
Stay positive: catalysis with 1,3,2-diazaphospholenes
John H.
Reed
,
Johannes
Klett
,
Craig
Steven
 and
Nicolai
Cramer
*
and
Nicolai
Cramer
*
Laboratory of Asymmetric Synthesis and Catalysis, Institute of Chemical Sciences and Chemical Engineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: nicolai.cramer@epfl.ch
First published on 3rd September 2020
Abstract
P-Hydrido 1,3,2-diazaphospholenes (DAPs) are a class of nucleophilic molecular hydrides capable of addition to a variety of substrates. They can be turned over with terminal reductants and emerged over the last few years as powerful and versatile catalysts. This review highlights developments made in understanding the chemistry of these unusual compounds and applying them in catalysis. An overview of the synthesis and important structural features of the DAPs is followed by a discussion of methodologies that exploit their chemistry in catalysis, including the rise of enantioselective reductions. We conclude with a brief analysis on the current limitations of the field and discuss how these might be overcome and which directions the field might take in the future.
Introduction
Phosphorus-containing compounds have enjoyed a privileged position in catalytic systems for organic synthesis. Their coordination chemistry has seen them extensively applied as ligands for transition-metal catalysts.1 Furthermore, they have also been used as catalysts in their own right: nucleophilic phosphines can induce C–C bond forming events through Morita–Baylis–Hillman and Rauhut–Currier type reactions;2 phosphoric acids promote a variety of polar and pericyclic reactions;3 while phosphonium salts have been widely applied as phase-transfer catalysts.4 1,3,2-Diazaphospholenes (DAPs) offer a reactivity profile orthogonal to these systems, as such they open up a completely new axis for phosphorus catalysis. These 5-membered heterocycles contain a phosphorus atom, two nitrogen atoms and a carbon–carbon double bond. They have emerged over the last decade as powerful catalysts in organic synthesis due to the unique reactivity of P-hydrido substituted members of this family.5Phosphorus-containing compounds with one or more P–H bonds are generally characterized by a partial negative charge on the phosphorous atom, and therefore a protic reactivity of the hydrogen.6 The electronegativities of phosphorus and hydrogen (χR(P) = 2.19, χR(H) = 2.20) are very closely matched. Consequently an Umpolung of the canonical reactivity is feasible.7 Indeed, the P-hydrido variants of 1,3,2-diazaphospholenes (DAPs), in which the electronic environment of the phosphorous is modulated by the surrounding atoms, exhibit a reversed polarity resulting in hydridic behavior of the P–H bond (Fig. 1).8 DAPs were first reported in the 1980s by the groups of Schmidpeter, Pudovik, and Hargis,9 but received little attention in the following decades. Gudat initiated their renaissance by investigating the structural and reactive properties of the P-hydrido substituted DAPs. DAPs exhibit markedly different properties to the compounds they most closely resemble, the saturated N-heterocyclic phosphines (NHPs), in that significant ionization of the exocyclic P–X bond is observed, even for substituents with relatively low electronegativity.10 Crucial to harnessing this unusual property was a better understanding of the electronic factors that contribute to it. To this end, the interactions between the π-electrons of the 5-membered ring (the two lone pairs on the nitrogen atoms, and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond) and the σ*-orbital of the exocyclic P–X bond were investigated both experimentally and computationally.11 The abnormally high degree of P–X bond polarization that results is the basis for the hydridic reactivity observed in DAPs.
C bond) and the σ*-orbital of the exocyclic P–X bond were investigated both experimentally and computationally.11 The abnormally high degree of P–X bond polarization that results is the basis for the hydridic reactivity observed in DAPs.
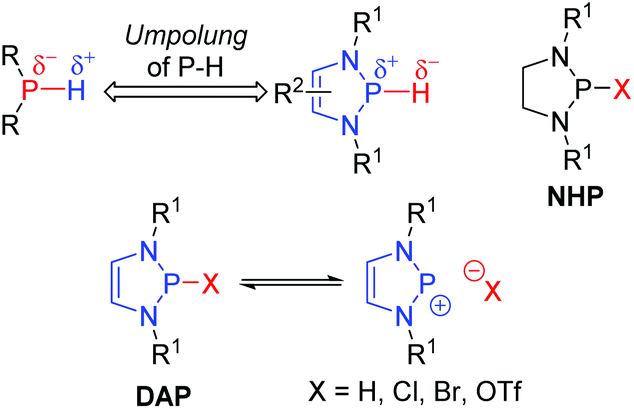 | ||
| Fig. 1 1,3,2-Diazaphospholenes exhibit an Umpolung of the P–H bond. | ||
The following review highlights the substantial progress made in understanding the chemistry of DAPs and harnessing these properties to develop synthetically useful catalytic transformations using their P-hydrido variants. We begin by discussing the synthesis of DAPs and their structural and electronic features. An overview of the catalytic applications that have since been developed is then presented. To date, both achiral and enantioselective transformations have been reported. Finally, an opinion on the future directions that should be taken within this research field is offered.
Synthesis and structural properties
The first reported syntheses of P-hydrido DAPs utilized diimines as convenient precursors (Scheme 1).8 Reduction of the diimines with lithium afforded the corresponding dilithiated ene-diamides, which upon protonation with trimethylamine hydrochloride tautomerized to the α-amino imines. These reacted with PCl3 through a double nucleophilic substitution at the phosphorus atom to give the P-chloro DAPs. Treatment of these with LiBEt3H or LiAlH4 then afforded the desired P-hydrido DAPs. Subsequent reports have detailed more streamlined protocols to access them via an aza-McCormack reaction between diimines and PBr3.12 In this case, the PBr3 undergoes a cheleotropic [4 + 2]-cycloaddition with the diimine resulting in a pentavalent phosphorus intermediate that spontaneously reductively eliminates Br2 to give the P-bromo DAP. Spectroscopic analysis of the P-hydrido DAPs provided immediate insights into their constitution and reactivity: the frequency of the P–H stretching vibration undergoes a red shift when compared to the saturated analogues (1: v = 2176 cm−1, 2: v = 2340 cm−1). Such an effect is consistent with a lengthening of the P–H bond and concomitant weakening thereof, an effect that was quantified after crystallographic determination of the solid state structure of 3. The heterocycle adopts an envelope conformation where the carbon and nitrogen atoms are all co-planar, while the phosphorus atom lies out of this plane. The hydrogen attached to the phosphorus adopts a flagpole position. Computational studies suggested a delocalization of the 6 π-electrons (two nitrogen lone pairs and the CC bond) through the σ*-orbital of the P–H bond. The increased electron density in the σ*-orbital weakens the P–H bond and thereby increases the charge density on the hydrogen, thus generating a hydridic species.
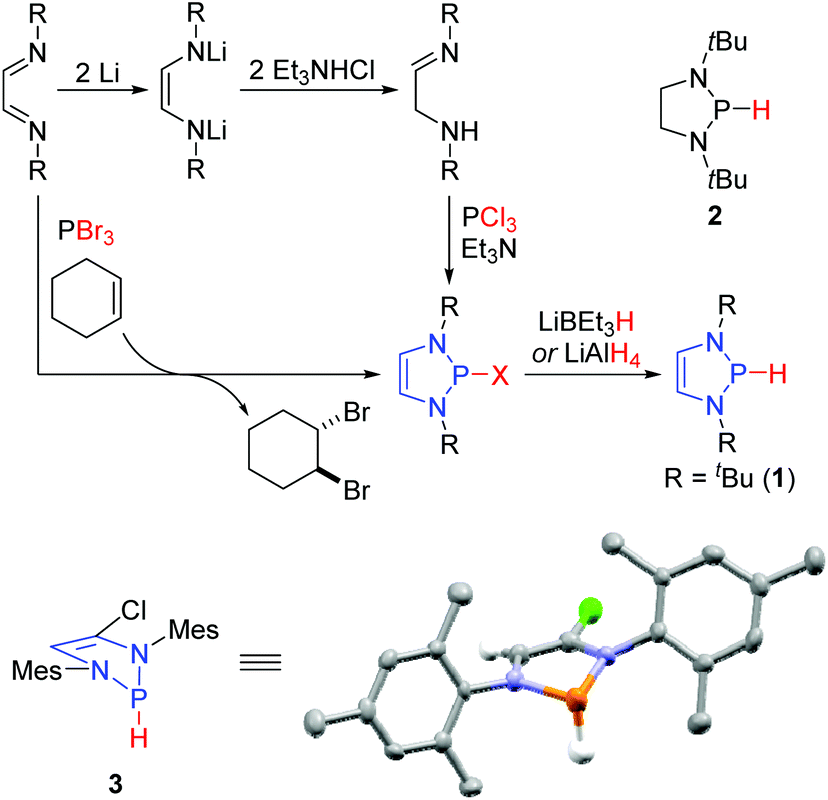 | ||
| Scheme 1 Routes to access DAPs using diimines. | ||
The hydridic nature of the DAPs was confirmed by a series of reactions with acids and electrophiles (Scheme 2).8 Compound 3 reacts with triflic acid to evolve hydrogen gas and form a diazaphosphenium triflate. Furthermore, compound 3 readily participates in hydride transfer reactions. It traps the triphenylcarbenium ion to give triphenylmethane, and it was also shown to reduce benzaldehyde to 4. Follow-up studies revealed that DAPs exhibit total 1,4-selectivity in the reduction of α,β-unsaturated carbonyl compounds such as cinnamaldehyde.13
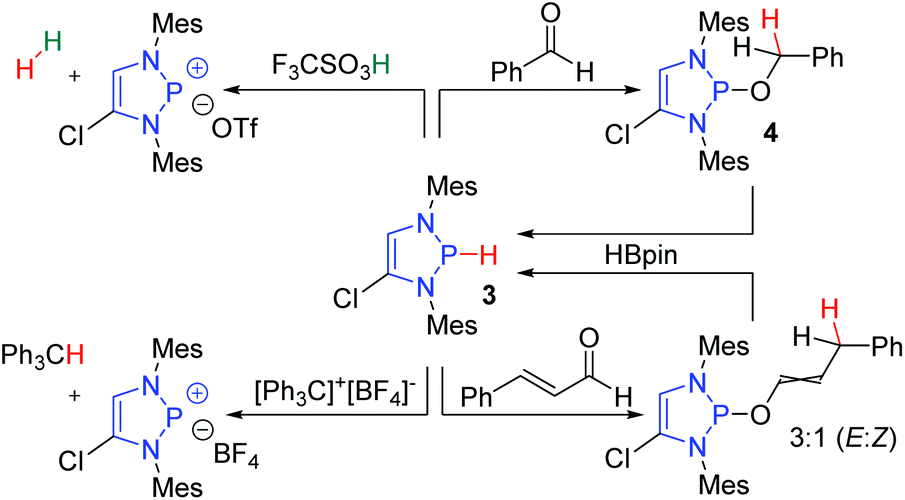 | ||
| Scheme 2 Reactions of DAPs with acids and electrophiles showcase its hydridic nature. | ||
An important contribution to access the P-hydrido DAPs was reported by Kinjo and co-workers.14 They demonstrated that P–O substituted DAPs undergo a σ-bond metathesis event with boranes such a pinacol borane (HBpin) thereby exchanging the P–O and B–H bonding pattern (Scheme 3). This remarkable discovery set the stage for the development of catalytic applications of DAPs as the σ-bond metathesis enables the in situ regeneration of the P-hydrido DAPs from P-alkoxy DAPs such as 5. It has been subsequently shown that secondary phosphine oxides (SPOs), such as 6, can also be converted to the P-hydrido congeners upon treatment with HBpin.15 This transformation presumably occurs through the thermodynamically less favored trivalent tautomer 6a and enabled the direct utilization of the air- and water stable SPOs as precatalysts for reduction reactions.
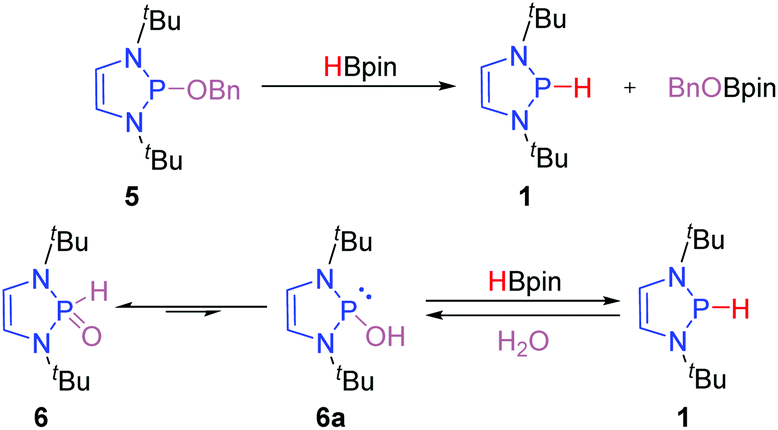 | ||
| Scheme 3 Access to DAP-hydrides by hydride transfer from boranes to P-alkoxy-DAPs and SPOs. | ||
Recently, the potent nucleophilicity of the DAP-hydrides was quantified according to the Mayr nucleophilicity scale by Cheng and co-workers.16 Their systematic study on the kinetics of hydride addition to several benchmark electrophiles established that 1 is, at the time of writing, the most nucleophilic hydride donor ever quantified, with a nucleophilicity parameter (N) of 25.54 (cf. for NaBH4, N = 14.74).
Catalytic reactions
The pioneering development by Gudat and co-workers of 1,3,2-diazaphospholenes as powerful molecular hydrides laid the groundwork for further realization of their synthetic utility.Building on the first catalytic application of DAPs to effect P–C bond formation,17 a number of protocols have been reported over the last ten years using DAP-hydrides in catalytic systems to effect reductive transformations. Such methodologies have helped to establish this field as an alternative to more developed metal-based catalytic systems that suffer from economic and ecological issues. To provide an overview of the variety available, a number of DAPs that have been applied in catalysis are presented (Fig. 2). The isolated P-hydrido DAP 1 has been used directly as a catalyst, while in some cases, more stable and user-friendly pre-catalysts have been used, which are converted to the active P-hydrido species in situ by the action of a stoichiometric reducing agent. These include the SPO 6. It has also been demonstrated that the P-neopentyloxy substituted DAP 7 exhibits greater tolerance to air and water than other alkoxy substituted DAPs such as 5.18 Upon treatment with HBpin, the strained triaminophosphine 8, reported by the Radosevich group, undergoes cleavage of one P–N bond to uncover the diazaphospholene unit.19 Efforts to fine-tune the chemical properties of these catalysts have resulted in the synthesis and application of DAPs bearing diverse structural modifications: the steric and electronic environment around the phosphorus center can be modulated by employing different amine components such as diisopropylaniline in the DAP synthesis (9).20 Conversely, benzannulated DAPs such as 10, 11, and the cationic diazaphosphenium triflate 12 use the carbon backbone as a means of introducing structural variance.21 Furthermore, recent interest in the development of enantioselective applications of DAP catalysis has seen the design of a growing family of chiral DAPs. First generation chiral scaffolds have been based around commercially-available chiral amines, giving quick and efficient access to the targeted DAPs 13–18.22,23 Subsequent chiral DAPs have employed annulated chiral manifolds to give more rigid DAP structures with conformationally locked stereochemical environments (19–29).20,24 The methodologies (both racemic and enantioselective) in which these catalysts and precatalysts have been utilized will now be systematically discussed.
 | ||
| Fig. 2 Representative examples of achiral (top) and chiral (bottom) 1,3,2-diazaphospholene catalysts and precatalysts. | ||
Reductions of azobenzenes and CO2
The first catalytic application of P-hydrido DAPs was reported in 2014, and involved the hydrogenation of azobenzenes (Scheme 4).25 The reaction proceeds from 1, which adds across the NN bond to give intermediate 30. H3NBH3 then effects hydrogenolysis of the P–N bond of this species, regenerating 1 and liberating the product. DFT calculations suggest the hydrogenolysis involves a concerted double hydrogen transfer proceeding via a 6-membered transition state.
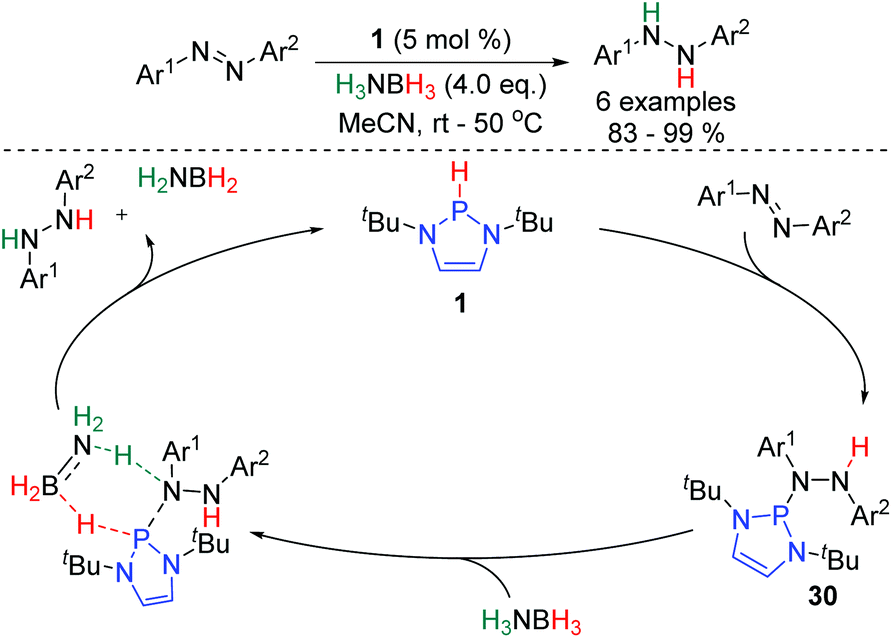 | ||
| Scheme 4 DAP-catalyzed hydrogenation of azobenzenes. | ||
Kinjo et al. have also reported a DAP-catalyzed reduction of CO2 and demonstrated that the reduced product could be trapped in a one-pot process with primary and secondary amines to efficiently access a range of formamides (Scheme 5).26 Interestingly, Ph2SiH2 was used as the terminal reductant, demonstrating that, in addition to boranes, certain silanes can also undergo a σ-bond metathesis with P-alkoxy substituted DAPs. The catalytic cycle commences with hydride addition from 1 to CO2, generating P-formyl substituted DAP 31. Consecutive hydride transfers from Ph2SiH2 lead to the formation of Ph2Si(OCHO)2 and the regeneration of 1. Acyl transfer from Ph2Si(OCHO)2 onto the amine component then completes the reaction by giving the desired formamide and the silanol byproduct. Primary and secondary amines, including a variety of electronically and sterically diverse anilines, engage in the coupling with the silanol, providing a glimpse into the remarkable tolerance of DAPs towards protic functionality.
 | ||
| Scheme 5 DAP-catalyzed reduction of CO2 to formamides. | ||
Reduction of pyridines
The scope of reactions catalyzed by DAPs has been extended to include the synthesis of dihydropyridines (Scheme 6).21 Using the benzannulated diazaphosphenium triflate 12, a variety of pyridines, substituted with both electron-withdrawing and electron-donating functionality at the C-2 and C-3 positions, were reduced to the corresponding dihydropyridines. Electron-withdrawing substituents greatly accelerated the reaction, while electron-donating groups necessitated the use of longer reaction times. Furthermore, the regioselectivity of the process was shown to be dependant on the substitution pattern of the substrate. Mostly, the 1,4-reduction was heavily favoured, however, the presence of strongly electron donating substituents at the C-3 position led to a switch in selectivity to favour the 1,2-reduction.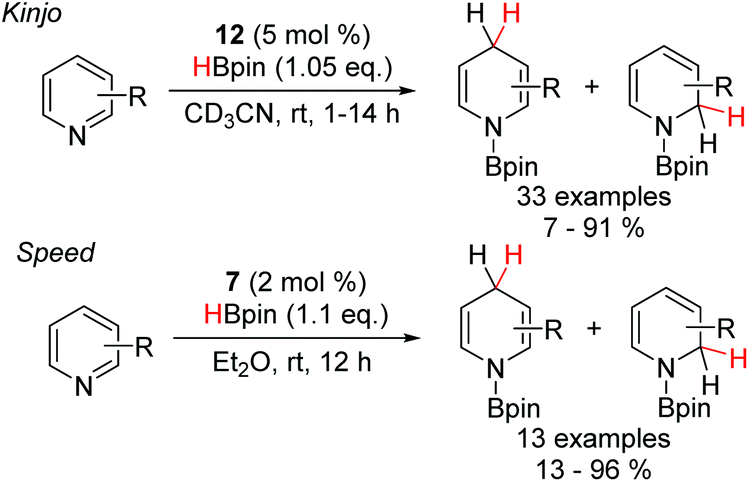 | ||
| Scheme 6 Pyridine hydroboration powered by DAP-catalysis. | ||
Similarly, the Speed group also showed that the neutral DAP 7 was also able to catalyze the hydroboration of pyridines.27 The majority of substrates were selectively reduced at the C-4 position, albeit with one substrate, bearing a CF3 group at the C-3 position resulting in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers, somewhat in contrast to the pattern observed by the Kinjo group. Furthermore, this catalytic system was unable to reduce more electron rich pyridine substrates that the cationic diazaphosphenium manifold could. This difference in reactivity is likely indicative of a slightly different mechanism (Scheme 7). Based on DFT studies, Kinjo and co-workers proposed that the interaction of the cationic 12 with pinacol borane and one equivalent of the pyridine substrate leads to formation of the Lewis adduct 32 with concomitant hydride transfer to generate the neutral P-hydrido DAP 33. Complexation of another pyridine unit to the boron gives the activated boronium species 34. 33 then effects the addition of hydride to one pyridine unit, while the other decomplexes to release the hydroborated product and regenerate the diazaphosphenium 12 and close the catalytic cycle. Based on NMR studies, Speed et al. proposed that the key boronium intermediate 34 of Kinjo's report does not form under the neutral manifold, which potentially explains the slight differences in reactivity and selectivity observed.
:1 mixture of regioisomers, somewhat in contrast to the pattern observed by the Kinjo group. Furthermore, this catalytic system was unable to reduce more electron rich pyridine substrates that the cationic diazaphosphenium manifold could. This difference in reactivity is likely indicative of a slightly different mechanism (Scheme 7). Based on DFT studies, Kinjo and co-workers proposed that the interaction of the cationic 12 with pinacol borane and one equivalent of the pyridine substrate leads to formation of the Lewis adduct 32 with concomitant hydride transfer to generate the neutral P-hydrido DAP 33. Complexation of another pyridine unit to the boron gives the activated boronium species 34. 33 then effects the addition of hydride to one pyridine unit, while the other decomplexes to release the hydroborated product and regenerate the diazaphosphenium 12 and close the catalytic cycle. Based on NMR studies, Speed et al. proposed that the key boronium intermediate 34 of Kinjo's report does not form under the neutral manifold, which potentially explains the slight differences in reactivity and selectivity observed.
 | ||
| Scheme 7 Diazaphosphenium catalyzed pyridine hydroboration proceeds through an activated boronium species. | ||
Carbonyl and imine reductions
In 2015, the Kinjo group reported that P-alkoxy substituted DAPs such as 5, the hydride transfer product between 1 and benzaldehyde, react with pinacol borane to regenerate the hydridic DAP 1 and release the hydroborated product (Scheme 8a).14 They exploited this reaction to develop a broadly applicable hydroboration of both aldehydes and ketones, catalyzed by 1 (Scheme 8b). Indeed, this was the first instance of a non-metal catalyzed hydroboration of carbonyl compounds. The reduction of aldehydes proceeded much faster than the reduction of ketones. Exclusive chemoselectivity towards the aldehyde was observed in substrates bearing both functional groups. In contrast to the mild conditions needed for the aldehyde reduction, higher temperatures and an increased catalyst loading were needed to convert the ketones. Nevertheless, in all cases, excellent yields were obtained for the hydroborated products.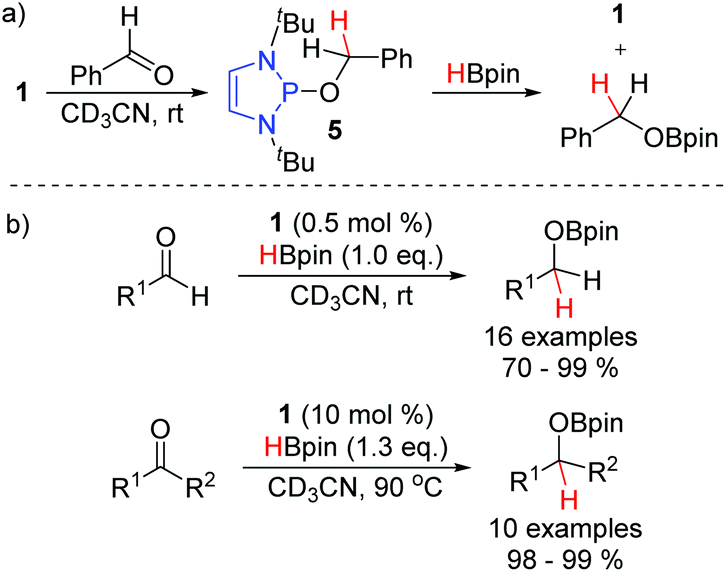 | ||
| Scheme 8 DAP-catalyzed hydroboration of aldehydes and ketones. | ||
Analogously to aldehydes and ketones, imines were demonstrated to be viable substrates independently by the groups of both Speed (Scheme 9, top) and Radosevich (bottom) in 2017.18,19 Rather than employing the isolated P-hydrido DAP, the two groups demonstrated that P-R and P-NR2 substituted DAPs could be used as pre-catalysts. In particular, it was demonstrated that 7, bearing a neopentyloxy substituent, displays significantly greater stability towards both air and moisture than the P-hydrido DAP 1 and other alkoxy substituted DAPs. Upon treatment with pinacol borane, conversion to 1 enables entry to the desired catalytic cycle. The Radosevich protocol employs the strained triaminophosphine 8 as the DAP catalyst. Activation with pinacol borane induce a σ-bond metathesis event with one of the flanking P–N bonds to generate a C1-symmetric P-hydrido species. In both reports, aldimines and ketimines could be efficiently converted to the corresponding amines at room temperature.
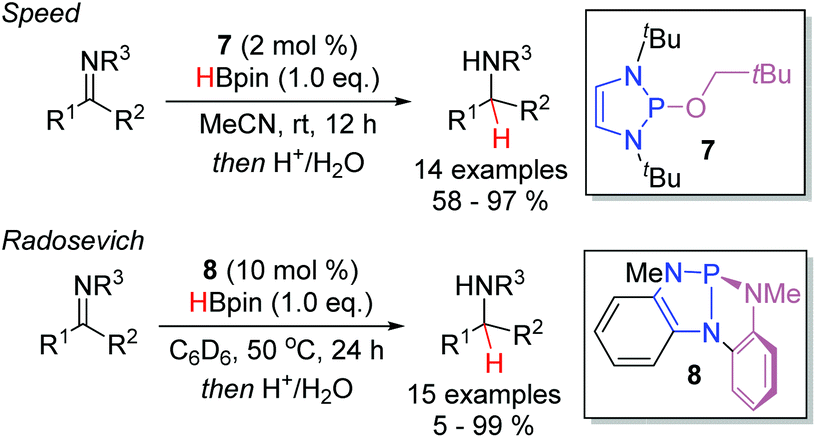 | ||
| Scheme 9 Related catalytic systems for imine hydroboration using DAPs. | ||
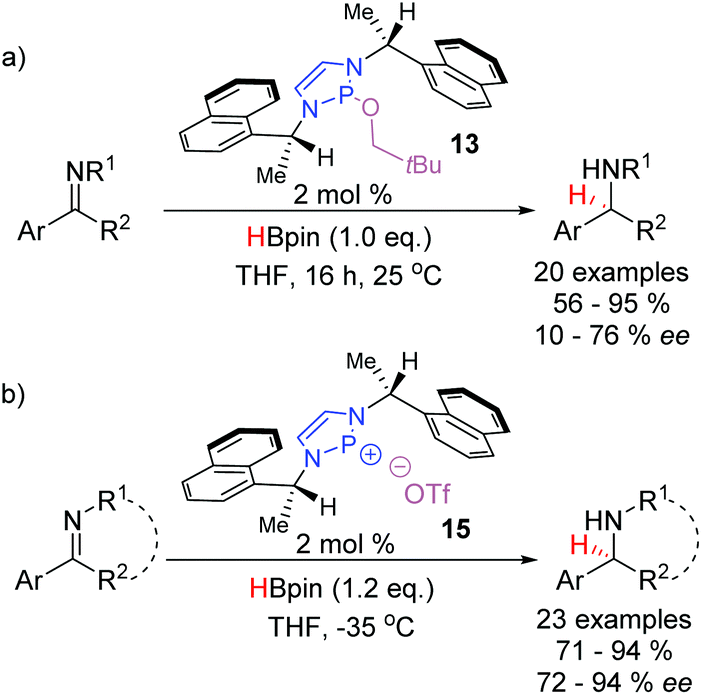
The DAP-catalyzed reduction of imines was extended by the Speed group in 2017 with the disclosure of a chiral C2-symmetric DAP precatalyst 13 capable of enantioselective hydride additions (Scheme 10a).22 This catalyst is based off a cheap, commercially-available chiral amine and is synthesized in three steps. The reaction proceeds under mild conditions providing access to a series of amines with low to good enantioselectivities. Aided by analysis of the crystal structure of 13, the authors rationalized the observed enantioselectivity based on a quadrant model. The approach of the substrate to the active catalytic species is dictated by the favorable placement of the large substituents in the relatively empty quadrants of the catalyst. Conversely, the smaller substituents on the substrate align with the quadrants occupied by the bulky naphthyl groups. As such, a sufficient difference in the steric bulk on either side of the imine was necessary for efficient transfer of chirality. A subsequent report in 2019 disclosed a more reactive catalytic system based on the chiral cationic diazaphosphenium triflate 15, that was able to reduce cyclic imines to the corresponding piperidines and pyrrolidines with good to excellent enantioselectivities (Scheme 10b).23 The chiral scaffold used in both catalytic systems is the same, however, the heightened activity of the cationic diazaphosphenium system enabled the reaction to proceed efficiently at −35 °C, giving an extra boost to the enantioselectivity.
 | ||
| Scheme 10 Chiral DAP manifolds for enantioselective imine reductions. | ||
Conjugate reductions of α,β-unsaturated carbonyl compounds
The profound regioselectivity of DAPs towards the 1,4-reduction of α,β-unsaturated carbonyl compounds has prompted the development of catalytic methodologies to effect this transformation (Scheme 11a). With 5 mol% of the precatalyst 7, α,β-unsaturated aldehydes, ketones, esters and an oxazolidinone were selectively reduced at 40 °C.18 A similar catalytic system has been reported by the Kinjo group. Using 1 mol % of 1 and 1.0 equivalent of H3NBH3 as the stoichiometric reductant, for the conjugate reduction of α,β-unsaturated esters (Scheme 11b).28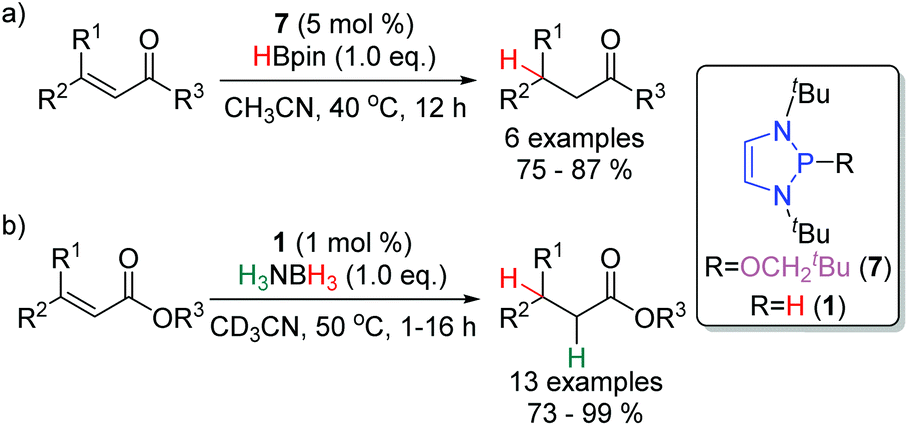 | ||
| Scheme 11 DAPs selectively catalyze the 1,4-reduction of α,β-unsaturated carbonyl compounds. | ||
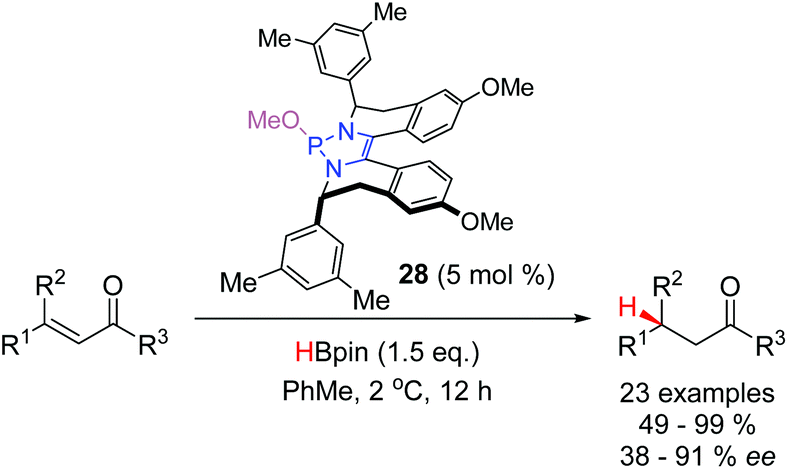
In 2018, Cramer and co-workers reported the first asymmetric conjugate reduction catalyzed by DAPs (Scheme 12).24 Catalysts based off simple, commercially-available chiral amines were examined, however, they did not display sufficient levels of enantioselectivity. It was hypothesized that free rotation about the exocyclic C–N bonds resulted in a poorly defined stereochemical environment around the reactive phosphorus centre. To achieve high enantioselectivities, it was necessary to design a new class of chiral DAPs that were locked into a more rigid conformation. Catalysts with chiral substituents that were annulated onto the backbone (19–28, Fig. 2) were synthesized and showed an improvement in selectivity. The best performing DAP from this class, 28, catalyzed the reduction of a range of substrates including α,β-unsaturated acyl pyrroles, ketones, and amides with enantioselectivities ranging from moderate to excellent.
 | ||
| Scheme 12 A designer chiral DAP enables excellent enantioinduction in conjugate reductions of α,β-unsaturated carbonyl compounds. | ||
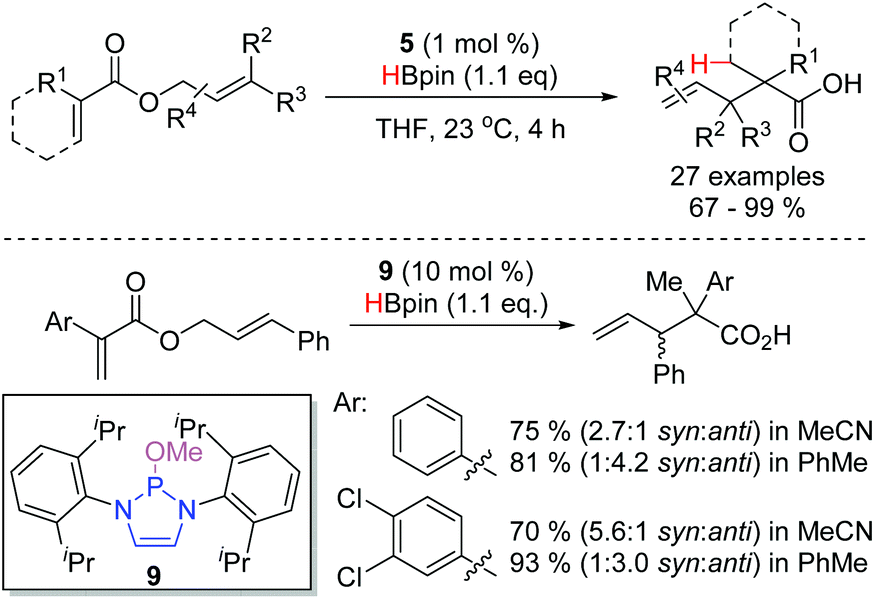
An interesting application of DAP-catalysis was disclosed in 2019: conjugate reduction of allyl-ester enoates, catalyzed by DAP 5, yielded allyl enolates, which then underwent [3,3]-sigmatropic rearrangements (Scheme 13).20 This is the first instance of a DAP-catalyzed process forming C–C bonds. A wide variety of substrates, many with sensitive functionality, could be engaged in the reaction. The diastereoselectivity of the reaction could be modulated with different catalysts, such as 9, and solvents. In some cases, this enabled access to either of the two possible product diastereomers with good selectivity.
 | ||
| Scheme 13 A DAP-catalyzed reductive claisen rearrangement forms C–C bonds under mild conditions. | ||
Using the chiral DAP 29 as the catalyst, a single example of an enantioselective rearrangement was described (Scheme 14). Hydride addition to substrate 35 generated the transient enolate 36 with a chiral DAP counterion. This species rearranged spontaneously to give the Claisen product 37 in excellent yield and diastereoselectivity, and with moderate enantioselectivity, proving that the chiral DAP counterion provides sufficient bias to favor one enantiomeric rearrangement product over the other. In doing so, this provided the first example of enantioselective α-carbonyl functionalization under DAP catalysis.
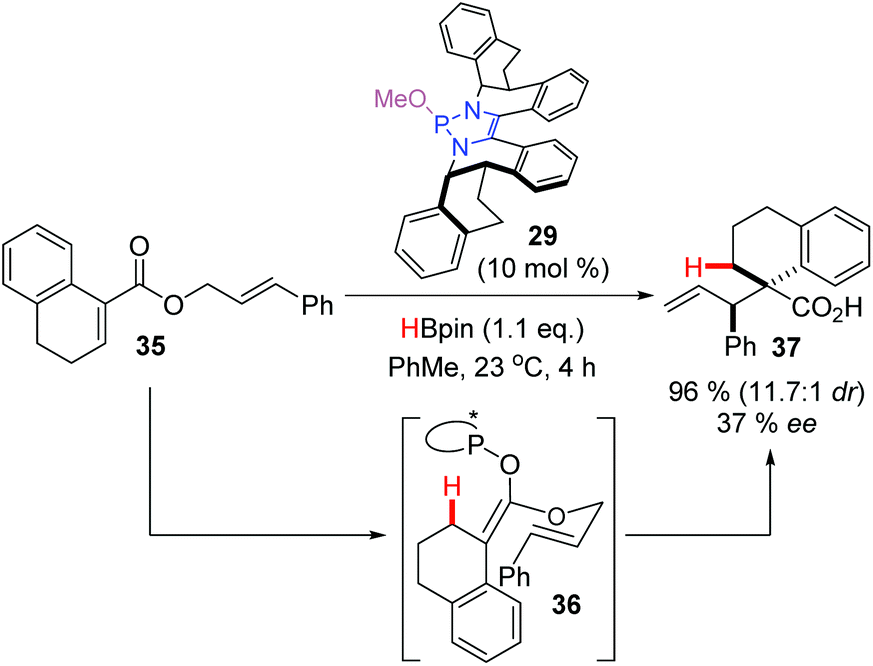 | ||
| Scheme 14 An enantioselective reductive claisen rearrangement under DAP-catalysis opens the door for enantioselective α-functionalization of enolates. | ||
The potent nucleophilic properties of DAP-hydrides, and their remarkably low basicity, was recently exploited by the Cramer group who showed that the combination of DAP-precatalyst 38 and phenylsilane (PhSiH3) as the terminal reductant can effect the selective 1,4-reduction of α,β-unsaturated carboxylic acids (Scheme 15).29 Despite the presence of the potentially problematic acidic proton, the reaction smoothly proceeds to completion with only 0.35 equivalents of PhSiH3, demonstrating that each hydride on the stoichiometric reducing agent is competent for catalyst turnover, and that any acid–base reactivity is virtually negligible under the reaction conditions and is dominated by the nucleophilic delivery of hydride to the β-position of the substrate. A wide variety of functional groups were well tolerated in the reaction, and when the chiral DAP 29 was employed with pinacol borane as the terminal reductant, 2-phenylpropionic acid was obtained with moderate enantioenrichment (64:36 er).
 | ||
| Scheme 15 The low basicity of diazaphospholenes enables the direct conjugate reduction of α,β-unsaturated carboxylic acids. | ||
Conclusions and future prospects
Over the last few years several seminal reports showcased the tremendous potential of DAP-hydrides in catalysis. DAPs have rapidly emerged from inorganic laboratories to a promising axis in catalysis, ready to further penetrate the standard toolbox of organic chemistry. We have identified several bottlenecks that should be addressed to further unlock the tremendous potential of the DAPs. These can be broadly classified into five areas:• Next-generation chiral DAP scaffolds.
• Parametrization of substituent effects.
• Hydrogen gas as stoichiometric reductant.
• New reduction-induced methodologies.
• Group transfers beyond hydride.
Progress within each area will facilitate a holistic development of the research field and will serve to accelerate the uptake of this chemistry. In first place, new structural designs of DAPs would provide an opportunity improving the structure – reactivity understanding. The development of powerful chiral DAPs able to induce high enantioselectivities on a variety of substrate types is still in its infancy. This leaves ample room for further catalyst development, ideally based on rapidly accessible or even commercially available chiral amines (Fig. 3). Further designs may take advantage of the potential for structural modifications on the carbon backbone. As more catalysts are reported and benchmarked, the extra data will permit a parametrization of the catalytic performance. A systematic analysis of the steric and electronic factors that affect such reactivity would then aid the rational design of subsequent catalysts.
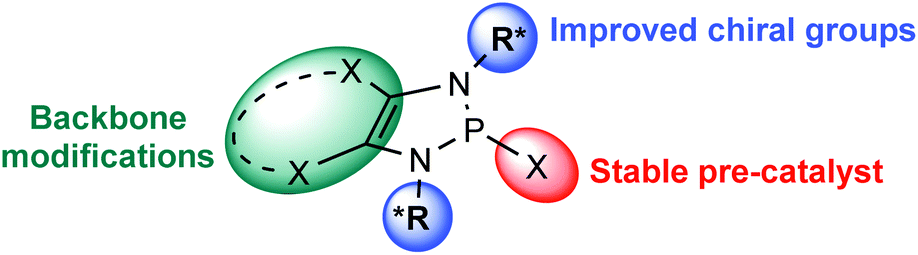 | ||
| Fig. 3 Opportunities for the next-generation DAP catalysts. | ||
To date, the terminal reductants used for catalyst turnover are limited to boranes and silanes. While these are very convenient and easy-to-handle solids or liquids in a laboratory setting, they have shortcomings in atom economy and price. In this respect, hydrogen gas would be a game changer and enable DAP-catalysis to compete with metal-catalysts in industrial scale applications. Accordingly, investigations towards the possibility of H2 splitting with DAPs are warranted. So far, DAP-catalyzed reductions have only been reported for polarized π-bonds. A significant step ahead would be an upgrade of its capabilities to engage in reductions off unbiased olefins putting it closer into competition with transition-metals based catalysts. To this end, the enantioselective reduction of heterocycles such as pyridines and quinolines would offer a highly sought-after entry point into chiral amine-containing building blocks. Moreover, DAP-catalyzed 1,4-reductions operate under very mild conditions without the need for strong bases and cryogenic temperatures, leading to phosphorus enolates as intermediates. Having a chiral DAP near, these intermediates may allow to tap into the very broad reactivity portfolio of transformation with nucleophilic enolates and render them enantioselective. Notably, during the drafting of this manuscript, two reports, detailing the use of DAP-hydrides in radical reactions appeared.30,31 These results open the possibility for a complementary reactivity mode involving hydrogen radicals instead of the hydride paradigm. Whereas both reactions utilise a stoichiometric amount of DAP-hydride, it remains to be investigated whether these processes can be affected using a catalytic amount of the DAP. The prospects of using them as an alternative and mild entry point to access radical-based transformations would open further versatility to the DAPs. While catalytic hydride transfer is unquestionably a cornerstone process, the transfer other groups would greatly extend the utility of DAPs in the future (Scheme 16). For example, it is well understood that silyl is an isolobal fragment to hydrogen. Additional elements or groups are conceivable to be transferred as well. With appropriate reagents, new catalyst designs may open the door to such transfer reactions.
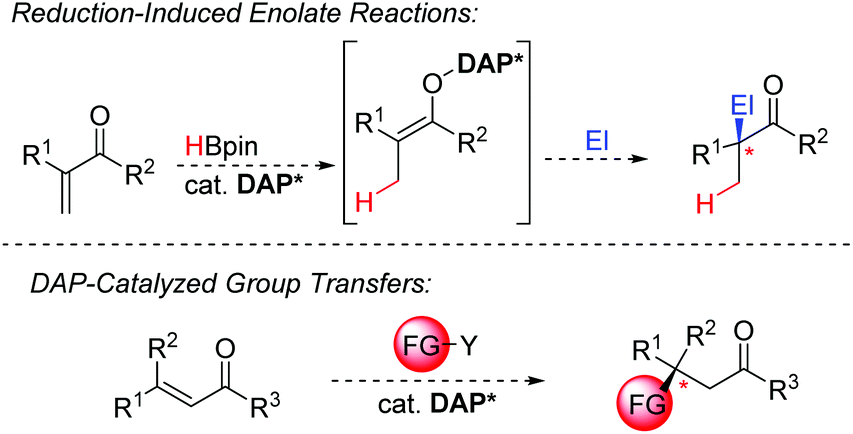 | ||
| Scheme 16 Potential expansion of DAP-catalyzed transformations. | ||
The emergence of DAP catalyzed reactions over the last few years impressively showed again the great importance of the element phosphorus for organic chemistry. We strongly believe, that DAP-catalysis is just at its beginning. It holds the promise of attractive reactivity with implications on many aspects of catalysis and synthetic organic chemistry just waiting to be unlocked.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPFL and the Swiss National Science Foundation (no. 157741) for financial support.References
-
(a)
Phosphorus Ligands in Asymmetric Catalysis, ed. A. Börner, Wiley-VCH, 2008 Search PubMed
; (b) L. A. Adrio and K. K. Hii, Application of phosphine ligands in organic synthesis, Organomet. Chem., 2009, 35, 62 CAS
- H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine Organocatalysis., Chem. Rev., 2018, 118, 10049 CrossRef CAS
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Phosphine Organocatalysis., Chem. Rev., 2014, 114, 9047 CrossRef CAS
- S. Liu, Y. Kumatabara and S. Shirakawa, Chiral quaternary phosphonium salts as phase-transfer catalysts for environmentally benign asymmetric transformations, Green Chem., 2016, 18, 331 RSC
-
(a) D. Gudat, Diazaphospholenes: N-Heterocyclic Phosphines between Molecules and Lewis Pairs, Acc. Chem. Res., 2010, 43, 1307 CrossRef CAS
- J.-N. Li, L. Liu, Y. Fu and Q.-X. Guo, What are the pKa values of organophosphorus compounds?, Tetrahedron, 2006, 62, 4453 CrossRef CAS
-
(a) F. Carré, C. Chuit, R. J. P. Corriu, A. Mehdi and C. Reyé, N–P intramolecular stabilization of phosphonium ions and preparation of hypercoordinated phosphanes with unusual properties, J. Organomet. Chem., 1997, 529, 59 CrossRef
- D. Gudat, A. Haghverdi and M. Nieger, Umpolung of P–H Bonds, Angew. Chem., Int. Ed., 2000, 39, 3084 CrossRef CAS
-
(a) A. Schmidpeter and K. Karaghiosoff, 4,5-Dicyano-1,3,2λ3-diazaphospholate – an Anionic 1,3,2-Diazaphosphole, Stable as a Monomer, Z. Naturforsch., B: J. Chem. Sci., 1981, 36, 1273 Search PubMed
-
(a) D. Gudat, Cationic low coordinated phosphorus compounds as ligands: Recent developments, Coord. Chem. Rev., 1997, 163, 71 CrossRef CAS
- D. Gudat, A. Haghverdi, H. Hupfer and M. Nieger, Stability and Electrophilicity of Phosphorus Analogues of Arduengo Carbenes - An Eperimental and Computational Study, Chem. – Eur. J., 2000, 6, 3414 CrossRef CAS
- D. Hermannsdörfer, M. Kaaz, O. Puntigam, J. Bender, M. Nieger and D. Gudat, The Reaction between Diazadienes and Element Tribromides EBr3 (E=P, B) Revisited: Metal-Free Synthesis of Halogenated N-Heterocyclic Phosphanes and Boranes, Eur. J. Inorg. Chem., 2015, 4819 CrossRef
- S. Burck, D. Gudat, M. Nieger and W.-W. Du Mont, P-Hydrogen-Substituted, 1,3,2-Diazaphospholenes: Molecular Hydrides, J. Am. Chem. Soc., 2006, 128, 3946 CrossRef CAS
- C. C. Chong, H. Hirao and R. Kinjo, Metal-Free σ-Bond Metathesis in 1,3,2-Diazaphospholene-Catalyzed Hydroboration of Carbonyl Compounds, Angew. Chem., Int. Ed., 2015, 54, 190 CrossRef CAS
- T. Lundrigan, C.-H. Tien, K. N. Robertson and A. W. H. Speed, Air and Water stable Secondary Phosphine Oxides as Diazaphospholene Precatalysts, Chem. Commun., 2020, 56, 8027 RSC
- J. Zhang, J.-D. Yang and J.-P. Cheng, A Nucleophilicity Scale for the Reactivity of Diazaphospholenium Hydrides: Structural Insights and Synthetic Applications, Angew. Chem., Int. Ed., 2019, 58, 5983 CrossRef CAS
- S. Burck, D. Förster and D. Gudat, Phosphorus-carbon bond formation catalysed by electrophilic N-heterocyclic phosphines, Chem. Commun., 2006, 2810 RSC
- M. R. Adams, C.-H. Tien, B. S. N. Huchenski, M. J. Ferguson and A. W. H. Speed, Diazaphospholene Precatalysts for Imine and Conjugate Reductions, Angew. Chem., Int. Ed., 2017, 56, 6268 CrossRef CAS
- Y.-C. Lin, E. Hatzakis, S. M. McCarthy, K. D. Reichl, T.-Y. Lai, H. P. Yennawar and A. T. Radosevich, P–N Cooperative Borane Activation and Catalytic Hydroboration by a Distorted Phosphorous Triamide Platform, J. Am. Chem. Soc., 2017, 139, 6008 CrossRef CAS
- J. H. Reed, P. A. Donets, S. Miaskiewicz and N. Cramer, A 1,3,2-Diazaphospholene-Catalyzed Reductive Claisen Rearrangement, Angew. Chem., Int. Ed., 2019, 58, 8893 CrossRef CAS
- B. Rao, C. C. Chong and R. Kinjo, Metal-Free Regio- and Chemoselective Hydroboration of Pyridines Catalyzed by 1,3,2-Diazaphosphenium Triflate, J. Am. Chem. Soc., 2018, 140, 652 CrossRef CAS
- M. R. Adams, C.-H. Tien, R. McDonald and A. W. H. Speed, Asymmetric Imine Hydroboration Catalyzed by Chiral Diazaphospholenes, Angew. Chem., Int. Ed., 2017, 56, 16660 CrossRef CAS
- T. Lundrigan, E. N. Welsh, T. Hynes, C.-H. Tien, M. Adams, K. R. Roy, K. N. Robertson and A. W. H. Speed, Enantioselective Imine Reduction Catalyzed by Phosphenium Ions, J. Am. Chem. Soc., 2019, 141, 14083 CrossRef CAS
- S. Miaskiewicz, J. H. Reed, P. A. Donets, C. C. Oliveira and N. Cramer, Chiral 1,3,2-Diazaphospholenes as Catalytic Molecular Hydrides for Enantioselective Conjugate Reductions, Angew. Chem., Int. Ed., 2018, 57, 4039 CrossRef CAS
- C. C. Chong, H. Hirao and R. Kinjo, A Concerted Transfer Hydrogenolysis: 1,3,2-Diazaphospholene-Catalyzed Hydrogenation of N=N Bond with Ammonia–Borane, Angew. Chem., Int. Ed., 2014, 53, 3342 CrossRef CAS
- C. C. Chong and R. Kinjo, Hydrophosphination of CO2 and Subsequent Formate Transfer in the 1,3,2-Diazaphospholene-Catalyzed N-Formylation of Amines, Angew. Chem., Int. Ed., 2015, 54, 12116 CrossRef CAS
- T. Hynes, E. N. Welsh, R. McDonald, M. J. Ferguson and A. W. H. Speed, Pyridine Hydroboration with a Diazaphospholene Precatalyst, Organometallics, 2018, 37, 841 CrossRef CAS
- C. C. Chong, B. Rao and R. Kinjo, Metal-Free Catalytic Reduction of α,β-Unsaturated Esters by 1,3,2-Diazaphospholene and Subsequent C–C Coupling with Nitriles, ACS Catal., 2017, 7, 5814 CrossRef CAS
- J. H. Reed and N. Cramer, 1,3,2-Diazaphospholenes Catalyze the Conjugate Reduction of Substituted Acrylic Acids, ChemCatChem, 2020, 12, 4262–4266 CAS
- J. Zhang, J.-D. Yang and J.-P. Cheng, Exploiting the Radical Reactivity of Diazaphosphinanes in Hydrodehalogenations and Cascade Cyclizations, Chem. Sci., 2020, 11, 4786 RSC
- B. S. N. Huchenski, K. N. Robertson and A. W. H. Speed, Functionalization of Bis-Diazaphospholene P-P Bonds with Diverse Electrophiles, Eur. J. Org. Chem., 2020, 5140–5144 CAS
| This journal is © the Partner Organisations 2020 |