 15.63 | 100 |
| DS1 | 15.63 | 110.1 | 62.50 | 109.5 | 125.0 | 104.2 | 15.63 | 113.1 |
| DS2 | 15.63 | 122.8 | 15.63 | 117.7 | 15.63 | 112.8 | 15.63 | 119.2 |
| DS3 | 15.63 | 103.0 | 15.63 | 98.0 | 31.25 | 95.2 | 15.63 | 99.3 |
| DS4 | 15.63 | 107.0 | 15.63 | 100.9 | 31.25 | 103.9 | 15.63 | 105.8 |
| DS5 | 15.63 | 116.4 | 15.63 | 110.2 | 31.25 | 108.6 | 15.63 | 112.4 |
| DS6 | 31.25 | 113.8 | 31.25 | 109.2 | 31.25 | 109.3 | 31.25 | 116.5 |
| TS1 | 15.63 | 109.4 | 15.63 | 101.6 | 31.25 | 103.3 | 15.63 | 100.7 |
| TS2 | 15.63 | 106.7 | 15.63 | 100.9 | 31.25 | 113.8 | 15.63 | 106.9 |
| TS3 | 15.63 | 109.1 | 15.63 | 101.3 | 62.50 | 102.9 | 15.63 | 101.7 |
| TS4 | 125.0 | 67.8 | 250.0 | 59.7 | 250.0 | 57.3 | 125.0 | 68.7 |
| TS5 | 15.63 | 117.1 | 15.63 | 109.2 | 31.25 | 109.3 | 15.63 | 116.5 |
| TS6 | 125.0 | 77.8 | 250.0 | 62.6 | 250.0 | 58.2 | 250.0 | 71.5 |
| Folic acid | >1000 | — | >1000 | — | >1000 | — | >1000 | — |
| p-Toluenesulfonyl chloride | >1000 | — | >1000 | — | >1000 | — | >1000 | — |
| Benzenesulfonyl chloride | >1000 | — | >1000 | — | > 1000 | — | >1000 | — |
| 2,4-Dibromo benzenesulfonyl chloride | >1000 | — | >1000 | — | >1000 | — | >1000 | — |
| aAmpicillin | 125.0 | 100.0 | 125.0 | 100.0 | 15.63 | 100.0 | 125.0 | 100.0 |
From a structure activity relationship point of view of the compounds, the compounds that showed the lowest MIC and highest inhibition are a result of the presence of electron withdrawing halogens (Br) attached to the aryl ring. In contrast, the compounds with an unsubstituted aryl moiety of sulfonamides also revealed excellent inhibition against bacterial strains compared to the methyl substitution on the aryl moiety, which is an electron donating group and decreases the inhibition effect.
3.3. FIC index calculation
The combination of two or more drugs could have more of a therapeutic effective than the sum of the independent effects caused by the individual component, as well as the toxicity and other side effects associated with high doses of a single drug. Medication compliance could also be improved by reducing the pill burden of patients with a better efficacy and smaller side effects compared to single drugs because the development and progression of systemic diseases often involves complex biological processes.47–49 The FIC index, as depicted in Table 3, combines the strong acting sulfonamide DS2 with ampicillin and revealed a broad antibacterial spectrum with a reduced dosage. Moreover, the FIC index was less than or equal to 1, which indicated that the combination system had a good additive effect. Notably, the combination of ampicillin (3.91 μg mL−1) with the compound DS2 (7.82 μg mL−1) inhibited the growth of E. coli, which was four-fold more potent than ampicillin alone, as described in Table 3.| Bacteria | Compounds | MIC | FIC index | Effect |
|---|---|---|---|---|
| a MIC = minimum inhibitory concentrations in μg mL−1. | ||||
| S. aureus | Ampicillin | 31.25 | 0.75 | Additive |
| DS2 | 7.82 | |||
| P. mirabilis | Ampicillin | 62.5 | 1.0 | Additive |
| DS2 | 7.82 | |||
| E. coli | Ampicillin | 3.91 | 0.75 | Additive |
| DS2 | 7.82 | |||
| P. aeruginosa | Ampicillin | 62.5 | 1.0 | Additive |
| DS2 | 7.82 | |||
3.4. Dihydrofolate reductase inhibitory activity
All of the FA conjugates were tested for inhibitory activity against bovine DHFR, the percentage inhibition values were calculated in triplicate and the average inhibitory value of each compound is presented in Table 4. Trimethoprim was used as a standard DHFR inhibitor. The results (mean ± standard error of the mean (SEM)) demonstrate that compound DS2 exhibited a 75.4 ± 0.12% inhibition, whereas the standard drug (trimethoprim as the reference DHFR inhibitor) showed a 74.6 ± 0.09% inhibition.| Compound | Glide score (kcal mol−1) | %DHFR inhibition at 10 μg mL−1 (mean ± SEM) |
|---|---|---|
| a Reference DHFR inhibitor. | ||
| MS1 | −6.4 | 65.5 ± 0.12 |
| MS2 | −7.1 | 61.4 ± 0.09 |
| MS3 | −6.7 | 62.3 ± 0.15 |
| DS1 | −5.7 | 68.7 ± 0.07 |
| DS2 | −6.7 | 75.4 ± 0.12 |
| DS3 | −6.9 | 63.4 ± 0.09 |
| DS4 | −7.5 | 64.6 ± 0.03 |
| DS5 | −6.3 | 68.9 ± 0.03 |
| DS6 | −7.2 | 71.5 ± 0.09 |
| TS1 | −5.5 | 65.6 ± 0.03 |
| TS2 | −5.6 | 69.5 ± 0.17 |
| TS3 | −7.7 | 67.8 ± 0.13 |
| TS4 | −6.2 | 59.6 ± 0.07 |
| TS5 | −7.9 | 64.8 ± 0.09 |
| TS6 | −4.7 | 58.6 ± 0.03 |
| aTrimethoprim | −5.9 | 74.6 ± 0.09 |
3.5. Molecular docking studies
Different sulfonamide containing compounds and folic acid derivatives inhibit the biological function of the DHFR enzyme. Therefore, synthesized FA sulfonamide conjugates were docked in Staphylococcus aureus DHFR enzyme active sites to determine the plausible binding modes of the novel synthesized compounds. The predicted glide scores of the synthesized compounds are in the range from −4.7 to −7.7 kcal mol−1, as given in Table 4. Fig. 1 shows that the best active compound binds well at the active site of the DHFR enzyme. The best binding pose of the DS2 compound is reasonably well superimposed on the cocrystal ligand (trimethoprim) as shown in Fig. 1a. The head part phenyl ring of the DS2 structure in the synthesized inhibitor series is in the same position as the trimethoprim head group.50 The DS2 also forms hydrogen bonding with the backbone carbonyl of Leu-20 as shown in Fig. 1b, and the residues point towards the solvent exposed site. Similarly, the Phe-92 residue forms π–π stacking with the head part phenyl ring of the DS2 compound in the binding site. The interatomic calculated distances of the DS2 compound atoms and the side chain atoms of the DHFR enzyme did not result in any bad clashes, which explain that the predicted binding pose can be accepted as the true binding mode. We have also observed in docking studies that the best docked pose of some of the novel synthesized compounds flipped their orientation in comparison to the co-crystal ligand. This might be due to the coupling of large moieties with the amino group of the pteridine ring present in the folic acid. In many compounds, the docking scores agreed well with the observed biological activities of the synthesized compounds, however those that did not exhibit correlation could be preferentially binding partially or fully at the NADPH substrate binding site.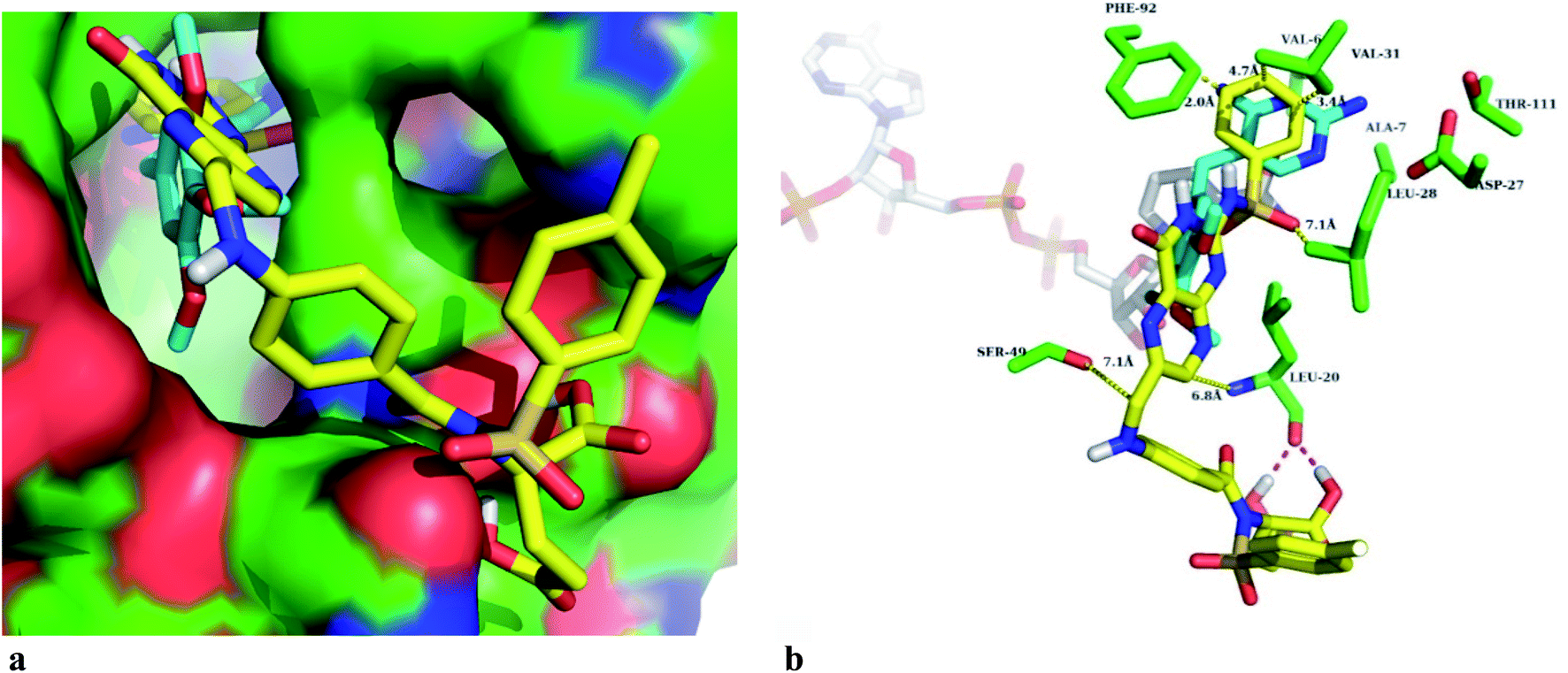 | ||
| Fig. 1 Probable binding mode of the most active compound DS2 in the DHFR active site. (a) Superposition of docked DS2 (yellow) in the active site of the DHFR surface containing trimethoprim (cyan color). (b) Binding site (green sticks) containing 4A residues around the docked pose of DS2 (yellow), the white sticks are the NADPH substrate and the dotted lines (magenta) represent hydrogen bonding. Interatomic distances are shown in Angstrom. | ||
4. Conclusion
In this study, folic acid sulfonamide conjugates were synthesized successfully and their structures were confirmed using FTIR, and 1H and 13C NMR spectroscopies. The combination of DS2 with ampicillin enhanced their antimicrobial activities compared to their individual use. Moreover, the FIC index was less than or equal to 1, which indicated that the combination system had a good additive effect. The majority of the conjugates have shown a similar or higher binding affinity with the DHFR enzyme as compared to the standard drug and thus can be used to design better antimicrobial agents. Further molecular docking studies demonstrated that the synthesized compounds are binding at the trimethoprim active site in the DHFR enzyme, which could help researchers to design more active molecules.Conflicts of interest
We wish to confirm that there are no known conflicts of interest associated with this publication.Acknowledgements
The authors are thankful to HEC-Pakistan for providing financial support to purchase the software and hardware for the computational studies vide Projects No. 6804/Federal/NRPU/R&D/HEC/2016 & 8094/Balochistan/NRPU/R&D/HEC/2017.References
- G. Nitulescu, I. M. Nicorescu, O. T. Olaru, A. Ungurianu, D. P. Mihai, A. Zanfirescu, G. M. Nitulescu and D. Margina, Int. J. Mol. Sci., 2017, 18, 2217 CrossRef CAS.
- M. V. Raimondi, O. Randazzo, M. La Franca, G. Barone, E. Vignoni, D. Rossi and S. Collina, Molecules, 2019, 24, 1140 CrossRef.
- A. Singh, N. Deshpande, N. Pramanik, S. Jhunjhunwala, A. Rangarajan and H. S. Atreya, Sci. Rep., 2018, 8, 3190 CrossRef.
- N. Wang, J.-X. Ren and Y. Xie, J. Biomol. Struct. Dyn., 2019, 37, 1054–1061 CrossRef CAS.
- M. A. Qadir, M. Ahmed and M. Iqbal, BioMed Res. Int., 2015, 2015, 7 Search PubMed.
- M. A. Qadir, M. Ahmed and A. Khaleeq, Lat. Am. J. Pharm., 2015, 34, 719–724 CAS.
- M. A. Qadir, M. Ahmed, H. Aslam, S. Waseem and M. I. Shafiq, J. Chem., 2015, 2015, 8 Search PubMed.
- M. A. Qadir, M. Ahmed, M. I. Shafiq, N. Mahmood, S. Anwar, T. Habib and S. Nosheen, Lat. Am. J. Pharm., 2015, 34, 1511–1515 CAS.
- A. Tačić, V. Nikolić, L. Nikolić and I. Savić, Advanced technologies, 2017, 6, 58–71 CrossRef.
- D. Diaconu, V. Mangalagiu, D. Amariucai-Mantu, V. Antoci, C. L. Giuroiu and I. I. Mangalagiu, Molecules, 2020, 25, 2946 CrossRef CAS.
- Y.-Y. Hu, R. R. Yadav Bheemanaboina, N. Battini and C.-H. Zhou, Mol. Pharm., 2019, 16, 1036–1052 CrossRef CAS.
- M. F. Hussein, Mediterr. J. Chem., 2018, 7, 370–385 CrossRef.
- J. Mareddy, S. B. Nallapati, J. Anireddy, Y. P. Devi, L. N. Mangamoori, R. Kapavarapu and S. Pal, Bioorg. Med. Chem. Lett., 2013, 23, 6721–6727 CrossRef CAS.
- L. Chen, D. Yang, Z. Pan, L. Lai, J. Liu, B. Fang and S. Shi, Chem. Biol. Drug Des., 2015, 86, 239–245 CrossRef CAS.
- J. Mareddy, N. Suresh, C. G. Kumar, R. Kapavarapu, A. Jayasree and S. Pal, Bioorg. Med. Chem. Lett., 2017, 27, 518–523 CrossRef CAS.
- J. Liu, C. Liu, X. Zhang, L. Yu, X. Gong and P. Wang, J. Enzyme Inhib. Med. Chem., 2019, 34, 1380–1387 CrossRef CAS.
- S. D. Firke and S. B. Bari, Bioorg. Med. Chem., 2015, 23, 5273–5281 CrossRef CAS.
- A. Abbas, S. Murtaza, M. N. Tahir, S. Shamim, M. Sirajuddin, U. A. Rana, K. Naseem and H. Rafique, J. Mol. Struct., 2016, 1117, 269–275 CrossRef CAS.
- N. U. H. Khan, S. Zaib, K. Sultana, I. Khan, B. Mougang-Soume, H. Nadeem, M. Hassan and J. Iqbal, RSC Adv., 2015, 5, 30125–30132 RSC.
- S. Mutahir, J. Jończyk, M. Bajda, I. U. Khan, M. A. Khan, N. Ullah, M. Ashraf, S. Riaz, S. Hussain and M. Yar, Bioorg. Chem., 2016, 64, 13–20 CrossRef CAS.
- M. Ahmed, M. A. Qadir, M. I. Shafiq, M. Muddassar, Z. Q. Samra and A. Hameed, Arabian J. Chem., 2019, 12, 41–53 CrossRef CAS.
- M. Ahmed, M. A. Qadir, A. Hameed, M. N. Arshad, A. M. Asiri and M. Muddassar, Biochem. Biophys. Res. Commun., 2017, 490, 434–440 CrossRef CAS.
- M. Ahmed, M. A. Qadir, A. Hameed, M. N. Arshad, A. M. Asiri and M. Muddassar, Bioorg. Chem., 2018, 76, 218–227 CrossRef CAS.
- R. A. Azzam, R. E. Elsayed and G. H. Elgemeie, ACS Omega, 2020, 5, 10401–10414 CrossRef CAS.
- C. Capasso and C. T. Supuran, Bacterial Resistance to Antibiotics–From Molecules to Man, 2019, pp. 163–172 Search PubMed.
- N. A. Al-Masoudi and Y. A. Al-Soud, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 1034–1044 CrossRef CAS.
- J. N. Domínguez, C. León, J. Rodrigues, N. G. de Domínguez, J. Gut and P. J. Rosenthal, Il Farmaco, 2005, 60, 307–311 CrossRef.
- Y. Ding, K. L. Smith, V. Prasad, V. Chamakura and B. Wang, Lett. Org. Chem., 2018, 15, 87–91 CAS.
- I. Ugwu David, C. Okoro Uchechukwu and D. Chukwurah Thompson, Med. Chem., 2014, 4, 357–360 Search PubMed.
- L. Novotny, P. Rauko and H. Schott, Anticancer Res., 2010, 30, 4891–4898 CAS.
- A. Thiry, J.-M. Dogne, C. T. Supuran and B. Masereel, Curr. Pharm. Des., 2008, 14, 661–671 CrossRef CAS.
- P. Selvam, N. Murugesh, M. Chandramohan, Z. Debyser and M. Witvrouw, Indian J. Pharm. Sci., 2008, 70, 779 CrossRef CAS.
- A. T. Hopper, A. Brockman, A. Wise, J. Gould, J. Barks, J. B. Radke, L. D. Sibley, Y. Zou and S. Thomas, J. Med. Chem., 2019, 62, 1562–1576 CrossRef CAS.
- A. Wróbel, K. Arciszewska, D. Maliszewski and D. Drozdowska, J. Antibiot., 2020, 73, 5–27 CrossRef.
- D. Fernández-Villa, M. R. Aguilar and L. Rojo, Int. J. Mol. Sci., 2019, 20, 4996 CrossRef.
- Y. Zhao, W. R. Shadrick, M. J. Wallace, Y. Wu, E. C. Griffith, J. Qi, M.-K. Yun, S. W. White and R. E. Lee, Bioorg. Med. Chem. Lett., 2016, 26, 3950–3954 CrossRef CAS.
- M. Tyers and G. D. Wright, Nat. Rev. Microbiol., 2019, 17, 141–155 CrossRef CAS.
- R. Capela, R. Moreira and F. Lopes, Int. J. Mol. Sci., 2019, 20, 5748 CrossRef CAS.
- X. Deng and N. S. Mani, Green Chem., 2006, 8, 835–838 RSC.
- R. A. Nadeem, M. Abdul Qadir, M. Ahmed and I. Sajid, Lett. Drug Des. Discovery, 2020, 17, 264–270 CrossRef CAS.
- M. Ahmed, M. A. Qadir and M. I. Shafiq, Lat. Am. J. Pharm., 2017, 36, 1789–1795 CAS.
- O. O. Olajuyigbe and A. J. Afolayan, Int. J. Mol. Sci., 2012, 13, 8915–8932 CrossRef CAS.
- M. Cammarata, R. Thyer, M. Lombardo, A. Anderson, D. Wright, A. Ellington and J. S. Brodbelt, Chem. Sci., 2017, 8, 4062–4072 RSC.
- M. R. Sawaya and J. Kraut, Biochemistry, 1997, 36, 586–603 CrossRef CAS.
- B. R. Bhawana, H. S. Buttar, V. Jain and N. Jain, J. Agric. Food Chem., 2011, 59, 2056–2061 CrossRef.
- X.-M. Wang, J. Xu, Y.-P. Li, H. Li, C.-S. Jiang, G.-D. Yang, S.-M. Lu and S.-Q. Zhang, Eur. J. Med. Chem., 2013, 67, 243–251 CrossRef CAS.
- J. Lehár, A. S. Krueger, W. Avery, A. M. Heilbut, L. M. Johansen, E. R. Price, R. J. Rickles, G. F. Short Iii, J. E. Staunton and X. Jin, Nat. Biotechnol., 2009, 27, 659–666 CrossRef.
- X. Li, G. Qin, Q. Yang, L. Chen and L. Xie, BioMed Res. Int., 2016, 2016, 8518945 Search PubMed.
- C. Zhang and G. Yan, Synth. Syst. Biotechnol., 2019, 4, 67–72 CrossRef.
- T. Khamkhenshorngphanuch, K. Kulkraisri, A. Janjamratsaeng, N. Plabutong, A. Thammahong, K. Manadee, S. Na Pombejra and T. Khotavivattana, Molecules, 2020, 25, 3059 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09051d |
| This journal is © The Royal Society of Chemistry 2020 |