
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDealkoxylation of N-alkoxyamides without an external reductant driven by Pd/Al cooperative catalysis†
Hirotsugu
Suzuki
 ,
Takahiro
Shiomi
,
Kenji
Yoneoka
and
Takanori
Matsuda
*
,
Takahiro
Shiomi
,
Kenji
Yoneoka
and
Takanori
Matsuda
*
Department of Applied Chemistry, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: mtd@rs.tus.ac.jp
First published on 15th September 2020
Abstract
Lewis acid-assisted palladium-catalysed dealkoxylation of N-alkoxyamides has been developed. This reaction proceeded smoothly with a range of N-alkoxyamides in the absence of an external reductant, thereby establishing a convenient and reductant-free protocol. In addition, a gram-scale reaction could be achieved. Preliminary mechanistic investigations indicated that β-hydrogen elimination from a palladium alkoxide intermediate generated an intramolecular hydride source.
N-Alkoxyamides are an important class of synthetic intermediates for a range of organic transformations.1 In particular, N-methoxy-N-methylamides, which are known as Weinreb amides, have unique properties as acylating reagents that suppress the overalkylation of reaction products by forming remarkably stable five-membered cyclic intermediates (Scheme 1a).2 This exceptional feature allows the transformation of readily available and stable N-alkoxyamides3 into useful aldehydes and ketones in a single step. Recently, N-alkoxyamides have emerged as versatile directing groups for C–H bond functionalisation, and various transformations employing N-alkoxyamides are currently available.4 While N-alkoxyamides are commonly used in various organic reactions, the dealkoxylation of N-alkoxyamides has not been explored enough yet (Scheme 1b).
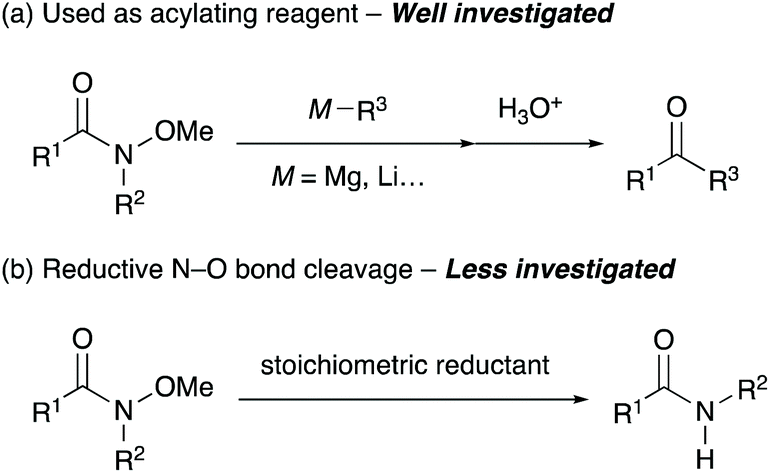 | ||
| Scheme 1 Transformation of N-alkoxyamides: (a) nucleophilic addition of organometallic reagents and (b) dealkoxylation of N-alkoxyamides. | ||
Conventional dealkoxylation of N-alkoxyamides requires stoichiometric metal-based reductants such as SmI2,5 Na/Hg6 and lithium powder7 (Scheme 2a). An organic, neutral super electron donor has been developed as a stoichiometric reductant, and it gives results comparable to those obtained using metal-based reductants.8 Base-mediated formal reduction of N-alkoxyamides has also evolved as a method for dealkoxylation.9 Treatment of N-alkoxyamides with lithium diisopropylamide,9a or tert-butyldimethylsilyl triflate and triethylamine9b resulted in the formal reduction of the amides, along with the formation of formaldehyde. Although these reductants and bases allow facile cleavage of the alkoxy groups from N-alkoxyamides under very mild conditions, excess amounts of reductants or bases are required for these reactions. In addition, these reducing reagents are sometimes expensive, difficult to handle, and hazardous. Ruthenium-catalysed dealkoxylation of N-alkoxyamides has been reported as an alternative protocol for avoiding the use of such stoichiometric reagents (Scheme 2b). Dealkoxylation proceeded in alcoholic solvents, which also behaved as a stoichiometric reductant.10 Although the catalytic reactions require only green and cheap alcohols as stoichiometric reductants, it is necessary to add a substoichiometric amount of Zn–Cu for activating the ruthenium catalyst. Herein, we report the palladium-catalysed dealkoxylation of N-alkoxyamides in the absence of an external reductant as a convenient and reductant-free protocol for dealkoxylation (Scheme 2c). To the best of our knowledge, this is the first report on the catalytic dealkoxylation of N-alkoxyamides without any external reductants.11
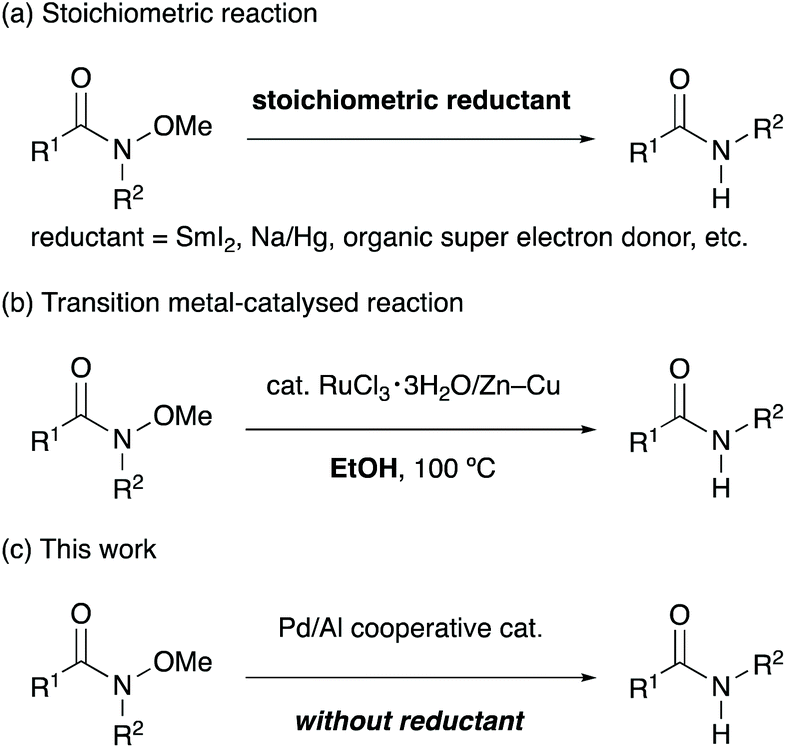 | ||
| Scheme 2 Dealkoxylation of N-alkoxyamides. | ||
We began our investigation using N-methoxy-N-methylbenzamide (1a) as a model substrate, which was heated in toluene at 150 °C in the presence of the Pd(dba)2/DPPBz catalyst (Table 1). After 6 h, the desired secondary amide 2a was formed in a moderate yield (entry 1). We then screened aluminium Lewis acids as co-catalysts in order to activate the N–O bond.12 The addition of aluminium(III) chloride (AlCl3) suppressed the reaction completely (entry 2). Trialkylaluminium or trialkoxyaluminium dramatically improved the yields (entries 3–6), and the best result was obtained when triisobutylaluminium (Ali-Bu3) was employed as a co-catalyst (entry 4).13 Solvent screening (entries 7–12) revealed that cyclopentyl methyl ether (CPME) was the optimal solvent for affording the desired product in an excellent yield (entry 9).14 In addition, this demethoxylation reaction could reach completion with reduced catalyst loadings (entry 13).
|
|
|||
|---|---|---|---|
| Entry | Lewis acid | Solvent | Yield (%) |
| a Reaction conditions: 1 (0.3 mmol), Pd(dba)2 (4 mol%), DPPBz (4 mol%) and Lewis acid (10 mol%) in CPME (0.3 M) at 150 °C for 6 h, unless otherwise noted. b Pd(dba)2/DPPBz (2 mol% each) and Ali-Bu3 (5 mol%) were used as catalysts. The reaction time was 20 h. | |||
| 1 | — | Toluene | 33 |
| 2 | AlCl3 | Toluene | NR |
| 3 | AlMe3 | Toluene | 87 |
| 4 | Ali-Bu3 | Toluene | 98 |
| 5 | Al(OEt)3 | Toluene | 83 |
| 6 | Al(Oi-Pr)3 | Toluene | 94 |
| 7 | Ali-Bu3 | p-Xylene | 86 |
| 8 | Ali-Bu3 | 1,4-Dioxane | 86 |
| 9 | Ali-Bu3 | CPME | 99 |
| 10 | Ali-Bu3 | Diglyme | 85 |
| 11 | Ali-Bu3 | DMF | 84 |
| 12 | Ali-Bu3 | DMSO | 61 |
| 13b | Ali-Bu3 | CPME | 99 |
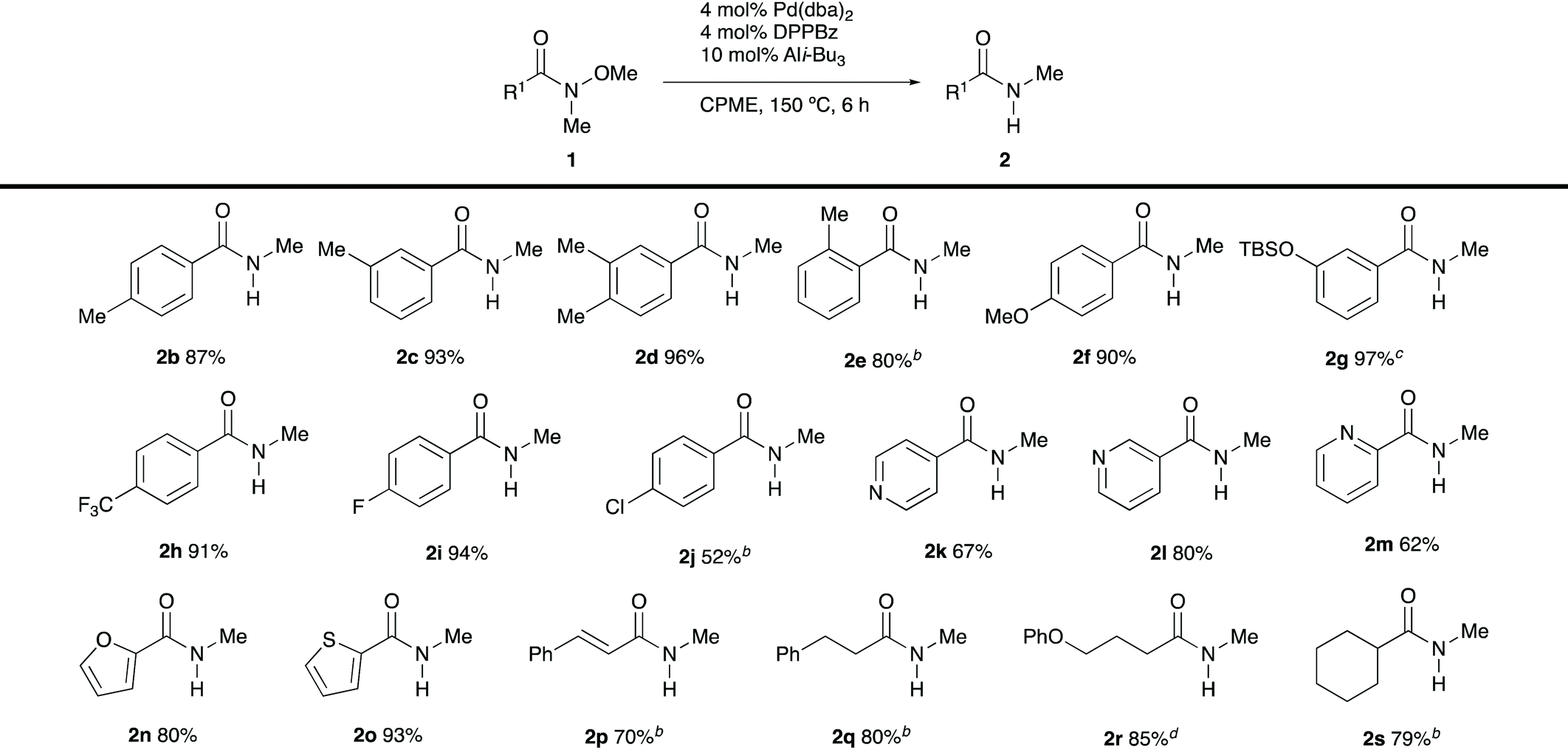
With the optimised reaction conditions in hand, we investigated the scope of demethoxylation (Table 2). Introduction of methyl groups at the para- and meta-positions of the benzene ring (1b–d) did not affect the efficiency of transformations. However, the reactivity decreased with o-methylbenzamide 1e, and increased catalyst loadings were required to obtain a reasonable yield of the desired secondary amide. Benzamides bearing electron-donating and electron-withdrawing groups 1f–j were well tolerated under the optimal conditions. Heteroaryl-substituted substrates 1k–o were also converted into the desired secondary amides with high efficiency. Cinnamamide 1p afforded the corresponding product without the reduction of the olefin moiety.10 It is worth noting that the demethoxylation of enolisable N-methyl-N-methoxyamides 1q–s proceeded smoothly, and no side reactions were observed.
| a Reaction conditions: 1 (0.3 mmol), Pd(dba)2 (4 mol%), DPPBz (4 mol%) and Ali-Bu3 (10 mol%) in CPME (0.3 M) at 150 °C for 6 h, unless otherwise noted. The yields represent the average yield of two reaction runs. b Pd(dba)2 (8 mol%), DPPBz (8 mol%) and Ali-Bu3 (20 mol%) were used (20 h). c The reaction time was 18 h. d Pd(dba)2 (8 mol%), DPPBz (8 mol%) and Ali-Bu3 (20 mol%) were used (18 h). |
|---|
|
To further demonstrate the applicability of demethoxylation, other alkoxyamides were examined. Under the optimal conditions, sulfonamide 3 gave the desired secondary sulfonamide 4 in a high yield (Scheme 3a). Moreover, phosphoramidate 5 was found to be a promising substrate for the demethoxylation to afford the corresponding product in a good yield (Scheme 3b). In both cases, an N–O bond was selectively cleaved, while the other heteroatom–heteroatom bonds remained intact. In contrast to previous studies, reductant-free demethoxylation was applicable to a wide range of N-methoxy-N-methylamides, without the occurrence of any side reactions or over-reactions. Furthermore, a gram-scale reaction was performed with 1a in diglyme, and the desired product 2a was obtained in 78% yield (Scheme 4).15
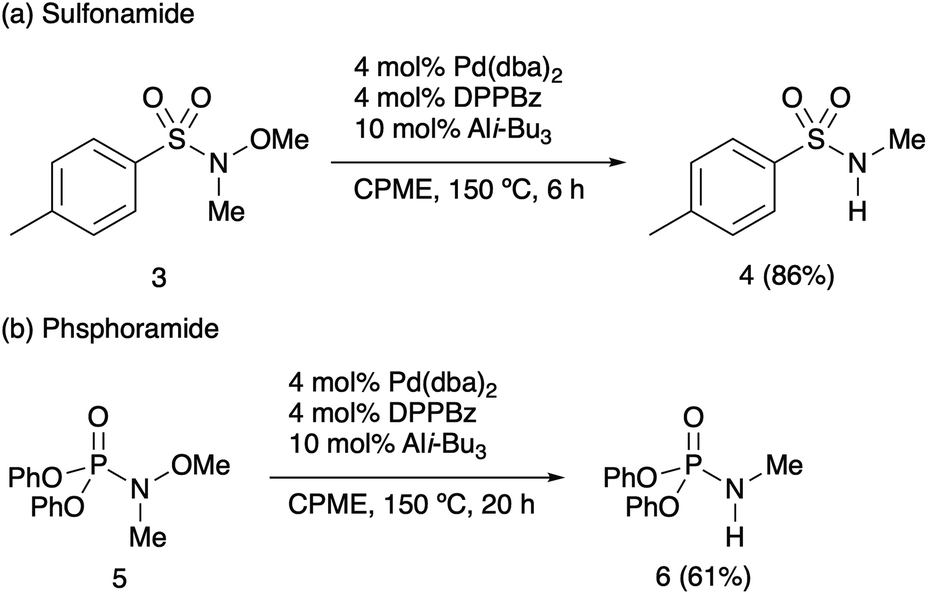 | ||
| Scheme 3 Palladium-catalysed demethoxylation of sulfonamide 3 and phosphoramidate 5. | ||
 | ||
| Scheme 4 A gram-scale reaction. | ||
To gain insight into the reaction mechanism, a control experiment was conducted (Scheme 5). When N-butoxy-N-methylbenzamide (1t) was subjected to the standard reaction conditions, butanal and its aldol condensation product were produced along with the desired secondary amide 2a. These byproducts may have been generated via the β-hydrogen elimination from a palladium alkoxide intermediate. The results reveal that an α-hydrogen atom with respect to the oxygen atom of the alkoxy group functions as a hydride source.11,16
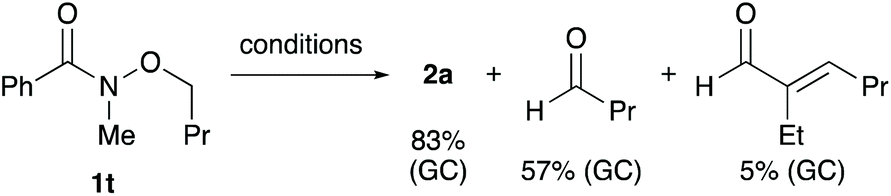 | ||
| Scheme 5 Palladium-catalysed debutoxylation of N-butoxy-N-methylbenzamide (1t) (conditions: 4 mol% Pd(dba)2, 4 mol% DPPBz, 10 mol% Ali-Bu3, CPME, 150 °C, 6 h). | ||
A plausible mechanism for the reductant-free demethoxylation is proposed on the basis of a previous report11 and our preliminary mechanistic investigations (Scheme 6). The carbonyl oxygen of alkoxyamide 1 coordinates to the aluminium Lewis acid to form A, thereby weakening the N–O bond. Subsequently, oxidative addition of the N–O bond to Pd(0) generates palladium alkoxide B, which undergoes β-hydrogen elimination to generate palladium hydride intermediate C and formaldehyde. Finally, reductive elimination from the intermediate C affords secondary amide 2 and simultaneously regenerates the catalytically active Pd(0) species.
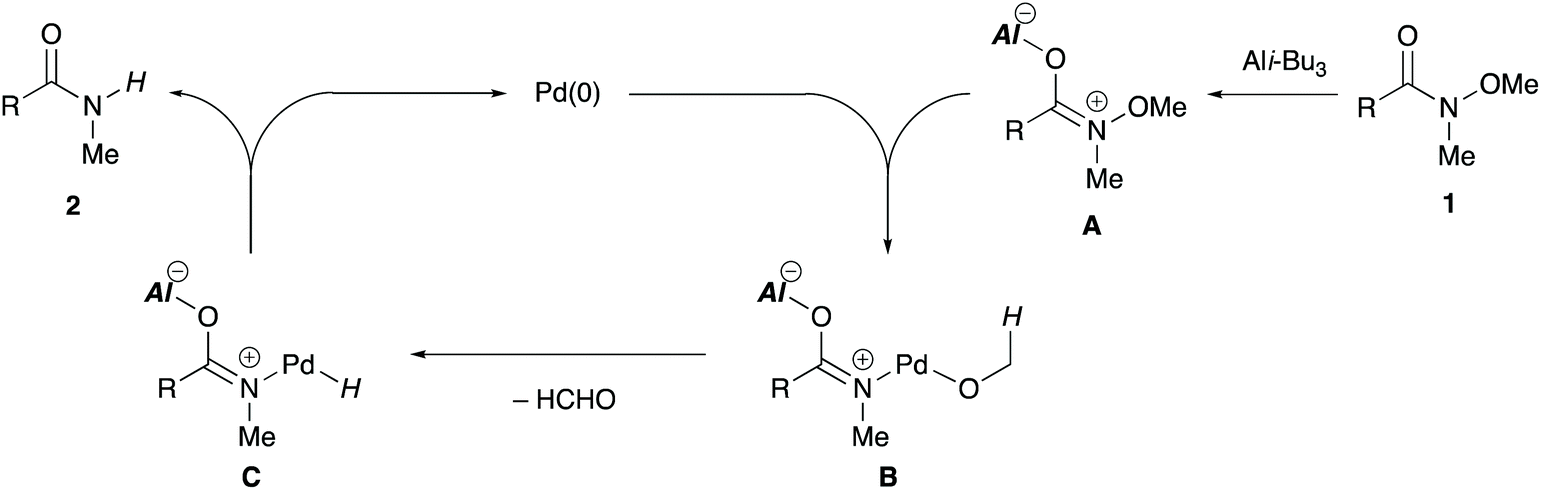 | ||
| Scheme 6 The plausible reaction mechanism for reductant-free demethoxylation. | ||
Conclusions
In summary, we achieved the demethoxylation of N-alkoxyamides in the presence of a Pd/Al cooperative catalytic system. The reaction proceeded with various N-alkoxyamides including a sulfonamide and a phosphoramide in the absence of an external reductant. The N–O bond was selectively reduced, and there were no side reactions or over-reactions. Preliminary mechanistic investigations revealed that β-hydrogen elimination of a palladium alkoxide intermediate generated an intramolecular hydride source. Further studies on palladium-catalysed reductant-free demethoxylation are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Balasubramaniam and I. S. Aidhen, Synthesis, 2008, 3707–3738 Search PubMed.
- (a) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 Search PubMed; (b) J. A. Murphy, A. G. J. Commeureuc, T. N. Snaddon, T. M. McGuire, T. A. Khan, K. Hisler, M. L. Dewis and R. Carling, Org. Lett., 2005, 7, 1427–1429 Search PubMed; (c) C. O. Kangani, D. E. Kelley and B. W. Day, Tetrahedron Lett., 2006, 47, 6289–6292 Search PubMed.
- (a) A. Basha, M. Lipton and S. M. Weinreb, Tetrahedron Lett., 1977, 18, 4171–4172 Search PubMed; (b) J. M. Williams, R. B. Jobson, N. Yasuda, G. Marchesini, U.-H. Dolling and E. J. J. Grabowski, Tetrahedron Lett., 1995, 36, 5461–5464 Search PubMed; (c) T. Shimizu, K. Osako and T. Nakata, Tetrahedron Lett., 1997, 38, 2685–2688 Search PubMed; (d) P.-Q. Huang, X. Zheng and X.-M. Deng, Tetrahedron Lett., 2001, 42, 9039–9041 Search PubMed.
- (a) F. Yang and L. Ackermann, Org. Lett., 2013, 15, 718–720 Search PubMed; (b) Y. Wang, C. Li, Y. Li, F. Yin and X.-S. Wang, Adv. Synth. Catal., 2013, 355, 1724–1728 Search PubMed; (c) G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 Search PubMed; (d) R. Das and M. Kapur, Chem. – Asian J., 2015, 10, 1505–1512 Search PubMed; (e) R. Das and M. Kapur, Chem. – Eur. J., 2016, 22, 16986–16990 Search PubMed; (f) K. Kawai, Y. Bunno, T. Yoshino and S. Matsunaga, Chem. – Eur. J., 2018, 24, 10231–10237 Search PubMed.
- (a) J. L. Chiara, C. Destabel, P. Gallego and J. Marco-Contelles, J. Org. Chem., 1996, 61, 359–360 Search PubMed; (b) G. E. Keck, S. F. McHardy and J. E. Murry, J. Am. Chem. Soc., 1995, 117, 7289–7290 Search PubMed.
- (a) R. Sword, S. O'Sullivan and J. A. Murphy, Aust. J. Chem., 2013, 66, 314–322 Search PubMed; (b) J. E. Jackson, B. N. O'Brien, S. K. Kedzior, G. R. Fryz, F. S. Jalloh, A. Banisafar, M. A. Caldwell, M. B. Braun, B. M. Dunyak and J. L. Dye, Tetrahedron Lett., 2015, 56, 6227–6230 Search PubMed.
- M. Yus, G. Radivoy and F. Alonso, Synthesis, 2001, 914–918 Search PubMed.
- S. P. Y. Cutulic, J. A. Murphy, H. Farwaha, S.-Z. Zhou and E. Chrystal, Synlett, 2008, 2132–2136 Search PubMed.
- (a) S. L. Graham and T. H. Scholz, Tetrahedron Lett., 1990, 31, 6269–6272 Search PubMed; (b) G. E. Keck, S. F. McHardy and J. A. Murry, Tetrahedron Lett., 1993, 34, 6215–6218 Search PubMed.
- H. Fukuzawa, Y. Ura and Y. Kataoka, J. Organomet. Chem., 2011, 696, 3643–3648 Search PubMed.
- For nickel-catalysed reductant-free dealkoxylation of aryl alkyl ethers, see: M. Tobisu, T. Morioka, A. Ohtsuki and N. Chatani, Chem. Sci., 2015, 6, 3410–3414 Search PubMed.
- For the decrease in the activation energy of an aryl C–O bond cleavage by aluminium Lewis acids, see: (a) X. Liu, C.-C. Hsiao, I. Kalvet, M. Leiendecker, L. Guo, F. Schoenebeck and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6093–6098 Search PubMed; (b) P. Kelley, G. A. Edouard, S. Lin and T. Agapie, Chem. – Eur. J., 2016, 22, 17173–17176 Search PubMed.
- For recent examples of transition metal/aluminium cooperative catalysis, see: (a) P. A. Donets and N. Cramer, J. Am. Chem. Soc., 2013, 135, 11772–11775 Search PubMed; (b) S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 Search PubMed; (c) S. Okumura and Y. Nakao, Org. Lett., 2017, 19, 584–587 Search PubMed; (d) F. Inoue, T. Saito, K. Semba and Y. Nakao, Chem. Commun., 2017, 53, 4497–4500 Search PubMed; (e) Q.-S. Liu, D.-Y. Wang, Z.-J. Yang, Y.-X. Luan, J.-F. Yang, J.-F. Li, Y.-G. Pu and M. Ye, J. Am. Chem. Soc., 2017, 139, 18150–18153 Search PubMed; (f) S. Okumura, T. Komine, E. Shigeki, K. Semba and Y. Nakao, Angew. Chem., Int. Ed., 2018, 57, 929–932 Search PubMed; (g) Y.-X. Wang, S.-L. Qi, Y.-X. Luan, X.-W. Han, S. Wang, H. Chen and M. Ye, J. Am. Chem. Soc., 2018, 140, 5360–5364 Search PubMed; (h) T. Zhang, Y.-X. Luan, S.-J. Zheng, Q. Peng and M. Ye, Angew. Chem., 2020, 59, 7439–7443 Search PubMed.
- The yield was dramatically decreased when the reaction was performed at 130 °C for 6 h (36% yield).
- The solvent was employed in order to perform the reaction in a flask.
- (a) X. Wu, B. P. Fors and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9943–9947 Search PubMed; (b) M. S. Mikus, C. Sanchez, C. Fridrich and J. F. Larrow, Adv. Synth. Catal., 2020, 362, 430–436 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for new compounds. See DOI: 10.1039/d0ob01815e |
| This journal is © The Royal Society of Chemistry 2020 |