
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecovery of yttrium and europium from spent fluorescent lamps using pure levulinic acid and the deep eutectic solvent levulinic acid–choline chloride†
Ioanna M. Pateli a,
Andrew P. Abbotta,
Koen Binnemansb and
Nerea Rodriguez Rodriguez*b
a,
Andrew P. Abbotta,
Koen Binnemansb and
Nerea Rodriguez Rodriguez*b
aUniversity of Leicester, Chemistry Department, Materials Centre, University Road, LE1 7RH Leicester, UK
bKU Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. Box 2404, B-3001 Leuven, Belgium. E-mail: nerea.rodriguezrodriguez@kuleuven.be; Tel: +32 16322711
First published on 4th August 2020
Abstract
A solvometallurgical approach for the recovery of rare-earth elements from lamp phosphor waste was developed. The solubility of individual phosphors in different deep-eutectic solvents (DESs) was measured. The DES levulinic acid–choline chloride (xChCl = 0.33) showed high solubility of the YOX phosphor (Y2O3:Eu3+) and low solubility of the HALO phosphor (Sr,Ca)10(PO4)(Cl,F)2:Sb3+,Mn2+, which does not contain any rare-earth element. This DES was selected for further investigation. When the DES was compared to pure levulinic acid, very similar leaching behaviour was observed, showing that the proton activity is more important than the chloride as a metal ligand. The leaching of YOX and HALO using levulinic acid–choline chloride (xChCl = 0.33) or pure levulinic acid was optimised in terms of water content, temperature and leaching time. The optimised parameters were validated in a synthetic mixture of phosphors and in real lamp phosphor waste. The co-dissolution of HALO is higher in the real waste than in the synthetic mixture. The real waste was also leached with an aqueous solution of hydrochloric acid, which was non-selective against dissolution of YOX, and with the functionalised ionic liquid betainium bis(trifluoromethylsulfonyl)imide. The ionic liquid gave a similar selectivity as levulinic acid, but is much more expensive. The recovery of the metals from the pregnant leach solution was tested via precipitation with oxalic acid and solvent extraction. Oxalic acid precipitation was not suitable for the DES system. The metals could be extracted via solvent extraction with the acidic extractant bis(2-ethylhexyl)phosphoric acid (D2EHPA) and stripped by an aqueous hydrochloric acid solution. Pure levulinic acid was found to be more suitable than the corresponding ChCl-based DES for the selective recovery of YOX.
Introduction
End-of-life fluorescent lamps are considered as alternative sources of rare earth elements (REEs) and are being collected in most countries because of their mercury content, a well-recognised environmental hazard.1,2 The residues are processed by specialised companies and separated into different fractions: (1) glass, to be used in the production of other glass products, (2) metals from the filaments and electrodes, (3) plastic parts, which are burnt for energy recovery, (4) lamp phosphor powder, and (5) mercury.The lamp phosphor waste is made up of phosphors containing REEs: the red phosphor Y2O3:Eu3+ (YOX); the green phosphors LaPO4:Ce3+,Tb3+ (LAP); (Ce,Tb) MgAl11O19 (CAT); and the blue phosphor BaMgAl10O17:Eu2+ (BAM). In addition, there is the halophosphate phosphor (Sr,Ca)10(PO4)(Cl,F)2:Sb3+,Mn2+ (HALO), which does not contain any REEs.2 The lamp phosphor waste can be considered as a valuable secondary resource of REEs, and especially of europium, terbium and yttrium.3 It is expected that by 2020 the stockpiled lamp phosphor waste will contain around 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes of REEs.3
000 tonnes of REEs.3
The extraction and recovery of REEs from lamp phosphor waste has been tested by different hydrometallurgical approaches.2–9 The solubility of the different phosphors in mineral acids or other chemicals differs greatly in the sequence HALO > YOX ≫ LAP > BAM, CAT. HALO dissolves in dilute HCl at room temperature, while YOX requires 1 mol L−1 HCl and 60–90 °C. The dissolution of LAP needs strong acidic conditions (18 M H2SO4, 120–130 °C), and CAT and BAM require alkaline conditions (35% NaOH, 150 °C) or molten alkali (Na2CO3, 1000 °C).10,11 Real lamp phosphor waste contains 50% of HALO phosphor. This phosphor is very easily dissolved compared to the other phosphors, but its dissolution consumes significant amounts of acid and introduces contaminants in the leachate. On the other hand, HALO is also a secondary resource for antimony.12 After the leaching of the lamp phosphor waste, the metals are recovered from the leachate solution either by oxalic acid precipitation or by solvent extraction. Different types of extractants and diluents have been tested for the recovery of REEs from acidic leachates.10
Solvometallurgy uses non-aqueous solvents, such as molecular organic solvents, ionic liquids (ILs), and deep-eutectic solvents (DESs) to extract metals. The main advantages of solvometallurgy are a reduced water consumption and an increased selectivity.13 Two solvometallurgical routes for the valorisation of REE phosphors have been reported: (1) the functionalised IL betainium bis(trifluoromethylsulfonyl)imide, [Hbet][Tf2N], showed good YOX selectivity when tested with a synthetic phosphor mixture.14 However, large liquid-to-solid ratios were required and [Hbet][Tf2N] is expensive. This process was never validated on a real waste. (2) The LAP phosphor could be recovered from the residue obtained from the hydrometallurgical leaching of real lamp phosphor waste using undiluted methanesulfonic acid at high temperatures. However, strong dilution with water was necessary for the recovery of the REEs from the leachate by solvent extraction.15
DESs are mixtures of hydrogen bond donors and acceptors with melting points around ambient temperature. DESs share most of their physicochemical properties with ILs, e.g., low volatility, wide liquid range and tunability, but they can be easily prepared by mixing readily available bulk components.16 DESs have been found to be useful solvents for the selective dissolution of metal oxides,17,18 as well as for the recovery of metals from industrial process residues and metallic ores.19–23 Therefore, DESs were tested for the recovery of REEs from lamp phosphor waste in this work. The solubility of four different phosphors in five representative choline chloride-based DESs was determined. Based on the high solubility of YOX, and the low solubility of HALO, the best performing DES was found to be levulinic acid–choline chloride (LevA–ChCl), which was selected for the rest of the work. The leaching of individual lamp phosphors, synthetic mixtures and real phosphor using LevA–ChCl has been studied and compared to the leaching using pure levulinic acid. Pure levulinic acid showed similar leaching behaviour compared to LevA–ChCl.
On top of the good performance of levulinic acid as leaching agent (both pure and as part of a DES), it also shows a green character since it can be produced in bulk amounts from renewable sources such as by-products of sugar industry (cellulose), starch rich wastes. In 2004, the US Department of Energy included levulinic acid in the top twelve building block chemicals that can be obtained by chemical or biological conversion of sugars.24 Many reports have been published for the production of levulinic acid from renewable sources.25–28
For comparison, the real lamp phosphor waste was also leached with hydrochloric acid and the IL [Hbet][Tf2N], which is one of the best performing ILs reported in the literature. The recovery of the metals from the pregnant leach solution (PLS) via precipitation by oxalic acid and solvent extraction was investigated.
Experimental
Products
The phosphors Y2O3:Eu3+ (YOX), LaPO4:Ce3+,Tb3+ (LAP), BaMg2Al16O27:Eu2+ (BAM), and (Sr,Ca)10(PO4)6(Cl,F)2:Sb3+,Mn2+ (HALO) were purchased from Nichia (Japan), and each has a purity of 99% or higher. The real lamp phosphor waste, obtained after the mercury removal, was provided by Relight Srl (Rho, Italy). Choline chloride (99%), ethylene glycol (99.5%), urea (99.5%), betaine hydrochloride (HbetCl, 99%), bis(2-ethylhexyl) phosphoric acid (D2EHPA, 95%), ethanol (99.8%) and 1-decanol (98%) were obtained from Acros Organics NV (Geel, Belgium). Levulinic acid (99%) and oxalic acid (99%) were purchased from J&K Scientific BVBA. Glycolic acid (99%) was purchased from Janssen Chimica (Beerse, Belgium). Nitric acid (>65 wt%) was purchased from Chem-Lab NV (Zedelgem, Belgium). Hydrochloric acid (>37 wt%) was purchased from VWR (Fontenay-sous-Bois, France). The standard solutions (1000 μg mL−1) of yttrium, europium, calcium, lanthanum, cerium, barium and antimony were purchased from Chem-Lab (Zedelgem, Belgium). Lithium bis(trifluoromethylsulfonyl)imide (LiTf2N, 99%) was purchased from IOLITEC (Heilbronn). Toluene (99%) and p-cymene (99%) were obtained from Sigma-Aldrich (Diegem, Belgium). The diluent Shell GTL Solvent GS190 was obtained from Shell Global Solutions (Amsterdam, The Netherlands). Water was deionized to a resistivity of 18.2 μS cm−1 with a Millipore Reference+ ultrapure water system. All chemicals were used as received without any further purification.Instrumentation
The real lamp phosphor waste was sieved using a vibratory sieve shaker Analysette 3 from Fritsch with a 125 μm sieve. A microwave (MW) digestion system (Berghof Speedwave Xpert) was used for the digestion of the solid samples. The metal content of the samples was determined via inductively coupled plasma optical emission spectroscopy (ICP-OES) using an Optima 8300 from PerkinElmer, with a 1-slot Hybrid XLT Ceramic-Quartz torch, also from PerkinElmer. A nuclear magnetic resonance (NMR) spectrometer (Bruker Ascend 300) operating at 300 MHz was used to record NMR spectra. A Heraeus Labofuge 200 centrifuge and/or an Eppendorf 5804 centrifuge were used to facilitate the phase separation. A benchtop TXRF spectrometer (Bruker S2 Picofox) with a molybdenum-anode X-ray source (50 keV) and a silicon drift detector was used for the determination of metals in the organic phase.Procedure
For the characterisation of the YOX and the HALO phosphors, 0.1 g of solid was dissolved using 4 mL of 6 mol L−1 HCl and stirred for 24 h at 80 °C and 500 rpm. The PLS was analysed via ICP-OES. The sample preparation for the ICP-OES analysis was as follows: the samples were diluted with 2 vol% HNO3 to have a final metal concentration lower than 50 ppm and indium (5 ppm) was used as the internal standard. The spectral lines (wavelengths in nm) selected for quantification were: yttrium 324.227; calcium 315.887; europium 381.967; iron 238.204; lanthanum 408.672; cerium 413.764; and barium 233.527. Since the solubility of LAP and BAM was found to be negligible at all the experimental conditions used in this work, they were not characterised. For the characterisation of the real lamp phosphor waste, approximately 50 mg of solid material was placed in the MW digestion vessels (DAK-100), and 10 mL of HCl was added. Each digestion was performed in quadruplicate. The digestion method was: (1) ramping from room temperature to 145 °C in 10 min and holding for 10 min, (2) ramping to 170 °C in 5 min and holding for 10 min, (3) ramping to 200 °C in 5 min and holding for 10 min, (4) cooling down to 50 °C and hold for 20 min. The digested sample was diluted with Milli-Q water to a final volume of 50 mL, and it was analysed for its metal content via ICP-OES.The DESs were prepared via the heating method, both components of the DES were placed in closed vials, and heated (50 °C) while stirring (500 rpm) using a magnetic stirrer (IKA RCT classic) with temperature controller (VWR VT-5) until a clear liquid was formed. The IL [Hbet][NTf2] was synthesised by metathesis of HbetCl and Li[NTf2], following a method previously reported in the literature.14
For the leaching experiments 0.1–0.3 g of material was placed in a glass vial, the lixiviant was added, and the closed vial was placed in a sand bath. The sand band was heated using a magnetic stirrer with a temperature controller inserted in a reference vial containing the lixiviant. After the leaching, the samples were centrifuged at 3600 rpm for 10 min and filtered using PET syringe filters (pore size 0.45 μm). The metal content of the PLS was analysed via ICP-OES. The leaching efficiency of the individual REEs (% L) was calculated according to eqn (1):
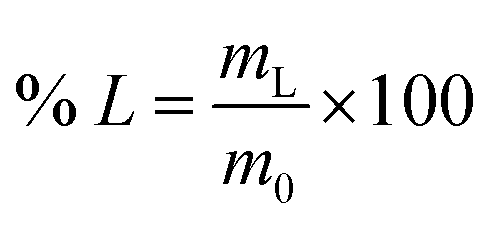 | (1) |
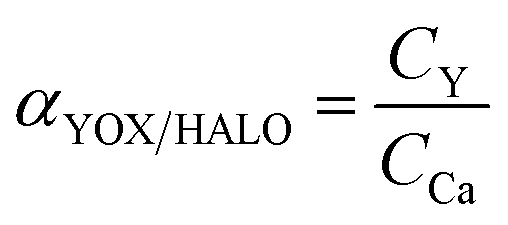 | (2) |
For the metal recovery via oxalic acid precipitation, pure oxalic acid was added to the PLS and mixed using the same stirrer and temperature controller as in the leaching experiments. The samples were centrifuged (3600 rpm, 10 min) to separate the oxalate precipitate. The lixiviant was analysed via ICP-OES to determine the remaining metal content. For the solvent extraction experiments, the aqueous and the organic phase (A/O ratio = 1) were placed in 4 mL glass vials and mixed using a magnetic stirrer with a temperature controller. Thereafter, the samples were centrifuged (4000 rpm, 5 min) to ensure phase separation. The metal content of the aqueous phase was analysed via ICP-OES, and that of the organic phase was analysed via TXRF. The organic samples were diluted 10 times in ethanol, and indium was used as internal standard. The concentration of the internal standard was selected to be similar to the analyte's concentration. The TXRF measurements were performed following an optimised methodology.29 The samples were measured on polished quartz glass disks. To prevent the pipetted sample droplet from moving and spreading on the carrier, 30 μL of a silicon solution in isopropanol (SERVA Electrophoresis GmbH, Heidelberg, Germany) was added on the carrier surface and dried for 5 min at 80 °C in a hot air oven. A small droplet (2.5 μL) of the prepared solution was added onto the hydrophobised carrier. Then, the carrier was dried in a hot air oven for 30 min at 80 °C.29
Every experiment was performed in duplicate, and the ICP/TXRF measurements were performed in triplicate. Standard deviations are included in every figure.
Results and discussion
Selection of DESs
The suitability of DESs as lixiviants for REEs phosphors was tested by measuring the solubility of the individual phosphors (YOX, HALO, CAT, and BAM) in a variety of DESs. The composition of the synthetic phosphors was determined via digestion followed by ICP-OES analysis. The obtained results are shown in the ESI, Table S1.† Previous literature showed that carboxylic acid-based DES can dissolve much more metal oxides than amide-based or polyol-based DESs.30 Therefore, different types of carboxylic acids were selected: dicarboxylic acid (oxalic acid), hydroxy acid (glycolic acid) and keto acid (levulinic acid). The obtained results were compared to other amide-based (urea) or polyol-based (ethylene glycol) DESs, to verify if that what it has been previously reported in the literature was also applicable to rare earth oxides. The hydrogen bond donor–choline chloride molar ratio was chosen to match the most widely studied compositions, which corresponds to what was believed to be the eutectic composition: two molecules of HBD were required to complex each chloride ion from the choline chloride, except for dicarboxylic acids where one hydrogen bond donor was sufficient.18,31,32 Recently, it was demonstrated that the eutectic composition is not related to complexation, but is merely thermodynamics.33 Nonetheless the real eutectic composition does not differ largely from the complexation hypothesis. Therefore, the selected DESs were: ethylene glycol–ChCl (xChCl = 0.33) (EtGly–ChCl), urea–ChCl (xChCl = 0.33) (urea–ChCl), oxalic acid–ChCl (xChCl = 0.5) (OxaA–ChCl), glycolic acid–ChCl (xChCl = 0.33) (GlyA–ChCl) and levulinic acid–ChCl (xChCl = 0.33) (LevA–ChCl). The chemical structure of the chemicals used in the preparation of the DESs are included in Fig. 1: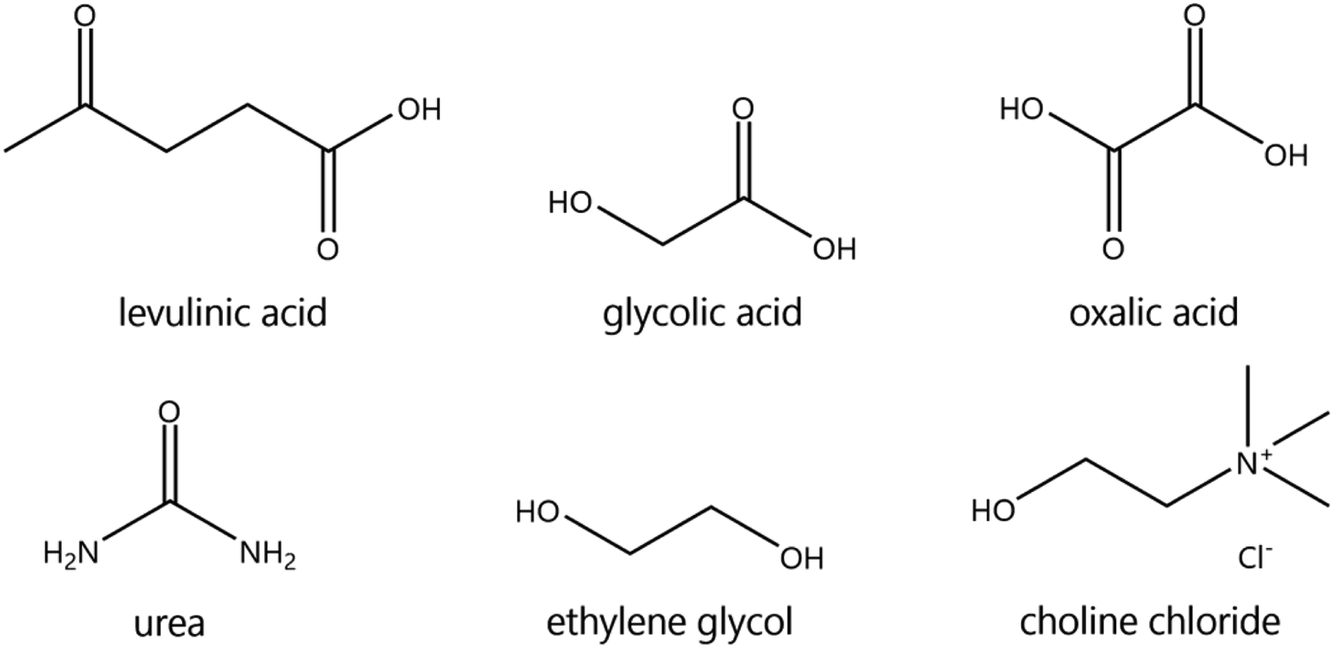 | ||
| Fig. 1 Chemical structure of the chemicals used in this work. | ||
The leaching efficiency of the individual phosphors in the selected DESs was measured under the following conditions: 80 °C, 48 h, liquid-to-solid ratio (L/S) of 10, 10 vol% H2O, and 500 rpm. The leaching efficiencies of CAT and BAM were found to be negligible in all the cases. The leaching efficiencies of YOX and HALO using different DESs are compared in Fig. 2, note the difference in scale of the Y-axes. The solubility of YOX was found be negligible in all the DESs (<1%), except for LevA–ChCl which was about 70%. Contrarily to what has been previously reported, carboxylic acid-based DESs did not necessary dissolve more metal oxides than amide-based or polyol-based. The unexpected low solubility of YOX in GlyA–ChCl and OxaA–ChCl compared to urea–ChCl and EtGly–ChCl will be further discussed in the next paragraphs. The leaching efficiency of HALO was very low in all the DESs, independently of the hydrogen bond donor used. Taking into account the high solubility of YOX and the low solubility of HALO, the DES LevA–ChCl was selected as a promising lixiviant for the selective recovery of YOX from the lamp phosphor waste.
 | ||
| Fig. 2 Effect of the hydrogen-bond donor type on the leaching efficiency of (a) YOX lamp phosphor and (b) HALO lamp phosphor. The leaching conditions are: H2O vol% = 10; T = 80 °C; t = 48 h; L/S = 10 and 500 rpm. | ||
When YOX was leached with GlyA–ChCl and OxaA–ChCl, it was noticed that the volume of solids after the leaching was larger than at the beginning. The first hypothesis was that the metals present in the phosphor were leached, and subsequently precipitated as metal glycolates or oxalates, respectively. The 13C and 1H NMR spectra of the GlyA–ChCl and the OxaA–ChCl were recorded before and after the leaching (ESI, Fig. S1 and S2†). The integration of the spectra showed that 45% of the glycolic acid and 41% of the oxalic acid were lost during the leaching. These losses are higher than what could be expected for the full transformation or the REEs into glycolates or oxalates (i.e., carboxylic acid/(Y, Eu) = 1.5). The following carboxylic acid/REE ratio was obtained: glycolic acid/(Y, Eu) = 3 and oxalic acid/(Y, Eu) = 2. This means that all the YOX was transformed into oxalates/glycolates. This result is very interesting from the theoretical point of view. However, during the leaching of real lamp phosphor waste the newly formed compound would be mixed with the remaining residue (HALO, silicates, LAP, and BAM). The separation of the glycolates/oxalates from the remaining leaching residue would be required. The 1H NMR spectra of LevA–ChCl, after leaching of YOX, was also recorded before and after the leaching (Fig. S3†) and it was found to be stable. Therefore, neither yttrium or europium precipitated as levulinates.
The high solubility of YOX in LevA–ChCl could be explained by ligation of the metal in a high chloride medium or by complexation with levulinic acid. Fig. 3 shows a comparison of both YOX and HALO leached with LevA–ChCl and levulinic acid (10 and 30H2O vol%). The leaching of YOX is similar in both lixiviants suggesting that the chloride (from the choline chloride) has negligible effect on metal solubility i.e. carboxylate complexes dominate speciation. Therefore, the addition of another chemical to the levulinic acid to form a DES might not be justified. To validate this assumption, subsequent studies compare the leaching with levulinic acid and LevA–ChCl, as well as the effect of both lixiviants on the metal recovery step.
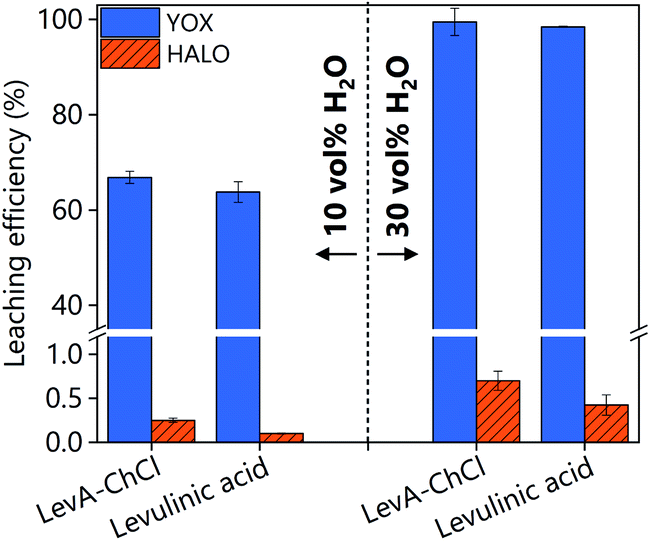 | ||
| Fig. 3 Comparison of leaching efficiency of YOX and HALO using LevA–ChCl and levulinic acid. The leaching conditions are: T = 80 °C, t = 48 h, L/S: 10 and 500 rpm. | ||
The effect of the water content on the leaching efficiency of YOX and HALO (Fig. 4) is similar for both lixiviants: increasing the water content significantly increases the solubility of YOX, while that of HALO is barely affected. A possible reason is that the presence of water facilitates the proton transfer between the carboxylic acid and the yttrium/europium oxide to form the carboxylate complex. The increase of the solubility of metal oxides as a function of the water content has been previously reported for IL systems.34 Another possible explanation is that by increasing the water content, the viscosity of the solvent decreases, leading to enhanced mass transport and thus augmented solubility.
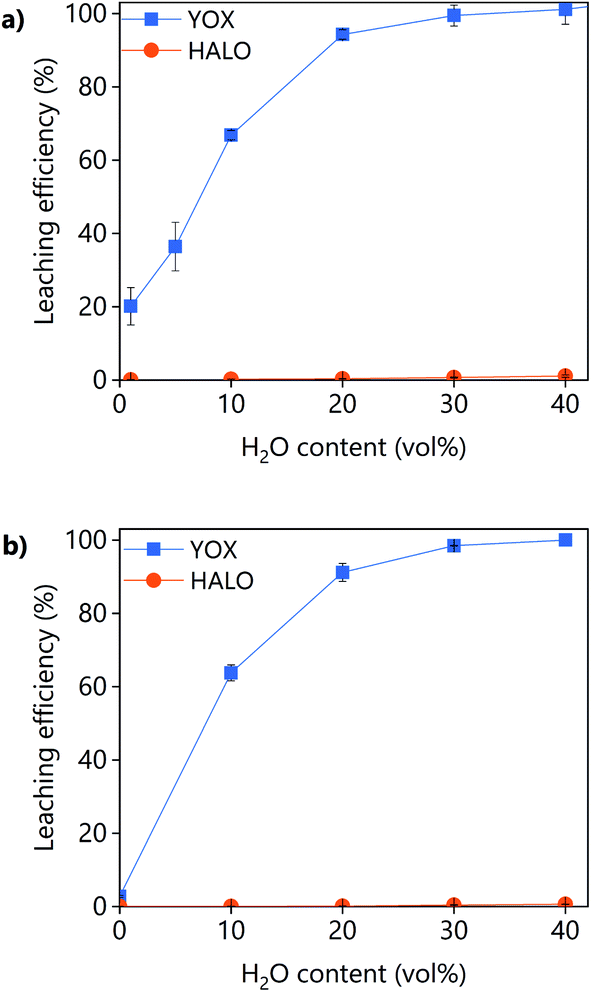 | ||
| Fig. 4 Effect of the water content on the leaching efficiency of YOX and HALO using (a) LevA–ChCl and (b) levulinic acid. The leaching conditions are: T = 80 °C, t = 48 h, L/S = 10 and 500 rpm. | ||
Without addition of water, the solubility of YOX is remarkably higher using LevA–ChCl (20%) than using levulinic acid (2.7%). This could be attributed to the higher water content of the LevA–ChCl, due to the hygroscopicity of the ChCl, and to the esterification reaction between ChCl and levulinic acid, which is enhanced by the high leaching temperatures (50–80 °C) and produces water.35 The addition of water prevents the esterification. In fact, the degree of esterification after the leaching with a water content of 30 vol% (calculated from Fig. S3b† following a procedure previously reported) only accounts for 2 mol%.35 A water content of 30 vol% was used for subsequent studies. A higher water content would decrease the cost of the lixiviant, but it has been reported that for DESs, the transition from an ionic mixture to an aqueous solution of its components is at around 40H2O vol%.36 In order to compare the behaviour of the DES acting as a pseudo-component to that of levulinic acid, the DES must be in the ionic mixture region. Another reason to work at low water content is to keep the selectivity towards HALO (Fig. 4).
The effect of the leaching time on the leaching efficiency of YOX and HALO are shown in Fig. 5. The leaching process was found to be relatively slow, independent of the lixiviant used, full dissolution of the YOX phosphor took 48 h. Although the leaching times are very long, they are in accordance to what has been previously reported in the literature for the leaching of YOX with mineral acids and ILs.6,14
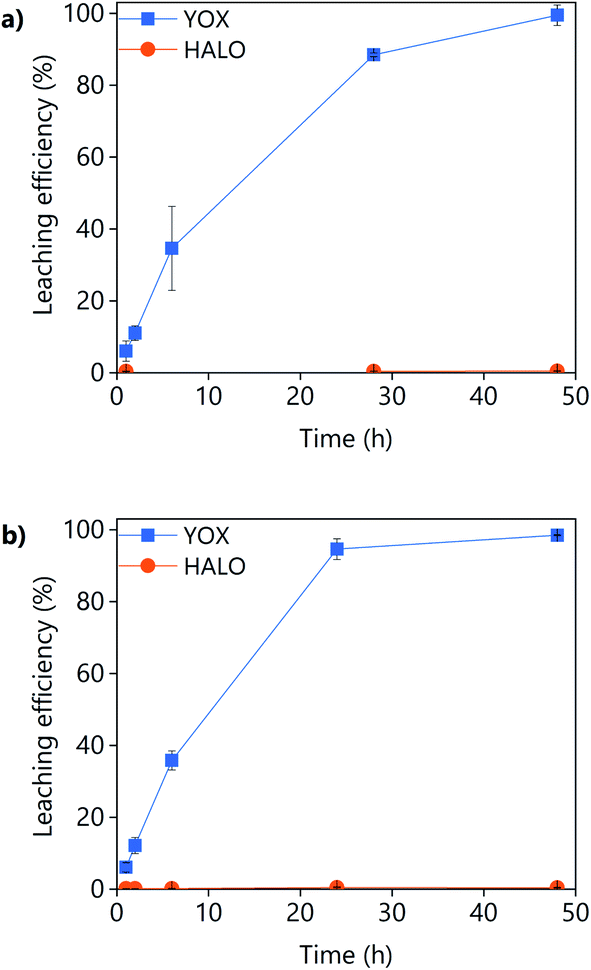 | ||
| Fig. 5 Effect of the leaching time on the leaching efficiency of YOX and HALO using (a) LevA–ChCl and (b) levulinic acid. The leaching conditions are: T = 80 °C; L/S = 10; H2O vol% = 30; and 500 rpm. | ||
The effect of the temperature on the leaching efficiency of YOX and HALO using levulinic acid or LevA–ChCl is shown in Fig. 6. For both lixiviants, increasing the leaching temperature significantly increases the leaching efficiency of YOX, while that of HALO is almost unaffected. Therefore, high temperatures would be preferred for this leaching process. The thermal decomposition of LevA–ChCl is 159 °C,35,37 while that of levulinic acid is 134 °C.27 The decomposition temperature is higher than the maximum operational conditions (boiling point of water). As previously shown in the literature, the temperature has a large effect on the esterification degree of carboxylic acid–ChCl DESs.35 The esterification reaction between the levulinic acid and the choline chloride will reduce the amount of available carboxyl groups for complexation, reducing the leaching efficiency. We have shown before that the presence of water inhibits the esterification; high leaching temperatures might reduce the leaching efficiency of the DES in the long term.
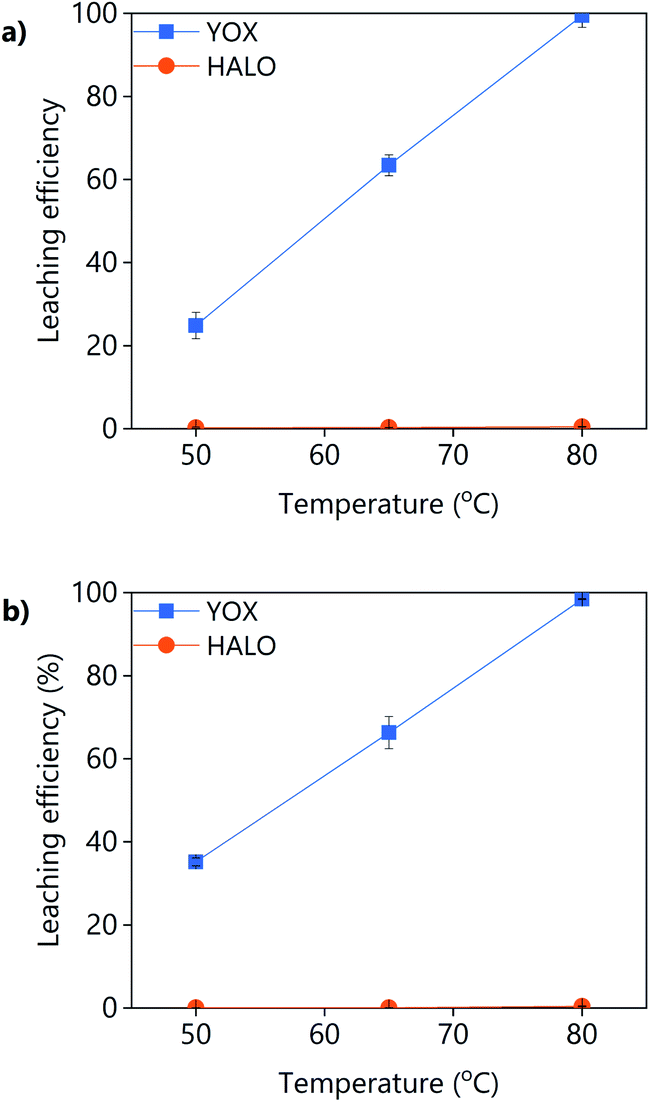 | ||
| Fig. 6 Effect of the temperature on the leaching efficiency of YOX and HALO using (a) LevA–ChCl and (b) levulinic acid. The leaching conditions are: H2O vol% = 30; t = 48 h; L/S = 10; 500 rpm. | ||
Leaching of synthetic lamp phosphor mixture
A synthetic mixture of the lamp phosphors HALO, YOX, BAM and LAP was leached with LevA–ChCl and with levulinic acid. The composition of the synthetic mixture was chosen to mimic the composition of the real lamp phosphor waste: 40–50 wt% HALO, 20 wt% YOX, 5 wt% BAM and 6–7wt% LAP and the remaining amount is composed of SiO2 as fine glass particles and Al2O3.14 The synthetic mixture used in this work included: 58 wt% HALO, 26 wt% YOX, 9 wt% LAP and 6 wt% BAM but without impurities such as SiO2 or Al2O3. The leaching efficiency as a function of the leaching time using LevA–ChCl or levulinic acid as lixiviants is shown in Fig. 7. The leaching of LAP and BAM was found to be below the detection limit of the ICP-OES; therefore, the results were not included in the figure and will not be discussed. The leaching behaviour of the synthetic mixture is similar to that of the individual lamp phosphors: high leaching efficiency of YOX and low leaching efficiency of HALO. However, the leaching efficiency of YOX was found to be lower when leached in a mixture of lamp phosphors than when leached individually. The decrease in the YOX solubility is much more noticeable for the LevA–ChCl than for the levulinic acid. A reason for this behaviour could be the higher viscosity of the mixture due to the presence of large amounts of insoluble HALO, which would hinder the solubility of YOX. This would also explain why the decrease in the YOX solubility is more pronounced in the case of LevA–ChCl which is more viscous than levulinic acid. Furthermore, the selectivity over HALO is lower when using LevA–ChCl (almost 5% of HALO is leached), which might explain the reduced leaching efficiency of YOX.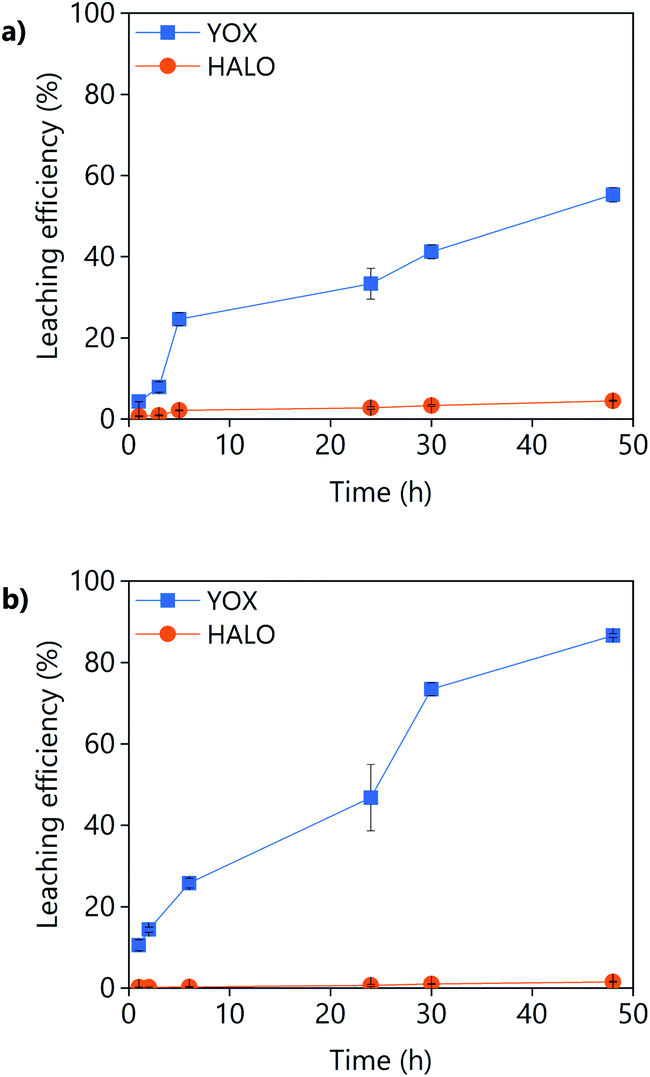 | ||
| Fig. 7 Effect of the mixing time on the leaching efficiency of a synthetic lamp phosphor mixture using (a) LevA–ChCl and (b) levulinic acid. The leaching conditions are: H2O vol% = 30; T = 80 °C; L/S = 10; 500 rpm. | ||
Leaching of real lamp phosphor waste
In order to validate the obtained results, the performance of LevA–ChCl and levulinic acid was tested on a real lamp phosphor waste. The real lamp phosphor waste was sieved, and only the fraction <125 μm was used. This fraction was selected to ensure the homogeneity of the solid samples (see Fig. S4†). The composition of the waste was determined according to the procedure reported in the Experimental section (Table 1).| Element | Composition (wt%) |
|---|---|
| Y | 10.1 ± 0.7 |
| Ca | 7.9 ± 0.7 |
| Al | 1.7 ± 0.3 |
| La | 1.47 ± 0.08 |
| Ba | 1.47 ± 0.15 |
| Ce | 0.97 ± 0.03 |
| Eu | 0.67 ± 0.05 |
| Tb | 0.48 ± 0.04 |
| Sr | 0.37 ± 0.04 |
| Mg | 0.19 ± 0.02 |
| Mn | 0.172 ± 0.014 |
| Sb | 0.110 ± 0.017 |
The effect of the leaching time on the leaching efficiency of the real lamp phosphor residue using LevA–ChCl and levulinic acid is shown in Fig. 8. The concentration of lanthanum, barium, and cerium from phosphors such as LAP, BAM and CAT was found to be negligible. For both lixiviants, the dissolution of YOX was lower than that obtained for the synthetic mixture while the co-dissolution of calcium (associated to the HALO phosphor) was found to be much higher. The higher calcium co-dissolution and its fast leaching suggest that the calcium in the real waste is present in a more accessible form than in the synthetic mixture. Levulinic acid could leach more YOX in a more selective way than the DES LevA–ChCl. With the DES, the maximum leaching efficiency of YOX was already reached after 24 h, but for levulinic acid the maximum leaching efficiency was not reached even after 48 h, so longer times were tested. Longer leaching times improve the leaching efficiency, but drastically decrease the selectivity against HALO.
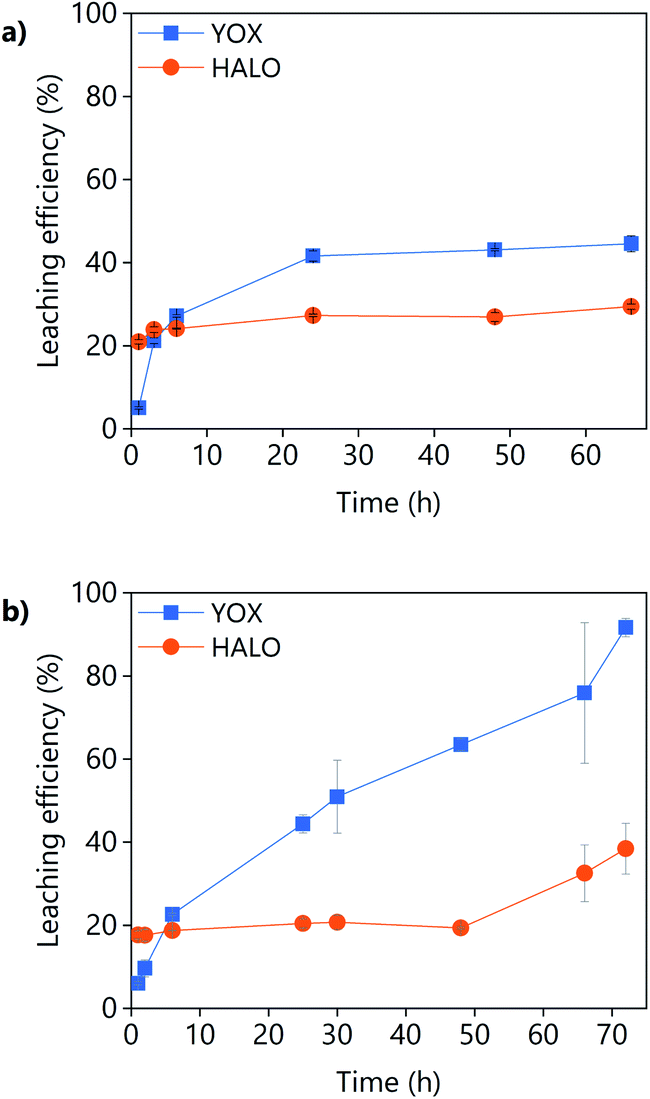 | ||
| Fig. 8 Effect of the leaching time on the leaching efficiency of a real lamp phosphor waste using (a) LevA–ChCl and (b) levulinic acid. The leaching conditions are: H2O vol% = 30; T = 80 °C; L/S = 10; and 500 rpm. | ||
In order to increase the YOX leaching efficiency, higher L/S ratios were tested (Fig. 9). For LevA–ChCl, the effect of the L/S on the leaching efficiency of YOX and HALO is quite small. For levulinic acid, increasing the L/S significantly increased the solubility of YOX, while the co-dissolution of HALO was almost unaffected. With L/S = 20, around 90% of the YOX could be leached in 48 h. With L/S = 30, all the YOX was leached in 48 h. However, some tests performed at a larger scale (120 mL), L/S = 30, higher mixing speed (1000 rpm) and a flask with a significantly larger diameter showed that a 100% leaching efficiency can also be obtained for the DES system. This confirms that the lower leaching efficiency obtained for the DES system is related to mass transfer limitations. The leaching residue obtained from the leaching of the lamp phosphor waste with levulinic acid was separated by filtration and dried at 80 °C. The composition of the leaching residue was determined via microwave digestion followed by ICP-OES analysis. The obtained results are included in Table S2.† The composition of the obtained residue is agreement with a near full dissolution of the YOX phosphor. Furthermore, the concentration of calcium decreased but to a much lower extent, which is consistent with the reported selectivity.
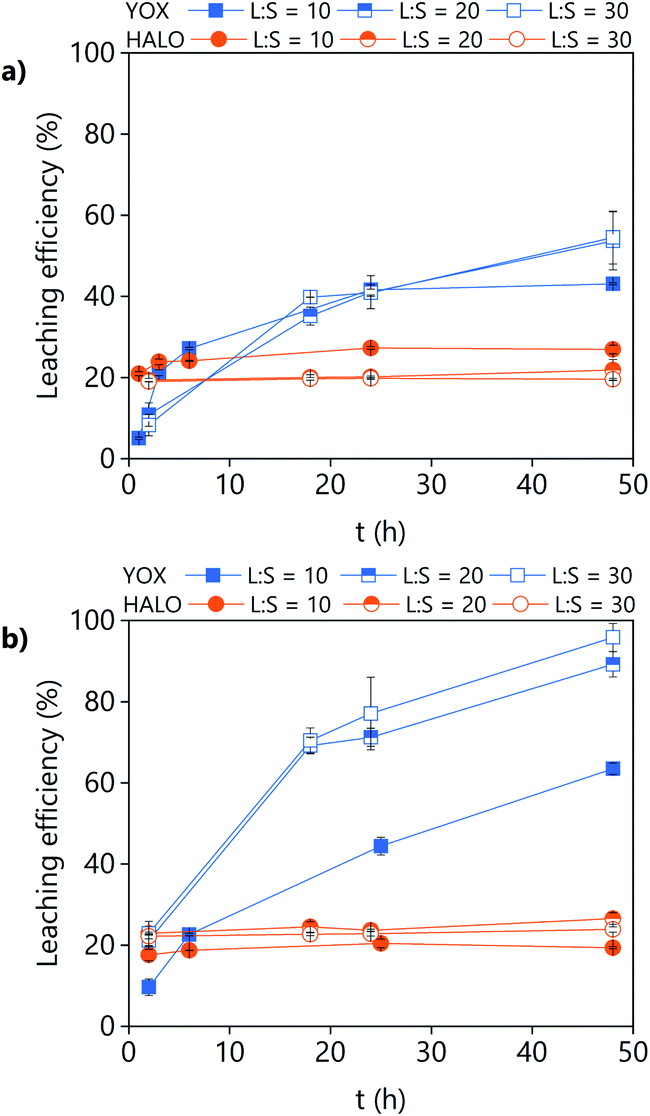 | ||
| Fig. 9 Effect of the L/S ratio and leaching time on the leaching efficiency of a real lamp phosphor waste using (a) LevA–ChCl and (b) levulinic acid. The leaching conditions are: H2O vol% = 30; T = 80 °C; 500 rpm. | ||
Based on the obtained results it became clear that the levulinic acid leaching is preferred over the LevA–ChCl leaching. Apart from higher leaching efficiencies, levulinic acid shows other benefits compared to the DES: (1) levulinic acid can be directly used without need of diluting in another organic solvent, which is cheaper and greener. (2) Levulinic acid is less viscous than the corresponding DES, which implies faster mass transfer and lower energy consumption. (3) Contrarily to carboxylic acid-based DESs, levulinic acid is not decomposed by esterification. (5) Levulinic acid is less corrosive than levulinic acid–ChCl because of the absence of chloride ions. Therefore, not only levulinic acid is greener, it also shows that the use of DESs over its individual components it is sometimes not justified.
Comparison of results
The IL [Hbet][NTf2] was reported to dissolve 100% YOX with a negligible co-dissolution of HALO from a synthetic mixture of phosphors.14 We have tested the performance of this IL in the leaching of the real lamp phosphor waste (Fig. 10). Similarly to our work, the co-dissolution of HALO increased when leaching real residue (up to 20%) while full dissolution of YOX could be obtained. Similar results were obtained for [Hmim][HSO4]:H2O in which nearly full dissolution of YOX was achieved with 25% co-dissolution of calcium.38 From these results, it can be concluded that pure levulinic acid is as efficient and selective as the most promising ILs reported in the literature. If we compare them from an economic point of view, the use of pure levulinic acid overcomes the main disadvantage of ILs, namely their high price. In terms of price, levulinic acid is clearly preferred over the ILs. Furthermore, not only the price is important, but the long-term availability of the chemicals. Contrarily to the ILs, levulinic acid can be prepared in bulk amounts from renewable bioresources. Both in terms of cost and availability levulinic acid is a better option than the ILs.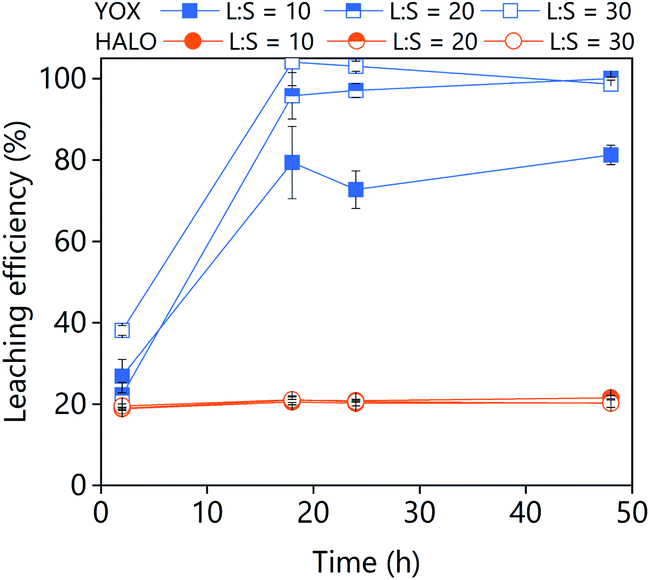 | ||
| Fig. 10 Effect of the L/S ratio and leaching time on the leaching efficiency of a real lamp phosphor waste using the ionic liquid [Hbet][NTf2]. The leaching conditions are: H2O vol% = 5; T = 80 °C; 500 rpm. | ||
To compare the obtained results, the real lamp phosphor waste was also leached with hydrochloric acid (Fig. 11). Mineral acids preferentially dissolve HALO instead of YOX, as expected based on the results previously reported in the literature.12,39 Contrarily, levulinic-acid-based lixiviants can dissolve 100% YOX with low HALO co-dissolution. These results illustrate the selectivity of solvometallurgical approaches. Increasing the acid concentration increases the leachability of both HALO and YOX. It has been reported that even with careful pH control the losses of yttrium and europium accounted for 11% of the mass.12 In addition, leaching of HALO produces phosphoric acid that may react with yttrium and europium to form insoluble phosphates. Therefore, even if hydrometallurgy is faster than solvometallurgy, it suffers from poor selectivity. Furthermore, solvometallurgy prevents the corrosive working environment caused by the evaporation of HCl from concentrated aqueous solutions because the DESs and its chloro species are not volatile.40
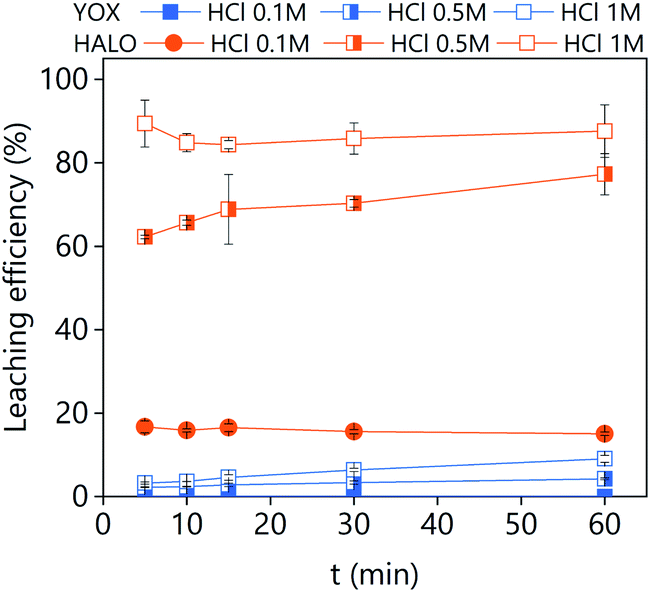 | ||
| Fig. 11 Effect of the leaching time and HCl concentration on the leaching efficiency of real lamp phosphor waste using an aqueous solution of HCl. The leaching conditions are: T = 25 °C; L/S = 10; 500 rpm. | ||
Recovery of metals from the PLS
| 2(Y3+,Eu3+) + 3H2C2O4 → (Y,Eu)2(C2O4)3 + 6H+ | (3) |
The addition of oxalic acid to precipitate yttrium and europium from a PLS obtained from the leaching YOX using LevA–ChCl or levulinic acid with 30 vol% H2O was studied (Fig. 12). For the levulinic acid-based system, a stoichiometric amount of oxalic acid was found to be sufficient for the full precipitation of the REEs if sufficient mixing time and temperature were applied, i.e., 20 min at 50 °C, or 15 min at 70 °C. For the LevA–ChCl system, a stoichiometric amount of oxalic acid was insufficient for the full precipitation of the REEs. Full recovery of REES was achieved only when 1.5 equivalents of oxalic acid were used, either after 5 min of mixing at 50 °C or 70 °C, or after 15 min of mixing at room temperature. Our first hypothesis was that, instead of forming oxalates with the REEs, some oxalic acid would become part of the DES structure (forming a ternary DES). However, from the 13C NMR no signs of oxalic acid were detected in the spectrum of LevA–ChCl after the addition of the stoichiometric amount or 1.5 times the stoichiometric amount of oxalic acid (Fig. S4†). This means that the oxalic acid had precipitated, but not in the form of a REE-oxalate. The analysis of the 1H NMR (Fig. S5†) showed that the LevA–ChCl ratio changed after oxalic acid addition. Originally, xChCl = 0.33, after addition of stoichiometric amount of oxalic acid was xChCl = 0.3 and after addition of 1.5 equivalents was xChCl = 0.27 (i.e., ≈10 and 20 mol% of ChCl losses, respectively). Those losses correspond to the formation (and precipitation) of oxalic acid–ChCl (xChCl = 0.3) and oxalic acid–ChCl (xChCl = 0.4), respectively. Those compositions are solids at the temperatures used for the precipitation experiments.42 Thus, it can be concluded that the recovery of REEs from DES-based PLSs using oxalic acid precipitation is not a suitable option due to the partial precipitation of the lixiviant and the contamination of the REE precipitate. Considering these results, levulinic acid would be preferred as lixiviant, reinforcing the conclusions of the leaching section.
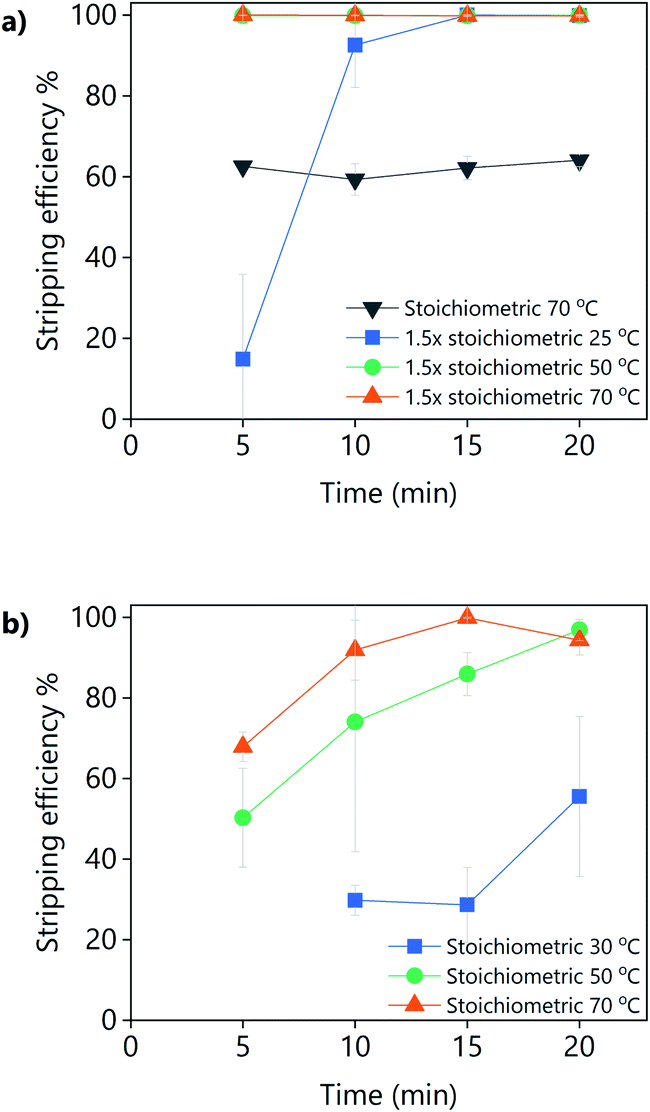 | ||
| Fig. 12 Stripping efficiency obtained by the addition of pure oxalic acid to (a) LevA–ChCl and (b) levulinic acid PLS, as a function of the amount of oxalic acid, mixing temperature and time. The leaching conditions are: H2O vol% = 30; T = 80 °C; t = 48 h; L/S = 10; 500 rpm. | ||
It can be noticed that experiments shown in Fig. 12 have much larger error bars compared to the rest of the experiments of this work. This is due to the very large concentration of REEs in the PLS when synthetic YOX is leached (Y = 74781 ppm and Eu = 5867 ppm), which lead to a very large volume of newly formed oxalates compared to the volume of the original liquid. Under those conditions, the mixing of the vial was not optimal. Furthermore, the precipitation of the oxalates is very fast, and it might continue to some extent during centrifugation.
The recovery of the REEs via oxalic acid precipitation from the PLS obtained from the levulinic acid leaching of real lamp phosphor waste was also investigated (Fig. 13). The composition of the PLS was: Y = 8772 ppm, Eu = 565 ppm and Ca = 1579 ppm. Fig. 13 shows that calcium is preferentially precipitated over yttrium and europium (which are equally extracted). This is why even if a stoichiometric amount of oxalic acid was added, not all the REEs were precipitated. Due to the calcium co-precipitation it is not possible to obtain a clean REE oxalate precipitate from a real levulinic acid-PLS. The precipitation from the PLS of the real waste is faster than from the PLS of the YOX leaching (Fig. 12), i.e., full precipitation in 5 min. The lower metal content of the real lamp phosphor residue PLS could explain this behaviour. Fig. 13 also shows that higher stripping efficiencies were obtained at lower temperatures (for both the calcium and the REEs). It has been previously reported that the co-precipitation of calcium can be reduced by low pH and/or high temperatures.43 This method could be used for the full stripping of the levulinic acid, so it can be reuse in subsequent leaching steps. However it is not suitable to produce a pure REEs oxalate residue.
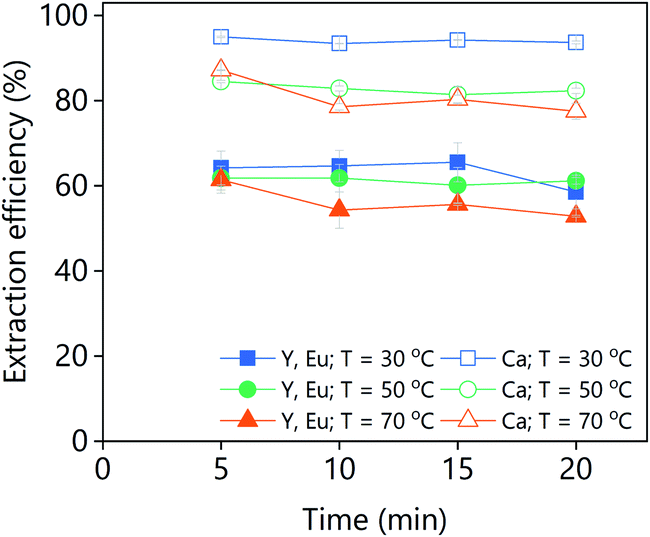 | ||
| Fig. 13 Stripping efficiency via oxalic acid addition (stoichiometric amount) as a function of time and temperature of a levulinic acid PLS obtained from the leaching of real lamp phosphor residue. The leaching conditions are: H2O vol% = 30; T = 80 °C; t = 48 h; L/S = 10; and 500 rpm. | ||
Solvent extraction
The recovery of the metals from the PLS via solvent extraction was investigated from both the levulinic acid-based and the DES-based PLS. In this work, the metals were extracted by the extractant bis(2-ethylhexyl)phosphoric acid (D2EHPA), which is a well-known extractant for REEs.44 The metals were subsequently stripped to an aqueous acidic solution. HCl has been selected in this work, but H2SO4 and HNO3 have also been reported as suitable options for stripping of REEs from a D2EHPA phase.43,45 Different diluents have been considered for the extraction, in presence or not for a modifier (Table 2). A third phase formation was observed if D2EHPA is diluted in aliphatic compounds, i.e., the aliphatic diluent GS190 or n-dodecane, even in presence of a modifier (1-decanol, 10–30 vol%). Although aliphatic diluents are preferred from an environmental perspective, the third phase formation forced us using an aromatic diluent. Two aromatic compounds were tested p-cymene and toluene. By using toluene or p-cymene as a diluent, the third phase formation was noticeably reduced. The addition of a modifier (1-decanol) avoided the third phase formation. The use of p-cymene over toluene is preferred for safety reasons, i.e. higher flash point. Furthermore, p-cymene is considered as a green aromatic solvent according to different criteria and it can be derived from renewable bioresources.46 Therefore, a mixture of D2EHPA, p-cymene and 1-decanol was selected as the organic phase.| Diluent | Modifier | Third phase? |
|---|---|---|
| n-Dodecane | No/yes | Yes |
| GS190 | No/yes | Yes |
| Toluene | No | Yes |
| Toluene | Yes (10 vol%) | No |
| p-Cymene | No | Yes |
| p-Cymene | Yes (10 vol%) | No |
The effect of the D2EHPA concentration on the extraction efficiency was investigated for both PLSs (Fig. 14). The concentration of D2EHPA was varied from 0–60 vol% and that of 1-decanol was kept constant at 10 vol%. For both PLSs, quantitative extraction of all the metals was achieved with D2EHPA concentrations ≥20 vol%. At D2EHPA concentrations <20 vol%, the REEs are preferentially extracted over calcium. No extraction was observed when no D2EHPA was used. The results showed that the extraction is more efficient for the DES-based PLS than for the levulinic acid PLS phase, which means that the DES–metal interactions are weaker. Those results are in agreement with the lower leaching efficiencies obtained for the DES. The 1H NMR spectra of both the D2EHPA phase and the depleted lixiviant phase were recorded (Fig. S6†). The absence of lixiviant in the organic phase, and the absent of extractant in the lixiviant phase were confirmed. The extraction of lanthanides from carboxylic-acid-containing solutions using D2EHPA has been reported previously.47,48
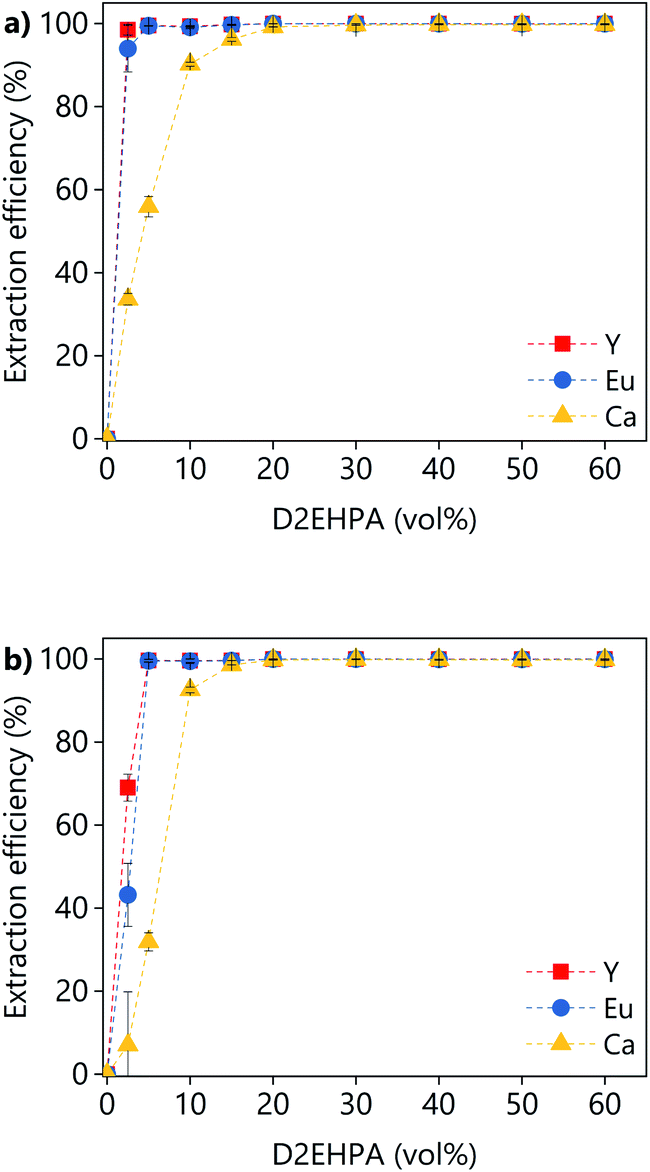 | ||
| Fig. 14 Effect of the D2EHPA concentration on the extraction efficiency from (a) LevA–ChCl-based PLS or (b) levulinic acid PLS. | ||
A large batch of loaded organic phase was prepared under the following conditions: A/O = 1; D2EHPA (vol%) = 40; p-cymene (vol%) = 50; 1-decanol (vol%) = 10. A D2EHPA concentration of 40 vol% was used because lower concentrations lead to a third phase formation when the extraction was performed at 50 mL scale. The third phase formation was not noticed when the extraction was performed at 1 mL scale, independently of the D2EHPA concentration. The stripping of the metals from the loaded organic phase to an aqueous HCl solution was investigated as a function of the HCl concentration (Fig. 15). Around 80% of yttrium an calcium could be stripped in single contact with low concentrations of HCl (1 M), while the stripping of europium requires higher concentrations (>3.5 M). Further increase of the HCl concentration did not increase the stripping efficiency.
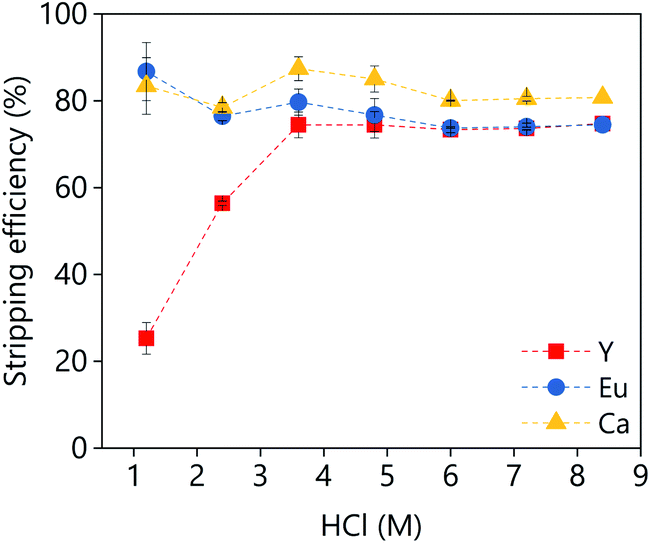 | ||
| Fig. 15 Effect of the HCl concentration on the stripping efficiency. The stripping conditions were: A/O = 1; mixing time = 30 min, T = 25 °C. | ||
In this work, a proof-of-principle is provided on how all the metals can be recovered without contamination of the lixiviant, which can be reused in subsequent leaching steps. Several procedures have been previously reported for the separation of the metals from loaded HCl solutions. However, the solvent extraction procedure should be investigated further, including direct separation of the metals from the PLS, instead of transferring all the metals to an aqueous phase before separation.
Conclusions
Both pure levulinic acid and LevA–ChCl showed high solubility of YOX while low solubility of HALO phosphor. The similar results obtained for both lixiviant showed that the chlorides from the choline chloride were not responsible for the high leaching efficiency of REEs. When leaching a synthetic mixture instead of the individual phosphors, lower YOX leaching efficiencies were obtained, but the leaching was still selective. In the leaching of real lamp phosphor waste, a further decrease of the dissolution of YOX and increase of co-dissolution of HALO was observed. Increasing the L/S ratio could increase the YOX dissolution from real lamp phosphor waste. The obtained results were compared to that of ([Hbet][NTf2]) and similar results in terms of leaching efficiency and selectivity were obtained, but both levulinic acid and LevA–ChCl are much cheaper alternatives. A simple hydrometallurgical approach was tested using different concentrations of HCl and full dissolution of HALO with certain YOX co-dissolution was obtained. This comparison emphasises the selectivity of solvometallurgical approaches. The recovery of the REEs via oxalic acid addition is possible in the levulinic acid-PLS, but when performed on a LevA–ChCl-PLS some ChCl precipitated together with oxalic acid and the REEs. Furthermore, due to the co-precipitation of calcium, this is not a suitable method for the direct recovery of REEs from a real waste PLS. The purification of the lixiviant could be achieved via solvent extraction, by extracting all the metals to a D2EHPA phase and stripping them to an aqueous HCl solution, from which they can be separated following existing methodologies. Throughout the entire work, levulinic acid was found to be equally suitable for the recovery of REEs from lamp phosphor waste than the corresponding ChCl-based DES. However, levulinic acid can be directly used without need of diluting in another organic solvent, which is cheaper and greener. Moreover, levulinic acid is a green solvent that can be produced from renewable resources.25–28Conflicts of interest
There are not conflicts to declare.Acknowledgements
Nerea Rodriguez Rodriguez acknowledges the financial support from the Research Foundation-Flanders (FWO, Grant no. 12X5119N, postdoctoral fellowship). This research received funding from the European Commission's H2020 – Marie Sklodowska Curie Actions (MSCA) – Innovative Training Networks within the SOCRATES project under the grant agreement no. 721385 (Project website: http://etn-socrates.eu). The authors acknowledge Relight Srl (Rho, Italy) for providing the lamp phosphor waste.References
- M. Aucott, M. McLinden and M. Winka, J. Air Waste Manage. Assoc., 2003, 53, 143–151 CrossRef CAS PubMed.
- K. Binnemans and P. T. Jones, J. Rare Earths, 2014, 32, 195–200 CrossRef CAS.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner Prod., 2013, 51, 1–22 CrossRef CAS.
- Q. Tan, J. Li and X. Zeng, Crit. Rev. Environ. Sci. Technol., 2015, 45, 749–776 CrossRef CAS.
- C. Tunsu, M. Petranikova, M. Gergorić, C. Ekberg and T. Retegan, Hydrometallurgy, 2015, 156, 239–258 CrossRef CAS.
- C. Tunsu, M. Petranikova, C. Ekberg and T. Retegan, Sep. Purif. Technol., 2016, 161, 172–186 CrossRef CAS.
- V. Innocenzi, N. M. Ippolito, I. De Michelis, F. Medici and F. Vegliò, J. Environ. Manage., 2016, 184, 552–559 CrossRef CAS PubMed.
- N. M. Ippolito, V. Innocenzi, I. De Michelis, F. Medici and F. Vegliò, J. Cleaner Prod., 2017, 153, 287–298 CrossRef CAS.
- L. Yurramendi, L. Gijsemans, F. Forte, J. L. Aldana, C. del Río and K. Binnemans, Hydrometallurgy, 2019, 187, 38–44 CrossRef CAS.
- V. Innocenzi, I. De Michelis, B. Kopacek and F. Vegliò, Waste Manage., 2014, 34, 1237–1250 CrossRef CAS PubMed.
- Y. Wu, X. Yin, Q. Zhang, W. Wang and X. Mu, Resour., Conserv. Recycl., 2014, 88, 21–31 CrossRef.
- D. Dupont and K. Binnemans, Green Chem., 2016, 18, 176–185 RSC.
- K. Binnemans and P. T. Jones, J. Sustain., 2017, 3, 570–600 Search PubMed.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
- L. Gijsemans, F. Forte, B. Onghena and K. Binnemans, RSC Adv., 2018, 8, 26349–26355 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060 CrossRef CAS PubMed.
- N. Rodriguez Rodriguez, L. Machiels and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 3940–3948 CrossRef CAS.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 47, 10031–10033 RSC.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC.
- A. P. Abbott, A. Z. Al-Bassam, A. Goddard, R. C. Harris, G. R. Jenkin, F. J. Nisbet and M. Wieland, Green Chem., 2017, 19, 2225–2233 RSC.
- A. P. Abbott, J. Collins, I. Dalrymple, R. C. Harris, R. Mistry, F. Qiu, J. Scheirer and W. R. Wise, Aust. J. Chem., 2009, 62, 341–347 CrossRef CAS.
- S. Anggara, F. Bevan, R. C. Harris, J. Hartley, G. Frisch, G. Jenkin and A. P. Abbott, Green Chem., 2019, 21, 6502–6512 RSC.
- T. Werpy and G. Petersen, Top value added chemicals from biomass: volume I--results of screening for potential candidates from sugars and synthesis gas, National Renewable Energy Lab., Golden, CO (US), 2004 Search PubMed.
- J. J. Bozell, L. Moens, D. Elliott, Y. Wang, G. Neuenscwander, S. Fitzpatrick, R. Bilski and J. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- A. Morone, M. Apte and R. Pandey, Renewable Sustainable Energy Rev., 2015, 51, 548–565 CrossRef CAS.
- L. Yan, Q. Yao and Y. Fu, Green Chem., 2017, 19, 5527–5547 RSC.
- F. Yu, R. Zhong, H. Chong, M. Smet, W. Dehaen and B. F. Sels, Green Chem., 2017, 19, 153–163 RSC.
- M. Regadío, S. Riaño, K. Binnemans and T. Vander Hoogerstraete, Anal. Chem., 2017, 89, 4595–4603 CrossRef PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, K. J. McKenzie and S. U. Obi, J. Chem. Eng. Data, 2006, 51, 1280–1282 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- K. Shahbaz, F. S. Mjalli, M. Hashim and I. M. ALNashef, J. Appl. Sci., 2010, 10, 3349–3354 CrossRef CAS.
- M. A. Martins, S. P. Pinho and J. A. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS.
- P. Nockemann, B. Thijs, S. Pittois, J. Thoen, C. Glorieux, K. Van Hecke, L. Van Meervelt, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2006, 110, 20978–20992 CrossRef CAS PubMed.
- N. Rodriguez Rodriguez, A. van den Bruinhorst, L. J. Kollau, M. C. Kroon and K. Binnemans, ACS Sustainable Chem. Eng., 2019, 7, 11521–11528 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS.
- N. Delgado-Mellado, M. Larriba, P. Navarro, V. Rigual, M. Ayuso, J. García and F. Rodríguez, J. Mol. Liq., 2018, 260, 37–43 CrossRef CAS.
- N. Schaeffer, X. Feng, S. Grimes and C. Cheeseman, J. Chem. Technol. Biotechnol., 2017, 92, 2731–2738 CrossRef CAS.
- S.-G. Zhang, M. Yang, H. Liu, D.-A. Pan and J.-J. Tian, Rare Met., 2013, 32, 609–615 CrossRef CAS.
- M. R. S. Foreman, Cogent Chem., 2016, 2, 1139289 Search PubMed.
- J. Brewer, Method for extraction and separation of rare earth elements, Rare Earth Salts Separation and Refining, Canada Pat., LLCCA2955313A1, 2017.
- C. G. Brown and L. G. Sherrington, J. Chem. Technol. Biotechnol., 1979, 29, 193–209 CrossRef CAS.
- D. Wu, X. Wang and D. Li, Chem. Eng. Process., 2007, 46, 17–24 CrossRef CAS.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10–28 CrossRef CAS.
- R. D. Abreu and C. A. Morais, Miner. Eng., 2014, 61, 82–87 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- J. C. Braley, T. S. Grimes and K. L. Nash, Ind. Eng. Chem. Res., 2012, 51, 629–638 CrossRef.
- S. Satpathy and S. Mishra, Sep. Purif. Technol., 2017, 179, 513–522 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 13C NMR spectra of OxaA–ChCl before and after the leaching. 1H NMR spectra of GlyA–ChCl before and after a leaching. 1H NMR spectra of LevA–ChCl before and after a leaching. Composition of the leaching residue. 13C NMR of LevA–ChCl before and after oxalic acid addition. 1H NMR of LevA–ChCl PLS before and after oxalic acid addition. 1H NMR spectra of the lixiviant and the extractant phases after solvent extraction experiments. See DOI: 10.1039/d0ra05508e |
| This journal is © The Royal Society of Chemistry 2020 |