
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceButein, isoliquiritigenin, and scopoletin attenuate neurodegeneration via antioxidant enzymes and SIRT1/ADAM10 signaling pathway
Naw Hser Gayae,
Wilasinee Suwanjang a,
Waralee Ruankhama,
Napat Songtaweec,
Prapimpun Wongchitrata,
Virapong Prachayasittikulb,
Supaluk Prachayasittikuld and
Kamonrat Phopin*ab
a,
Waralee Ruankhama,
Napat Songtaweec,
Prapimpun Wongchitrata,
Virapong Prachayasittikulb,
Supaluk Prachayasittikuld and
Kamonrat Phopin*ab
aCenter for Research and Innovation, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand. E-mail: kamonrat.php@mahidol.ac.th; Fax: +66 2 441 4380; Tel: +66 2 441 4376
bDepartment of Clinical Microbiology and Applied Technology, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand
cDepartment of Clinical Chemistry, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand
dCenter of Data Mining and Biomedical Informatics, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand
eDepartment of Medical Laboratory Technology, University of Medical Technology, Yangon 11012, Myanmar
First published on 27th April 2020
Abstract
Neuronal cell death is a key feature of neurodegenerative disorders such as Parkinson's and Alzheimer's diseases. Plant polyphenols, namely butein, isoliquiritigenin, and scopoletin, have been shown to exhibit various biological activities including anti-inflammatory, antimicrobial, and antioxidant activities. Herein, butein, isoliquiritigenin, and scopoletin were explored for their neuroprotective properties against oxidative stress-induced human dopaminergic SH-SY5Y cell death. The cells exposed to hydrogen peroxide (H2O2) revealed a reduction in cell viability and increases in apoptosis and levels of reactive oxygen species (ROS). Interestingly, pretreatment of SH-SY5Y cells with 5 μM of butein, isoliquiritigenin, or scopoletin protected against the cell death induced by H2O2, and decreased the levels of apoptotic cells and ROS. In addition, the levels of SIRT1, FoxO3a, ADAM10, BCL-2, and antioxidant enzymes (catalase and SOD2) were maintained in the cells pretreated with butein, isoliquiritigenin, or scopoletin before H2O2 treatment compared to cells without pretreatment and the reference (resveratrol). Molecular docking analysis revealed that the interactions between the activator-binding sites of SIRT1 and the phenolic compounds were similar to those of resveratrol. Taken together, the data suggest that these polyphenolic compounds could be potential candidates for prevention and/or treatment of neurodegeneration.
1. Introduction
A neurodegenerative disorder refers to the progressive loss of functions and structures, and neuronal cell death caused by various conditions including genetic and environmental factors. Presently, the prevalence of neurodegenerative diseases is markedly increasing worldwide, and tends to increase each year. This situation is one of the major problems that is a primary concern for the healthcare of people. Normally, the antioxidant defense system maintains redox state and prevents cells from oxidative stress. On the other hand, reactive oxygen species (ROS) can be excessively generated when the antioxidant defense mechanism in our body works improperly leading to the oxidative damage.1,2 ROS can be free radicals including hydroxyl radical (·OH), superoxide anion radical (O2˙−), or non-radicals like hydrogen peroxide (H2O2). H2O2 is an oxidizing compound, which is generated through dismutation of superoxide. Under physiological condition, H2O2 is then converted into ·OH or water. ·OH is extremely reactive and propagates the formation of ROS in a chain reaction. ROS may damage DNA, proteins, and lipids provoking a cascade of events leading to cell death.3 Previous studies have reported that under physiological conditions retinal pigment epithelium (RPE) cells generate H2O2 during phagocytosis and degradation of photoreceptor outer segments. The generation of H2O2 during phagocytosis may act as an intracellular signal in RPE cells that leads to increase levels of key antioxidant catalase and metallothionein gene expression for preventing the progression of age-related macular degeneration (AMD).4 In contrast, H2O2 is mostly used as an exogenous source of ROS. It has been reported that exogenously applied H2O2 in rat brain astrocytes induced mitochondrial swelling, plasma membrane blebs, and loss of retained mitochondrial function, resulting in cell death.5 Additionally, exposure of neuronal cells to H2O2 significantly increased the levels of ROS, B-cell lymphoma 2-associated X protein (BAX), poly (ADP-ribose), cleaved poly (ADP-ribose) polymerase, cytochrome c, apoptosis-inducing factor, cleaved caspase-9, and cleaved caspase-3, which are associated with apoptosis signalling and cell death.6 It was also reported that H2O2 can induce nuclear or mitochondrial DNA damage in various cell types, including neurons,7 which is one of the contributing factors of neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's diseases.8 Especially, Alzheimer's disease (AD) is the common form of dementia and accounts for about 70% of the dementia patients.9 Thus, the interest on antioxidant therapy due to its remarkable effects on controlling antioxidant defense system and subsequently protection of neurodegeneration has also increased.8,10,11Plant polyphenols with antioxidant properties are ubiquitously found in natural sources. There are more than 8000 different polyphenolic compounds identified to date. A polyphenol can be chemically classified as a substance having one or more hydroxyl substituents attached to its aromatic rings,12 one of which is represented by flavonoids. These plant phenolics can be synthesized from cinnamic acid via phenylalanine which is deaminated by phenylalanine ammonia. Polyphenolic molecules have their therapeutic and preventive potentials for neurodegenerative diseases. They contribute substantial neuroprotection through their phenoxy radical formations and their effects on cellular signal transduction pathways.13–15 Several lines of evidence have revealed that polyphenols contain multiple biological actions such as antioxidant, antimicrobial, anti-inflammatory, anticancer, and metal chelating effects as well as anti-Aβ aggregation.16
Butein and isoliquiritigenin (2′,4,4′-trihydroxychalcone) are chalcone compounds identified in 2008 from the ant plant, tuber of Hydnophytum formicarum Jack.17,18 Chalcones are an aromatic ketone and considered to be precursors of flavonoids and isoflavonoids. They consist of two benzene rings linked by three carbons of α,β-unsaurated carbonyl group.19,20 The metabolism of isoliquiritigenin in vitro has been evaluated by HPLC-MS analysis in several studies, where the Phase I metabolites of isoliquiritigenin including butein were confirmed in human and rat liver microsomes. The specific chemical structure of chalcones makes butein act as a powerful radical scavenger. Furthermore, both butein and isoliquiritigenin have been found to possess anti-inflammatory, antimicrobial and antioxidant properties.21–24 In 2013, a study demonstrated that butein from ethyl acetate fraction of the bark of Rhus verniciflua has neuroprotective and anti-inflammatory effects in vitro and cognitive enhancing effects in vivo by maintaining SOD, glutathione reductase, and glutathione peroxidase as well as by restoring the content of glutathione.25 Thus, biosynthetic and structural relationships of these two compounds and many previous reports about antioxidant properties of these compounds prompted us to further investigate their anti-neurodegenerative activities. Another one, scopoletin (7-hydroxy-6-methoxy-2H-1-benzopyran-2-one), was identified from the aerial part of Spilanthes acmella Murr.26,27 Scopoletin is a coumarin derivative derived from a common carbon skeleton building block (C6–C3) and contains a backbone of 1,2-benzopyrone, which contains the core skeleton of flavonoid compounds. The hydroxyl groups of coumarin are good hydrogen donors and have high possibilities to scavenge free radicals and terminate redox reactions that damage cells.28 Scopoletin has been reported to exert antioxidant, anti-inflammatory, anti-acetylcholinesterase,29 antifungal,30 and anticancer activities.31 In the presence of scopoletin, increased expression of transcription factors such as Nrf-2 and p-FoxO1 related to anti-aging was documented.22 Additionally, scopoletin was one of the potent antioxidants against Aβ25–35 induced cytotoxicity in Neuro-2A cells32 and monoamine oxidase inhibitor.33 In this context, scopoletin also prompted us to further study the protective effect of this compound on oxidative stress-induced neuronal cells.
With regards to polyphenols, although their mechanisms of action and molecular targets in neurodegenerative diseases are still unanswered, the phenyl ring structure is believed to increase beneficial effects of polyphenols. As mentioned earlier, butein, isoliquiritigenin, and scopoletin (Fig. 1) have been reported to exert diverse biological properties,21–24 but the molecular mechanisms which underlie these activities have not been fully unveiled. This research explored the underlying mechanisms of these antioxidants against H2O2-induced neuronal cell death in SH-SY5Y cells and addressed how chalcones (butein and isoliquiritigenin) and coumarin (scopoletin) affect protein markers associated with amyloid beta production, as well as H2O2-induced neurotoxicity in human neuronal cells.
 | ||
| Fig. 1 Chemical structures of butein, isoliquiritigenin, and scopoletin. | ||
2. Materials and methods
2.1. Chemicals and reagents
Dulbecco's Modified Eagle's Medium (DMEM), 1% penicillin/streptomycin, and fetal bovine serum (FBS) were purchased from Gibco BRL (Gaithersburg, MD, USA). Butein (purity: 98%, melting point: 216 °C (216 ± 6 °C), geometry: E), isoliquiritigenin (purity: 98%, melting point: 185 °C to 188 °C), and resveratrol (purity: 99%, melting point: 261 °C to 263 °C) were received from Sigma-Aldrich (St. Louis, Missouri, USA). Additionally, scopoletin (purity: 99%, melting point: 228 °C to 234 °C) was obtained from Acros Organics (New Jersey, USA). For western blotting, the following antibodies were obtained from Cell Signalling Technology (Beverly, MA, USA): primary antibodies (anti-ADAM10, anti-FoxO3a, anti-SIRT1, anti-SOD2, anti-BCL-2, anti-catalase, and anti-β-actin) and secondary antibodies (horseradish peroxidase-conjugated anti-rabbit IgG and anti-mouse IgG). Human dopaminergic SH-SY5Y cells were received from American Type Culture Collection (ATCC, VA, USA). ECL plus western blot detecting reagent was obtained from Amersham Biosciences (Piscataway, NJ, USA). Analytical grade reagents used in this work were all obtained from Sigma-Aldrich.2.2. Cell culture and polyphenolic treatments
SH-SY5Y cells were seeded in 75 cm2 flasks containing DMEM, 10% heat-inactivated FBS, and 1% streptomycin and penicillin, followed by incubation at 37 °C and 5% CO2. The cells were grown in the medium refreshed every three days until 80% confluence. The polyphenolics, butein, isoliquiritigenin, scopoletin, and resveratrol, were dissolved in DMSO and diluted with DMEM containing 10% FBS to various concentrations. Following seeding for 24 h, the cells were incubated for 3 h with 5 μM of one of the three compounds or 1 μM of resveratrol before exposure to 400 μM H2O2 for 24 h. Untreated SH-SY5Y cells were used as a control.2.3. Cell viability detection
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) method was used to detect cell viability. Thus, the absorbance of formazan is directly proportional to the number of viable cells. SH-SY5Y cells (1.0 × 105 cells per mL) were seeded onto 96-well plates. Butein, isoliquiritigenin, and scopoletin were used in various final concentrations (1, 5, 10, and 50 μM) followed by 3 h incubation prior to the 24 h incubation with 400 μM H2O2. MTT solution (5 mg mL−1) was loaded into each well with 37 °C incubation for 3 h in the dark. The culture medium was discarded, and 0.04 N HCl in isopropanol was added to dissolve the formazan crystals. Cell viability was measured at 570 nm by microplate detection (Bio Tek Instruments, Inc, Winooski, VT, USA). The viability of cells was calculated as a percentage compared to the untreated cells.2.4. Cell apoptosis assay
To characterize apoptotic cell ratios, cells were stained with annexin V (annexin V-fluorescein isothiocyanate) using annexin V and a dead cell assay kit. Annexin V was used to detect the membrane phosphatidylserine of apoptotic cells. Additionally, the cells were incubated with 7-amino-actinomycin D (7-AAD), a specific marker of death cell. Briefly, SH-SY5Y cells (1.0 × 105 cells per mL) were seeded in 6-well plates for 24 h. After the incubation, the cells were exposed to 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM of resveratrol for 3 h prior to incubation with 400 μM H2O2 for 24 h. Both floating and adherent cells were collected and centrifuged at 1000 RPM for 5 min. The fluorescent solution was mixed with 100 μL of cell suspension and held in the dark for 20 min. The quantitation evaluation of live, apoptotic, and dead cells was conducted by Muse Cell Analyzer (EMD Millipore, Billerica, MA, USA).2.5. Carboxy-DCFDA assay
Intracellular ROS levels in the neuronal cells were measured by carboxy-DCFDA assay. In the presence of ROS, this reagent is converted to a highly green fluorescent dichlorofluorescein (DCF). Cells (1.0 × 105 cells per mL) were cultured for 24 h in 96-well plates, and treated with 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM of resveratrol for 3 h prior to incubation for 24 h with 400 μM H2O2. After the incubation, the culture medium was discarded and rinsed with phosphate-buffered saline (PBS). Under dark conditions, 10 μL of 25 μM carboxy-DCFDA was loaded into each well and incubated at 37 °C for 30 min. ROS levels were detected immediately on a fluorescence plate reader at 485 nm and 528 nm for excitation and emission wavelengths, respectively.2.6. Protein expression assay by western blotting
SH-SY5Y cells (1.0 × 105 cells per mL) were plated onto 6-well plates for 24 h at 37 °C. Then, pretreatment of the cells was performed with 5 μM of butein, isoliquiritigenin, scopoletin or 1 μM of resveratrol for 3 h prior to 24 h incubation with 400 μM H2O2. RIPA lysis buffer with protease inhibitors was added into the cell suspension followed by sonication for 10 s and centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 20 min at 4 °C. Bradford protein assays were conducted to measure total protein levels. The protein lysates were resolved by 10% SDS-PAGE and moved onto nitrocellulose membrane. Next, 5% skim milk in 1× Tris-buffered with Tween-20 (TBST) was used for blocking non-specific sites on the membrane for 1 h at room temperature (RT) and rinsed with TBST. The membrane was incubated with specific primary antibodies overnight at 4 °C followed by incubating with HRP-conjugated secondary antibodies for 1.5 h. Lastly, the blotted membrane was developed by ECL before being captured by chemiluminescent signals in ChemiDoc™ MP imager and the protein levels were quantitated using densitometry analysis with Image Lab software (Bio-Rad, Hercules, CA).
000 g for 20 min at 4 °C. Bradford protein assays were conducted to measure total protein levels. The protein lysates were resolved by 10% SDS-PAGE and moved onto nitrocellulose membrane. Next, 5% skim milk in 1× Tris-buffered with Tween-20 (TBST) was used for blocking non-specific sites on the membrane for 1 h at room temperature (RT) and rinsed with TBST. The membrane was incubated with specific primary antibodies overnight at 4 °C followed by incubating with HRP-conjugated secondary antibodies for 1.5 h. Lastly, the blotted membrane was developed by ECL before being captured by chemiluminescent signals in ChemiDoc™ MP imager and the protein levels were quantitated using densitometry analysis with Image Lab software (Bio-Rad, Hercules, CA).
2.7. Immunocytochemical analysis for SIRT1 localization
SH-SY5Y cells (5 × 104 cells per mL) were grown on a slide (SPL Life Sciences Co., Ltd., Korea) at 37 °C for 24 h. Butein, isoliquiritigenin, and scopoletin at the concentration of 5 μM and 1 μM of resveratrol were used to pretreat the cells for 3 h followed by 24 h incubation with 400 μM H2O2. After the incubation, the cells were fixed with 4% paraformaldehyde for 10 min at RT followed by 10 min permeabilization at RT with 1% Triton X-100 in PBS. Following washing with PBS, the cells were incubated with 1% BSA for 90 min at RT to block non-specific binding areas. Primary antibody against SIRT1 (1:1000 in PBS containing 0.1% BSA) was incubated with the cells overnight at 4 °C followed by 2 h incubation at RT with 1:400 of Alexa 488-labelled, goat anti-rabbit IgG in PBS containing 0.1% BSA. Lastly, the cells were stained with DAPI for 10 min, and mounted using an anti-fade reagent (Vector Laboratories, Inc, Burlingame, CA, USA). Stained slides were visualized with a confocal microscope (Fluoview 1000, Olympus, Tokyo, Japan).
2.8. Molecular docking analysis
Interactions between the activator-binding site of SIRT1 and the phenolic compounds (butein, isoliquiritigenin, and scopoletin) were determined by a molecular docking analysis. Crystal structure of SIRT1 co-crystalized with three resveratrol molecules and the fluorophore-containing, acetylated p53 peptide (PDB code 5BTR)37 was used as the search model. Structural domain of the PDB consists of the catalytic domain (CD) connected with the extended N-terminal domain (NTD). Atomic coordinates of butein, isoliquiritigenin, and scopoletin were taken from the PubChem database (pubchem.ncbi.nlm.nih.gov). Before docking calculation, three resveratrol molecules were removed from the PDB before the docking calculation, and polar hydrogen atoms were then added into the compounds and protein. AutoDock 4.2.6 program was utilized to perform molecular docking calculations.34 The rotational bonds of the protein structure were considered as rigid, while those of the compound structures were treated as flexible. The activator-binding site of the enzyme structure, which covers the NTD/CD interface, was used as the center of the grid box with the grid spacing 0.375 Å. Lamarckian Genetic Algorithm was applied as the search parameter, in which the maximum number of energy evaluations was set to the medium level.35 Discovery Studio Visualizer 2016 was utilized for analyzing and visualizing the intermolecular interactions between SIRT1 and each compound.2.9. Statistical analysis
The results from three independent experiments were expressed as mean ± S.E.M. GraphPad Prism 6 scientific software (Graph Pad Software, La Jolla, CA, USA) was used to perform One-Way Analysis of Variance (ANOVA) with Tukey's test to calculate the statistical significance of differences. A p-value less than 0.05 was defined as statistically significant.3. Results
3.1. Effects of butein, isoliquiritigenin, and scopoletin on SH-SY5Y cell viability
To explore the effects of butein, isoliquiritigenin, and scopoletin against H2O2-induced cytoxicity, the cells were pretreated with different concentrations (1, 5, 10, and 50 μM) of butein, isoliquiritigenin, scopoletin, or resveratrol and also exposed to various concentrations of H2O2 (200, 300, 400, and 500 μM) for 24 h, and then assessed with the MTT assay. In this work, cell viabilities when treated with 1, 5, 10, and 50 μM of butein were 98.5 ± 1.1%, 104.4 ± 1.2%, 103.3 ± 2.58%, and 92.6 ± 2.1%, respectively. 95.8 ± 2.6%, 103.2 ± 1.8%, 104.5 ± 0.4%, and 91.1 ± 1.7% of cell viabilities were found in the cells treated with isoliquiritigenin at 1, 5, 10, and 50 μM, respectively, scopoletin treatments were 96.4 ± 3.2%, 103.5 ± 3.2%, 101.2 ± 3.8%, and 97.3 ± 4.5%, respectively, and resveratrol treatments were 95.1 ± 4.3%, 97.6 ± 1.6%, 91.5 ± 6.1%, and 82.5 ± 7.6%, respectively compared to the control (Fig. 2A, B, C, and D, respectively). In contrast, H2O2 was found to induce dose-dependent cytotoxicity in concentrations of 200–500 μM (78.1 ± 4.2%, 78.2 ± 4.8%, 65.5 ± 0.4%, and 59.6 ± 3.4%) compared to the control. 400 μM H2O2 induced 34.4 ± 0.4% cell death (Fig. 2E) and was chosen for use in subsequent experiments.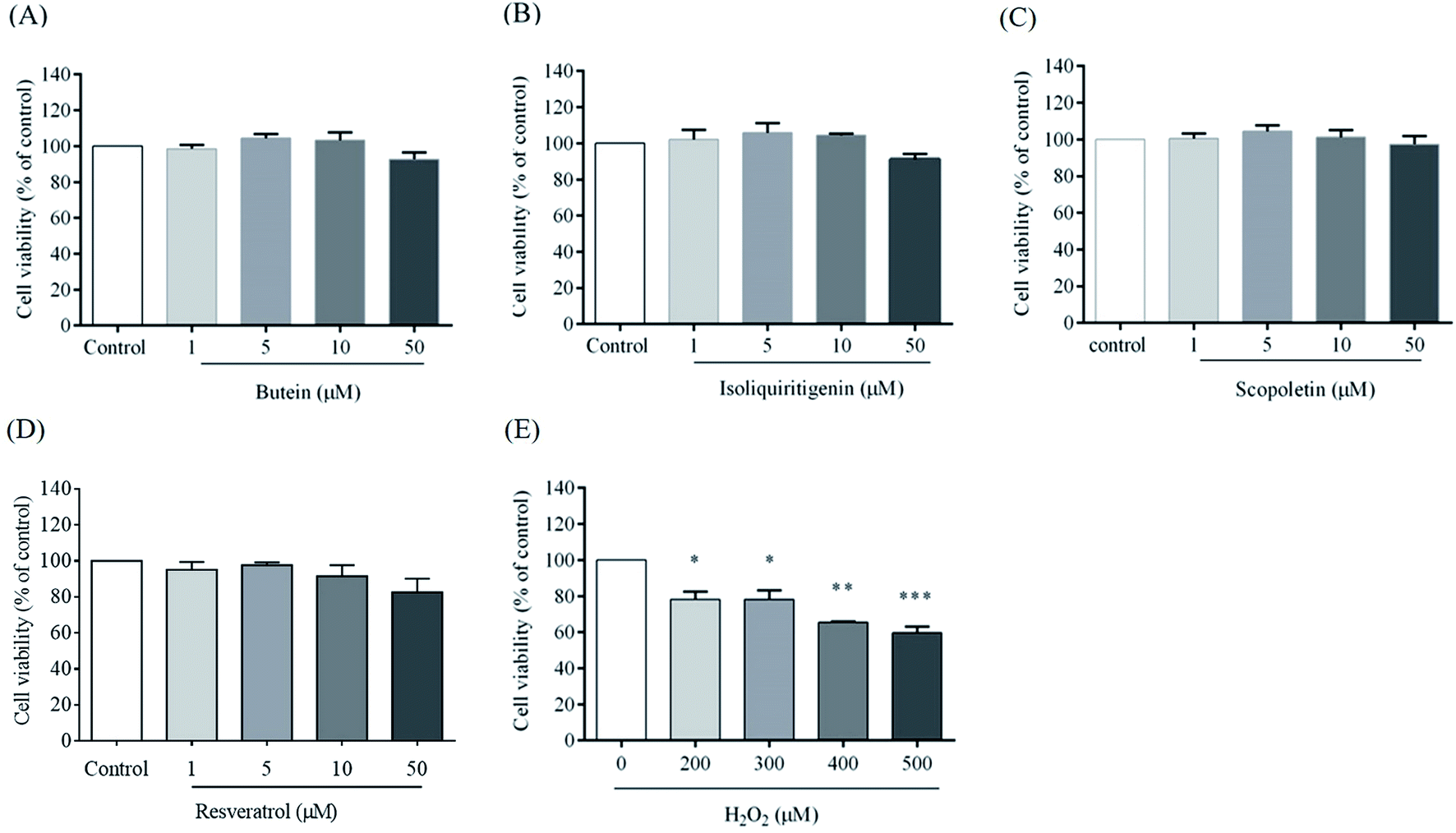 | ||
| Fig. 2 Cell viability of SH-SY5Y cells treated with 1, 5, 10, and 50 μM of butein, isoliquiritigenin, scopoletin, or resveratrol for 24 h (A, B, C, and D, respectively). Neurotoxicity induced by H2O2 on SH-SY5Y cells shown as % cell viability (E). The data are expressed as mean ± S.E.M. of three independent experiments. Statistical analysis was performed using ANOVA and Tukey–Kramer post hoc test (*p < 0.05, **p < 0.01, and ***p < 0.001 vs. control). | ||
The protective effects of butein, isoliquiritigenin, and scopoletin against 400 μM H2O2-induced neurotoxicity were studied. The cell viability after 24 h exposure to 400 μM H2O2 was recovered in 5 μM of both isoliquiritigenin (94.9 ± 3.9%) and scopoletin (95.1 ± 4.7%). Additionally, butein (5 μM) showed an even stronger protective effect with cell viability of 98.2 ± 4.0% in the face of H2O2 exposure. All of these compounds revealed higher cell viabilities than resveratrol (93.7 ± 3.2%) (Fig. 3A). Cell losses and morphological changes including cell shrinkage and floating cells, caused by the 400 μM H2O2 were noted. Pretreatment of cells with 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM of resveratrol resulting in cell morphology similar to that of control cells not exposed to H2O2 (Fig. 3B). Thus, butein, isoliquiritigenin, and scopoletin were able to prevent the neuronal cell death induced by H2O2.
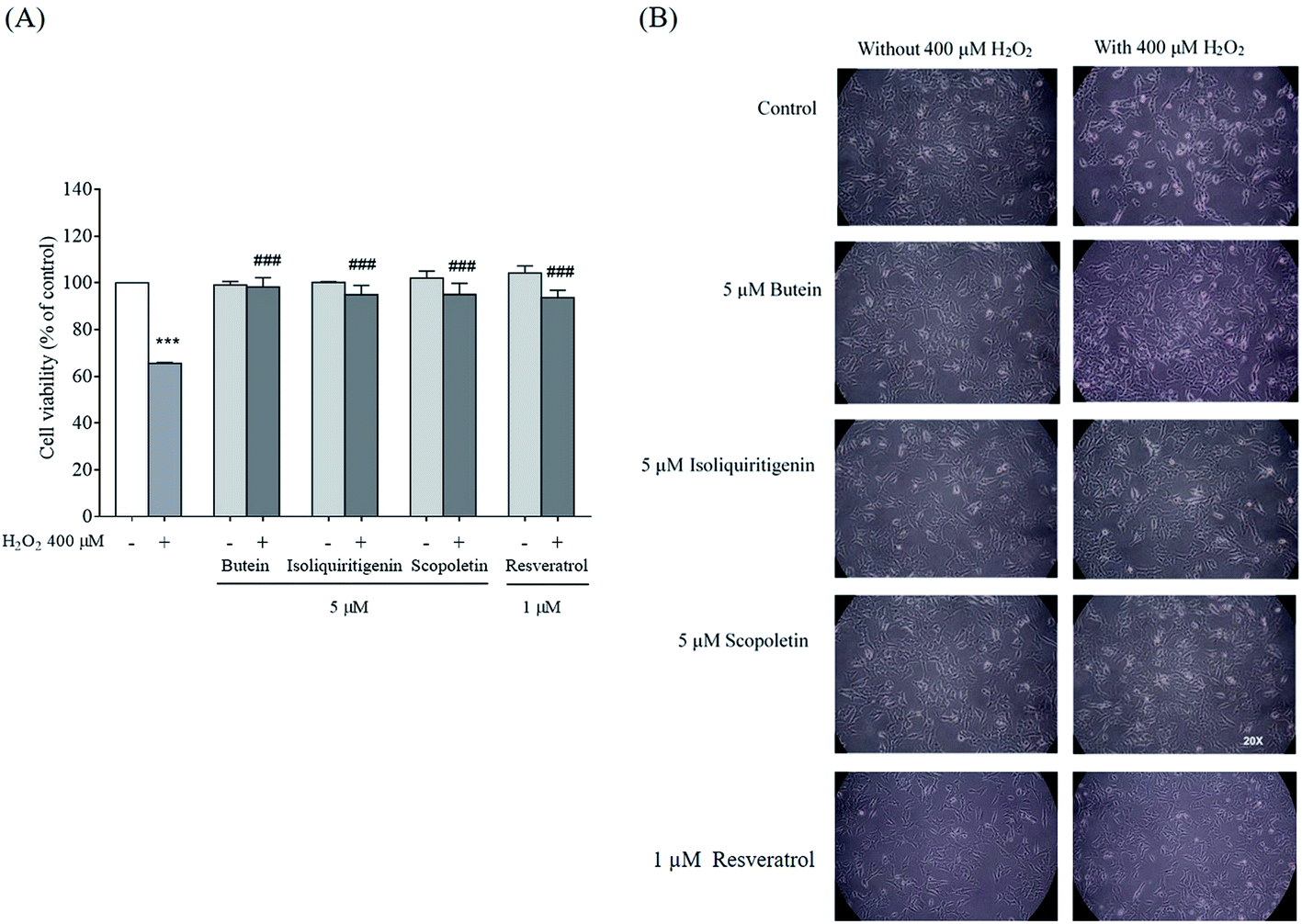 | ||
| Fig. 3 (A) Effects of 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM resveratrol on H2O2-induced cell viability (***p < 0.001 vs. control; ###p < 0.001 vs. 400 μM H2O2-treated group). (B) Morphology of SH-SY5Y cells in the presence of 400 μM H2O2 and the cells pretreated with 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM resveratrol following exposure to 400 μM H2O2. Observations were made under light microscopy at 20× magnification. | ||
3.2. Effects of butein, isoliquiritigenin, and scopoletin on H2O2-induced ROS levels in SH-SY5Y cells
Exposure to H2O2 induces a rapid increase in ROS production in neuronal cells which plays a major role in cell death. To provide further evidence that polyphenolics (butein, isoliquiritigenin, and scopoletin) are responsible for radical scavenging, the fluorescence intensity of DCF as a correlation of ROS levels was determined. The fluorescence intensity significantly increased (129.8 ± 1.0%) following the exposure of cells to H2O2 at the concentration of 400 μM for 24 h compared to that of unexposed cells. However, the intensities of the cells pre-treated with the compounds mentioned above were reduced to 96.2 ± 1.4%, 99.1 ± 1.0%, and 100.0 ± 1.4%, respectively similar to resveratrol (100.1 ± 0.5%) (Fig. 4). The findings indicate that these polyphenolics inhibited H2O2-induced ROS production.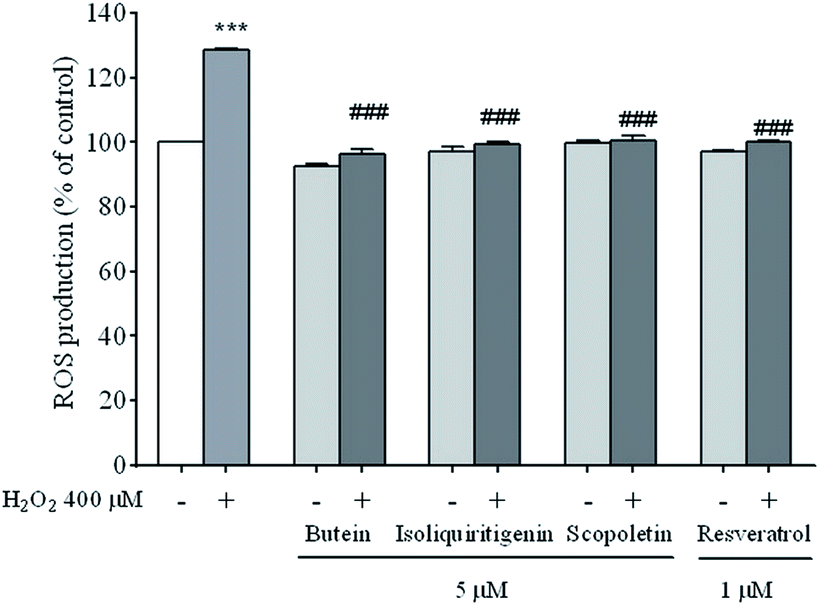 | ||
| Fig. 4 The protective effects of 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM resveratrol on H2O2-induced ROS production. The data are shown as mean ± S.E.M. of three independent experiments (***p < 0.001 vs. control; ###p < 0.001 vs. 400 μM H2O2-treated group). | ||
3.3. Effects of butein, isoliquiritigenin, and scopoletin on SH-SY5Y cell apoptosis induced by H2O2
The protective effects of butein, isoliquiritigenin, and scopoletin upon SH-SY5Y cell apoptosis were investigated. The number of apoptotic cells increased significantly following 400 μM H2O2 treatment. Percentages of the apoptotic cells after pretreatment with 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM of resveratrol were decreased to 11.7 ± 1.0%, 12.2 ± 1.0%, 13.3 ± 1.2%, and 7.9 ± 0.2%, respectively compared to those of cells without pretreatment, which were exposed to 400 μM H2O2 (36.1 ± 0.9%). Additionally, treatment of cells with butein, isoliquiritigenin, or scopoletin alone did not induce increases in the levels of apoptotic cells (Fig. 5). The data suggested that attenuation of H2O2-induced apoptosis was affected by pretreatment with these phenolic compounds. Therefore, we further investigated potential protective mechanisms of each compound by western blotting.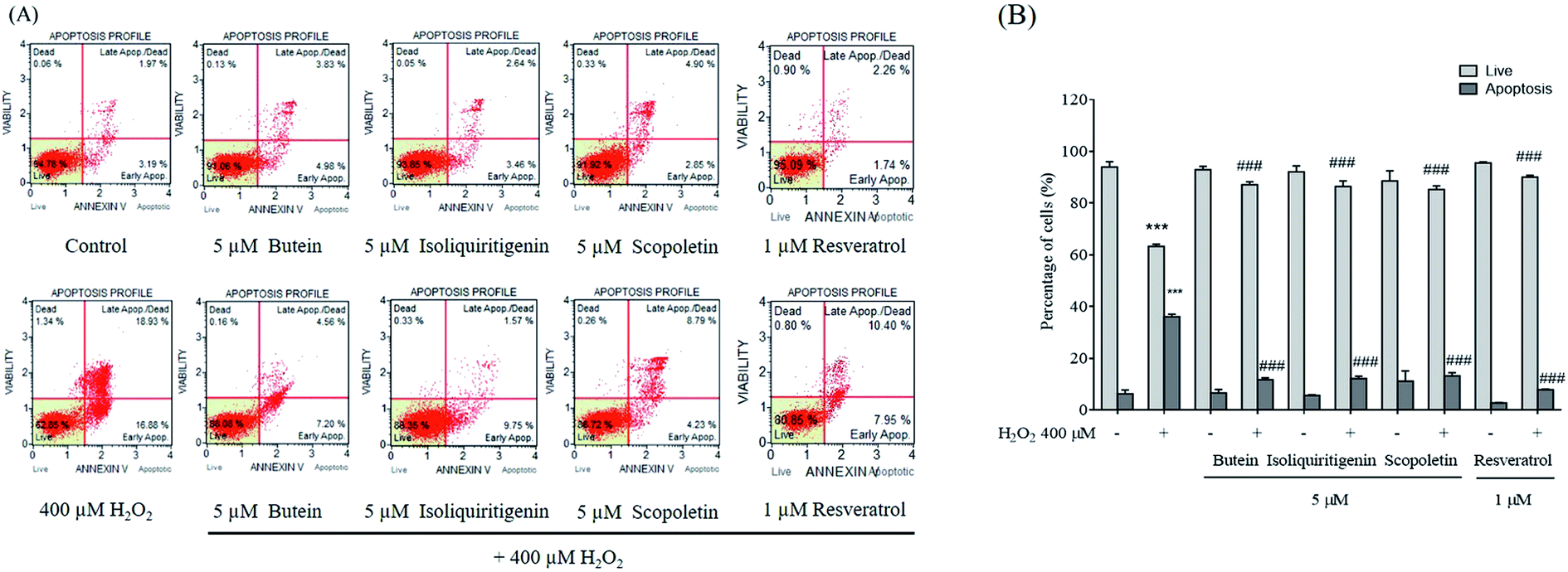 | ||
| Fig. 5 Flow cytometric measurement of live and apoptotic cells given pretreatment with 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM resveratrol and exposed to 400 μM H2O2 for 24 h. (A) Histograms of live and apoptotic cells stained by annexin V. (B) Bar diagrams of percentage live and total apoptotic cells. The data are shown as mean ± S.E.M. of three independent experiments (***p < 0.001 vs. control; ###p < 0.001 vs. 400 μM H2O2-treated group). | ||
3.4. Effects of butein, isoliquiritigenin, and scopoletin on anti-apoptotic, antioxidant, and FoxO3a protein levels
To determine antioxidant capacities, protein levels of FoxO3a, BCL-2, SOD2, and catalase, in the presence and absence of butein, isoliquiritigenin, and scopoletin in the experiments, were assessed using a western blot assay. Since BCL-2 expression plays a key role in neuronal survival; thus, BCL-2 levels were determined. Catalase and SOD, active scavengers of H2O2 and O2˙−, are important players that are regulated by FoxO3a. Hence, the removal of ROS is achieved by an increase in FoxO3a and these two enzymes. Following pretreatment with 5 μM of butein, isoliquiritigenin, or scopoletin, the decrease in the levels of BCL-2, FoxO3a, SOD2, and catalase (62–65%) after induction by H2O2 was significantly reversed (BCL-2: 109.7 ± 5.0%, 116.3 ± 5.5%, and 118.4 ± 6.8%; FoxO3a: 114.1 ± 3.2%, 110.9 ± 4.8%, and 109.4 ± 6.7%; SOD2: 104.0 ± 4.4%, 109.3 ± 6.5%, and 116.3 ± 6.9%; catalase: 109.0 ± 4.3%, 107.4 ± 6.0%, and 119.2 ± 2.9%, respectively) (Fig. 6). These results suggested that butein, isoliquiritigenin, and scopoletin suppressed the oxidative stress-induced neurotoxicity by augmentation of antiapoptotic and antioxidant protein levels in the same manner as resveratrol (BCL-2: 103.7 ± 5.9%; FoxO3a: 108.5 ± 8.7%; SOD2: 99.8 ± 11.3%; catalase: 101.6 ± 7.8%).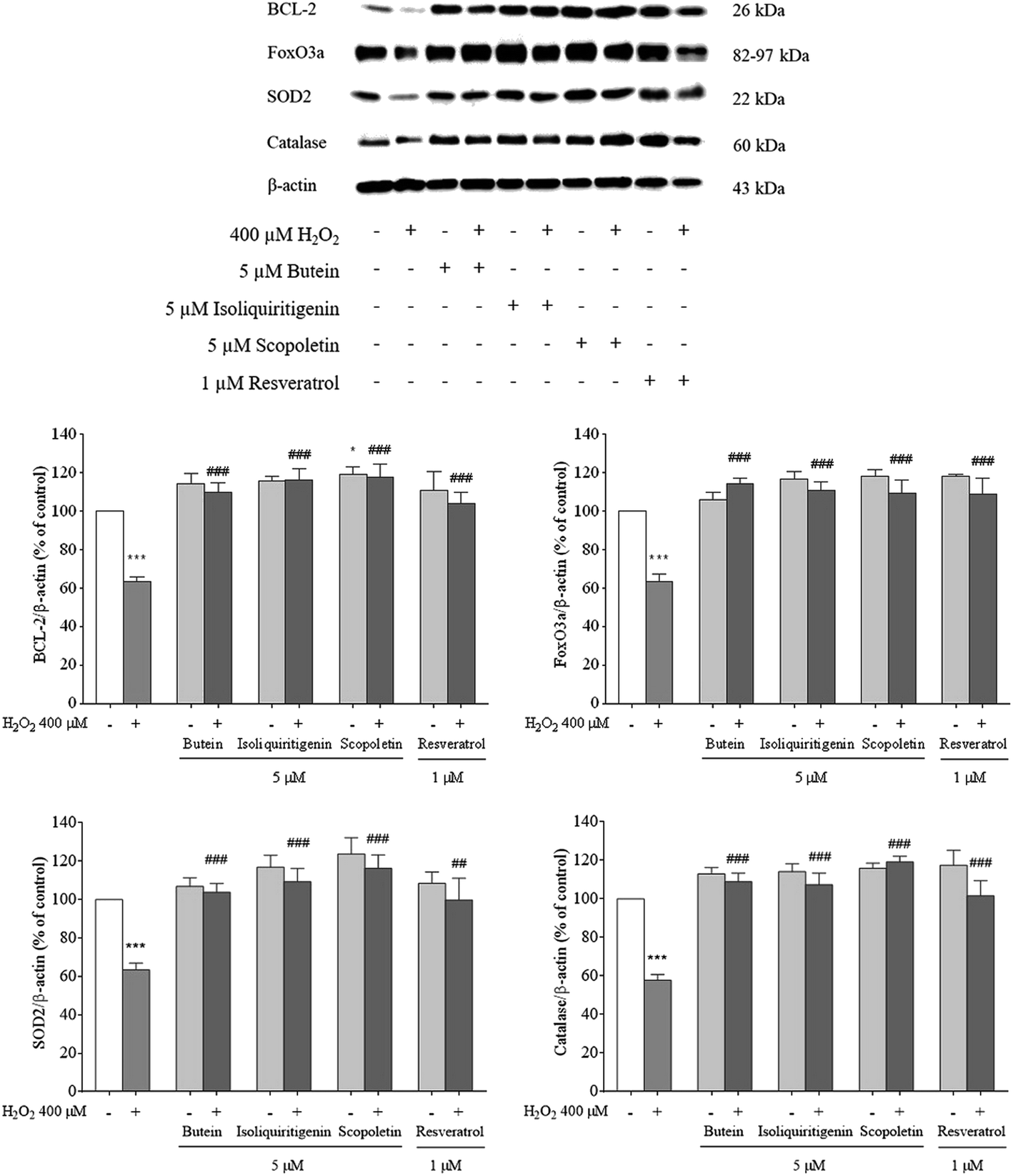 | ||
| Fig. 6 Effects of 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM resveratrol against 400 μM H2O2-induced decreases in BCL-2, FoxO3a, SOD2, and catalase proteins were determined by western blot analysis. Densitometric data of protein bands are presented in the graph as a ratio to β-actin. The results are expressed as mean ± S.E.M. of three independent experiments (*p < 0.05 and ***p < 0.001 vs. control; ##p < 0.01 and ###p < 0.001 vs. 400 μM H2O2-treated group). | ||
3.5. Effects of butein, isoliquiritigenin, and scopoletin on SIRT1 and ADAM10
It was reported that overexpression of either ADAM10 or SIRT1-coupled ADAM10 was linked to a reduction in Aβ production and full length amyloid precursor protein (APP) cleavage.36 Thus, SIRT1 and ADAM10 proteins were assessed to explore the possible neuroprotective effects of butein, isoliquiritigenin, and scopoletin. The results showed that decreased levels of SIRT1 and ADAM10 proteins induced by 400 μM H2O2 (64–65%) were dramatically restored by 5 μM of butein, isoliquiritigenin, scopoletin, and 1 μM of resveratrol pretreatment (SIRT1: 106.4 ± 4.5%, 104.7 ± 6.7%, 114.2 ± 6.8%, and 102.2 ± 2.6%, respectively; ADAM10: 108.3 ± 3.3%, 92.4 ± 0.9%, 105.3 ± 2.2%, and 93.9 ± 7.1%, respectively) (Fig. 7).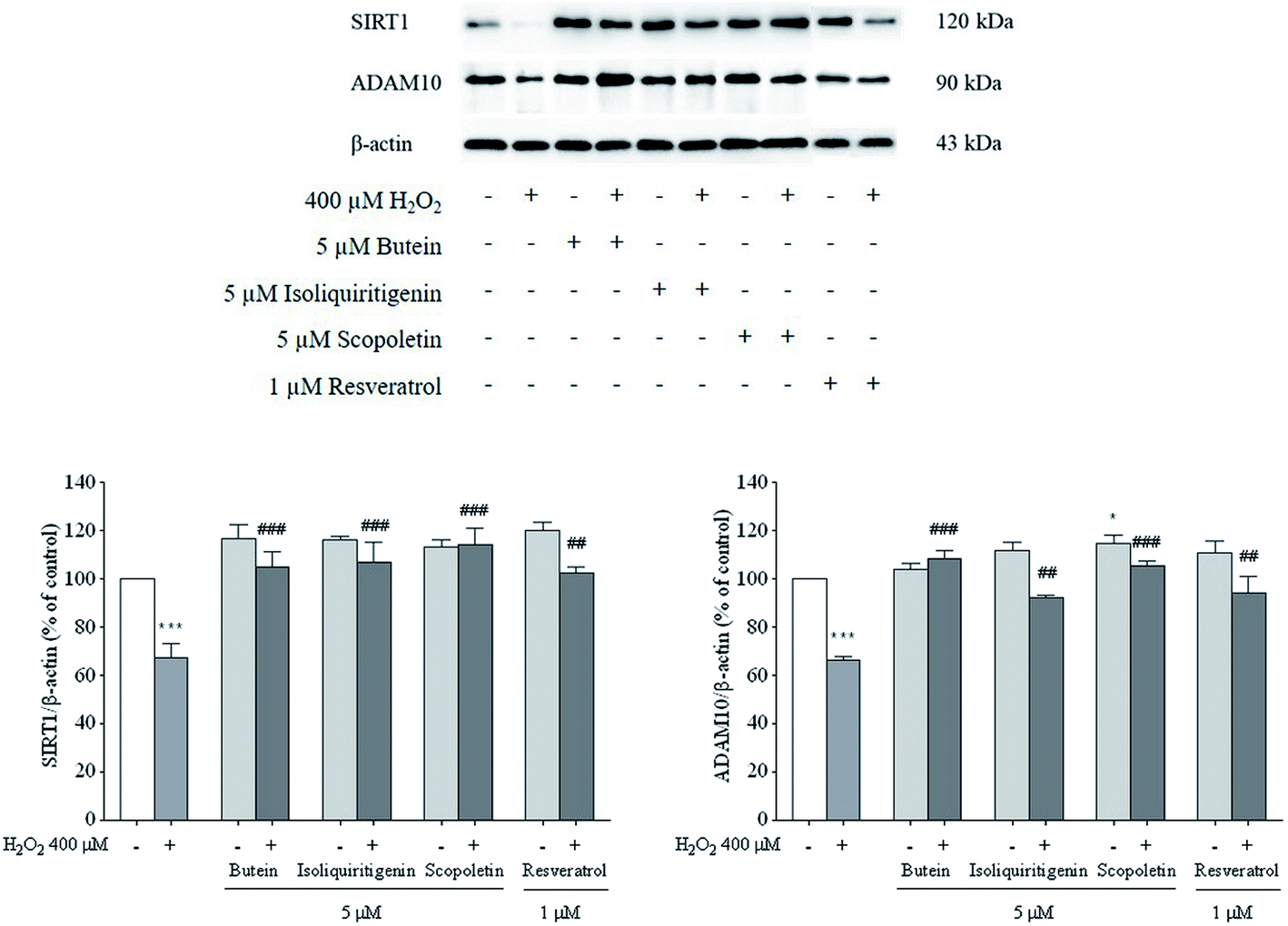 | ||
| Fig. 7 Effects of 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM resveratrol against 400 μM H2O2-induced decreases in SIRT1 and ADAM10 proteins. β-Actin was used as a loading control. The results are expressed as mean ± S.E.M. of three independent experiments (*p < 0.05 and ***p < 0.001 vs. control; ##p < 0.01 and ###p < 0.001 vs. 400 μM H2O2-treated group). | ||
To confirm the effect of these investigated compounds on the nuclear enzyme SIRT1, immunofluorescence analysis was performed. The previous results showed that H2O2 was involved in neuronal cell toxicity demonstrated in the in vitro oxidative stress model. As expected, our results (Fig. 8) showed a similar effect of H2O2 in reducing SIRT1 expression. However, SIRT1 fluorescence signals were recovered when the cells were pretreated with 5 μM of butein, isoliquiritigenin, or scopoletin similar to 1 μM of resveratrol.
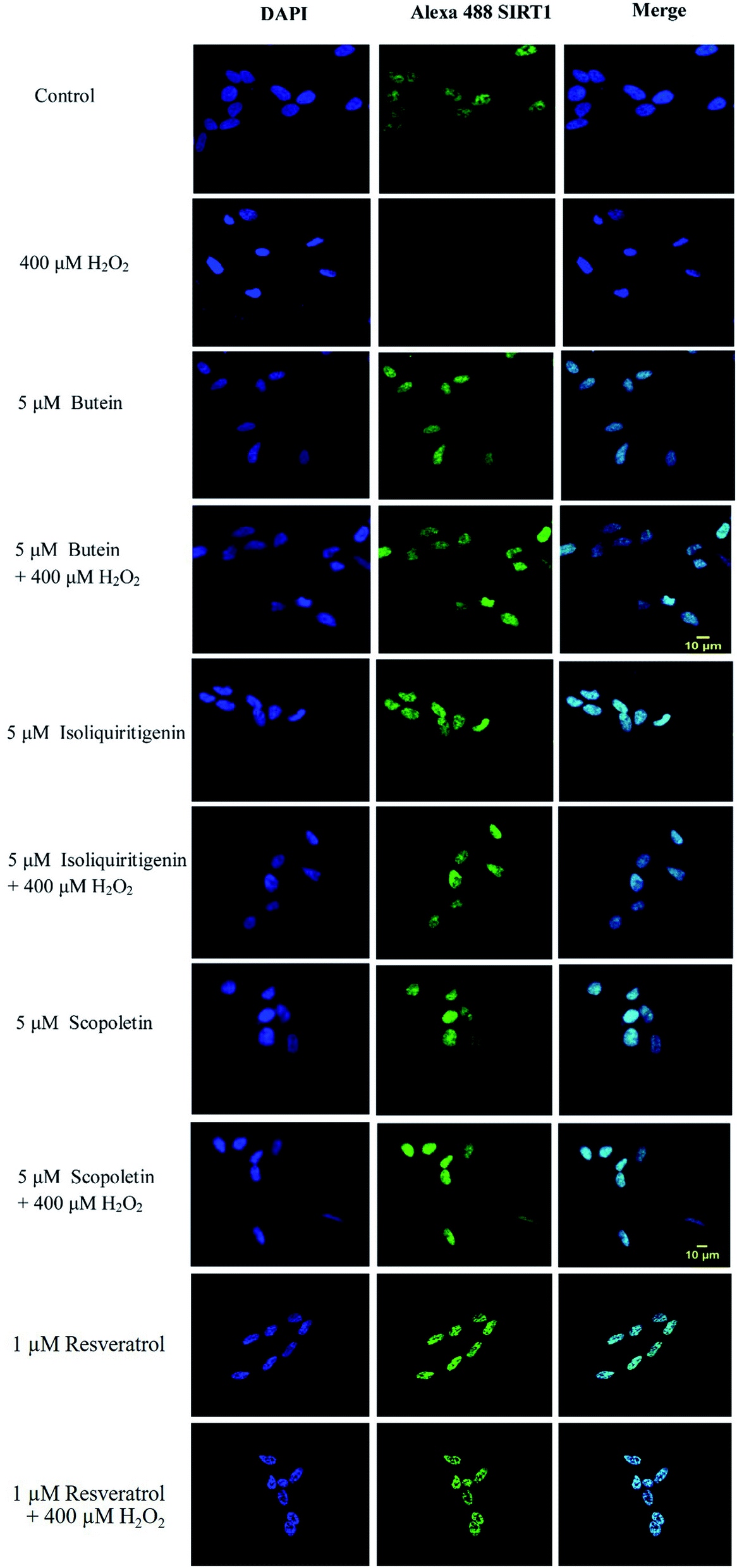 | ||
| Fig. 8 Confocal microscopic images of SH-SY5Y cells demonstrating localization of SIRT1 in butein, isoliquiritigenin, scopoletin, and resveratrol pretreated cells. The cells were incubated with 5 μM of butein, isoliquiritigenin, scopoletin, or 1 μM of resveratrol for 3 h followed by exposure to 400 μM H2O2. The cells were stained with mouse monoclonal anti-SIRT1. Green color indicated SIRT1 immunostaining [using Alexa 488 (green)-conjugated goat anti-rabbit IgG], and blue DAPI counter-staining was used to show nuclei. Scale bar = 10 μm. | ||
3.6. Docking of butein, isoliquiritigenin, and scopoletin to SIRT1 activator binding site
Molecular docking analysis on the SIRT1 structure revealed that butein, isoliquiritigenin, and scopoletin could bind to the activator-binding sites of the enzyme (Fig. 9A–C) with their estimated binding free energy of −7.76, −8.27, and −5.95 kcal mol−1, respectively. Such binding site accommodated the same pocket, in which two of the three resveratrol molecules (Resv1 and Resv2) binded to SIRT1 (Fig. 9D) as observed in the crystal structure.37 With our self-docking, the estimated binding free energies for Resv1 and Resv2 were −7.72 and −7.52 kcal mol−1, respectively. Several residues in the catalytic and N-terminal domains of SIRT1, as well as in the fluorogenic peptide were shown to make several π-type interactions, hydrogen bonds, and van der Waals with the three compounds (Fig. 10). These include the ASN226, GLU230 (on the NTD), ARG446 (on the CD) of SIRT1 and ARG1, LYS3, coumarin ring of the fluorogenic peptide, which also contributed to SIRT1-resveratrol binding (Fig. 9 and 11).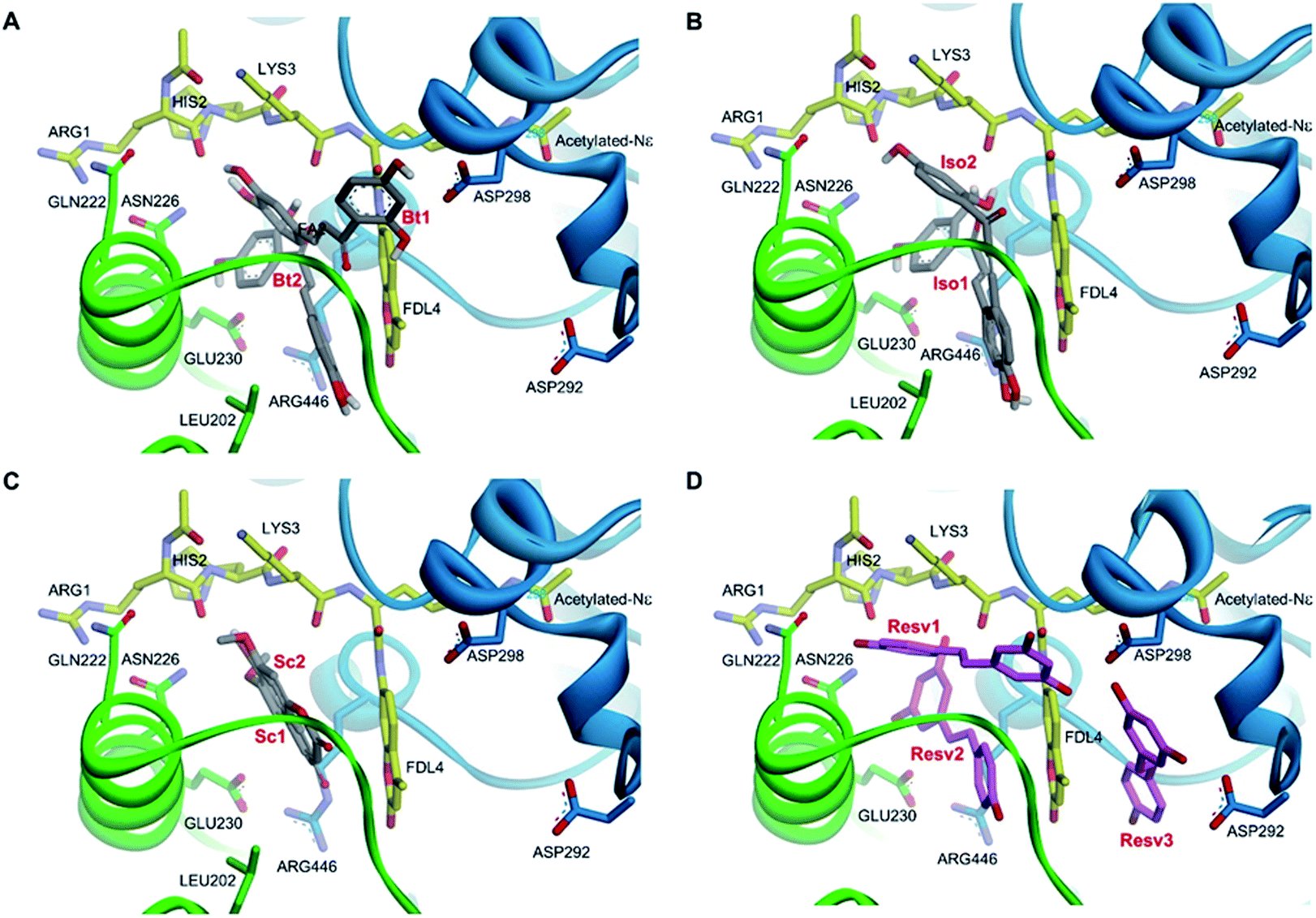 | ||
| Fig. 9 Three-dimensional illustrations of two different binding modes of butein (A), isoliquiritigenin (B), and scopoletin (C), compared to three different binding modes of resveratrol (D) in the SIRT1 activator-binding site (PDB code 5BTR).37 The catalytic and N-terminal domains of SIRT1 are shown in blue and green ribbon representations, respectively. The fluorogenic acetylated peptides (carbon colored yellow), hydrogen bond interacting amino acid residues (carbon colored green and blue), resveratrol (carbon colored magenta), and polyphenolic compounds (carbon colored dark gray) are shown in stick models. (Butein; Bt, isoliquiritigenin; Iso, scopoletin; Sc, resveratrol; Resv.) | ||
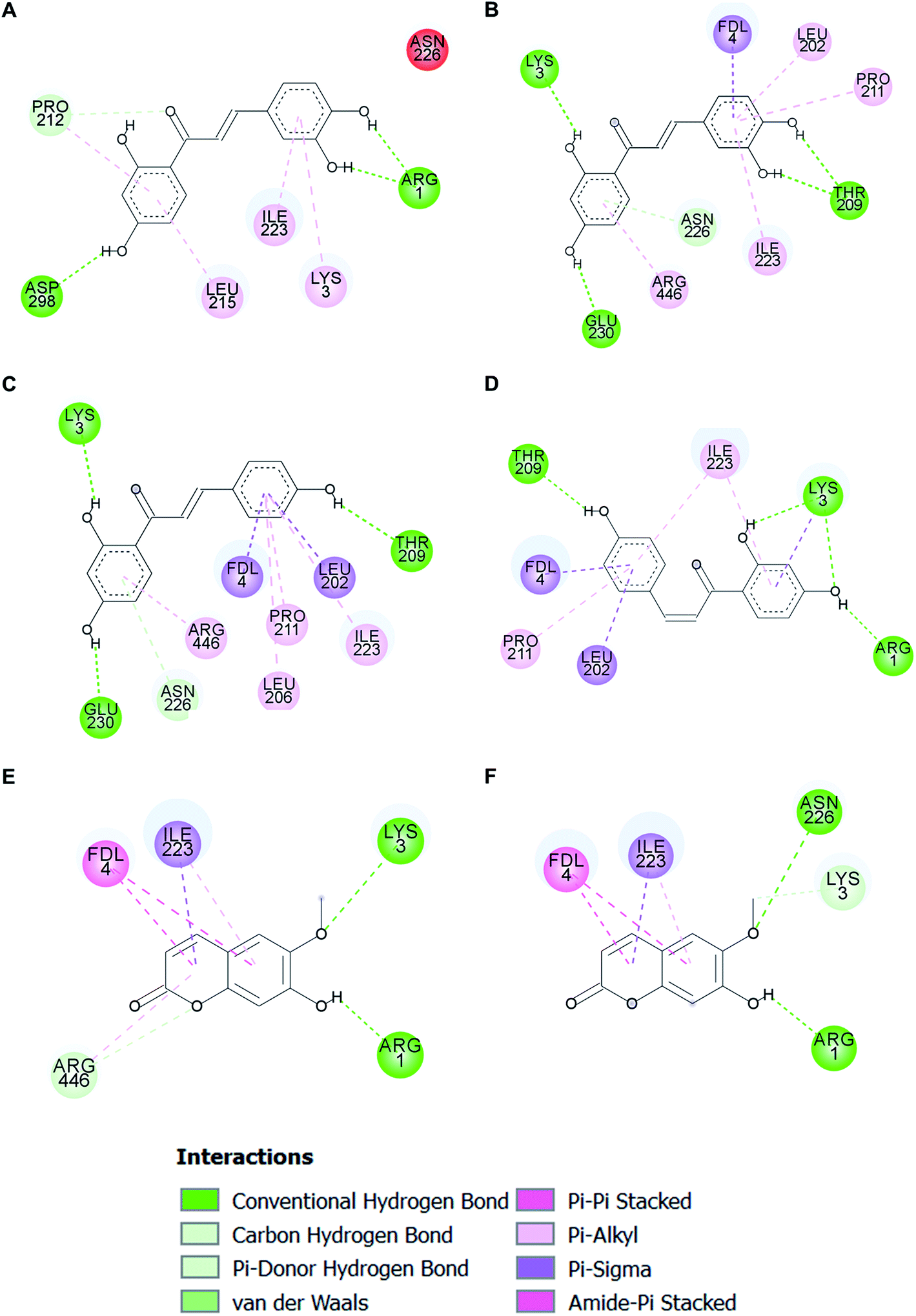 | ||
| Fig. 10 Two-dimensional illustrations of two different binding modes of butein (A and B), isoliquiritigenin (C and D), and scopoletin (E and F) in the SIRT1 activator-binding site. The two-dimensional structures of the compounds are drawn as black lines. Interacting residues are represented as balls that are colored according to the type of interaction. A red ball indicates the residue contributing to an unfavorable interaction. | ||
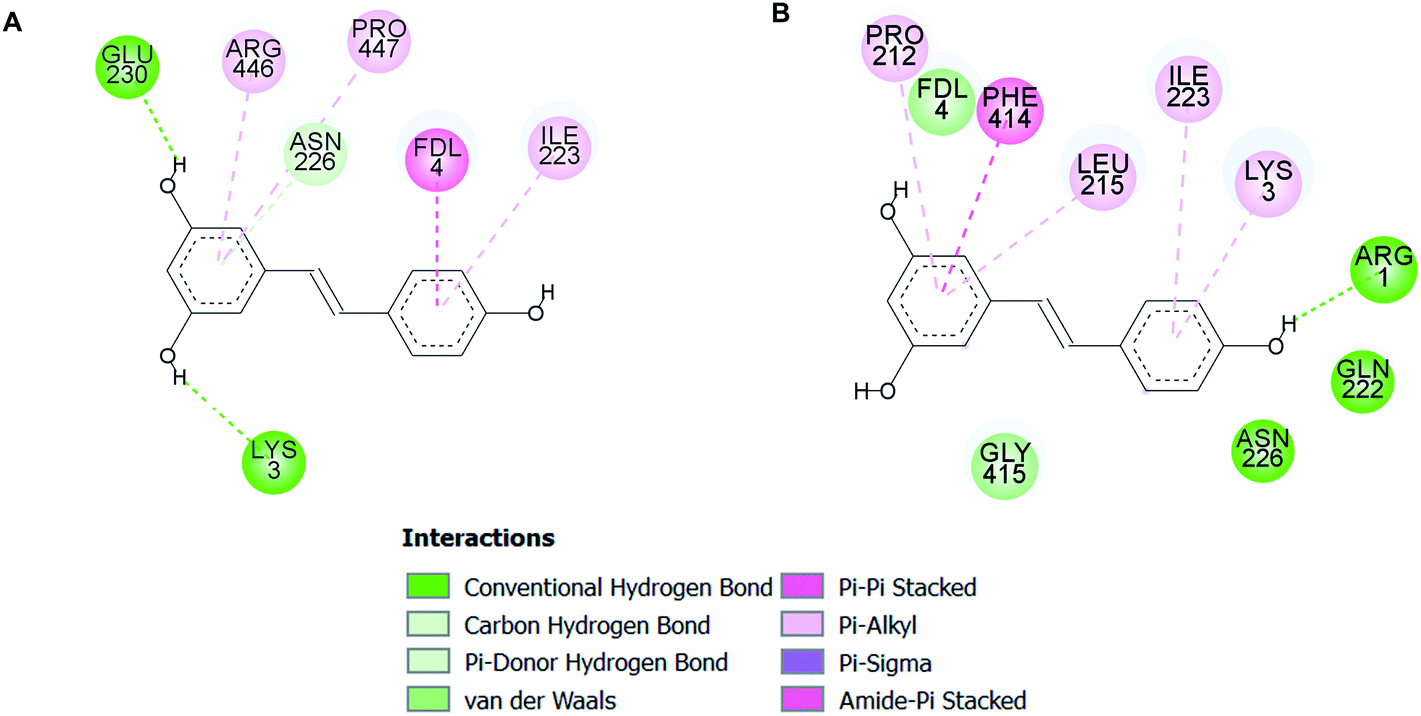 | ||
| Fig. 11 Two-dimensional illustrations of binding mode 1 (A) and mode 2 (B) of resveratrol molecules in the SIRT1 activator-binding site (PDB code 5BTR).37 The two-dimensional structure of the resveratrol is drawn as a black line. Interacting residues are represented as balls that are color-coded according to the type of interaction. | ||
4. Discussion
Oxidative stress plays significant roles in the development of chronic neurological diseases such as PD and AD. It is well known that the central nervous system is one of the targets for oxidative stress-generating free radicals, where they attack proteins resulting in structural changes and loss of enzymatic activity.8 Similarly, the interactions between free radicals and cellular components, including lipids and DNA, amplify the neuronal loss.38,39 Due to the deleterious effects of free radicals, antioxidant defenses for neutralizing and scavenging free radicals are crucial to prevent and cope with advanced neuronal damage.40 This study revealed the neurocytotoxic effects of H2O2-induced protein expression, as well as the protective effects of butein, isoliquiritigenin, and scopoletin on neuronal cells. H2O2 is a mild oxidant that is commonly used to model induction of oxidative stress in neuronal cells. H2O2 toxicity to neurons is mediated through covalent cross-linking of Aβ.41,42 An imbalance between oxidants and antioxidants can lead to physiological dysfunction, neurodegenerative conditions, and neuronal cell death. Therefore, the enhancement of cellular antioxidant defenses can decrease neuronal losses.The potential use of polyphenols to alleviate the burden of oxidative stress is widely disputed.43,44 Polyphenols in human diets are well known for their antioxidant properties. For example, flavonoid is the most important phenolic compound in foods; it possesses high antioxidant capability due to the structure of its phenolic hydroxyl groups. Coumarin (namely scopoletin) was isolated from the herb, S. acmella Murr.26 Two others, butein and isoliquiritigenin, containing abundant bioactive ingredients come from H. formicarum Jack.17 These compounds exert diverse biological activities including anti-inflammatory, antimicrobial, and antioxidant activities.25,29
Our studies showed that some bioactive compounds, including chalcones (butein and isoliquiritigenin) and a coumarin derivative (scopoletin), display neuroprotective activities. Interestingly, strong protective effects of these phenolics (butein, isoliquiritigenin, and scopoletin) in SH-SY5Y cells were noted when the cells were incubated with H2O2. This investigation supports the view that these compounds mitigate the morphological changes induced in cells by H2O2 as documented in the microscopy (Fig. 3). The intracellular ROS accumulation resulted from H2O2 exposure was also documented with the DCFDA assay. It was found that pretreatment of the cells with butein, isoliquiritigenin, and scopoletin significantly reduced the ROS levels. Moreover, the effects of butein, isoliquiritigenin, and scopoletin on reducing apoptosis were elucidated using flow cytometric analysis. The results confirmed that these phenolics ultimately protected against cellular apoptosis induced by H2O2. Specifically, pretreatment of the cells with all three phenolics showed lower apoptotic cell numbers, but this decline was greater with butein than with isoliquiritigenin and scopoletin as shown in Fig. 5. Thus, the better protective effects of butein against H2O2-induced neuronal cell death appeared to be caused by effects on both ROS generation and apoptosis.
Since the family of mammalian forkhead box O (FoxO) transcription factors is an important regulator of cellular responses to stress stimuli and promote the cellular antioxidant activity, FoxOs activation upon a series of target genes involved in the cellular stress responses can lead to induction of manganese-containing superoxide dismutase (SOD2)45 and catalase46,47 that control detoxification of ROS. SOD2 catalyzes dismutation of the oxygen reduction product, O2˙−, to generate oxygen and neurodamaging agent H2O2. H2O2, a stable form of ROS, is further decomposed to water and oxygen in a reaction accelerated by catalase whose generation was regulated by FoxO3a.48,49 The present study revealed that butein, isoliquiritigenin, and scopoletin effectively prevented H2O2-induced neuronal cell death by restoring the expression of the antioxidant enzymes (SOD2 and catalase). In addition to their antioxidant activities, polyphenols have been coupled with the increased expression of FoxO3a protein, a key molecule for ROS detoxification (Fig. 6), assisting oxidative stress resistance. The results from this studies match the data from in vivo studies of cognitive enhancing effects50 and hold promising implications for butein, isoliquiritigenin, and scopoletin. Consequently, the butein, isoliquiritigenin, and scopoletin pretreatments also activated the anti-apoptotic protein BCL-2, a critical protein for survival of neurons (Fig. 6). Thus, the neuroprotection resulted from the decrease of oxidative stress and attenuation of apoptosis.
SIRT1, the human sirtuin protein family, is a deacetylase protein which removes acetyl groups from many histone and non-histone proteins. A variety of substrates are deacetylated by SIRT1. Thus, SIRT1 is linked to numerous protein players to provide plausible mechanisms for control of gene expression, metabolism, and aging.51 Interestingly, an increased ROS level as well as other stressful stimuli that elicit the formation of ROS, may modulate FoxO transcriptional activity and subcellular localization of FOXO transcription factors via phosphorylation, acetylation, methylation, and ubiquitination.52 Under conditions of oxidative stress, FoxO3a has been shown to translocate from the cytosol into the nucleus, where it forms a protein complex with the SIRT1 that contributes to deacetylation of FoxO3. By deacetylating FOXO transcription factors, SIRT1 might mediate FOXO-dependent responses toward stress resistance because of the differential effects of SIRT1 on FoxO3 function in potentiating its level on cell cycle arrest, DNA repair, and genes coding for antioxidant enzymes such as SOD2 and catalase.46,53 Furthermore, a role of SIRT1 in the modulation of neurodegeneration by extending the survival of neuronal cells has been reported.54,55
Plant polyphenols including resveratrol have been reported to activate SIRT1.56,57 We assessed the binding of SIRT1 with the investigated compounds (butein, isoliquiritigenin, and scopoletin) using a molecular docking method. The crystal structure of the SIRT1 complex, containing the N-terminal domain with the fluorophore-labelled peptide and resveratrol, was used as a search model. Our docking results showed that the compounds were able to bind to the activator-binding sites of SIRT1, mediating the association between the fluorophore-attached peptide and the N-terminal domain of SIRT1. Recent crystallographic and biochemical studies suggest that two resveratrol molecules (Resv1 and Resv2) accommodate such binding sites and result in lowering the Km value of the peptide to SIRT1.37 The ARG446 residue positioned in the catalytic domain, GLU230 and ASN226 situated in the N-terminal domain of SIRT1, and LYS3, ARG1, and the coumarin moiety located on the fluorogenic peptide were revealed to associate with Resv1 and Resv2.37 These residues were also shown to interact with our three phenolic compounds (Fig. 9). Since evidence suggested that the deacetylation rate of SIRT1 with either GLU230 or ASN226 mutations was reduced when stimulated with resveratrol.37 Thus, the stimulatory effects of butein, isoliquiritigenin, and scopoletin on SIRT1 activity, which come about by promoting tight binding between the peptide substrate and enzyme, need to be further investigated.
Our data demonstrated that SIRT1 expression was increased by pretreatment with the tested compounds. Furthermore, following butein, isoliquiritigenin, and scopoletin pretreatment, SIRT1 protein was observed to have a similar trend in expression levels as those of FoxO3a, SOD2, and catalase proteins (Fig. 7). However, the protein expression levels differed among the tested compounds at the concentrations assessed. Therefore, the SIRT1/FOXO axis has been known as evolutionarily well conserved survival pathway that could be involved in regulation of cellular responses to both metabolic changes and inhibition of oxidative stress.
Proteolytic processing of APP to form Aβ peptides are contributed by enzymes known as secretases. ADAM10, a disintegrin and metalloprotease (ADAM) family of protease was identified as the main α secretase for APP. The ADAM family is also involved in the processing of several proteins substrates such as tumor necrosis factor (TNF) α, Notch, Delta, and others.58 In the non-amyloidogenic pathway, APP is consecutively cleaved by α-secretase to generate secreted amyloid precursor protein α (sAPPα), and clevages of APP by α- and γ-secretases produce sAPPα, p3, and APP intracellular domain (AICD).59,60 All of which contribute to constitutive sAPPα production, a protein that elicits neuroprotective properties.61,62 This is considered to be an important mechanism preventing the generation of Aβ. Mechanisms governing ADAM10 activation are still elusive, and studies implicate that proprotein convertase PC7 is a mandatory for endoproteolytic activation of ADAM10.63 Furthermore, SIRT1 deacetylase enhances the transcription of ADAM10 by deacetylating and activating the retinoic acid receptor β.64 A study demonstrated that oleuropein, a polyphenol and constituent of olive, leads to markedly elevated levels of sAPPα and to significant reduction of Aβ oligomers in HEK293 cells stably transfected with the isoform 695 of human AbPP (APP695).65 Moreover, either overexpression of ADAM10 or SIRT1-coupled ADAM10 was linked to both a reduction in Aβ production and full length APP cleavage.66 This could reflect the insight into the use of SIRT1-ADAM10 as a target for AD, and this is considered to use bioactive compounds that promote ADAM10 activity in modulation of Aβ shedding. Notably, butein, isoliquiritigenin, and scopoletin maintain SIRT1 and ADAM10 levels as shown in Fig. 7. Under oxidative damage, the possible mechanism of these compounds in the attenuation of neuronal cell death is revealed in Fig. 12.
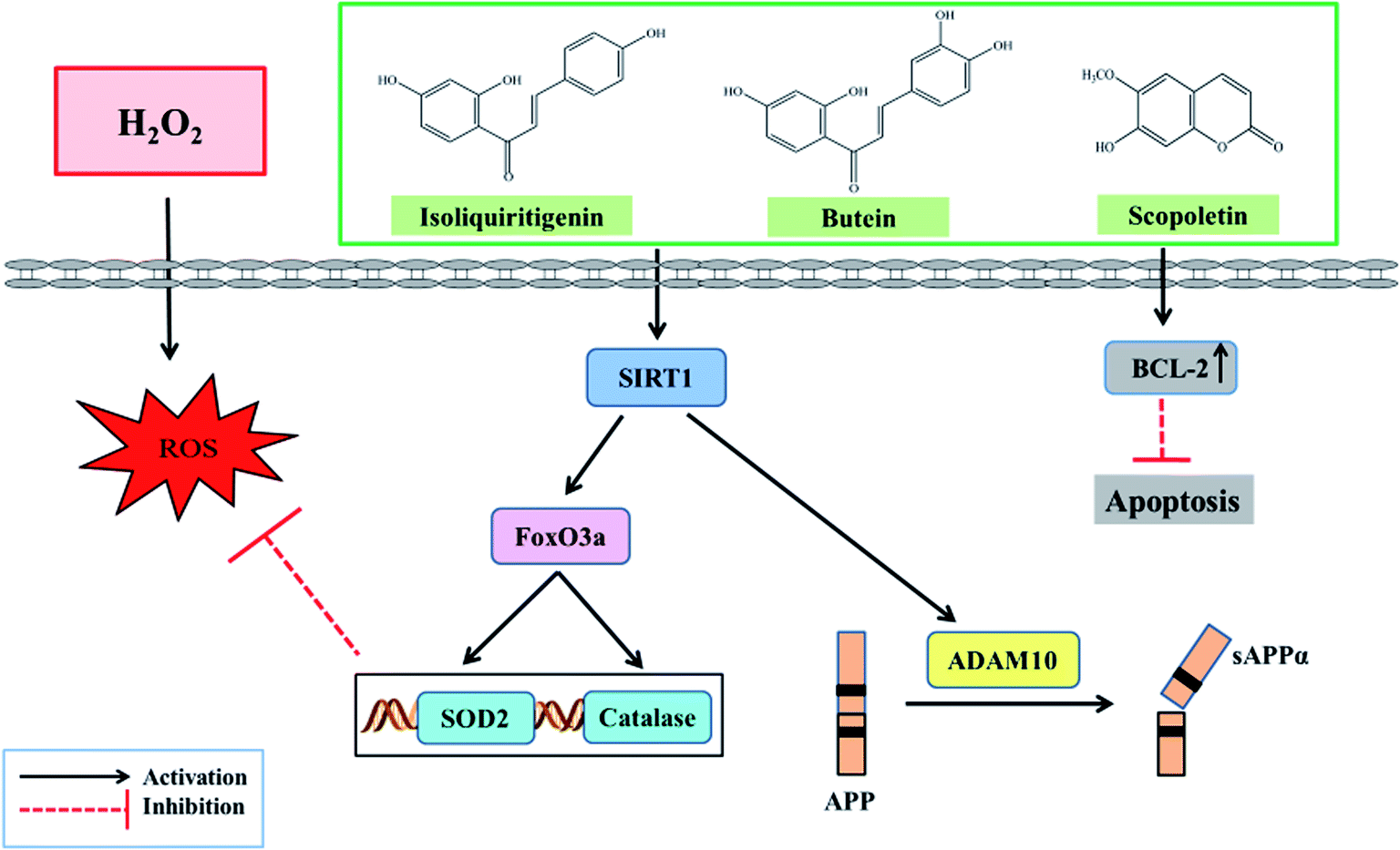 | ||
| Fig. 12 Possible neuroprotective effects of butein, isoliquiritigenin, and scopoletin on neuronal cells. Butein, isoliquiritigenin, and scopoletin attenuated H2O2-induced oxidative stress by decreasing ROS, balancing the antioxidant system, upregulating the SIRT1-FoxO3a-ADAM10 signaling pathway, and preventing cell apoptosis. | ||
5. Conclusion
This study revealed that butein, isoliquiritigenin, and scopoletin play crucial roles in neuroprotection by maintaining the antioxidant status. This may critically support neuronal cell survival in the face of H2O2-induced neurotoxicity. Such action may prevent AD via reducing toxic Aβ shedding, although it remains unclear exactly how these compounds ameliorate the neurotoxicity. The findings suggest that these polyphenolic compounds are potential candidates for prevention and/or treatment of neurodegeneration in the future.Abbreviations
| Aβ | β-Amyloid |
| AD | Alzheimer's disease |
| ADAM | A disintegrin and metalloproteinases |
| ADAM10 | A disintegrin and metalloproteinases 10 |
| AICD | APP intracellular domain |
| APP | Amyloid precursor protein |
| ARG446 | Arginine |
| ARG1 | Arginine |
| ASN226 | Asparagine |
| ATCC | American type culture collection |
| BCL-2 | B-cell lymphoma 2 |
| BSA | Bovine serum albumin |
| CD | Catalytic domain |
| DCF | Dichlorofluorescein |
| DCFDA | 2′,7′-Dichlorofluorescin diacetate |
| ECL | Enhanced chemiluminescence |
| FOXO | Forkhead box-O transcription factor |
| FoxO3a | Forkhead box O3a |
| GLU230 | Glutamic acid |
| H2O2 | Hydrogen peroxide |
| LYS3 | Lysine |
| MTT | 3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide |
| NTD | N-terminal domain |
| p3 | Short peptide containing the C-terminal region of Aβ |
| PD | Parkinson's disease |
| ROS | Reactive oxygen species |
| RPM | Revolutions per minute |
| sAPPα | Soluble APPα |
| PBS | Phosphate buffered saline |
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This project is supported by Mahidol University and the Office of the Higher Education Commission, Mahidol University under the National Research Universities Initiative.References
- D. J. Betteridge, Metabolism, 2000, 49, 3–8 CrossRef CAS.
- M. Ramalingam and S. J. Kim, J. Neural Transm., 2012, 119, 891–910 CrossRef CAS.
- S. Fahn and G. Cohen, Ann. Neurol., 1992, 32, 804–812 CrossRef CAS.
- D. J. Tate Jr, M. V. Miceli and D. A. Newsome, Invest. Ophthalmol. Visual Sci., 1995, 36, 1271–1279 CAS.
- B. Chetsawang, C. Putthaprasart, P. Phansuwan-Pujito and P. Govitrapong, J. Pineal Res., 2006, 41, 116–123 CrossRef CAS.
- J. H. Jeong, M. Y. Noh, J. H. Choi, H. Lee and S. H. Kim, Exp. Ther. Med., 2016, 11, 1201–1210 CrossRef CAS.
- J. A. Klein and S. L. Ackerman, J. Clin. Invest., 2003, 111, 785–793 CrossRef CAS.
- B. Uttara, A. V. Singh, P. Zamboni and R. T. Mahajan, Curr. Neuropharmacol., 2009, 7, 65–74 CrossRef CAS.
- C. K. Glass, K. Saijo, B. Winner, M. C. Marchetto and F. H. Gage, Cell, 2010, 140, 918–934 CrossRef CAS.
- M. Sano, C. Ernesto, R. G. Thomas, M. R. Klauber, K. Schafer, M. Grundman, P. Woodbury, J. Growdon, C. W. Cotman, E. Pfeiffer, L. S. Schneider and L. J. Thal, N. Engl. J. Med., 1997, 336, 1216–1222 CrossRef CAS.
- S. G. Yang, W. Y. Wang, T. J. Ling, Y. Feng, X. T. Du, X. Zhang, X. X. Sun, M. Zhao, D. Xue, Y. Yang and R. T. Liu, Neurochem. Int., 2010, 57, 914–922 CrossRef CAS.
- M. Caruana and N. Vassallo, Adv. Exp. Med. Biol., 2015, 863, 117–137 CrossRef CAS.
- A. Ataie, M. Shadifar and R. Ataee, Basic Clin. Neurosci., 2016, 7, 81–90 CAS.
- D. O. Kennedy, Adv. Nutr., 2014, 5, 515–533 CrossRef CAS.
- S. Quideau, ChemBioChem, 2004, 5, 427–430 CrossRef CAS.
- C. Ramassamy, Eur. J. Pharmacol., 2006, 545, 51–64 CrossRef CAS.
- S. Prachayasittikul, P. Buraparuangsang, A. Worachartcheewan, C. Isarankura-Na-Ayudhya, S. Ruchirawat and V. Prachayasittikul, Molecules, 2008, 13, 904–921 CrossRef CAS.
- S. Prachayasittikul, R. Pingaew, V. Yamkamon, A. Worachartcheewan, S. Wanwimolruk, S. Ruchirawat and V. Prachayasittikul, Int. J. Pharmacol., 2012, 8, 440–444 CAS.
- B. B. Chavan, A. S. Gadekar, P. P. Mehta, P. K. Vawhal, A. K. Kolsure and A. R. Chabukswar, Asian J. Biomed. Pharm. Sci., 2016, 6, 01–07 CrossRef CAS.
- R. R. Ternavisk, A. J. Camargo, F. B. Machado, J. A. Rocco, G. L. Aquino, V. H. Silva and H. B. Napolitano, Journal of molecular modeling, 2014, 20, 2526 CrossRef.
- J. Y. Kim, S. J. Park, K. J. Yun, Y. W. Cho, H. J. Park and K. T. Lee, Eur. J. Pharmacol., 2008, 584, 175–184 CrossRef CAS.
- H. Nam and M. M. Kim, Phytomedicine, 2015, 22, 362–368 CrossRef CAS.
- V. Prachayasittikul, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, EXCLI J., 2013, 12, 291–312 Search PubMed.
- J. Sung and J. Lee, J. Med. Food, 2015, 18, 557–564 CrossRef CAS.
- N. Cho, K. Y. Lee, J. Huh, J. H. Choi, H. Yang, E. J. Jeong, H. P. Kim and S. H. Sung, Food Chem. Toxicol., 2013, 58, 355–361 CrossRef CAS.
- S. Prachayasittikul, S. Suphapong, A. Worachartcheewan, R. Lawung, S. Ruchirawat and V. Prachayasittikul, Molecules, 2009, 14, 850–867 CrossRef CAS.
- O. Wongsawatkul, S. Prachayasittikul, C. Isarankura-Na-Ayudhya, J. Satayavivad, S. Ruchirawat and V. Prachayasittikul, Int. J. Mol. Sci., 2008, 9, 2724–2744 CrossRef CAS.
- F. Sersen and M. Lácová, Acta Fac. Pharm. Univ. Comenianae, 2015, 62, 41–45 CAS.
- R. Mogana, K. Teng-Jin and C. Wiart, J. Evidence-Based Complementary Altern. Med., 2013, 2013, 734824 CAS.
- M. C. Carpinella, C. G. Ferrayoli and S. M. Palacios, J. Agric. Food Chem., 2005, 53, 2922–2927 CrossRef CAS.
- E. J. Seo, M. Saeed, B. Y. Law, A. G. Wu, O. Kadioglu, H. J. Greten and T. Efferth, Molecules, 2016, 21, 496 CrossRef.
- H. C. Lin, S. H. Tsai, C. S. Chen, Y. C. Chang, C. M. Lee, Z. Y. Lai and C. M. Lin, Biochem. Pharmacol., 2008, 75, 1416–1425 CrossRef CAS.
- B. S. Yun, I. K. Lee, I. J. Ryoo and I. D. Yoo, J. Nat. Prod., 2001, 64, 1238–1240 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- H. R. Lee, H. K. Shin, S. Y. Park, H. Y. Kim, W. S. Lee, B. Y. Rhim, K. W. Hong and C. D. Kim, J. Neurosci. Res., 2014, 92, 1581–1590 CrossRef CAS.
- D. Cao, M. Wang, X. Qiu, D. Liu, H. Jiang, N. Yang and R. M. Xu, Genes Dev., 2015, 29, 1316–1325 CrossRef CAS.
- L. A. Pham-Huy, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2008, 4, 89–96 CAS.
- K. Rahman, Clin. Interventions Aging, 2007, 2, 219–236 CAS.
- C. Guo, L. Sun, X. Chen and D. Zhang, Neural Regener. Res., 2013, 8, 2003–2014 CAS.
- M. Guglielmotto, L. Giliberto, E. Tamagno and M. Tabaton, Front. Aging Neurosci., 2010, 2, 3 CAS.
- W. Hu, G. Wang, P. Li, Y. Wang, C.-L. Si, J. He, W. Long, Y. Bai, Z. Feng and X. Wang, Chem.-Biol. Interact., 2014, 224, 108–116 CrossRef CAS.
- A. Thapa and E. Y. Chi, Adv. Exp. Med. Biol., 2015, 863, 55–77 CrossRef CAS.
- S. Yu, X. Wang, X. He, Y. Wang, S. Gao, L. Ren and Y. Shi, Cell Stress Chaperones, 2016, 21, 697–705 CrossRef CAS.
- G. J. Kops, T. B. Dansen, P. E. Polderman, I. Saarloos, K. W. Wirtz, P. J. Coffer, T. T. Huang, J. L. Bos, R. H. Medema and B. M. Burgering, Nature, 2002, 419, 316–321 CrossRef CAS.
- A. Brunet, L. B. Sweeney, J. F. Sturgill, K. F. Chua, P. L. Greer, Y. Lin, H. Tran, S. E. Ross, R. Mostoslavsky, H. Y. Cohen, L. S. Hu, H. L. Cheng, M. P. Jedrychowski, S. P. Gygi, D. A. Sinclair, F. W. Alt and M. E. Greenberg, Science, 2004, 303, 2011–2015 CrossRef CAS.
- W. Q. Tan, K. Wang, D. Y. Lv and P. F. Li, J. Biol. Chem., 2008, 283, 29730–29739 CrossRef CAS.
- L. O. Klotz, C. Sanchez-Ramos, I. Prieto-Arroyo, P. Urbanek, H. Steinbrenner and M. Monsalve, Redox Biol., 2015, 6, 51–72 CrossRef CAS.
- S. Nemoto and T. Finkel, Science, 2002, 295, 2450–2452 CrossRef CAS.
- N. Cho, J. H. Choi, H. Yang, E. J. Jeong, K. Y. Lee, Y. C. Kim and S. H. Sung, Food Chem. Toxicol., 2012, 50, 1940–1945 CrossRef CAS.
- S. Rahman and R. Islam, Cell Commun. Signaling, 2011, 9, 11 CrossRef CAS.
- H. Daitoku, J. Sakamaki and A. Fukamizu, Biochim. Biophys. Acta, 2011, 1813, 1954–1960 CrossRef CAS.
- R. R. Alcendor, S. Gao, P. Zhai, D. Zablocki, E. Holle, X. Yu, B. Tian, T. Wagner, S. F. Vatner and J. Sadoshima, Circ. Res., 2007, 100, 1512–1521 CrossRef CAS.
- B. P. Hubbard and D. A. Sinclair, Trends Pharmacol. Sci., 2014, 35, 146–154 CrossRef CAS.
- D. Kim, M. D. Nguyen, M. M. Dobbin, A. Fischer, F. Sananbenesi, J. T. Rodgers, I. Delalle, J. A. Baur, G. Sui, S. M. Armour, P. Puigserver, D. A. Sinclair and L. H. Tsai, EMBO J., 2007, 26, 3169–3179 CrossRef CAS.
- J. S. Allard, E. Perez, S. Zou and R. de Cabo, Mol. Cell. Endocrinol., 2009, 299, 58–63 CrossRef CAS.
- K. T. Howitz, K. J. Bitterman, H. Y. Cohen, D. W. Lamming, S. Lavu, J. G. Wood, R. E. Zipkin, P. Chung, A. Kisielewski, L. L. Zhang, B. Scherer and D. A. Sinclair, Nature, 2003, 425, 191–196 CrossRef CAS.
- W. Annaert and B. De Strooper, Annu. Rev. Cell Dev. Biol., 2002, 18, 25–51 CrossRef CAS.
- A. E. Moneim, Curr. Alzheimer Res., 2015, 12, 335–349 CrossRef.
- G. P. Morris, I. A. Clark and B. Vissel, Acta Neuropathol. Commun., 2014, 2, 135 Search PubMed.
- E. Lopez-Perez, Y. Zhang, S. J. Frank, J. Creemers, N. Seidah and F. Checler, J. Neurochem., 2001, 76, 1532–1539 CrossRef CAS.
- D. F. Obregon, K. Rezai-Zadeh, Y. Bai, N. Sun, H. Hou, J. Ehrhart, J. Zeng, T. Mori, G. W. Arendash, D. Shytle, T. Town and J. Tan, J. Biol. Chem., 2006, 281, 16419–16427 CrossRef CAS.
- A. Anders, S. Gilbert, W. Garten, R. Postina and F. Fahrenholz, FASEB J., 2001, 15, 1837–1839 CrossRef CAS.
- S. W. Min, P. D. Sohn, S. H. Cho, R. A. Swanson and L. Gan, Front. Aging Neurosci., 2013, 5, 53 Search PubMed.
- M. Kostomoiri, A. Fragkouli, M. Sagnou, L. A. Skaltsounis, M. Pelecanou, E. C. Tsilibary and A. K. Tzinia, Cell. Mol. Neurobiol., 2013, 33, 147–154 CrossRef CAS.
- H. R. Lee, H. K. Shin, S. Y. Park, H. Y. Kim, W. S. Lee, B. Y. Rhim, K. W. Hong and C. D. Kim, J. Neurosci. Res., 2014, 92, 1581–1590 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |