
Recent advances in engineering active sites for photocatalytic CO2 reduction
Yanan
Bo
,
Chao
Gao
 * and
Yujie
Xiong
*
* and
Yujie
Xiong
*
Hefei National Laboratory for Physical Sciences at the Microscale, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), National Synchrotron Radiation Laboratory, and School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: yjxiong@ustc.edu.cn; gaoc@ustc.edu.cn
First published on 12th May 2020
Abstract
The photocatalytic conversion of green-house gas CO2 into high value-added carbonaceous fuels and chemicals through harvesting solar energy is a great promising strategy for simultaneously tackling global environmental issues and the energy crisis. Considering the vital role of active sites in determining the activity and selectivity in photocatalytic CO2 reduction reactions, great efforts have been directed toward engineering active sites for fabricating efficient photocatalysts. This review highlights recent advances in the strategies for engineering active sites on surfaces and in open frameworks toward photocatalytic CO2 reduction, referring to surface vacancies, doped heteroatoms, functional groups, loaded metal nanoparticles, crystal facets, heterogeneous/homogeneous single-site catalysts and metal nodes/organic linkers in metal organic frameworks.
 Yanan Bo | Yanan Bo received her B.S. in chemical engineering in 2016 from Southeast University. Since then she has been studying as a graduate student under the tutelage of Professor Yujie Xiong at the University of Science and Technology of China. Her research interests are focused on the design of photocatalysts and photoelectrodes for carbon dioxide reduction. |
 Chao Gao | Chao Gao received his B.S. in chemistry in 2010 from Anhui Normal University, and Ph.D. in inorganic chemistry in 2015 (with Professors Xingjiu Huang and Zhiyong Tang) from the University of Science and Technology of China (USTC). During his Ph.D. research, he had two-year training (2013–2015) with Professor Zhiyong Tang at the National Center for Nanoscience and Technology (NCNST). He is currently an Associate Professor at USTC. His current research interests are focused on the design and synthesis of photocatalysts and photoelectrodes for carbon dioxide reduction, nitrogen fixation and methane conversion. |
 Yujie Xiong | Yujie Xiong received his B.S. in chemical physics in 2000 and Ph.D. in inorganic chemistry in 2004, both from the University of Science and Technology of China (USTC). From 2004 to 2009, he worked as a Postdoctoral Fellow at the University of Washington in Seattle and as a Research Associate at the University of Illinois at Urbana-Champaign. He was Principal Scientist of the National Nanotechnology Infrastructure Network (NSF-NNIN) site at Washington University in St. Louis between 2009 and 2011. He joined the USTC faculty in 2011, and currently is Chair Professor of Chemistry. His research interests include inorganic materials and devices for carbon dioxide reduction, nitrogen fixation, methane conversion, water splitting and chemical production. |
1. Introduction
With increasing energy demand and extensive emission of green-house gases, it becomes more critical to develop sustainable energy systems.1–3 One of the best solutions is to convert CO2 into high value-added carbonaceous fuels and chemical building blocks by means of solar energy.4 To realize such CO2 conversion practicably, there is an urgent need to develop highly efficient photocatalysts.The capacity of photocatalysts to drive CO2 conversion largely depends on three crucial factors: (i) light harvesting; (ii) charge separation and transfer; (iii) surface reactions. Numerous studies have endeavored to improve light utilization, charge separation and transfer efficiency during the past decades.1,3 Beyond that, surface reactions referring to effective CO2 adsorption and activation play a critical role in enhancing the performance for CO2 conversion.5,6 More specifically, the adsorption and activation of CO2 molecules exactly occur at the active sites on surfaces. As CO2 conversion involves multi-electron reduction processes, various carbonaceous reduction products could be obtained, accompanied by competitive hydrogen evolution in the presence of water on account of its lower redox potential.7 In this regard, engineering the active sites is vital to regulate the reaction pathways through altering the adsorption behavior of intermediates and suppressing the undesired hydrogen evolution, achieving a high selectivity for specific reduction products. Moreover, in addition to affecting surface reactions, engineering the active sites can also influence the efficiencies of charge separation and light harvesting, both of which will influence the whole performance of photocatalytic CO2 reduction. Toward this end, great research efforts have been devoted to engineering active sites aiming at high activity and selectivity in photocatalytic CO2 reduction.
The photocatalytic system can be considered to consist of two parts: light absorbers and active sites.5 Generally, the light absorbers are semiconductors, quantum dots and organic photosensitizers, while the active sites exist on the surface or in the porous matrix of these light absorbers.1,6,8,9 The active sites can be engineered in the form of surface vacancies,10–12 doped heteroatoms, functional groups,13,14 loaded metal nanoparticles, crystal facets, heterogeneous single-site catalysts,15–17 homogeneous single-site catalysts18–23 and metal nodes/organic linkers in metal–organic frameworks (MOFs), by which the coordinately unsaturated state, electronic structure, acidity/alkalinity, and local environment of the active sites can be adjusted for enhancing activity and selectivity in photocatalytic CO2 reduction.
Taken together, the advancements of photocatalytic CO2 reduction essentially profit from the development of well-defined highly active sites in photocatalysts. In this review, we will focus on recent advances in rationally engineering active sites for photocatalytic CO2 reduction. Firstly, the basic principles of photocatalytic CO2 reduction will be briefly discussed based on the currently developed mechanisms, from which we can outline the critical parameters for engineering active sites. Secondly, the developed strategies for engineering active sites on surfaces and in open frameworks are systematically summarized, including creating surface vacancies, doping heteroatoms, modifying functional groups, loading metal nanoparticles, engineering crystal facets, dispersing heterogeneous single-site catalysts, anchoring homogeneous single-site catalysts and modulating metal nodes/organic linkers in MOFs. Finally, the existing challenges and future prospects are presented.
2. Basic principles of photocatalytic CO2 reduction
The photocatalytic reduction of CO2 into fuels with H2O driven by solar energy has drawn much attention since the investigation by Fujishima in 1979.2 The overall process of photocatalytic CO2 reduction involves the following three critical steps (Fig. 1): (i) the photocatalyst absorbs the incident photons with an energy equal to or higher than the bandgap energy and generates electron–hole pairs; (ii) the photogenerated electron–hole pairs are then separated and electrons migrate to the active sites where the CO2 molecules are adsorbed; (iii) the photogenerated electrons are transferred to the adsorbed CO2 molecules and initiate CO2 reduction. Simultaneously, the photogenerated holes are consumed to oxidize H2O into O2. In the first step, the key to the full utilization of solar light is to absorb visible/infrared light, as natural solar light consists of about 95% visible and infrared light. In the second step, a large fraction of electrons and holes may be consumed through recombination during migration. To facilitate charge separation and make full use of the photogenerated electrons, the rational design of active sites, such as defect creation, facet engineering and cocatalyst loading, is commonly applied. Otherwise, when photogenerated carriers are not consumed timely, taking an example of Cu2O, accumulation of photogenerated holes on Cu2O would lead to its photocorrosion.24,25 In this regard, inorganic scavengers such as Na2SO3, and organic scavengers such as methanol and triethanolamine (TEOA) are commonly used as electron donors to consume the holes, which is obviously not economical. In the third step, CO2 molecule adsorption at the active sites is the prerequisite for subsequent reduction reactions.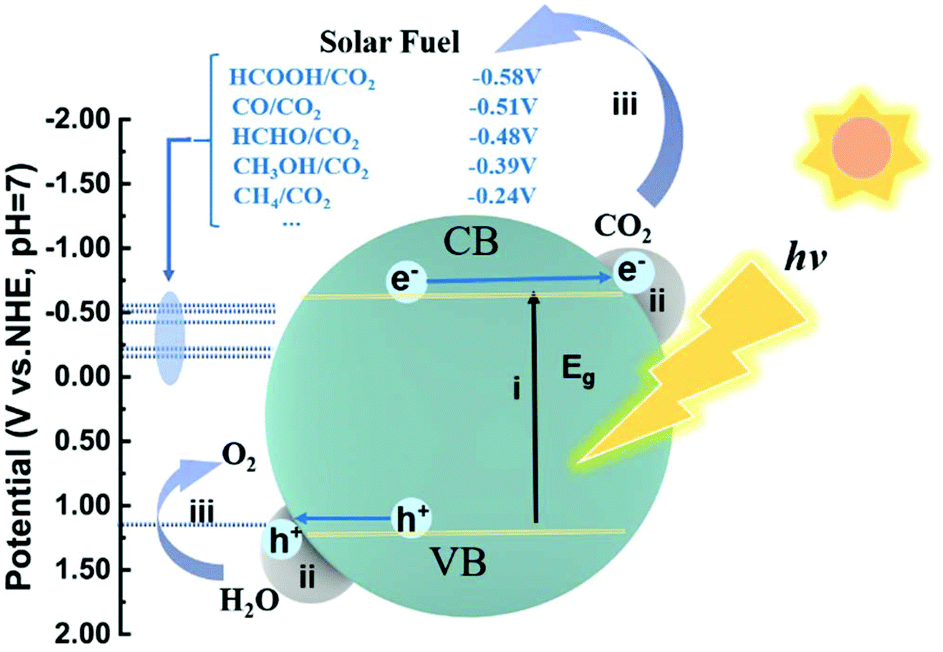 | ||
| Fig. 1 Schematic illustration of the overall process for photocatalytic CO2 reduction with H2O over a semiconductor photocatalyst. The dotted lines indicate the thermodynamic potentials for water oxidation into O2 and CO2 reduction into CH4, CH3OH, HCHO, CO and HCOOH, respectively. The numbers i, ii and iii represent the three critical steps in photocatalytic CO2 reduction. | ||
When a CO2 molecule is adsorbed on a surface, the CO2 molecule can act as an electron donor and acceptor simultaneously.1 Each of the oxygen atoms with a lone pair of electrons can coordinate with surface Lewis acid sites, donating the electrons to these Lewis acid sites. Inversely, the carbon atom could gain electrons from Lewis base sites, forming a carbonate-like species. Thus, intentionally modifying the electronic properties of surface active sites can substantially tackle its dilemma of adsorption. To this end, surface acidity and alkalinity can significantly influence CO2 adsorption. Once the CO2 molecule interacts with the surface active site, the CO2 molecule can be polarized resulting in bent bonding for electron repulsion. The bent CO2 molecule can lower its LUMO (lowest unoccupied molecular orbital) level and thus make it easier for electron acceptance.26 Subsequently, multi-electron transfer together with the multi-step reduction of CO2, including cleavage of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds and formation of C–H bonds, leads to several different products (as summarized in the Table 1) depending on the specific active sites. Furthermore, since CO2 reduction is commonly performed in aqueous solution, water can be used as the electron donor. Thus the reduction of water into H2 often occurs as a competing reaction, resulting in electron consumption and decreased product selectivity.27 An anhydrous solvent (typically acetonitrile) may prevent H2 evolution; however, it is undoubtedly not an economical strategy, and the lack of a hydrogen source also prohibits hydrocarbon products. As such, engineering active sites is the essential solution to obtain desired products with high activity and selectivity. Generally, the active sites are mainly closely related to the surface reactions, but also may influence charge transfer and light harvesting. In the following section, we will discuss in detail the developed strategies for engineering active sites and how they impact the key steps in improving the performance in photocatalytic CO2 reduction.
O bonds and formation of C–H bonds, leads to several different products (as summarized in the Table 1) depending on the specific active sites. Furthermore, since CO2 reduction is commonly performed in aqueous solution, water can be used as the electron donor. Thus the reduction of water into H2 often occurs as a competing reaction, resulting in electron consumption and decreased product selectivity.27 An anhydrous solvent (typically acetonitrile) may prevent H2 evolution; however, it is undoubtedly not an economical strategy, and the lack of a hydrogen source also prohibits hydrocarbon products. As such, engineering active sites is the essential solution to obtain desired products with high activity and selectivity. Generally, the active sites are mainly closely related to the surface reactions, but also may influence charge transfer and light harvesting. In the following section, we will discuss in detail the developed strategies for engineering active sites and how they impact the key steps in improving the performance in photocatalytic CO2 reduction.
| Product | Reaction | E° (V vs. NHE) |
|---|---|---|
| Hydrogen | 2H2O + 2e− → 2OH− + H2 | −0.41 |
| Methane | CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 |
| Methanol | CO2 + 6H+ + 6e− → CH3OH + H2O | −0.39 |
| Carbon monoxide | CO2 + 2H+ + 2e− → CO + H2O | −0.51 |
| Formic acid | CO2 + 2H+ + 2e− → HCOOH + H2O | −0.58 |
| Ethane | 2CO2 + 14H+ + 14e− → C2H6 + 4H2O | −0.27 |
| Ethanol | 2CO2 + 6H+ + 6e− → C2H5OH + H2O | −0.33 |
| Oxalate | 2CO2 + 2H + 2e− → H2C2O4 | −0.87 |
3. Engineering active sites
To achieve high activity and selectivity in photocatalytic CO2 reduction, the key point is to construct specific and effective active sites for facilitating the adsorption and activation of CO2 molecules. Generally, light absorbers lack efficient active sites. For this reason, various strategies have been developed for engineering active sites on the surface of diverse light absorbers, including creating surface vacancies, doping heteroatoms, modifying functional groups, loading metal nanoparticle cocatalysts, engineering crystal facets, dispersing heterogeneous single-site catalysts, anchoring homogeneous single-site catalysts, and modulating metal nodes/organic linkers in MOFs. Whatever the strategies are, the essence of engineering active sites is to adjust the localized electronic structure and environment to facilitate the adsorption and activation of CO2 molecules.3.1 Surface vacancies
Perfect lattices or saturated coordination of intrinsic surfaces usually exhibit poor activity. Surface vacancies such as oxygen vacancy and metal vacancy have been proven to participate in many chemical reactions and also can serve as active sites playing an essential role in governing CO2 adsorption and activation.10,28–31 Enormous efforts have been dedicated to creating oxygen vacancies on the surface of light-harvesting semiconductors.32 Lee et al. have investigated CO2 adsorption at oxygen vacancies on the (110) facet of TiO2 by scanning tunneling microscopy (STM).33 After exposure to CO2 at 55 K, as shown in Fig. 2a, the oxygen vacancy sites occupied by the CO2 features (marked with squares) are brighter than the bridging hydroxyl features (OHb) (marked with circles), suggesting the interaction between the CO2 molecule and vacancy sites. The corresponding schematic of a CO2 molecule adsorbed at an oxygen vacancy (VO) site is shown in Fig. 2b. The adsorption and activation of CO2 on the surface oxygen vacancy sites also can be revealed by the calculated adsorption energy (Eads).32 The existence of oxygen vacancies greatly promoted CO2 adsorption with −0.377 eV for Sr2Bi2Nb2TiO12 with abundant oxygen vacancies in contrast to −0.125 eV for the sample without oxygen vacancies. Such a strong covalent interaction between an oxygen vacancy and CO2 molecule is confirmed by the charge difference distribution and electronic location function (Fig. 2c–f). Moreover, the oxygen vacancies not only can serve as active sites to enhance CO2 adsorption but also broaden light absorption. For example, the presence of oxygen vacancies in the Bi2WO6 photocatalyst can extend the light-harvesting from ultraviolet to the near-infrared region for photocatalytic CO2 reduction, which is attributed to the newly generated in-gap defect band.34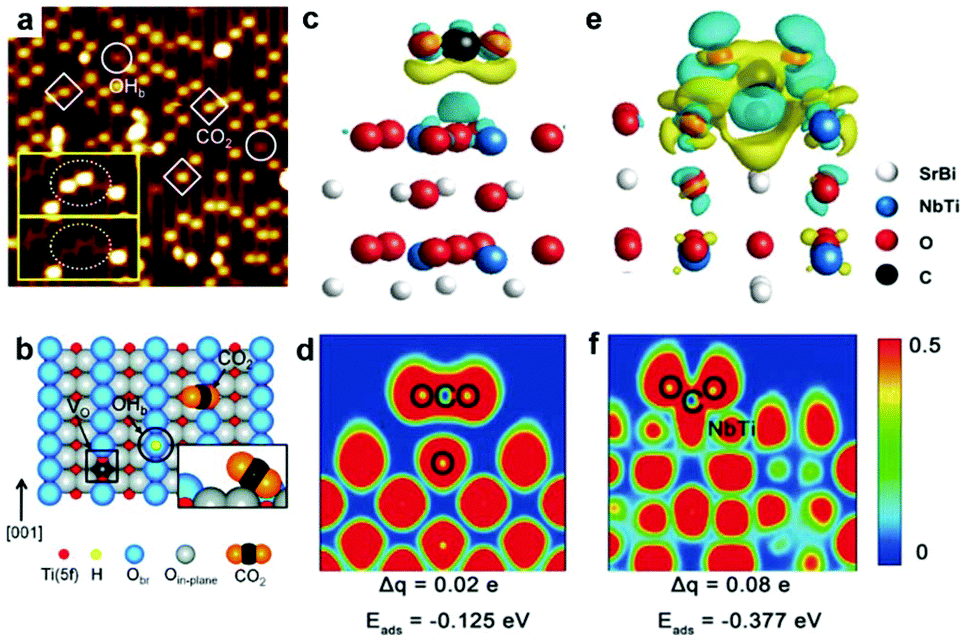 | ||
| Fig. 2 (a) STM image (1.5 V, 5 pA, 15 nm × 15 nm) of the TiO2 (110) surface after adsorption of CO2 at 55 K. The CO2 and OHb features are marked with squares and circles, respectively. (b) Schematic illustration of a bridging hydroxyl (OHb) (black circle), an oxygen vacancy defect (VO) (black square), and a CO2 molecule adsorbed at a VO site on the reduced TiO2 (110) surface. Reproduced with permission from ref. 33, copyright 2011 American Chemical Society. (c) Differential charge distribution and (d) electronic location function for the oxygen-free sample. (e) Differential charge distribution and (f) electronic location function for the oxygen-abundant sample. Reproduced with permission from ref. 32, copyright 2019 John Wiley and Sons. | ||
Although the oxygen vacancies have shown good performance in photocatalytic CO2 reduction, the regeneration of the oxygen vacancies is still a tough problem since pristine oxygen vacancies are easily filled up by the O atom from CO2 during the reaction process and thus are gradually inactivated with poor stability. To address this issue, Zhang et al. reported a highly efficient BiOCl nanoplate with reproducible oxygen vacancies for the photocatalytic reduction of CO2 to CO and CH4.31 The photoreduction-induced oxygen vacancy can be easily regenerated by light irradiation due to the high density of the oxygen atom and the low energy of the Bi–O bond. As schematically illustrated in Fig. 3a, oxygen regeneration is induced by the process that outer Bi can easily escape from the lattice after illumination and then the high density surface O atoms with a low formation energy are readily released to balance the charge, rendering the formation of oxygen vacancies. The oxygen vacancies recovered in light-treated BiOCl (denoted as BiOCl-LT) can be further confirmed by XPS (Fig. 3b and c), in which the valence change from Bi3+ (partially converted) to Bi0 and Bi3+x after light irradiation promotes the high binding energy component in the O 1s XPS spectra.
 | ||
| Fig. 3 (a) Possible reaction pathways for CO2 photoreduction with H2O on BiOCl nanoplates. (b) XPS spectra of the Bi4f region and (c) the O1s region for BiOCl and BiOCl-LT nanoplates. Reproduced with permission from ref. 31, copyright 2015 Springer. | ||
In addition to oxygen vacancy, metal vacancies also can serve as excellent active sites for photocatalytic CO2 reduction. Xie's group have built an ideal model of two-dimensional ZnInS2 atomic layers with a tunable concentration of Zn vacancies to elucidate the relationship between defect sites and photocatalytic CO2 reduction.35 Aberration-corrected scanning transmission electron microscopy (Fig. 4a–e) directly manifests the distinct zinc vacancy concentrations at the atomic level, which is also confirmed by electron spin resonance (ESR) analysis (Fig. 4f). The abundant Zn vacancies not only act as active sites for higher CO2 adsorption capacity, but also provide increased photo-absorption, stronger surface hydrophilicity and higher efficiency of electron–hole separation. As a result, ultrathin ZnIn2S4 nanosheets with rich zinc vacancies exhibit a CO yield rate of 33.2 μmol g−1 h−1, around 3.6 times higher than that of the ultrathin ZnIn2S4 nanosheets with limited zinc vacancies.
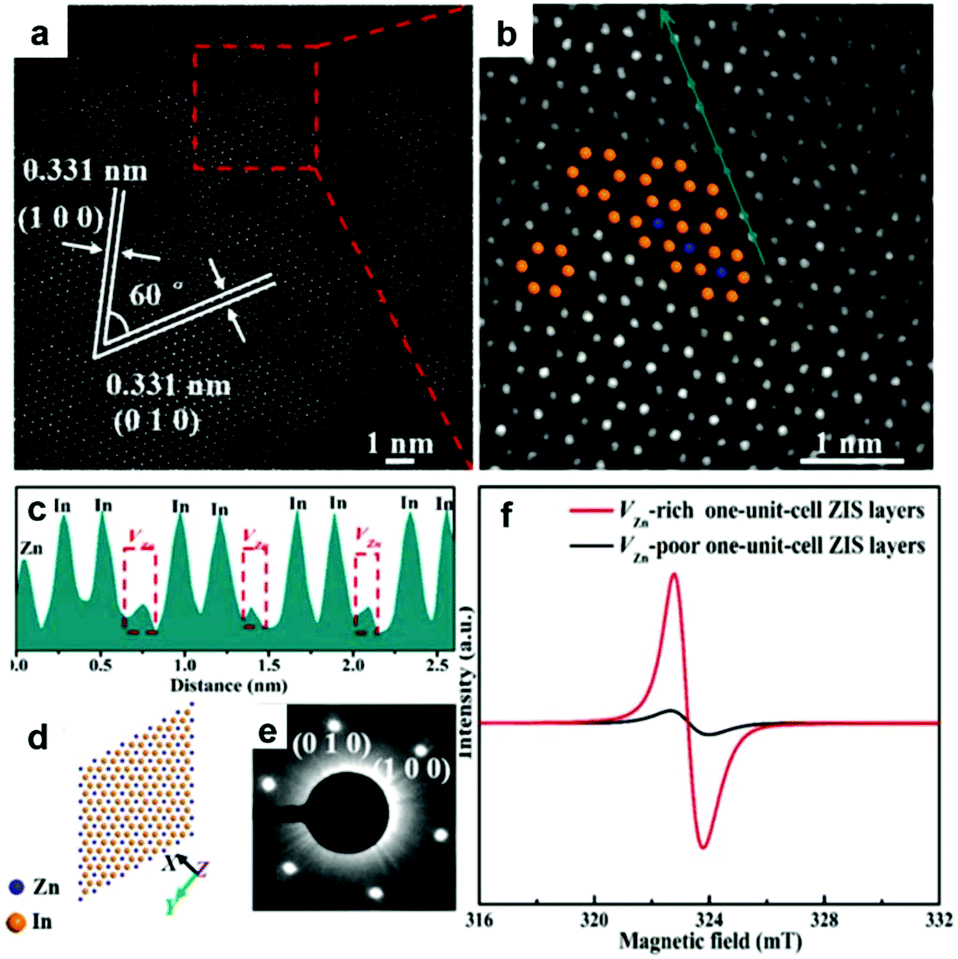 | ||
| Fig. 4 Characterization of VZn-rich one-unit-cell ZnIn2S4 (ZIS) layers obtained at 200 °C. HAADF-STEM images (a, b) and intensity profile (c) corresponding to the dark cyan arrow in panel b. The corresponding crystal structures (d) and SAED patterns (e). (f) EPR spectra of VZn-rich and VZn-poor one-unit-cell ZIS layers. Reproduced with permission from ref. 35. copyright 2017 American Chemical Society. | ||
Overall, engineering surface vacancies is an important strategy for tuning photocatalytic CO2 reduction. This strategy provides abundant adsorption sites, promotes electron–hole pair separation efficiency and fulfils broad-spectrum light harvesting, thus leading to excellent activity for photocatalytic CO2 reduction. However, the density and stability of the vacancies still need to be optimized for more active and durable photocatalytic CO2 reduction given that oxygen vacancy can be filled up leading to downward activity during the CO2 dissociation process.31
3.2 Heteroatom doping
The local electronic structure of the active sites is a critical factor determining their activity and selectivity in photocatalytic CO2 reduction. Introducing heteroatoms into semiconductors or quantum dots as active sites or nearby the active sites is an alternative approach to modify their local electronic structure for governing activity and selectivity in photocatalytic CO2 reduction. To date, various metal cations (i.e., Fe,36 Co,37 Ni,38,39 Cu,40 In,41 Zr42) and nonmetal atoms (i.e., S,43 P,44 B,45 N,46 F,47 I48) have been investigated as dopants in semiconductor lattices to modulate electronic structures or serve as active sites for photocatalytic CO2 reduction. Wang et al. have doped Co into BiVO4 to increase the electron density around O anions, serving as a Lewis base active site to facilitate CO2 activation and electron transfer to CO2 molecules.37 In addition to activity, the correlation between elemental doping and selectivity regulation for photocatalytic CO2 reduction has also been established. Xie's group have reported that Ni-doped ZnCo2O4 atomic layers exhibit a 3.5-time higher CO selectivity than undoped ZnCo2O4 atomic layers for photocatalytic CO2 reduction to CO and CH4.38 Moreover, the competing H2 evolution can be significantly suppressed by heteroatom doping. Our research group has developed Ni-doped CdS quantum dots for CO2 reduction into CO and CH4.39 The Ni2+ dopants serving as active sites can substantially suppress H2 evolution as compared with pristine CdS (Fig. 5a). In addition, the photoexcited electrons can be effectively trapped at the doped Ni atoms in sulfur vacancies and relax to the shallow charge trap state, thus substantially promoting charge separation, as confirmed by the reduced intensity of the signal for Ni2+ upon light irradiation in ESR (Fig. 5b). This argument was further confirmed by the enhanced photoluminescence (PL) around 600 nm (Fig. 5c), and supported by the reduced PL at 500–550 nm for radiative recombination in the presence of methyl viologen (MV2+) (Fig. 5d), an agent to sacrifice surface electrons of QDs. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) suggests that the Ni2+ active sites capture photo-generated electrons and adsorb CO2 molecules to form the Ni-COOH complex (Fig. 5e and f), as indicated by the peaks located at 1305 cm−1, 1412 cm−1, and 1640 cm−1 corresponding to C–OH, C–O, and CO stretching, respectively. Subsequently, the Ni–COOH complex dehydrates to the Ni–CO complex as indicated by two stretching bands at 1903 and 1982 cm−1, which accounts for the different selectivity.
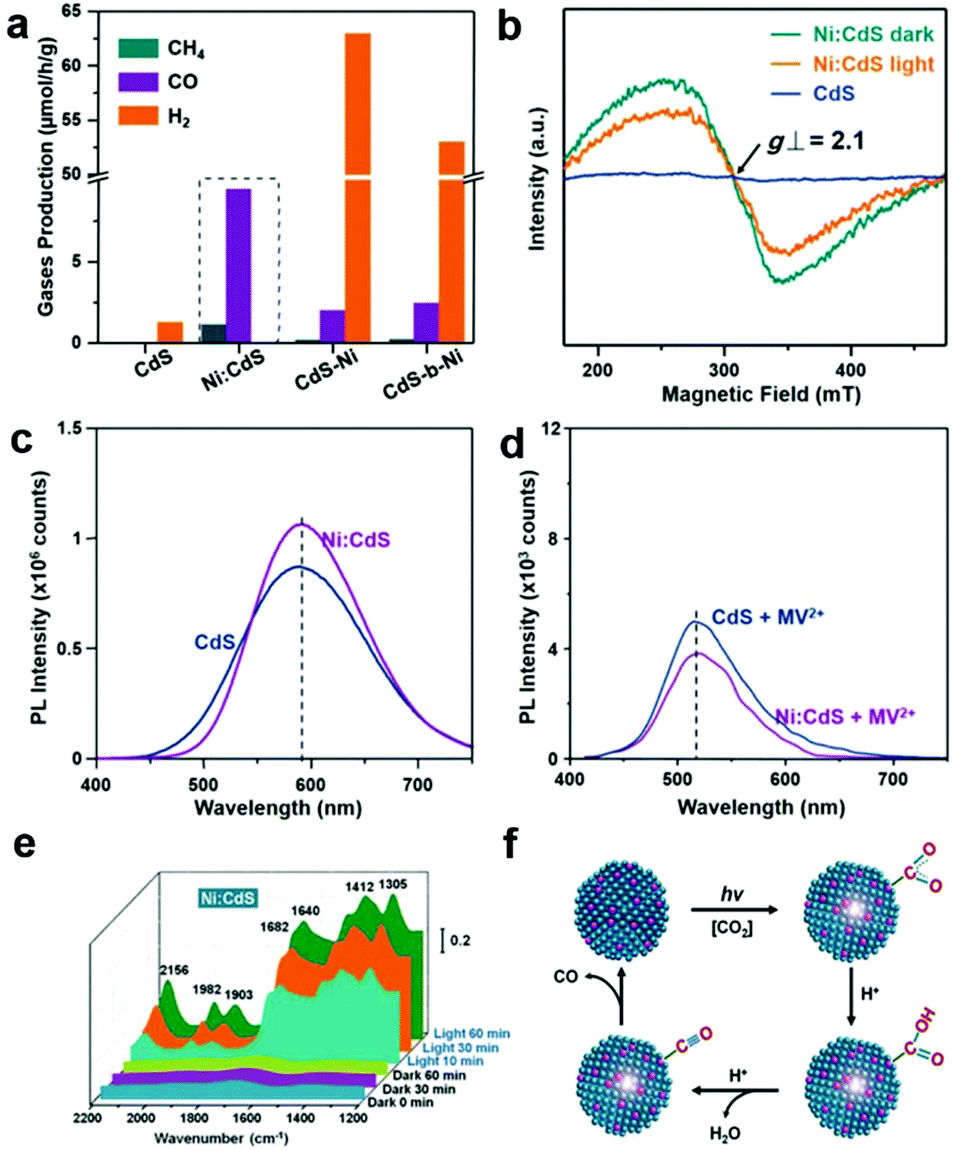 | ||
Fig. 5 Gaseous products by Ni-CdS (0.26%) QDs in photocatalytic CO2 reduction. (b) EPR spectra of CdS QDs and Ni-CdS (0.26%) QDs before and after illumination. (c) PL emission spectra of bare CdS and Ni(0.26%):CdS QDs. (d) PL emission spectra of bare CdS and Ni(0.26%):CdS QDs in the presence of MV2+. (e) In situ FTIR spectra. (f) Proposed mechanism for photocatalytic CO2 reduction using Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CdS QDs. Reproduced with permission from ref. 39, copyright 2018 John Wiley and Sons. :CdS QDs. Reproduced with permission from ref. 39, copyright 2018 John Wiley and Sons. | ||
Apart from metal dopants, non-metals also can be introduced into light-absorption substrates. Non-metal doping into graphitic carbon nitride (g-C3N4) to modulate coordination moieties and local electronic structures is widely investigated.49 Fu et al. have doped B into the g-C3N4 scaffold between adjacent tri-s-triazine units by coordination with two-coordinated N atoms.45 The B dopants serving as active sites can change the adsorption of the CO intermediate to promote CH4 production. Furthermore, theoretical calculations reveal that B atom doping can make the electron excitation much easier and improve the charge transfer and localization as well as reaction dynamics, thus significantly enhancing the catalytic performance. Shown et al. have fabricated a carbon-doped SnS2 nanostructure for photocatalytic CO2 reduction, in which the carbon dopants are incorporated as excellent electron acceptor centers to suppress the charge recombination during the electron transfer process.50 Furthermore, carbon doping shrank the band gap from 2.54 eV to 2.43 eV, and significantly enlarged the light absorption range from a visible-light wavelength of 530 nm toward a longer wavelength, which is the most intense region in the solar spectrum. Thus the synthesized SnS2-C photocatalyst significantly enhanced the activity in selective photocatalytic CO2 reduction to acetaldehyde under visible light with a photochemical quantum efficiency of above 0.7%.
Overall, doping metal and nonmetal atoms into light-harvesting quantum dots and semiconductors can significantly impact the CO2 adsorption state as the addition of the heteroatoms can induce increased localized electron density, in favor of transferring electrons to CO2 molecules. Meanwhile, dopants in a pristine lattice can generate a new impurity level below the maximum conduction band where photo-generated electrons relax to, which is beneficial for photo-generated electron–hole separation and broad-spectrum light absorption.28,51 Therefore, heteroatom doping is an alternative strategy for integrating active sites to bind CO2 molecules, modulating the local electronic structure nearby adsorbed CO2 molecules to regulate selectivity, and generating defect states or impurity energy levels so as to improve the light utilization efficiency and electron–hole separation efficiency.
3.3 Surface acidity and alkalinity
Lewis alkalinity refers to the ability of giving an electron pair to a Lewis acid molecule.52 Since CO2 is a weak acidic molecule, Lewis basic sites possess the capacity of capturing and adsorbing CO2 molecules. As such, constructing Lewis basic sites is an alternative method of generating CO2 binding sites. As discussed above, it is a versatile method to engineer oxygen vacancies on the surface of metal oxide semiconductors serving as active sites. The hydroxyl groups tend to produce on oxygen vacancies in the presence of water forming Lewis basic sites, which are beneficial for the adsorption of weak acidic CO2 molecules.53 Some solid basic hydroxides or metal oxides such as NaOH and MgO can interact with the carbon atom in CO2 because of their basic nature.52,54,55 Damien et al. have investigated the actual structure of the CO2 species adsorbed on the magnesium oxide surface by combining experimental infrared spectroscopy and density functional theory (DFT) study.52 In addition, modifying a surface with hydroxyl groups can be realized by alkali treatment such as NaOH.13 Meng et al. have modified TiO2 with NaOH to promote CO2 chemisorption.55 Increasing the loading amount of NaOH can facilitate the adsorption of CO2 on a surface with enhanced CH4 production (Fig. 6a and b). However, excessive NaOH loading leads to surface encapsulation and induces the aggregation of TiO2 nanoparticles, which hinders the CH4 production. Apart from basic hydroxides and metal oxides as common Lewis bases, surface functionalization with a basic group such as an amino group has also been widely employed for CO2 capture.56 Liu et al. have reported a one-step preparation of P-doped C3N4 nanocrystals by the thermal annealing method.44 The interstitial doping of phosphorus creates an amino-rich surface confirmed by the Fourier transform infrared spectrum (Fig. 6c), which is conducive to the adsorption of CO2 and conversion into CO and CH4 (Fig. 6d).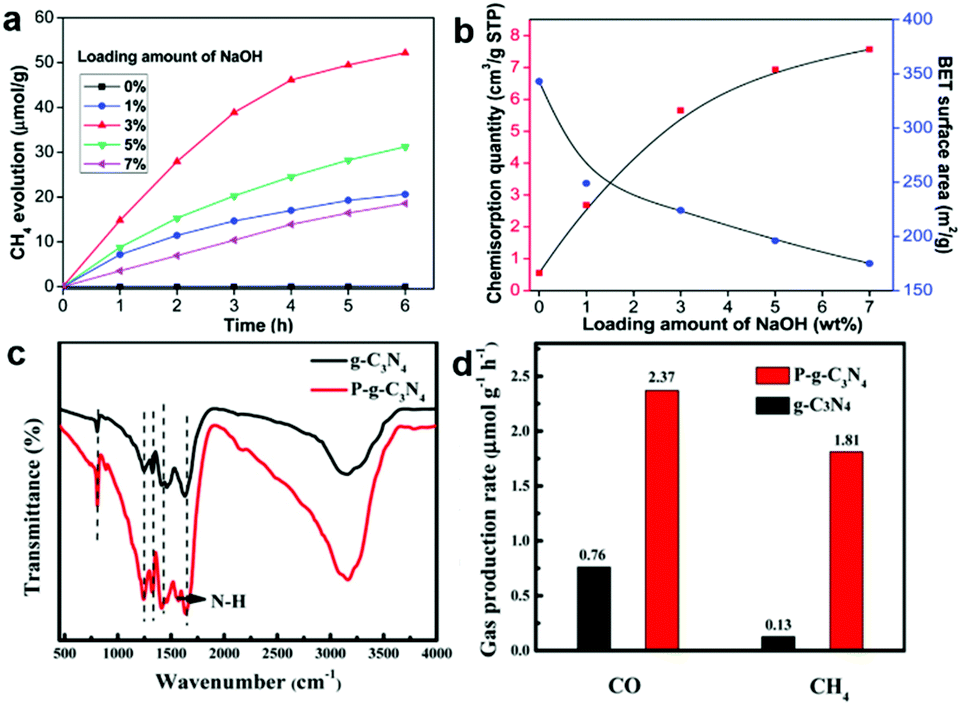 | ||
| Fig. 6 (a) Time dependence of CH4 evolution. (b) Cumulative CO2 pulse chemisorption quantity and BET surface area of NaOH-TiO2 composites. Reproduced with permission from ref. 55, copyright 2014 Royal Society of Chemistry. (c) FT-IR spectra and (d) CO and CH4 yield. Reproduced with permission from ref. 44, copyright 2018 American Chemical Society. | ||
Furthermore, frustrated Lewis pairs (FLPs) consist of sterically hindered Lewis base–acid pair, which can serve as active sites for both binding CO2 and heterolytically dissociating H2. In view of this, FLPs provide a promising approach for CO2 activation and subsequent stepwise hydrogenation to produce hydrocarbon fuels. Surface modification of hydroxyl groups on metal oxides to form surface FLPs, a Lewis basic surface hydroxide site and a Lewis acidic coordinatively unsaturated surface metal site nearby an oxygen vacancy, have been reported as active sites to effectively improve CO2 adsorption and activation.57,58 However, FLPs require significant steric hindrance to prevent the formation of inactive Lewis base-Lewis acid adducts in solution.59 The bulky substituents in FLPs may increase the energy required to activate the CO2. Thus it is important to explore other possibilities to stabilize Lewis pairs (LPs) that do not involve steric hindrance. To address this issue, Johnson's group has proposed a strategy for incorporating LPs into porous MOFs for the catalytic reduction of CO2 with H2.59 As shown in Fig. 7a–c, Lewis pairs (LPs) possessing both Lewis acid (B) and base (Nb) sites within a single molecule have been successfully incorporated into UiO-66 MOFs as excellent candidates for both CO2 adsorption and subsequent stepwise hydrogenation to produce formic acid. The LPs retain their chemical activity being able to facilely bind CO2. According to the calculation, CO2 can be reduced to HCOOH via a lower energy barrier pathway in contrast to the sample without LP functionalization (Fig. 7d).
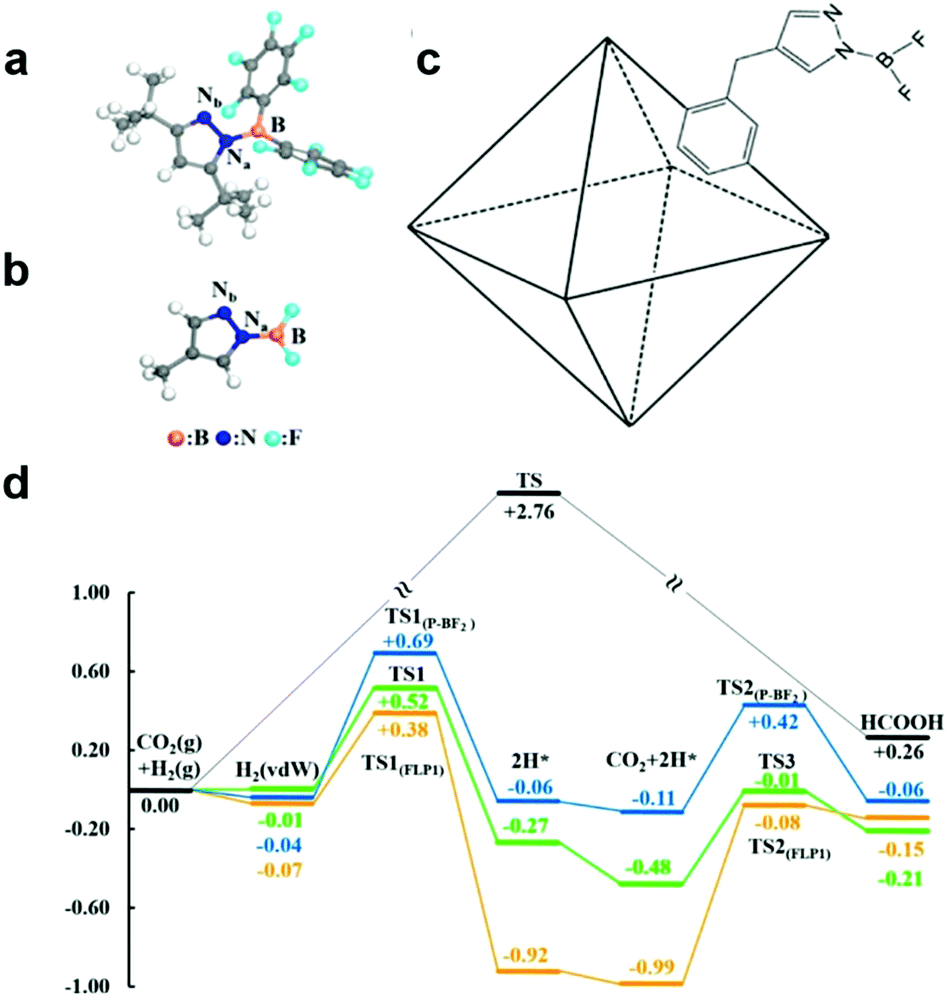 | ||
| Fig. 7 Structure of frustrated Lewis pairs (FLP1) (a) and P-BF2 (b). (c) Schematic of a UiO-66 primitive cell functionalized with P-BF2. (d) Relative potential energy surfaces for gas phase CO2 hydrogenation without a catalyst (black), catalyzed by FLP1 (orange), catalyzed by P-BF2 (blue), and catalyzed by UiO-66-P-BF2 (green). Reproduced with permission from ref. 59, copyright 2015 American Chemical Society. | ||
In brief, surface acidity and alkalinity significantly impact the adsorption and activation of CO2 molecules. Building Lewis basic sites as well as constructing FLPs or LPs on surfaces via modifying the basic group/amphiphilic molecule is a promising approach to engineer active sites for favourable CO2 adsorption and lower energy barriers in subsequent steps. However, characterization of the Lewis basic sites is vital for the deeper understanding between catalytic activity and surface state, to which more attention should be paid.
3.4 Metal nanoparticle cocatalysts
Incorporation of metal nanoparticle cocatalysts providing active sites can make great contribution in enhancing the surface reaction kinetics through lowering the activation energy, which greatly improves the activity and selectivity of photocatalysts for CO2 reduction. Beyond that, metal nanoparticle cocatalysts can significantly improve the charge separation efficiency by the formation of a Schottky junction especially for noble metals.6,60 Engineering mono-component metal nanoparticles, such as Pt, Pd, Au, Ag, Rh, Cu, Ni, and Co, as cocatalysts supported on semiconductors is a simple and effective way to construct active sites.61–64 Constructing metal nanoparticle cocatalysts could induce charge redistribution on the surface after photoexcitation, resulting in discrepant electronic acceptance ability for adsorbed intermediates. As a result, we can integrate metal nanoparticle cocatalysts to regulate product selectivity.Among all the noble metals, Pt possesses the highest work function (5.6 eV), implying strong electron-extracting ability.65 Therefore, Pt as a cocatalyst has been extensively investigated for loading onto various semiconductors.54,64,66 Xie et al. have examined a series of noble metal nanoparticle cocatalysts loaded on TiO2 for photocatalytic CO2 reduction.54 Interestingly, the yield of CH4 formation increases in the sequence of Ag < Rh < Au < Pd < Pt, in good accordance with the increase in work function Ag (4.26 eV) < Rh (4.98 eV) < Au (5.10 eV) < Pd (5.12 eV) < Pt (5.65 eV), as well as the efficiency of carrier separation. This result indicates that the strongest capacity of Pt for extracting photogenerated electrons gives the enhanced selectivity of CH4 in CO2 reduction. However, the severe competition of H2 evolution occurring likewise to Pt and Pd would consume a large quantity of the photoexcited electrons and newly released H atoms, both of which are necessary for the follow-up reduction of the CO intermediate into target CH4. Thus, the target CH4 product will be sacrificed with low selectivity inevitably. In this regard, our research group has proposed a hydridation strategy to incorporate H atoms into the lattice of metal cocatalysts prior to the photocatalytic CO2 reduction reaction.27 The hydride PdH0.43 obtained through hydriding Pd cocatalysts significantly suppressed competing H2 revolution and facilitated the conversion of the CO intermediate into the final CH4 product. As a result, the selectivity for CH4 production is increased from 3.2% to 63.6% on Pd{100} and from 15.6% to 73.4% on Pd{111}, with excellent photocatalytic durability being maintained in successive cycles (Fig. 8a–d). The enhanced performance is attributed to the fact that hydriding promotes the electron trapping on PdH0.43 sites and improves the charge separation, which ensures adequate electrons and H atoms for the reduction of CO2 into CH4 (Fig. 8e).
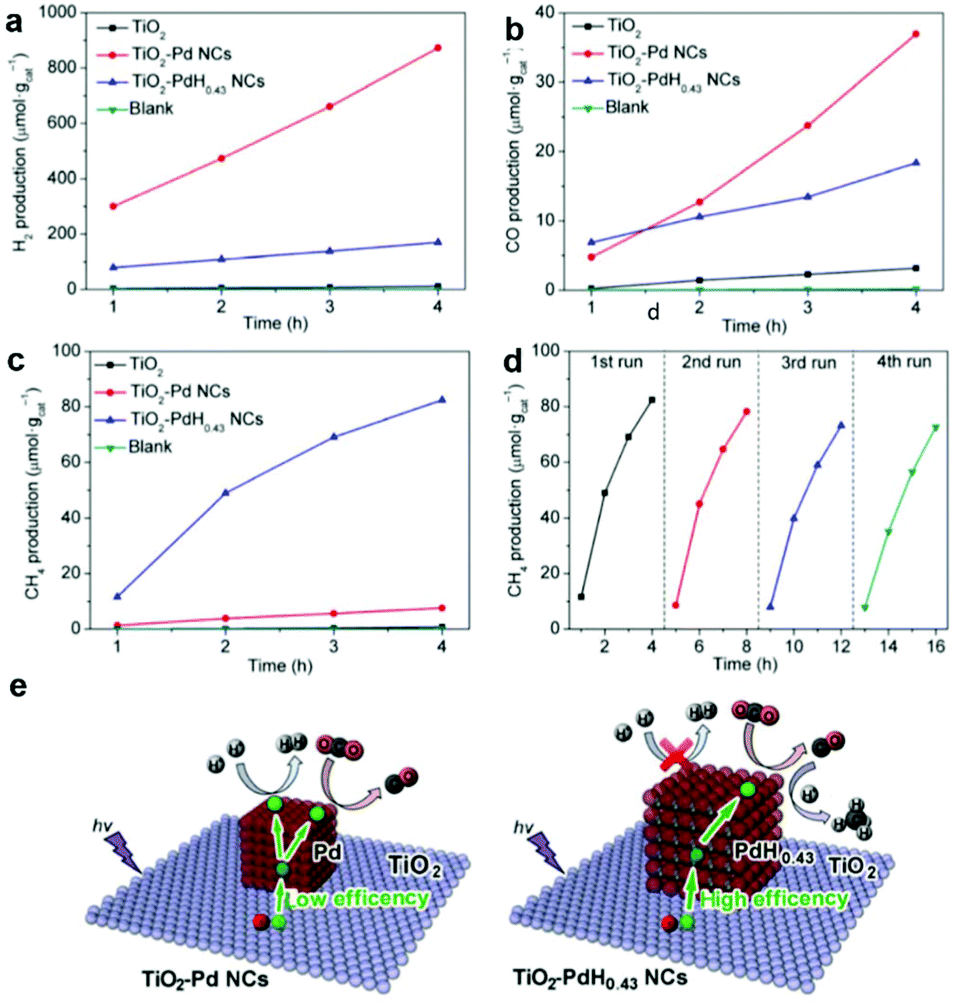 | ||
| Fig. 8 Time-dependent production of (a) H2, (b) CO, and (c) CH4 in photocatalytic CO2 reduction with bare TiO2, TiO2-Pd NCs, and TiO2-PdH0.43 NCs under UV (λ < 400 nm) irradiation. (d) Cycling CH4 evolution with TiO2-PdH0.43 NCs in photocatalytic CO2 reduction. (e) Schematics illustrating the photocatalytic reactions between CO2 and H2O for the TiO2-Pd NCs and TiO2-PdH0.43 NCs. Reproduced with permission from ref. 27, copyright 2017 Springer. | ||
The composition of the cocatalyst could play an essential role in regulating selectivity. Alloying is a promising approach to adjust the surface composition of mono-component metal cocatalysts. Binary alloy cocatalysts (i.e., Au/Cu,67 Au/Ag68) have been reported to exhibit a synergistic effect on promoting the photocatalytic CO2 reduction. Ye's group revealed that the Au–Cu bimetallic alloy nanoparticles as a cocatalyst in photocatalytic CO2 reduction exhibited significantly improved selectivity for hydrocarbon species production instead of CO evolution owing to the synergetic effect, in which Au atoms favour CO desorption and Cu atoms boost the formation of hydrogenation species.67 Given the abovementioned facts, the composition of the cocatalyst can be regulated to affect the local charge density at the adsorption site, which would influence the adsorption of CO2 molecules as well as intermediates. Based on their intrinsic nature, various metals could display different abilities for the absorption of intermediates. For instance, there is strong interaction between Pt and CO whereas only weak interaction takes places between Ag and CO.65 Thus understanding the inherent electronic properties of different metals is instructional to construct suitable metal nanoparticle cocatalysts in terms of regulating activity and selectivity in photocatalytic CO2 reduction.
In general, a semiconductor serving as a light absorber requires a suitable band structure, in which both the conduction band and valence band can have sufficient redox potentials to drive the corresponding reduction/oxidation reactions.69 In photocatalytic reactions, tuning reduction activity based on the band bending near the semiconductor surface can be achieved by loading metal nanoparticle cocatalysts.70 When the semiconductor contacts with metal nanoparticles forming an interface, their different Fermi level (Ef) and work function will result in the formation of an internal field that drives electrons to flow toward the more positive Ef side until achieving an equilibrium state, leading to an upward bending of the band edge for the n-type semiconductor, but a downward band bending for the p-type semiconductor.3,71 The surface with upward band bending exhibits higher oxidation but lower reduction activities, while the surface with downward band bending results in the reverse outcome.
3.5 Facet engineering
Diverse morphologies of metallic cocatalysts and semiconductors have been fabricated for photocatalytic CO2 reduction and exhibited different performance. Substantially, the morphology is determined by the proportion of miscellaneous exposed facets. A chosen facet can exhibit a unique electronic structure owing to its specific atomic arrangement and coordination, which is crucial to the adsorption of reactant molecules and desorption of intermediates.8,72 In this regard, facet engineering by altering the proportion of specific exposed facets can substantially tune the ratio of different coordination atoms on the surface and regulate surface charge density. As such, selectively exposing specific active facets would make a great contribution to the enhancement of activity.23,73,74 In contrast to low index facets, high index facets generally possess a higher density of low-coordinated atoms (e.g., steps, terraces and kinks) serving as more efficient active sites for surface catalytic reactions.75 Bai's research group has compared the performance of high-index (730) facet-covered PtCu concave nanocubes and (100) facet-enclosed PtCu nanocubes as cocatalysts loading on light-harvesting C3N4 for the photocatalytic reduction of CO2 to CH4.75 In comparison with low-index (110) facets, high-index (730) facets showed a higher yield of CO and CH4 on account of their higher density of low-coordinated Pt atoms as catalytically active sites and higher CO2 adsorption energy. In parallel, Yu's group has demonstrated that tetrahedral Pd nanoparticles with exposed {111} facets functioned as a more efficient cocatalyst loading on g-C3N4 for photocatalytic CO2 reduction, as compared to cubic Pd nanoparticles with exposed {100} facets.76 DFT calculations confirmed that the Pd{111} surface showed more preferable CO2 adsorption capability and CH3OH desorption capability as compared to the Pd{100} surface.In addition to metallic cocatalysts, facet engineering also works for metal oxide semiconductors. Gao et al. have reported that hexagonal platelet Co3O4 exposing {112} facets enable superior activity to {110} facets for the photocatalytic reduction of CO2 to CO along with a visible-light photo-sensitizer.77 DFT calculations (Fig. 9a) further confirmed that the exposed {112} facets favour the adsorption of CO2 and the subsequent reduction to CO via COOH, which accounts for the facet effect on photocatalytic CO2 reduction. Constructing co-exposed crystal facets within a single crystal is regarded as an attractive strategy for realizing efficient surface charge separation, in which the photo-generated electrons and holes would transfer to anisotropic facets for spatial separation. Toward this strategy, Yu's group has fabricated and optimized the ratio of coexposed {101} and {001} facets within a single TiO2 particle, forming a “surface heterojunction” toward enhanced activity in the photocatalytic reduction of CO2 to CH4.73 DFT calculations for the electronic structure (Fig. 9b and c) reveal that the Fermi levels of {001} facets and {101} facets are hauled to equal due to their contact, forming facet heterojunctions to facilitate the transfer and separation of photogenerated electrons and holes. Similarly, Chen et al. have synthesized BiOIO3 nanoplates with an adjustable proportion of exposed {010} top facets to {100} lateral facets by altering the thickness.78 The decrease in thickness of BiOIO3 nanoplates tuned the ratio of {010}/{100} facet junctions (Fig. 9d–h), which significantly shortened the diffusion pathway for charge carriers and allowed the efficient separation of photogenerated electrons and holes on the anisotropic facets. As a result, the activity for the photocatalytic reduction of CO2 to CO was greatly improved.
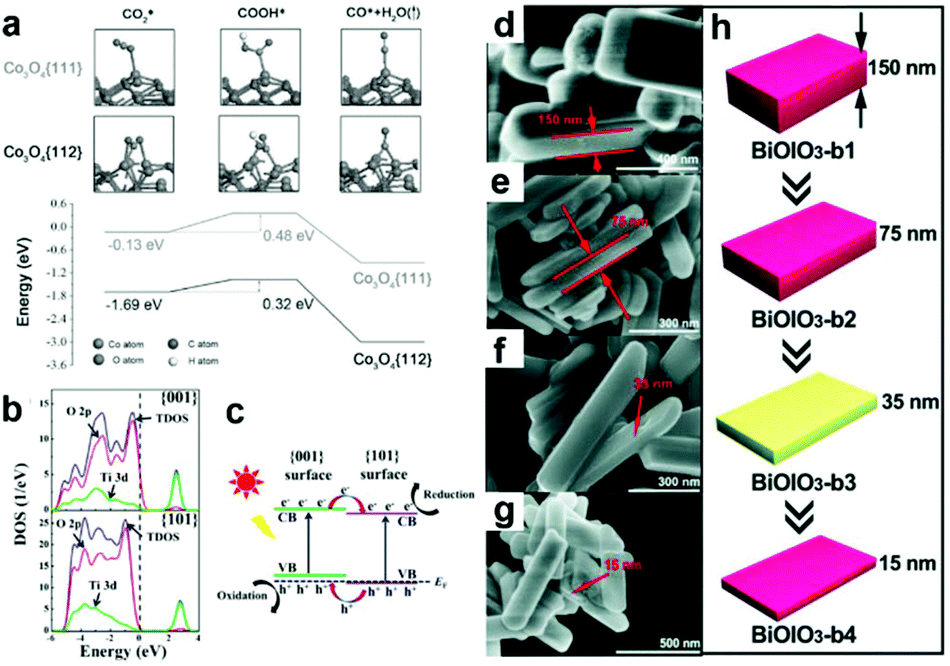 | ||
| Fig. 9 (a) DFT calculation of adsorption and reduction of CO on Co3O4 surfaces. Free energy diagram describes the COOH* intermediate from CO2 reduction to CO on {111} and {112} surfaces of Co3O4 hexagonal platelets. Reproduced with permission from ref. 77, copyright 2016 John Wiley and Sons. (b) Density of states (DOS) plots for {101} and {001} surfaces of anatase TiO2. (c) Illustration of {001} and {101} surface heterojunctions. Reproduced with permission from ref. 73, copyright 2014 American Chemical Society. (d and e) SEM images of BiOIO3 with various thickness. (h) A schematic illustration of the morphology evolution for BiOIO3 with various thicknesses. Reproduced with permission from ref. 78, copyright 2018 John Wiley and Sons. | ||
Overall, facet engineering is an effective strategy for selectively exposing catalytically active sites by modulating the atomic arrangement on surfaces, which has a great impact on molecular adsorption and desorption of intermediates. Furthermore, co-exposure of multi-facets allows efficient charger transfer and separation by spatially separating the photogenerated electrons and holes on the anisotropic facets.
3.6 Heterogeneous single metal sites
Generally in heterogeneous catalysts, high dispersion of the active sites results in good catalytic performance. In this regard, great efforts have been focused on highly dispersed catalysts with ultrasmall sizes, even at the atomic scale. Fabrication of isolated metal active sites plays an unprecedented role in governing the activity and selectivity of heterogeneous metal catalysts.15–17,79,80 As we have discussed in the section above, the highly efficient cocatalysts in photocatalytic CO2 reduction are commonly noble metals. To maximize the efficiency of each active site and minimize the cost, single-site catalysts are intriguing and have attracted widespread interest in photocatalytic CO2 reduction considering their merits of high atomic utilization and easy modification of both the spatial and electronic environment of active sites. To this end, a variety of single metal sites, including isolated ions, atoms, and molecular complexes, have been engineered on light-harvesting supports to explore their potential in photocatalytic CO2 reduction.One goal for solar-driven CO2 conversion is to develop visible-light photocatalysts. Huang et al. have engineered single Co2+ sites on carbon nitride (C3N4) by a simple microwave-assistant deposition method, in which framework nitrogen atoms can coordinate with and activate Co2+ sites demonstrating excellent activity and selectivity toward visible-light photocatalytic CO2 reduction into CO.81 With the same cobalt loading, the amount of CO produced by single-site Co2+@C3N4 is almost 5 times higher than that produced by the mixture of CoCl2 and C3N4, suggesting that single Co2+ site coordinated with N on C3N4 could be activated to exhibit superior activity. Beyond semiconductors, Au nanoclusters (NCs) can serve as a potential light absorber to harvest visible light but are inert for photocatalytic CO2 reduction due to the lack of catalytic sites. In this regard, our research group has developed an effective strategy to graft various metal cations on Au NCs through bridging ligands.82 The grafted isolated metal cations (Fe2+, Co2+, Ni2+, and Cu2+) can accept photogenerated electrons from Au NCs through the bridging ligands (L-cysteine) and serve as catalytic sites for CO2 reduction. Demonstrated by DFT calculations (Fig. 10), different single metal sites possess varied d-band centers and binding energies with CO2 molecules, which allow for tuning electron transfer efficiency and CO2 activation energy to control the activity and selectivity. This work provides an effective strategy for engineering single active sites on inert light-harvesting components through bridging ligands.
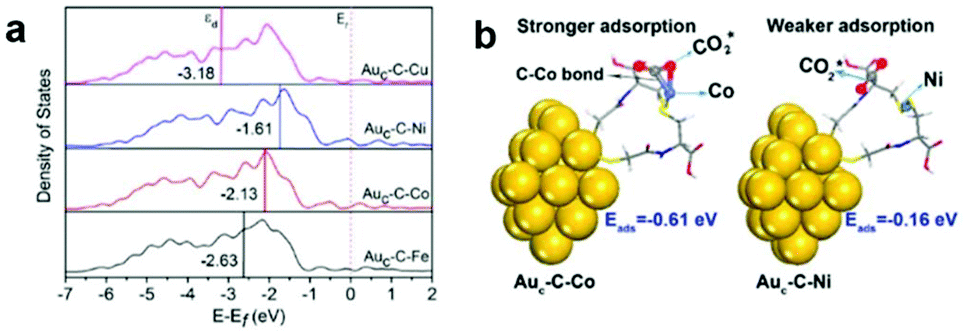 | ||
| Fig. 10 (a) Projected density of states (PDOS) of the d-orbitals of surface atoms for Auc–C–Fe, Auc–C–Co, Auc–C–Ni, and Auc–C–Cu. (b) Models for CO2 adsorption on the Auc–C–Co surface (left) and Auc–C–Ni surface (right). Reproduced with permission from ref. 82, copyright 2018 American Chemical Society. | ||
In addition to metal ions, various isolated single metal atoms also have been engineered on semiconductors for photocatalytic CO2 reduction, which enables the regulation of selectivity for high-valued hydrocarbon products such as methane. Our research group has designed highly selective active sites for the photocatalytic reduction of CO2 to CH4via isolating Cu atom in the Pd lattice supported on a TiO2 semiconductor.80 The d-band centers of isolated Cu atoms were elevated through the electronic structure coupling with surrounding Pd atoms, which enhanced the adsorption and activation of CO2, as well as suppressed competing H2 evolution. As a result, the obtained photocatalyst exhibited a high selectivity of 96% for CO2 reduction to CH4, with a production rate of 19.6 μmol gcat−1 h−1.
Compared with heterogeneous catalysts, the metal centers in homogeneous catalysts are well separated by organic ligands, which can serve as highly efficient single active sites. Engineering the active sites in homogeneous systems could be easily modulated through adjusting the metal centers with various linked organic ligands. Benefiting from the isolated catalytic sites and versatile coordination forms, homogeneous photocatalysts have shown excellent capacity in light absorption as well as high activity and selectivity for CO2 reduction.18,19,21,22,83–90 Considering that homogeneous and heterogeneous photocatalysts both have their own strengths and weaknesses, it is a promising strategy to combine together the merits of each. To this end, our research group has developed a highly efficient photocatalytic system for CO2 reduction by bridging the heterogeneous single-atom Co as an active site with a homogeneous light absorber [Ru(bpy)3]Cl2 through graphene.79 The individual and isolated Co atoms coordinated on the partially oxidized graphene nanosheets can be regarded as homogeneous catalyst analogues, which achieved an outstanding turnover number (TON) of 678 for CO production with an high turnover frequency (TOF) of 3.77 min−1. Inversely, metal molecular complexes also can be anchored on various semiconductor supports as heterogeneous single active sites. Such photocatalysts possess the merits of high TON and good durability provided by the homogeneous catalyst and heterogeneous catalyst, respectively. Kuriki et al. have designed a Ru complex anchored on C3N4 as the catalytic sites for the photocatalytic reduction of CO2 into formic acid, achieving a high TON more than 1000 (by 20 h) and an apparent quantum yield of 5.7% (under irradiation light of λ = 400 nm).91 Considering the complicated preparation process of the Ru complex and the rarity of Ru, Ye's group has designed a covalently linked hybrid photocatalyst composed of a non-noble metal based Co-porphyrin molecule catalyst and oligomers of melems (Co-POM) for photocatalytic CO2 reduction.92 The high affinity of Co active sites for CO2 and efficient electron trapping by the Co active sites contributed to the enhanced activity for CO2 reduction.
Overall, the engineering of atomically dispersed active sites in a heterogeneous catalysis system offers unique opportunities for combining the advantages of homogeneous and heterogeneous catalysis, thereby building a bridge between them. Single-atom photocatalysts exhibit high atomic utilization, low cost and high activity, which well meets the original intention for catalyst design. However, there still remain challenges in the fabrication of spatially uniform, highly dispersed and stable single-atom photocatalysts, due to that the extremely active metal single-atoms favourably aggregate or react with the surrounding reactants to form undesired species during fabrication and reaction. For this reason, seeking and developing novel strategies to controllably synthesize single-atom photocatalysts with a high metal content and robustness is of great significance to fully show their capability in practical application.
3.7 Active sites in metal–organic frameworks
Metal–organic frameworks (MOFs) are springing up as a new class of crystalline porous materials consisting of metal nodes and organic linkers.93 By functionalizing metal nodes and organic linkers to tailor their local environments, the physical and chemical properties of MOFs can be finely tuned, making them as promising materials for small molecule adsorption and conversion.93,94 With regard to CO2 capture and conversion, MOFs can provide excellent active sites for adsorption and activation based on their unique advantages: (i) ultrahigh surface area and porous structure for gaseous uptake; (ii) diversiform metal centers and organic ligands for engineering active sites.1,95–97 Therefore, modulating different metal centers and organic linkers in MOFs provides an alternative strategy for engineering active sites toward photocatalytic CO2 reduction.Open metal nodes in MOFs bridging with ligands can serve as isolated active sites for CO2 reduction.98 Wang et al. reported a series of Fe-containing MOFs (MIL-101(Fe), MIL-53(Fe), MIL-88B(Fe)) for photocatalytic CO2 reduction to formate under visible light irradiation.99 Upon light irradiation, the electron transferred from O2− to Fe3+ center to form Fe2+, and then the unsaturated coordination of Fe nodes endowed open active sites for CO2 adsorption and activation. Beyond metal nodes, another important component of MOFs, organic linkers, also show potential for engineering active sites for facilitating CO2 adsorption and activation. The aforementioned strategy of introducing Lewis base sites on surfaces also works for MOFs. Incorporation of organic linkers bearing Lewis base sites into MOFs has been proven to greatly improve their binding affinities for CO2. For instance, organic linkers having uncoordinated amine-containing functional groups in MOFs can provide the binding sites for CO2. Such binding sites nearby the metal active sites significantly enhance the efficiency for CO2 reduction.100,101 Fu et al. have reported amine-functionalized titanium MOFs (NH2-MIL-125(Ti)) for the photocatalytic reduction of CO2 to formic acid under visible light irradiation.100 The presence of the amino functional group enhanced the interaction with CO2 molecules and also extended light absorption from 350 nm (UV-responsive MIL-125(Ti)) to 550 nm (NH2-MIL-125(Ti)) (Fig. 11a). During light irradiation, the photogenerated electrons transfer from the organic ligand to the Ti4+ node in NH2-MIL-125(Ti) generating Ti3+, which is the active site for CO2 adsorption and reduction (Fig. 11b). Apart from amine-functionalized ligands, other nitrogen containing ligands including pyrimidines, azoles and triazines, could also be incorporated into MOFs as Lewis base sites for CO2 capture.
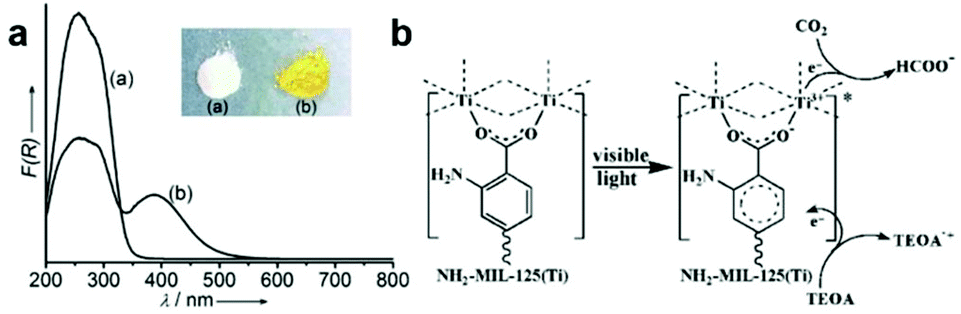 | ||
| Fig. 11 (a) UV-vis spectra of MIL-125(Ti) and NH2-MIL-125(Ti). (b) Proposed mechanism of CO2 reduction over NH2-MIL-125(Ti) under visible light irradiation. Reproduced with permission from ref. 100, copyright 2012 John Wiley and Sons. | ||
The active sites in MOFs are not limited to their own components. In addition to the original metal nodes and organic linkers in MOFs, the single sites or metal complexes introduced into the MOF structure could also serve as active sites, providing an alternative strategy to engineer active sites in MOFs for CO2 conversion.59,97,102–104 Lin and coworkers have reported the integration of different molecular complexes in UiO-67(Zr) for photocatalytic CO2 reduction under visible light irradiation.102 Beyond that, Yaghi’ s group has simultaneously encapsulated Ag nanocube and Re molecular complexes into a zirconium MOF.104 Utilizing intensified near-surface electric fields at the surface of Ag nanocubes, the spatially confined photoactive Re centers resulted in a 7-fold improvement of CO2-to-CO conversion under visible light.
In brief, designing appropriate metal nodes and organic linkers, as well as incorporating additional functional moieties and metal sites into MOFs, opens up a promising method of engineering active sites for photocatalytic CO2 reduction. However, one main challenge to be resolved is that most of the MOFs suffer from inferior stability under long-time light irradiation in aqueous solutions.94
4. Summary and outlook
In this review, enormous achievements have been witnessed in the photocatalytic conversion of CO2 by taking advantage of engineering active sites (Table 2). Engineering active sites can dramatically improve catalytic activity, and regulate the reaction pathway achieving high selectivity for target products. The developed strategies for engineering active sites on surfaces and in open frameworks have been systematically summarized, including creating surface vacancies, doping heteroatoms, modifying functional groups, loading metal nanoparticles, engineering crystal facets, dispersing heterogeneous single-site catalysts, anchoring homogeneous single-site catalysts, and modulating metal nodes or organic linkers in MOFs. Whatever their existing forms are, the essential feature of engineering active sites is to tune the surface state and local electronic structure of the active sites, which significantly influences the adsorption and activation of the CO2 molecule, broadens light absorption and promotes photogenerated charge separation and transfer.| Method | Catalyst | Products | Reaction conditions | Ref. | |
|---|---|---|---|---|---|
| a LDH: layered double hydroxides; b QDs: quantum dots; c TEOA: triethanolamine; d MeCN: acetonitrile; e Au-GSH NCs: glutathione-protected Au nanoclusters. | |||||
| Surface vacancy | Oxygen vacancy | Sr2Bi2Nb2TiO12 | CO, 17.11 μmol g−1 h−1 | NaHCO3, H2SO4 (4 M), 300 W Xe lamp | 32 |
| Oxygen vacancy | ZnAl-LDH | CO, 7.6 μmol g−1 h−1 | H2O, 300 W Xe lamp | 10 | |
| Oxygen vacancy | Bi2WO6 | CH4, 13.94 μmol g−1 | 500 W Xe lamp | 34 | |
| Oxygen vacancy | Bi12O17Cl2 | CO, 48.6 μmol g−1 h−1 | H2O, 300 W Xe lamp | 30 | |
| Zn vacancy | ZnIn2S4 | CO, 33.2 μmol g−1 h−1 | H2O, 300 W Xe lamp (AM 1.5 filter) | 35 | |
| Heteroatom doping | Ni doping | Ni-ZnCo2O4 | CO, 31.4 μmol g−1 h−1 | H2O, 300 W Xe lamp (AM 1.5 filter) | 38 |
| CH4, 20.2 μmol g−1 h−1 | |||||
| Ni doping | Ni:CdS QDs | CO, ∼9 μmol g−1 h−1 | H2O, cTEOA, 300 W Xe lamp (λ ≥ 400 nm) | 39 | |
| CH4, ∼1 μmol g−1 h−1 | |||||
| Co doping | Co-BiVO4 | CH4, 3.57 μmol (4 h) | H2O, 25 W ultraviolet lamp (λ = 284 nm) | 37 | |
| B doping | B-C3N4 | CH4, 0.16 μmol g−1 h−1 | 300 W Xe lamp | 45 | |
| I doping | I-TiO2 | CO, 2.4 μmol g−1 h−1 | H2O vapor, 450 W Xe lamp (λ ≥ 400 nm) | 48 | |
| F doping | F-TiO2−x | CH4, 5.57 μmol g−1 h−1 | 300 W Xe lamp (AM 1.5 filter) | 47 | |
| S doping | S-g-C3N4 | CH3OH, 1.12 μmol g−1 h−1 | NaHCO3, HCl (4 M), 300 W Xe lamp | 43 | |
| Surface basic site | NaOH | TiO2-NaOH | CH4, 52 μmol g−1 (6 h) | H2O vapour, 300 W Xe lamp | 55 |
| Metal nanoparticle cocatalyst | Pt | Pt-TiO2 | CH4, 60.1 μmol g−1 h−1 | H2O, 500 W Xe lamp | 66 |
| AuAg | AuAg-TiO2 | CO, 1813 μmol g−1 h−1 | 200 W Hg lamp | 68 | |
| CH4, 35 μmol g−1 h−1 | |||||
| AuCu | Au3Cu@SrTiO3/TiO2 | CH4, 421.2 μmol g−1 h−1 | N2H4·H2O, 300 W Xe lamp | 67 | |
| PdH0.43 | PdH0.43-TiO2 | CH4, 82.4 μmol g−1 (4 h) | H2O, 300 W Xe lamp | 27 | |
| Facet engineering | {112} | Co3O4{112} | CO, 2003 μmol g−1 h−1 | [Ru(bpy)3]Cl2, dMeCN, TEOA, H2O, | 77 |
| 300 W Xe lamp (λ > 420 nm) | |||||
| {111} | Pd{111}-C3N4 | CH3OH, 3.17 μmol g−1 h−1 | NaHCO3, H2SO4 (2 M), 300 W Xe lamp | 76 | |
| {101}/{001} | TiO2{101}/{001} | CH4, 1.35 μmol g−1 h−1 | NaHCO3, HCl (4 M), 300 W Xe lamp | 73 | |
| (730) | PtCu(730)-C3N4 | CO, 0.046 μmol h−1 | H2O, 300 W Xe lamp (780 nm - 400 nm) | 75 | |
| CH4, 0.112 μmol h−1 | |||||
| Single metal site | Co | Co1-graphene | CO, TON 678TOF, 3.77 min−1 | [Ru(bpy)3]Cl2, MeCN, TEOA, H2O, | 79 |
| 300 W Xe lamp (λ ≥ 400 nm) | |||||
| Au | Au-GSH NCs | CO, 3.45 μmol g−1 h−1 | TEOA, 300 W Xe lamp (λ ≥ 420 nm) | 82 | |
| Cu | Pd7Cu1-TiO2 | CH4, 19.6 μmol g−1 h−1 | 300 W Xe lamp (λ ≥ 400 nm) | 80 | |
| Co | Co-C3N4 | CO, 1.056 μmol g−1 (2 h) | MeCN, TEOA, Halogen lamp | 81 | |
| MOFs | Ti node/-NH2 | NH2-MIL-125(Ti) | HCOOH, 8.14 μmol (10 h) | MeCN, TEOA, 500 W Xe lamp (λ ≥ 420 nm) | 100 |
| Fe node/-NH2 | NH2-MIL-101(Fe) | HCOOH, 178 μmol g−1 h−1 | MeCN, TEOA, 300 W Xe lamp (λ ≥ 420 nm) | 99 | |
| NH2-MIL-53(Fe) | 46.5 μmol g−1 h−1 | ||||
| NH2-MIL-88B(Fe) | 30 μmol g−1 h−1 | ||||
| Metal and molecular modification | Ag-Re-based molecular-MOF | CO, 7-fold enhancement | MeCN, TEOA, 300 W Xe lamp (400-700 nm) | 104 | |
Although remarkable progress has been made in engineering active sites on surfaces and in open frameworks, there remain grand challenges and several issues that should be taken into consideration. Firstly, these photocatalysts are far from being put to practical use. The development of photocatalysts with broad-spectrum light absorption is in high demand to enhance the efficiency of light utilization as the UV region only accounts for a limited portion (5%) of natural sunlight. Thus continuous research efforts should be devoted to increase the efficiency of photocatalytic CO2 reduction by taking advantage of engineering active sites and efficiently cooperating with broad-spectrum light-harvesting units. Secondly, the bottlenecks of CO2 reduction are the competing H2 evolution and the limited selectivity toward certain products. Meanwhile, the major carbon products from photocatalytic CO2 reduction are mono-carbon products, while energy-rich multi-carbon products are greatly pursued. In this regard, the local electronic structure of the active sites should be further finely tuned to achieve real artificial photosynthesis. Thirdly, the structure-performance relationship remains ambiguous thus far. For this reason, advanced in situ characterization techniques should be applied to resolve the detailed structure and catalytic process during photocatalytic reactions, which would provide instructional guidance for designing and constructing active sites. Inspired by multidisciplinary cooperation, integrating active sites at the atomic level and resolving the underlying mechanisms at the atom/molecular level will pave the way for identifying efficient catalytic systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFA0207301), NSFC (21725102, 91961106, 91963108, and 21703220), CAS Key Research Program of Frontier Sciences (QYZDB-SSW-SLH018), and CAS Interdisciplinary Innovation Team, DNL Cooperation Fund, CAS (DNL201922).Notes and references
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637 CrossRef CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- Z. Chen, H. Zhang, P. Guo, J. Zhang, G. Tira, Y. J. Kim, Y. A. Wu, Y. Liu, J. Wen, T. Rajh, J. Niklas, O. G. Poluektov, P. D. Laible and E. A. Rozhkova, J. Am. Chem. Soc., 2019, 141, 11811–11815 CrossRef CAS PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- S. Bai, W. Yin, L. Wang, Z. Li and Y. Xiong, RSC Adv., 2016, 6, 57446–57463 RSC.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- S. Bai, L. Wang, Z. Li and Y. Xiong, Adv. Sci., 2017, 4, 1600216 CrossRef PubMed.
- C. Gao, J. Wang, H. Xu and Y. Xiong, Chem. Soc. Rev., 2017, 46, 2799–2823 RSC.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, L. J. Smith, D. O'Hare and T. Zhang, Adv. Mater., 2015, 27, 7824–7831 CrossRef CAS PubMed.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X.-D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2016, 16, 466–470 CrossRef CAS PubMed.
- L. Liu, F. Gao, H. Zhao and Y. Li, Appl. Catal., B, 2013, 134–135, 349–358 CrossRef CAS.
- W. Deng, L. Zhang, L. Li, S. Chen, C. Hu, Z.-J. Zhao, T. Wang and J. Gong, J. Am. Chem. Soc., 2019, 141, 2911–2915 CrossRef CAS PubMed.
- Z. He, J. Tang, J. Shen, J. Chen and S. Song, Appl. Surf. Sci., 2016, 364, 416–427 CrossRef CAS.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- A. Nakada, R. Kuriki, K. Sekizawa, S. Nishioka, J. J. M. Vequizo, T. Uchiyama, N. Kawakami, D. Lu, A. Yamakata, Y. Uchimoto, O. Ishitani and K. Maeda, ACS Catal., 2018, 8, 9744–9754 CrossRef CAS.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef CAS PubMed.
- C. Y. Toe, J. Scott, R. Amal and Y. H. Ng, J. Photochem. Photobiol., C, 2019, 40, 191–211 CrossRef CAS.
- C. Y. Toe, Z. Zheng, H. Wu, J. Scott, R. Amal and Y. H. Ng, Angew. Chem., Int. Ed., 2018, 57, 13613–13617 CrossRef CAS PubMed.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- Y. Zhu, C. Gao, S. Bai, S. Chen, R. Long, L. Song, Z. Li and Y. Xiong, Nano Res., 2017, 10, 3396–3406 CrossRef CAS.
- S. Bai, N. Zhang, C. Gao and Y. Xiong, Nano Energy, 2018, 53, 296–336 CrossRef CAS.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- J. Di, C. Zhu, M. Ji, M. Duan, R. Long, C. Yan, K. Gu, J. Xiong, Y. She, J. Xia, H. Li and Z. Liu, Angew. Chem., Int. Ed., 2018, 57, 14847–14851 CrossRef CAS PubMed.
- L. Zhang, W. Wang, D. Jiang, E. Gao and S. Sun, Nano Res., 2015, 8, 821–831 CrossRef CAS.
- H. Yu, J. Li, Y. Zhang, S. Yang, K. Han, F. Dong, T. Ma and H. Huang, Angew. Chem., Int. Ed., 2019, 58, 3880–3884 CrossRef CAS PubMed.
- J. Lee, D. C. Sorescu and X. Deng, J. Am. Chem. Soc., 2011, 133, 10066–10069 CrossRef CAS PubMed.
- X. Y. Kong, Y. Y. Choo, S.-P. Chai, A. K. Soh and A. R. Mohamed, Chem. Commun., 2016, 52, 14242–14245 RSC.
- X. Jiao, Z. Chen, X. Li, Y. Sun, S. Gao, W. Yan, C. Wang, Q. Zhang, Y. Lin, Y. Luo and Y. Xie, J. Am. Chem. Soc., 2017, 139, 7586–7594 CrossRef CAS PubMed.
- Y. Wang, F. Wang, Y. Chen, D. Zhang, B. Li, S. Kang, X. Li and L. Cui, Appl. Catal., B, 2014, 147, 602–609 CrossRef CAS.
- K. Wang, L. Zhang, Y. Su, S. Sun, Q. Wang, H. Wang and W. Wang, Catal. Sci. Technol., 2018, 8, 3115–3122 RSC.
- K. Liu, X. Li, L. Liang, J. Wu, X. Jiao, J. Xu, Y. Sun and Y. Xie, Nano Res., 2018, 11, 2897–2908 CrossRef CAS.
- J. Wang, T. Xia, L. Wang, X. Zheng, Z. Qi, C. Gao, J. Zhu, Z. Li, H. Xu and Y. Xiong, Angew. Chem., Int. Ed., 2018, 57, 16447–16451 CrossRef CAS PubMed.
- Slamet, H. W. Nasution, E. Purnama, S. Kosela and J. Gunlazuardi, Catal. Commun., 2005, 6, 313–319 CrossRef CAS.
- M. Tahir and N. S. Amin, Appl. Catal., B, 2015, 162, 98–109 CrossRef CAS.
- L. Li, P. Li, Y. Wang, L. Lin, A. H. Shah and T. He, Appl. Surf. Sci., 2018, 452, 498–506 CrossRef CAS.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176–177, 44–52 CAS.
- B. Liu, L. Ye, R. Wang, J. Yang, Y. Zhang, R. Guan, L. Tian and X. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 4001–4009 CrossRef CAS PubMed.
- J. Fu, K. Liu, K. Jiang, H. Li, P. An, W. Li, N. Zhang, H. Li, X. Xu, H. Zhou, D. Tang, X. Wang, X. Qiu and M. Liu, Adv. Sci., 2019, 6, 1900796 CrossRef CAS PubMed.
- J. W. F. To, J. He, J. Mei, R. Haghpanah, Z. Chen, T. Kurosawa, S. Chen, W.-G. Bae, L. Pan, J. B. H. Tok, J. Wilcox and Z. Bao, J. Am. Chem. Soc., 2016, 138, 1001–1009 CrossRef CAS PubMed.
- M. Xing, Y. Zhou, C. Dong, L. Cai, L. Zeng, B. Shen, L. Pan, C. Dong, Y. Chai, J. Zhang and Y. Yin, Nano Lett., 2018, 18, 3384–3390 CrossRef CAS PubMed.
- Q. Zhang, Y. Li, E. A. Ackerman, M. Gajdardziska-Josifovska and H. Li, Appl. Catal., A, 2011, 400, 195–202 CrossRef CAS.
- Y. Li, C. Gao, R. Long and Y. Xiong, Mater. Today Chem., 2019, 11, 197–216 CrossRef CAS.
- I. Shown, S. Samireddi, Y. C. Chang, R. Putikam, P. H. Chang, A. Sabbah, F. Y. Fu, W. F. Chen, C. I. Wu, T. Y. Yu, P. W. Chung, M. C. Lin, L. C. Chen and K. H. Chen, Nat. Commun., 2018, 9, 169 CrossRef PubMed.
- X. Pan, M.-Q. Yang, X. Fu, N. Zhang and Y.-J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- D. Cornu, H. Guesmi, J.-M. Krafft and H. Lauron-Pernot, J. Phys. Chem. C, 2012, 116, 6645–6654 CrossRef CAS.
- H. Yu, S. Yan, P. Zhou and Z. Zou, Appl. Surf. Sci., 2018, 427, 603–607 CrossRef CAS.
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644–3653 CrossRef CAS.
- X. Meng, S. Ouyang, T. Kako, P. Li, Q. Yu, T. Wang and J. Ye, Chem. Commun., 2014, 50, 11517–11519 RSC.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem. - Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- M. Ghoussoub, S. Yadav, K. K. Ghuman, G. A. Ozin and C. V. Singh, ACS Catal., 2016, 6, 7109–7117 CrossRef CAS.
- K. K. Ghuman, L. B. Hoch, P. Szymanski, J. Y. Y. Loh, N. P. Kherani, M. A. El-Sayed, G. A. Ozin and C. V. Singh, J. Am. Chem. Soc., 2016, 138, 1206–1214 CrossRef CAS PubMed.
- J. Ye and J. K. Johnson, ACS Catal., 2015, 5, 2921–2928 CrossRef CAS.
- W. Ye, R. Long, H. Huang and Y. Xiong, J. Mater. Chem. C, 2017, 5, 1008–1021 RSC.
- D. Liu, Y. Fernández, O. Ola, S. Mackintosh, M. Maroto-Valer, C. M. A. Parlett, A. F. Lee and J. C. S. Wu, Catal. Commun., 2012, 25, 78–82 CrossRef CAS.
- I. Shown, H.-C. Hsu, Y.-C. Chang, C.-H. Lin, P. K. Roy, A. Ganguly, C.-H. Wang, J.-K. Chang, C.-I. Wu, L.-C. Chen and K.-H. Chen, Nano Lett., 2014, 14, 6097–6103 CrossRef CAS PubMed.
- M. Tasbihi, M. Schwarze, M. Edelmannová, C. Spöri, P. Strasser and R. Schomäcker, Catal. Today, 2019, 328, 8–14 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632–2636 CrossRef CAS.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- Y. Wang, Q. Lai, F. Zhang, X. Shen, M. Fan, Y. He and S. Ren, RSC Adv., 2014, 4, 44442–44451 RSC.
- Q. Kang, T. Wang, P. Li, L. Liu, K. Chang, M. Li and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 841–845 CrossRef CAS PubMed.
- M. Tahir, B. Tahir and N. A. S. Amin, Appl. Catal., B, 2017, 204, 548–560 CrossRef CAS.
- H. Wu, H. L. Tan, C. Y. Toe, J. Scott, L. Wang, R. Amal and Y. H. Ng, Adv. Mater., 2019, 1904717 Search PubMed.
- M. G. Kibria, S. Zhao, F. A. Chowdhury, Q. Wang, H. P. Nguyen, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2014, 5, 3825 CrossRef CAS PubMed.
- H. L. Tan, F. F. Abdi and Y. H. Ng, Chem. Soc. Rev., 2019, 48, 1255–1271 RSC.
- S. Bai, C. Gao, J. Low and Y. Xiong, Nano Res., 2018, 12, 2031–2054 CrossRef.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- H. L. Tan, X. Wen, R. Amal and Y. H. Ng, J. Phys. Chem. Lett., 2016, 7, 1400–1405 CrossRef CAS PubMed.
- Q. Lang, Y. Yang, Y. Zhu, W. Hu, W. Jiang, S. Zhong, P. Gong, B. Teng, L. Zhao and S. Bai, J. Mater. Chem. A, 2017, 5, 6686–6694 RSC.
- S. Cao, Y. Li, B. Zhu, M. Jaroniec and J. Yu, J. Catal., 2017, 349, 208–217 CrossRef CAS.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li, H. Zhao, X. Huang, Y. Gao and Z. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS PubMed.
- F. Chen, H. Huang, L. Ye, T. Zhang, Y. Zhang, X. Han and T. Ma, Adv. Funct. Mater., 2018, 28, 1804284 CrossRef.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- R. Long, Y. Li, Y. Liu, S. Chen, X. Zheng, C. Gao, C. He, N. Chen, Z. Qi, L. Song, J. Jiang, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2017, 139, 4486–4492 CrossRef CAS PubMed.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS PubMed.
- X. Cui, J. Wang, B. Liu, S. Ling, R. Long and Y. Xiong, J. Am. Chem. Soc., 2018, 140, 16514–16520 CrossRef CAS PubMed.
- J. D. Froehlich and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 3565–3573 CrossRef CAS PubMed.
- D. Hong, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2017, 139, 6538–6541 CrossRef CAS PubMed.
- T. Fogeron, P. Retailleau, L. M. Chamoreau, Y. Li and M. Fontecave, Angew. Chem., Int. Ed., 2018, 57, 17033–17037 CrossRef CAS PubMed.
- Z. Guo, S. Cheng, C. Cometto, E. Anxolabéhère-Mallart, S.-M. Ng, C.-C. Ko, G. Liu, L. Chen, M. Robert and T.-C. Lau, J. Am. Chem. Soc., 2016, 138, 9413–9416 CrossRef CAS PubMed.
- R. Kuriki, M. Yamamoto, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2017, 56, 4867–4871 CrossRef CAS PubMed.
- D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed.
- C.-H. Lim, A. M. Holder and C. B. Musgrave, J. Am. Chem. Soc., 2013, 135, 142–154 CrossRef CAS PubMed.
- C. Liu, T. R. Cundari and A. K. Wilson, J. Phys. Chem. C, 2012, 116, 5681–5688 CrossRef CAS.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- G. Zhao, H. Pang, G. Liu, P. Li, H. Liu, H. Zhang, L. Shi and J. Ye, Appl. Catal., B, 2017, 200, 141–149 CrossRef CAS.
- M. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- R. W. Flaig, T. M. O. Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS PubMed.
- J. Wang, T. Xia, L. Wang, X. Zheng, Z. Qi, C. Gao, J. Zhu, Z. Li, H. Xu and Y. Xiong, Angew. Chem., Int. Ed., 2018, 57, 16447–16451 CrossRef CAS PubMed.
- Z. Liang, C. Qu, D. Xia, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2018, 57, 9604–9633 CrossRef CAS PubMed.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS PubMed.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254–4260 CrossRef CAS.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- M. Anpo, H. Yamashita, K. Ikeue, Y. Fujii, S. G. Zhang, Y. Ichihashi, D. R. Park, Y. Suzuki, K. Koyano and T. Tatsumi, Catal. Today, 1998, 44, 327–332 CrossRef CAS.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |