DOI:
10.1039/D0RA03424J
(Paper)
RSC Adv., 2020,
10, 21839-21844
Fundamental properties of TEMPO-based catholytes for aqueous redox flow batteries: effects of substituent groups and electrolytes on electrochemical properties, solubilities and battery performance†
Received
17th April 2020
, Accepted 26th May 2020
First published on 8th June 2020
Abstract
Water-soluble 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) derivatives have been frequently utilized as catholytes for aqueous redox flow batteries to achieve cost-effective renewable energy storage. However, fundamental knowledge of TEMPO derivatives is still largely underdeveloped. Herein, a comprehensive study on the properties of TEMPO derivatives has been conducted in aqueous electrolytes. The results confirm that the redox potential, diffusion coefficient, electron transfer rate constant and solubility are clearly influenced by functional groups of TEMPO derivatives and supporting electrolytes. The charge–discharge cycling performance is evaluated using a symmetric redox flow battery configuration. The capacity decay for TEMPO-based catholytes is mainly derived from the crossover of the oxidized state. The presented study not only advances an in-depth understanding of TEMPO-based RFB applications, but also highlights the challenge of crossover of redox-active TEMPO derivative molecules applied in aqueous RFBs.
Introduction
Redox flow batteries (RFBs) have demonstrated a reliable ability to provide large scale electrical energy storage, owing to charge-storing redox species existing in the liquid form and complete decoupling of power and energy density.1,2 To achieve widespread implementation of RFBs, it is necessary to develop cost-efficient, highly stable and safe electro-active materials.3–6 Recently, organic and organometallic redox-active molecules whose structure can be effectively tuned have been explored for RFB systems.1,2,5–12 For example, many derivatives of viologen, anthraquinone and phenazine were employed as anolytes for aqueous and nonaqueous RFBs because of their negative equilibrium potential and high redox reversibility.1,2,6,8 Redox-reversible nitroxyl radicals (e.g. 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and its derivatives7,9,10) and ferrocene derivatives (e.g. (ferrocenylmethyl)trimethylammonium chloride (FcNCl) and N1-ferrocenylmethyl-N1,N1,N2,N2,N2-pentamethylpropane-1,2-diaminium dibromide, (FcN2Br2)11) were used as high potential catholytes.
As catholyte candidate materials of aqueous RFBs, TEMPO derivatives are especially attractive due to their considerable positive redox potential, rapid charge transfer kinetics, high water-solubility (high storage capacity) and excellent structural stability.7,9,13–16 However, the properties of TEMPO derivatives have not yet been systematically studied, not only due to structural diversity of TEMPO derivatives, but also because their electrochemical and physicochemical properties are highly dependent on structural differences. In this paper, we have first examined systematically the effects of different substituent groups of TEMPO derivatives and supporting electrolytes on electrochemical properties and solubilities. Employing a symmetric RFB configuration, the charge–discharge cycling performance was evaluated. The mechanism of performance degradation is revealed by post-cycling investigations.
Experimental
Chemicals
4-Hydroxy-TEMPO (4HT), 4-oxo-TEMPO (4OT), 4-amino-TEMPO (4AT), 4-cyano-TEMPO (4CyT) and 4-carboxy-TEMPO (4CaT) were purchased from Tokyo Chemical Industry (TCI) and used directly, whose chemical structures were presented in Scheme 1. All other reagents such as inorganic acids, bases and salts were purchased from Sinopharm Chemical Reagent Co. Ltd. Deionized water (Milli-Q, 18.2 MΩ) was used for all experiments.
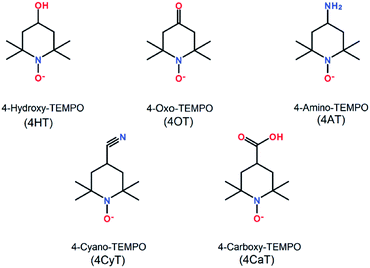 |
| | Scheme 1 Chemical structures of five TEMPO derivatives. | |
Solubility studies
The solubility of TEMPO derivatives was determined by UV-vis absorption spectra. Before the solubility test, excess sample was equilibrated for 48 h in pure water or 0.5 M aqueous electrolyte solution at room temperature. After the obtained saturated solution was diluted to the desired concentration, the concentration was evaluated on UV-vis (Shimadzu UV mini 1240) spectrophotometer.
Electrochemical studies
All electrochemical studies were conducted on an Interface 1000 electrochemical working station (Gamry) with a conventional three-electrode cell at room temperature. The glassy carbon rotating-disk electrode (RDE, 5 mm diameter), Pt foil and saturated calomel electrode (SCE) were used as the working electrode, counter electrode and reference electrode, respectively. The potential was reported relative to the standard hydrogen electrode (SHE). The TEMPO derivative (1 mM) was dissolved in a 0.5 M aqueous electrolyte, served as the electrolyte solution. Prior to measurements, the electrolyte solution was purged with nitrogen gas for at least 20 min to remove any dissolved oxygen. During the electrochemical tests, nitrogen gas was purged continuously over the surface of electrolyte solutions.
The Pourbaix diagram (E0 versus pH) was constructed using the TEMPO derivative (1 mM) dissolved in aqueous electrolyte solutions of different pH. The concentration of the supporting electrolyte is 0.5 M for each electrolyte solution. The pH of solution was adjusted with dilute KOH or HCl solution and measured with a PHS-3C pH meter (Shanghai Instrument Electric Science Instrument Ltd.).
Symmetric RFB tests
Symmetric RFB tests were carried out on an Interface 1000 electrochemical working station (Gamry) using a commercial flow cell (Fuel Cell Technologies) that was comprised of aluminium end plates, current collectors, graphite flow field plates and two stacked (×2) carbon paper electrodes (Toray TGPH060) with an active area of 5 cm2. Cationic exchange membrane (N112, 50 μm thick, DuPont) and anion exchange membrane (A201, 25 μm thick, Tokuyama) pre-treated by immersion in 2 M KCl solution for 2 h at 60 °C, were utilized as a separator for the symmetric RFB test, respectively. Both anolyte and catholyte were circulated between the flow cell and reservoir at a flow rate of 60 mL min−1 through a peristaltic pump (BT-600EA, Chongqing Jieheng). 0.2 M 4HT (or 4AT) + 2 M KCl electrolyte (10 mL) and 0.2 M [4HT]+Cl− (or [4AT]+Cl−) (the oxidation state) + 2 M KCl electrolyte (10 mL) were used as anolyte and catholyte, respectively. The [4HT]+Cl− (or [4AT]+Cl−) catholyte was obtained by electrochemically reduced 4HT (or 4AT) in a 4HT (or 4AT)-ferricyanide RFB. The specific capacity was calculated based on all the anolyte and catholyte. Nitrogen gas was purged continuously over the surface of electrolytes to remove oxygen during battery cycling.
Results and discussion
Electrochemical properties and solubility
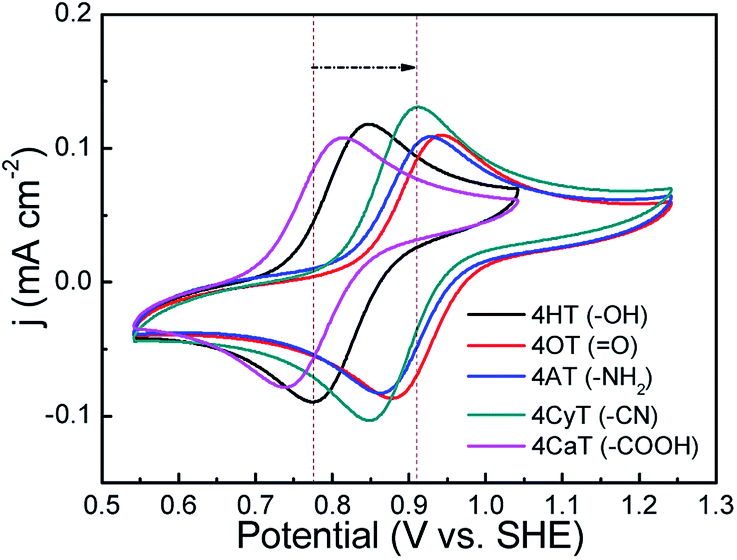
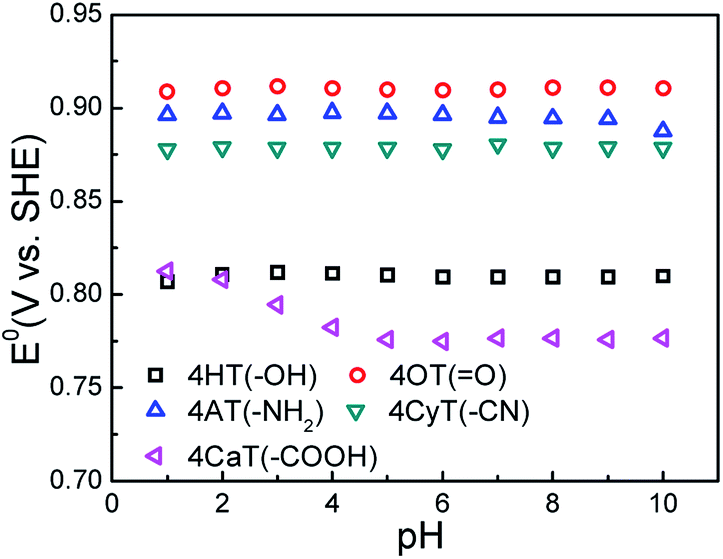
The functional substituent groups play a key role on tuning the electrochemical properties and water solubility of redox-active organic molecules.17,18 Thus, the structure–activity relationship for TEMPO derivatives was first examined by cyclic voltammograms (CVs). Fig. 1 presents CVs of five redox-active TEMPO derivatives, all of which display well-defined redox peaks, representing the one-electron transfer reactions. The formal potential (E0) is found to be 0.812 V for 4HT (–OH), 0.910 V for 4OT (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 0.895 V for 4AT (–NH2), 0.881 V for 4CyT (–CN), and 0.776 V for 4CaT (–COOH), respectively. The redox potentials are strongly influenced by electron-donating or electron-withdrawing substituents for electro-active organic molecules. The presence of electron-donating groups lowers the redox potential while the presence of electron-withdrawing groups may improve the redox potential.17,19 Therefore, it is plausible that The E0 values for 4AT and 4CyT are observed to be much higher than that of 4HT because of electron-withdrawing characteristics of O and –CN groups. However, although –NH2 is a typical electron-donating group, 4AT shows a relatively high redox potential, which is due to the fact that amino groups of 4AT can react with hydrogen ions in aqueous solution to form electron-withdrawing –NH3+ cations.20 Then, the effect of pH on the formal potential for TEMPO derivatives was studied by CV measurements (Fig. 2). Except for that of CaT, the E0 values of the other four TEMPO derivatives are all pH-independent in the pH range of 1.0–10.0. The equilibrium potential of CaT in the pH range of 1.0–5.0 is pH-dependent with a E0–pH slope of approximately 10 mV pH−1. When the pH is greater than 5.0, the CaT exhibits a pH-independent feature, similar to the other TEMPO derivatives.
O), 0.895 V for 4AT (–NH2), 0.881 V for 4CyT (–CN), and 0.776 V for 4CaT (–COOH), respectively. The redox potentials are strongly influenced by electron-donating or electron-withdrawing substituents for electro-active organic molecules. The presence of electron-donating groups lowers the redox potential while the presence of electron-withdrawing groups may improve the redox potential.17,19 Therefore, it is plausible that The E0 values for 4AT and 4CyT are observed to be much higher than that of 4HT because of electron-withdrawing characteristics of O and –CN groups. However, although –NH2 is a typical electron-donating group, 4AT shows a relatively high redox potential, which is due to the fact that amino groups of 4AT can react with hydrogen ions in aqueous solution to form electron-withdrawing –NH3+ cations.20 Then, the effect of pH on the formal potential for TEMPO derivatives was studied by CV measurements (Fig. 2). Except for that of CaT, the E0 values of the other four TEMPO derivatives are all pH-independent in the pH range of 1.0–10.0. The equilibrium potential of CaT in the pH range of 1.0–5.0 is pH-dependent with a E0–pH slope of approximately 10 mV pH−1. When the pH is greater than 5.0, the CaT exhibits a pH-independent feature, similar to the other TEMPO derivatives.
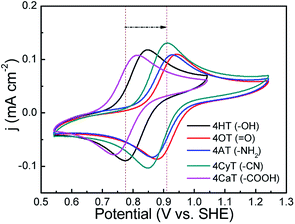 |
| | Fig. 1 CV curves of five TEMPO derivatives (1 mM) in 0.5 M KCl supporting electrolyte (pH = 7) at a scan rate of 25 mV s−1 at 25 °C. Before CV measurements, the pH values of the electrolyte solutions were adjusted to 7 for 4AT and 4CaT by using 0.01 M HCl and KOH solution, respectively, because 4AT is weakly alkaline and 4CaT is weakly acidic. | |
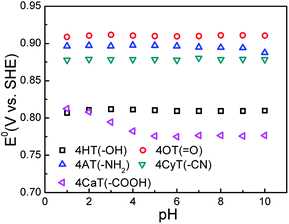 |
| | Fig. 2 The effect of pH on equilibrium potential (E0) of five TEMPO derivatives in 0.5 M KCl supporting electrolyte at 25 °C. | |
The electron transfer rate constant and diffusion coefficient for redox-active materials are two fundamental properties which have impacts on charge/mass transfer and further efficiency and power of RFBs. Koutecký–Levich analyses of RDE voltammetry data were applied to determine the electron transfer rate constants (k0) of five redox-active TEMPO derivatives. The rotation speed increased from 200 to 900 rpm to gain different mass transport-limited currents (Fig. S1†). The Koutecký–Levich plots whose intercepts represent the reciprocal of the kinetic current (ik) exhibit good linearity at different overpotentials. And then, based on the relationship between overpotential and log(ik), k0 can be calculated using the slope of the overpotential versus log(ik) plot. The diffusion coefficients for oxidized state (DO) and reduced state (DR) were measured by cyclic voltammetry (CV) at variable scan rate (Fig. S2†) and then calculated from the Randles–Sevcik equation. The k0 and D values of five TEMPO derivatives are summarized in Table 1. The 4HT, 4AT and 4CyT show similar electron transfer constants in KCl electrolyte within a range from 4.01 × 10−3 to 4.45 × 10−3 cm s−1, which is obviously slower than that of 4OT (1.05 × 10−2 cm s−1), but higher than that of 4CaT (2.25 × 10−3 cm s−1). The significant difference in electron transfer rates for TEMPO derivatives may be attributed to different inductive substituent constants of functional groups at 4-position and accessibility of the nitroxide group.21 Considering the inductive substituent constants are not easy to measure, we have investigated the effect of redox potentials on the reaction rate constants for five TEMPO derivatives since the redox reaction of organic molecules is governed by the charge state of electroactive units.17,19 It can be seen that the standard potentials roughly correlate with logarithms of the rate constants (Fig. S3†), indicating that a low electron density on alicyclic ring of nitroxyl radicals probably accelerate the charge transfer of redox reaction. In addition, the D values for 4HT, 4OT, 4AT, 4CyT and 4CaT in aqueous KCl electrolyte are 3.14 × 10−6, 4.61 × 10−6, 3.77 × 10−6, 6.23 × 10−6 and 2.64 × 10−6 cm2 s−1, respectively. The 4CyT shows a fastest diffusion among five derivatives.
Table 1 Solubility, theoretical capacity, equilibrium potential and electrochemical kinetic parameters of different TEMPO derivatives in KCl aqueous solutions at room temperature
| Sample |
Solubility in pure water (M) |
Solubility in 0.5 M KCl (M) |
CTheoa (Ah L−1) |
E0b (V) |
Dc (cm2 s−1) |
k0d (cm s−1) |
| Theoretical capacity (CTheo) were calculated based solubility of materials in 0.5 M KCl solution. Equilibrium potential (E0) was obtained from CV measurements in 0.5 M KCl solution. Diffusion coefficient (D) that is an average value of DO and DR. Electron transfer constant (k0) was measured from RDE measurements in 0.5 M KCl solution. |
| 4HT |
2.11 |
1.25 |
33.5 |
0.812 |
3.14 × 10−6 |
4.45 × 10−3 |
| 4OT |
1.22 |
0.79 |
21.2 |
0.910 |
4.61 × 10−6 |
1.05 × 10−2 |
| 4AT |
5.79 |
4.80 |
128.6 |
0.895 |
3.77 × 10−6 |
4.01 × 10−3 |
| 4CyT |
2.6 × 10−2 |
NA |
NA |
0.881 |
6.23 × 10−6 |
4.29 × 10−3 |
| 4CaT |
2.1 × 10−2 |
NA |
NA |
0.776 |
2.64 × 10−6 |
2.25 × 10−3 |
The electrochemical stability of TEMPO derivatives was further studied. As presented in Fig. S4,† the 4HT, 4OT and 4AT exhibit high electrochemical stability while the stability of the 4CyT and 4CaT is slightly worse after 100 CV cycles. The subtle difference in electrochemical stability is likely to originate from different chemical stability of nitroxyl radicals and/or the corresponding oxoammonium cations.
As shown in Table 1 and Fig. S5,† the 4AT shows a highest concentration in pure water among five TEMPO derivatives, which reaches ca. 5.79 M, followed by 2.11 M of the 4HT and 1.22 M of the 4OT. However, the 4CyT and 4CaT display poor solubility in water, demonstrating that they are not suitable as electroactive catholytes for aqueous redox flow battery applications. Theoretical capacity for TEMPO derivatives were calculated based on their solubilities in 0.5 M KCl solution. It can be seen that the theoretical capacity of 4AT is about 128.6 Ah L−1, much higher than those of 4HT and 4OT (33.5 and 21.2 Ah L−1, respectively). These experimental data clearly indicate that electrochemical property and solubility are greatly influenced by functional groups of TEMPO derivatives.
Effects of supporting electrolytes
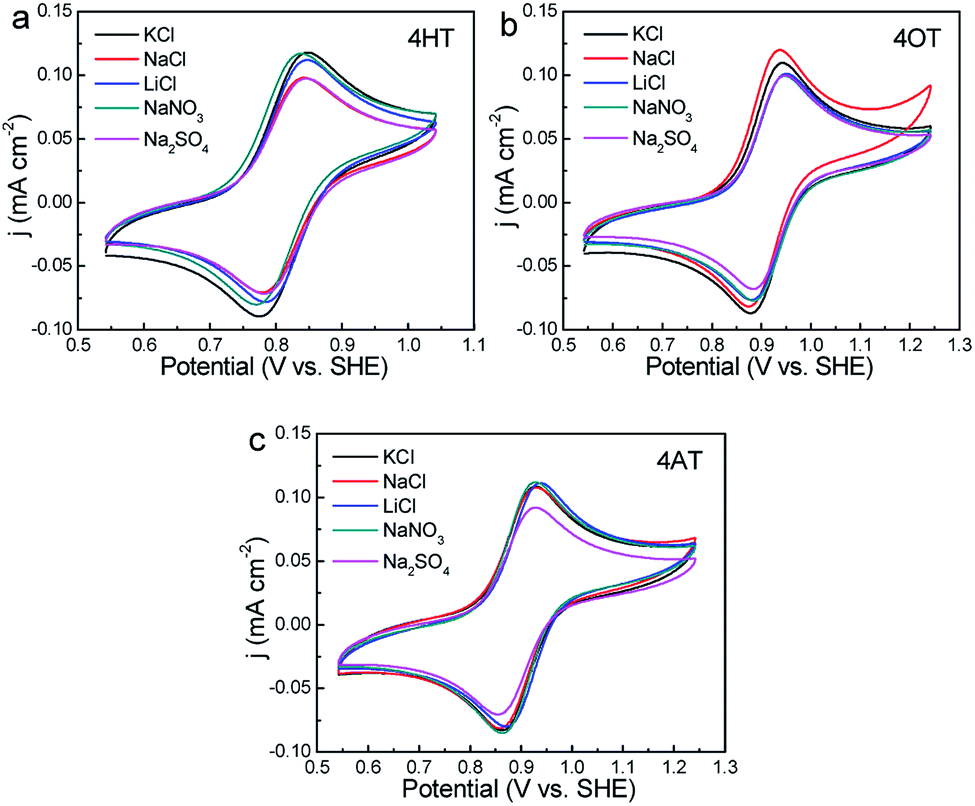
The effects of supporting electrolytes were evaluated for three TEMPO derivatives with high water solubility (that is, 4HT, 4OT and 4AT). The CV curves in different supporting electrolytes were shown in Fig. 3. Although the supporting electrolyte has little effect on the peak potential of three peak current. For example, the 4HT shows significantly higher peak current in LiCl, KCl and NaNO3 solution than in NaCl and Na2SO4 solution. The 4OT exhibits the largest peak current in NaCl solution, followed by in KCl solution. In addition, the peak current of 4AT in Na2SO4 solution is obviously lower than in the other four electrolyte solutions.
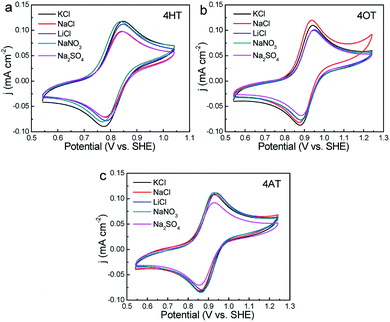 |
| | Fig. 3 CVs of 1 mM 4HT (a), 4OT (b) and 4AT (c) in different supporting electrolytes (0.5 M) at a scan rate of 25 mV s−1, respectively. | |
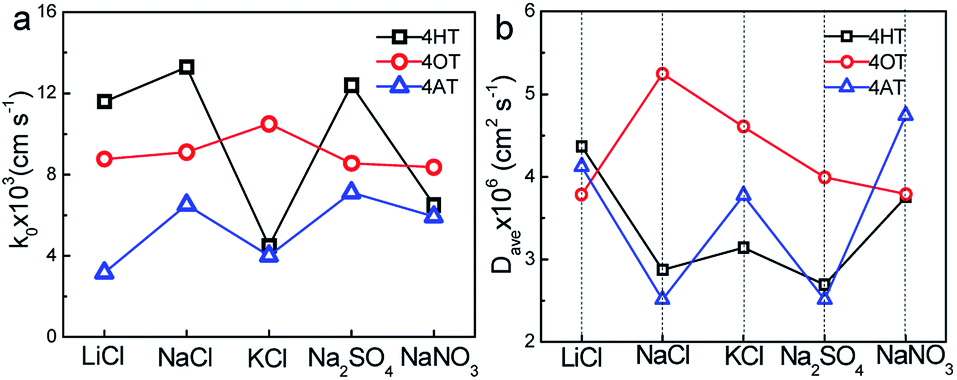
The electron transfer constants of 4HT, 4OT and 4AT in different supporting electrolytes were shown in Fig. 4a. In LiCl, NaCl and Na2SO4 solution, the 4HT shows the fastest electrochemical kinetics among three samples which are 1.16 × 10−2, 1.33 × 10−2 and 1.24 × 10−2 cm s−1, respectively. However, in KCl and NaNO3 solution the 4OT displays faster redox reaction mechanism compared with the 4HT and 4AT. In addition, the rate constants of 4-OT in different electrolytes are close to each other, indicating that the redox kinetics of 4-OT is almost independent of the electrolyte ions. The diffusion coefficients in different aqueous supporting electrolytes were shown in Fig. 4b. The 4HT and 4AT possess relatively faster diffusion in LiCl and NaNO3 solution than in other supporting electrolyte solutions. Different from them, the 4OT exhibits a fastest diffusion rate in NaCl solution. The solubilities of 4HT, 4OT and 4AT in different electrolyte solutions are determined using UV-vis absorption spectra, which are shown in Table 2 and Figs. S6–S8.† All three TEMPO derivatives have lower solubility in supporting electrolyte solution than their solubility in pure water because of the salting out effect, which means reduced theoretical capacity of the flow batteries. For instance, the 4OT exhibits solubility of 0.83 M in LiCl, 0.89 M in NaCl, 0.79 M in KCl, 0.87 M in NaNO3 and 0.78 M in Na2SO4, respectively. The 4AT has solubility of 3.92 M in LiCl, 5.23 M in NaCl, 4.80 M in KCl, 5.06 M in NaNO3 and 2.98 M in Na2SO4, respectively. The solubilities of 4-OT and 4-AT in NaCl aqueous solution are higher than those in LiCl and KCl. This means that the salting out effect of sodium ion is relatively weak. Similarly, compared with NO3− and SO42−, Cl− shows a weaker salting out effect on 4-OT and 4-AT. Therefore, NaCl is probably a best choice for TEMPO-based catholytes among five supporting electrolytes.
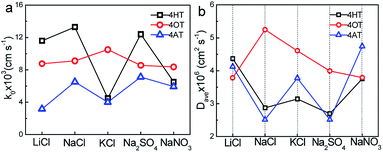 |
| | Fig. 4 Effect of supporting electrolytes (0.5 M) on electron transfer constant (k0) and diffusion coefficient (D). | |
Table 2 Solubility of TEMPO derivatives in different supporting electrolyte solutions (0.5 M) at room temperature
| Sample |
LiCl |
NaCl |
KCl |
NaNO3 |
Na2SO4 |
| 4HT |
0.69 M |
1.26 M |
1.25 M |
1.38 M |
0.33 M |
| 4OT |
0.83 M |
0.89 M |
0.79 M |
0.87 M |
0.78 M |
| 4AT |
3.92 M |
5.23 M |
4.80 M |
5.06 M |
2.98 M |
Symmetric RFB performance
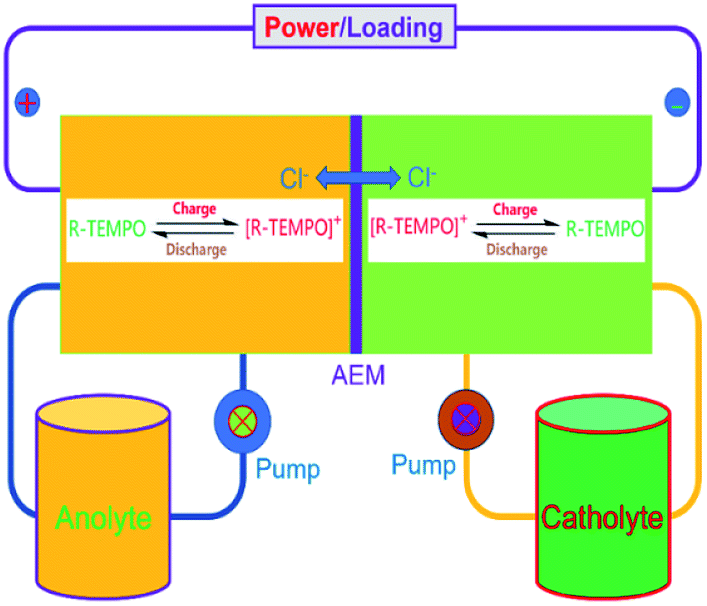
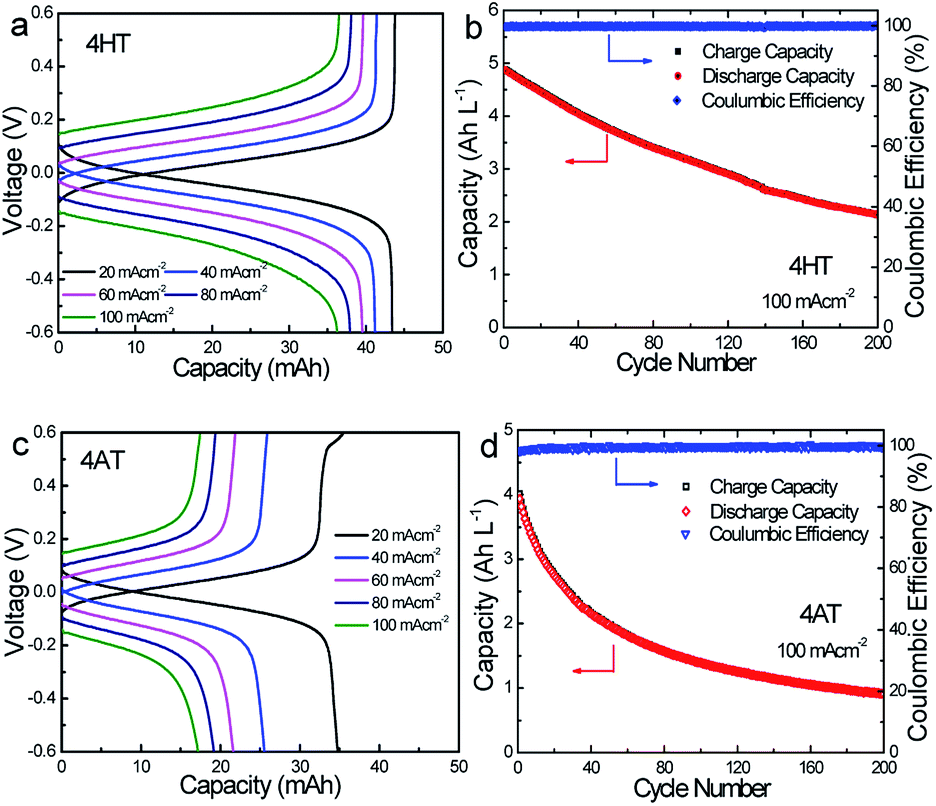
To verify the availability of TEMPO derivatives for aqueous RFBs, two symmetric RFBs utilized 4HT and 4AT, respectively, were fabricated and examined. As show in Fig. 5, a TEMPO derivative (the reduced state) and its oxidized state are employed in the symmetric RFB configuration as anolyte and catholyte, respectively.22,23 The rate performance is examined from 20 to 100 mA cm−2. The capacities of the symmetric 4HT and 4AT RFBs at 20 mA cm−2 are observed to be 43.5 and 34.8 mA h, 81.2% and 64.9% of the theoretical capacity (53.6 mA h), respectively. The increase of the current density leads to decreased capacity outputs, which is attributed to the increased ohmic loss as indicated by the charge–discharge voltage profiles at different current densities (Fig. 6a and c). During the charge–discharge cycling, the coulombic efficiency for two symmetric RFBs is nearly 100%. After 200 cycles, the discharge capacity of 4HT decreased from 4.88 to 2.13 Ah L−1 and that of 4AT decreased from 3.94 to 0.91 Ah L−1 with a significant capacity loss for each cycle (Fig. 6b and d).
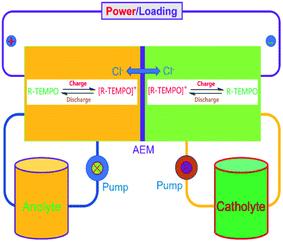 |
| | Fig. 5 Schematic diagram of the symmetric TEMPO derivative redox flow battery. | |
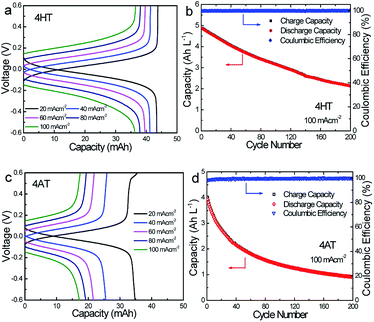 |
| | Fig. 6 Representative charge–discharge profiles of the symmetric 4HT (a) and 4AT (b) RFBs at different current densities. Cycling capacities and efficiencies of the symmetric 4HT (c) and 4AT (d) RFBs over 200 cycles at 100 mA cm−2. The concentration of TEMPO-based radicals is 0.2 M and an anion exchange membrane (AEM, A201) is used as the separator. | |
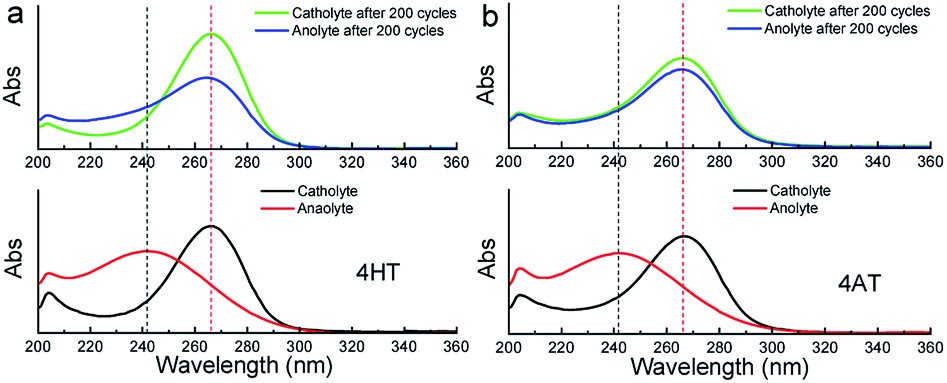
To clarify the possible degradation mechanism, ultraviolet spectra of the anolyte (reduced state) and catholyte (oxidation state) before and after 200 charge–discharge cycles were measured. As shown in Fig. 7, for the symmetric 4HT and 4AT RFBs, the fresh anolyte shows an absorption peak at ca. 242 nm while the fresh catholyte has the absorption peak at about 266 nm. After 200 cycles, the absorption peak position of the anolyte shifts from around 242 to 266 nm whereas the absorption peak of the catholyte is observed to be still at ∼266 nm, clearly indicating that the permeability of the oxidation state (oxoammonium cation) is the main cause of performance degradation of two symmetric RFBs. Furthermore, we investigated the effect of different types of ion exchange membranes on the battery performance and the permeability of the oxidation state. Compared with those with cation exchange membrane (N112), the symmetric RFBs with anion exchange membrane (A201) exhibit obviously higher practical remaining capacity after 200 charge–discharge cycles (Fig. S9†). It can be deduced that the anion exchange membrane possesses higher resistance to permeation since performance degradation is mainly due to the penetration of oxoammonium cation in the catholyte into the anolyte. The presented research reveals unambiguously that the crossover of oxoammonium cation is a major challenge for redox-active TEMPO derivatives applied in aqueous RFBs. In addition, it should also be noted that uncharged TEMPO derivatives can easily pass through membranes. Some researchers designed to introduce ionic groups (e.g. sulfonate9 or quaternary ammonium2,24) to the molecular structure of TEMPO derivatives and utilized the electrostatic exclusion between redox-active species and ion exchange membranes to reduce the penetration across membranes. However, other groups constructed soluble polymers containing redox-active units14,16,25 to lower this crossover. Therefore, in the future more attention needs to be paid to the compatibility between different redox-active organic molecules and membrane materials, and the development of high-performance membrane materials that can limit effectively the crossover of redox-active species.
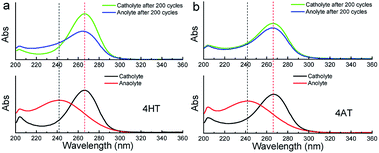 |
| | Fig. 7 Ultraviolet spectra of the anolyte and catholyte of the symmetric 4HT (a) and 4AT (b) RFBs, in 2 M KCl solution before and after 200 charge–discharge cycles. | |
Conclusions
In summary, we have examined the potential, redox kinetics, electrochemical stability and solubility of TEMPO derivatives in aqueous supporting electrolytes. The redox potential, diffusion coefficient, electron transfer rate constant and solubility are clearly influenced by functional groups of TEMPO derivatives and supporting electrolytes. Although their excellent electrochemical stability, the 4-HO-TEMPO and 4-H2N-TEMPO exhibit considerable performance degradation in the symmetric RFB configuration due to the permeability of oxoammonium cations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to acknowledge this research was supported by the National Natural Science Foundation of China (21573025 and 21773018), the program of China Scholarships Council (No. 201808320086), Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX19_0684) and Qing Lan project of Jiangsu Province.
Notes and references
- T. Liu, X. Wei, Z. Nie, V. Sprenkle and W. Wang, Adv. Energy Mater., 2016, 6, 1501449 CrossRef.
- T. Janoschka, N. Martin, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2016, 55, 14427–14430 CrossRef CAS PubMed.
- K. J. Kim, M.-S. Park, Y.-J. Kim, J. H. Kim, S. X. Dou and M. Skyllas-Kazacos, J. Mater. Chem. A, 2015, 3, 16913–16933 RSC.
- M. Skyllas-Kazacos, L. Cao, M. Kazacos, N. Kausar and A. Mousa, ChemSusChem, 2016, 9, 1521–1543 CrossRef CAS PubMed.
- C. Wang, Z. Yang, Y. Wang, P. Zhao, W. Yan, G. Zhu, L. Ma, B. Yu, L. Wang, G. Li, J. Liu and Z. Jin, ACS Energy Lett., 2018, 3, 2404–2409 CrossRef CAS.
- K. Lin, Q. Chen, M. R. Gerhardt, L. C. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 CrossRef CAS PubMed.
- Y. Liu, M.-A. Goulet, L. Tong, Y. Liu, Y. Ji, L. Wu, R. G. Gordon, M. J. Aziz, Z. Yang and T. Xu, Chem, 2019, 7, 1861–1870 Search PubMed.
- A. Hollas, X. Wei, V. Murugesan, Z. Nie, B. Li, D. Reed, J. Liu, V. Sprenkle and W. Wang, Nat. Energy, 2018, 3, 508–514 CrossRef CAS.
- J. Winsberg, C. Stolze, A. Schwenke, S. Muench, M. D. Hager and U. S. Schubert, ACS Energy Lett., 2017, 2, 411–416 CrossRef CAS.
- J. Luo, B. Hu, C. Debruler and T. L. Liu, Angew. Chem., Int. Ed., 2018, 57, 231–235 CrossRef CAS PubMed.
- B. Hu, C. DeBruler, Z. Rhodes and T. L. Liu, J. Am. Chem. Soc., 2017, 139, 1207–1214 CrossRef CAS PubMed.
- E. S. Beh, D. De Porcellinis, R. L. Gracia, K. T. Xia, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2017, 2, 639–644 CrossRef CAS.
- Z. Li, S. Li, S. Liu, K. Huang, D. Fang, F. Wang and S. Peng, Electrochem. Solid-State Lett., 2011, 14, A171–A173 CrossRef CAS.
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- J. Winsberg, C. Stolze, S. Muench, F. Liedl, M. D. Hager and U. S. Schubert, ACS Energy Lett., 2016, 1, 976–980 CrossRef CAS.
- J. Winsberg, T. Janoschka, S. Morgenstern, T. Hagemann, S. Muench, G. Hauffman, J.-F. Gohy, M. D. Hager and U. S. Schubert, Adv. Mater., 2016, 28, 2238–2243 CrossRef CAS PubMed.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- J. Cao, M. Tao, H. Chen, J. Xu and Z. Chen, J. Power Sources, 2018, 386, 40–46 CrossRef CAS.
- K. Lin, R. Gómez-Bombarelli, E. S. Beh, L. Tong, Q. Chen, A. Valle, A. Aspuru-Guzik, M. J. Aziz and R. G. Gordon, Nat. Energy, 2016, 1, 16102 CrossRef CAS.
- M. N. Paddon-Row, C. Santiago and K. N. Houk, J. Am. Chem. Soc., 1980, 102, 6561–6563 CrossRef CAS.
- S. Morris, G. Sosnovsky, B. Hui, C. O. Huber, N. U. M. Rao and H. M. Swartz, J. Pharm. Sci., 1991, 80, 149 CrossRef CAS PubMed.
- J. Luo, A. Sam, B. Hu, C. DeBruler, X. Wei, W. Wang and T. L. Liu, Nano Energy, 2017, 42, 215–221 CrossRef CAS.
- C. Zhang, Z. Niu, S. Peng, Y. Ding, L. Zhang, X. Guo, Y. Zhao and G. Yu, Adv. Mater., 2019, 1901052 CrossRef PubMed.
- Z. Chang, D. Henkensmeier and R. Chen, ChemSusChem, 2017, 10, 3193–3197 CrossRef CAS PubMed.
- T. Janoschka, S. Morgenstern, H. Hiller, C. Friebe, K. Wolkersdörfer, B. Häupler, M. D. Hager and U. S. Schubert, Polym. Chem., 2015, 6, 7801–7811 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03424j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Juan Xu*a,
Jianyu Cao
a,
Juan Xu*a,
Jianyu Cao