
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanomaterials for molecular signal amplification in electrochemical nucleic acid biosensing: recent advances and future prospects for point-of-care diagnostics
Léonard
Bezinge
 ,
Akkapol
Suea-Ngam
,
Andrew J.
deMello
* and
Chih-Jen
Shih
*
,
Akkapol
Suea-Ngam
,
Andrew J.
deMello
* and
Chih-Jen
Shih
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, 8093 Zürich, Switzerland. E-mail: andrew.demello@chem.ethz.ch; chih-jen.shih@chem.ethz.ch
First published on 8th November 2019
Abstract
With the increasing importance of personalized medicine and genomics, biosensors have received considerable attention in various fields, including clinical diagnostics, food inspection and environmental monitoring. In particular, advances in electrochemical biosensing technologies for clinical diagnostics pave the way for ultrasensitive detection of nucleic acid sequences using miniaturized and low-cost devices suitable for point-of-use applications. This account reviews three major amplification strategies utilizing nanomaterials as (i) ‘nanocatalysts’ in electrocatalysis, (ii) redox active reporters (‘nanoreporters’) and (iii) cargos for redox markers (‘nanocarriers’). In addition, and motivated by the need for reproducible and robust sensing, the integration of biosensors with microfluidic tools, including paper-based devices, are also covered.
 Léonard Bezinge | Léonard Bezinge obtained his BSc and MSc in Chemical and Bioengineering from ETH Zurich in 2017, with a master thesis on the synthesis of fluorescent nanoparticles in microreactors. He then did an internship at the London Center for Nanotechnology (UCL) as part of the i-sense research program on the development of nanomaterials for ultrasensitive point-of-care tests. In 2019, he started his PhD studies under the supervision of Prof. Shih and Prof. deMello at ETH Zurich. His current research interests focus on the development of paper-based electrochemical biosensors for rapid diagnostic testing. |
 Akkapol Suea-Ngam | Akkapol Suea-Ngam completed his undergraduate degree in chemistry from Chulalongkorn University, receiving the best Bachelor thesis and the Hitachi Tophy 2014 awards. He received his Masters degree in Analytical Chemistry from same department, having also spent a five month period in the Furutani group at the IMS in Japan, and was the recipient of the best Master Research Award. Thereafter, he also received Ratchdapiseksompotch as the best Master Thesis Award from Chulalongkorn University. Akkapol joined the deMello group in 2016, and was the recipient of a Swiss Government Excellence Scholarship. He has experience in developing electrochemical detectors for droplet-based and paper-based microfluidic systems. |
 Andrew J. deMello | Andrew J. deMello is Professor of Biochemical Engineering in the Department of Chemistry and Applied Biosciences at ETH Zürich and Head of the Institute for Chemical and Bioengineering. Previously he was Professor of Chemical Nanosciences in the Chemistry Department at Imperial College London. His group's research interests cover a broad range of activities in the general area of microfluidics and nanoscale science. Primary specializations include the development of microfluidic devices for high-throughput biological and chemical analysis, ultra-sensitive optical detection techniques, imaging flow cytometry, novel methods for nanoparticle synthesis, the exploitation of semiconducting materials in diagnostic applications and the development of intelligent microfluidic systems. |
 Chih-Jen Shih | Chih-Jen Shih has been a tenure-track assistant professor of chemical engineering at the Department of Chemistry and Applied Biosciences, ETH Zurich since 2015. He was trained in Stanford University (Postdoc, 2014–2015) and Massachusetts Institute of Technology (PhD, 2009–2014). His research group focuses on morphology, dynamics, molecular forces, and transport phenomena at nanomaterials interfaces. His interest ranges from fundamental understanding of how dielectric screening of atomically thin nanomaterials influences the movement and interactions of charges, excitons, and molecules near interfaces, to application-motivated studies aimed at developing new engineering strategies to control over the interplay of these mechanisms, towards new technological opportunities in optoelectronics, sensors, and actuators. His research has been recognized by the Victor K. LaMer Award from the American Chemical Society, the Ruzicka Prize from the Swiss Chemical Society, and the ERC Starting Grant from the European Union. |
Design, System, ApplicationElectrochemical biosensors most usually leverage the detection of an electroactive reporter molecule to generate an analytically useful electrical signal. The sensitivity and detection limits of such sensors directly depend on the number of redox reporters that can be detected at the electrode. To detect the low (mass and concentration) levels of biomarkers in clinical samples, amplification strategies are used to increase the number of detectable molecules. In this respect, nanomaterials, due to their small size, can be used to label biomolecules and amplify the number of redox reporters. In particular, functional nanomaterials can be engineered to interact with specific nucleic acid strands and subsequently amplify associated electrochemical signals, paving the way towards robust and ultrasensitive methods for pathogen detection and disease diagnostics. |
1. Introduction
Advances in nanotechnology have defined a step change in analytical chemistry. Owing to their high surface-to-volume ratios, nanomaterials exhibit unique physiochemical properties that differ from their bulk counterparts, providing opportunities for improved biosensing.1 For example, gold nanoparticles, which appear intense red due to the plasmonic effects, have found significant commercial success as colorimetric labels in one of the most established diagnostic tests: the home urinary pregnancy test.2 In many ways, they represent the ‘gold standard’ in rapid diagnostic tests with colorimetric readouts, and have also been used to detect various infectious diseases, such as malaria; with over 276 million tests being sold in 2017.3 However, such tests provide qualitative or at best semi-quantitative readouts. Accordingly, electrochemical biosensors have emerged as promising platforms for miniaturized devices and quantitative analyte detection. Since the first demonstration of an electrochemical glucose sensor in 1962 by Clark and Lyons,4 glucose sensors have become the tool of reference for quantitative and low-cost diabetes monitoring. Indeed, as the field of diagnostics moves towards personalized medicine and molecular diagnostics, biosensors with defined specificity and high analytical sensitivity are essential to detect nucleic acids (NAs) at femto- and atto-molar concentrations in clinical samples. To this end, the development of amplification methods, involving either the multiplication of the target NAs (as in the case of polymerase chain reaction – PCR) or the amplification of biosensor signal itself, becomes critical.Nanomaterials, due to their multifunctional nature, have facilitated the improvement of several key features in electrochemical bioassays, including sample preatrement,5 analyte capture,6 signal amplification7 and transduction.8 Widely investigated as electrode modifiers to improve bioreceptor immobilization and charge transport properties,9 nanomaterials have also demonstrated their potential to amplify electrochemical signals by generating increased numbers of electroactive reporters. Herein, we review recent advances in nanomaterial-based amplification strategies for electrochemical signaling, with a focus on nucleic acid detection for point-of-care diagnosis.
2. Nucleic acids as biorecognition elements
Bacterial diseases (such as tuberculosis) and their antimicrobial resistance to common antibiotics, such as penicillin, amoxicillin and erythromycin, represent a critical threat to global health10 and can effectively be identified by genetically detecting the antibiotic resistome (antibiotic resistance genes).11 Aside from the direct genomic detection of pathogens, microRNAs (miRNAs), the short non-coding RNAs involved in gene regulation, have also been found to be promising biomarkers for cancers and infectious diseases, through their role in cellular expression during infection.12–14 The highly specific properties of NA hybridization constitute a fundamental principle of DNA biosensors (genosensors), and as such enable the detection of sequence-specific NA probes with high selectivity.15 In addition to DNA and RNA, other synthetic NA structures with distinct backbone properties – such as peptide nucleic acids (PNA) that possess amino acid-based backbones with neutral overall net charge and locked nucleic acid (LNA) containing a methylene bridge – have shown improvements in hybridization with target (natural) nucleic acids.16,17 Synthetic NAs not only possess high binding affinities to DNA and RNA (quantified by KD), but are robust and allow the detection of single-base-pair mismatches.18 Accordingly, the development of DNA-based sensors benefits a broad spectrum of analytical applications, including the detection of small molecules and proteins via SELEX-based aptamer recognition,19,20 DNA-processing enzyme activity,21 DNA damage22 and heavy metals ion analysis.23It is well-recognized that PCR remains the gold-standard method for DNA amplification in both laboratory and clinical settings, allowing exponential amplification of DNA via well-established protocols. More recently, isothermal amplification methods, such as loop mediated isothermal amplification (LAMP), recombinase polymerase amplification (RPA) and rolling circle amplification (RCA), have become increasingly important, and facilitate the development of NA assays applicable for remote-field testing.24 Each of these methods has particular advantages and drawbacks, with all requiring bespoke optimization (of primer sequences, reaction concentration and reaction temperatures) to minimize non-specific amplification. Moreover, the use of NA amplification methods for diagnostics requires that biosensors can determine the presence or concentration of amplicons, either in real time or at the endpoint. Additionally, the integration of signal amplification strategies further improves the sensitivity of the assays.7 Put simply, the direct detection of NA in clinical samples demands not only high sensitivity but also stringent specificity, to ensure discrimination of the target from a heterogeneous collection of other sequences present in the sample matrix. To this end, recent efforts have focused on the development of ultrasensitive biosensors that offer robust yet flexible approaches for NA detection in resource-limited settings.25
3. Electrochemical sensors for nucleic acid detection
3.1 Electrochemical methods
In recent decades, we have witnessed tremendous development of electrochemical biosensors as miniaturized and low-cost platforms for the rapid and sensitive detection of biomolecules. The basic principle of electrochemical detection relies on the transduction of an electrical signal, via a change in current, potential, accumulated charge or impedance, triggered by the recognition of a target biomolecular analyte (Fig. 1).26 Accordingly, a number of electrochemical techniques have been employed for the signal characterization and quantification; predominantly amperometry, linear sweep voltammetry (LSV), cyclic voltammetry (CV), differential pulse voltammetry (DPV), square wave voltammetry (SWV), and electrochemical impedance spectroscopy (EIS). In particular, pulse methods based on voltammetry, such as DPV and SWV, have been recognized to be amongst the most sensitive approaches for quantitative analysis, allowing the differentiation of both promptly decayed charging current and the faradic current with short measurement times.27,28 These methods however require system-specific optimization to maximize the signal gain, by tuning the pulse amplitude, frequency, and sampling time.29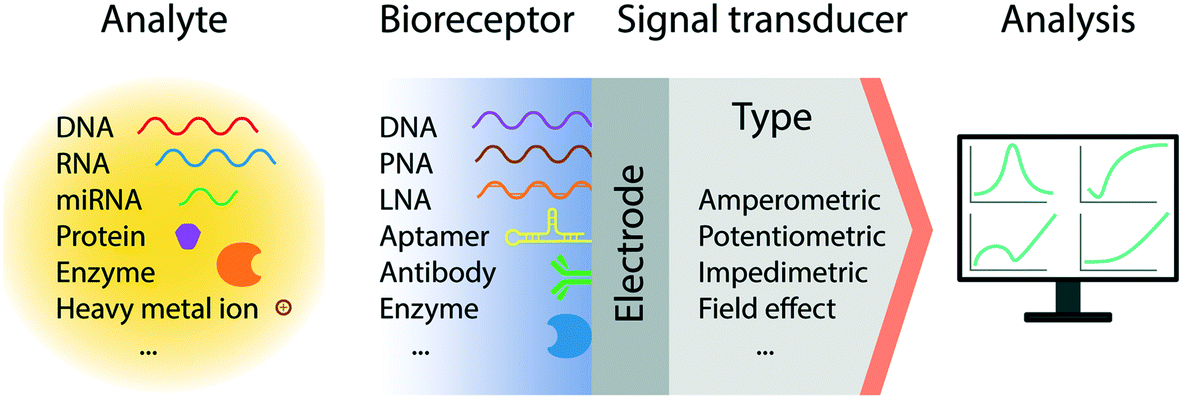 | ||
| Fig. 1 Schematic representation of the general architecture of an electrochemical biosensor. The sensor consists of an integrated receptor–transducer device capable of providing analytical information based on a biological recognition element. | ||
3.2 Electron transport through DNA
The detection of NA sequences generally relies on the hybridization of a target sequence with an immobilized capture probe. For example, the self-assembly of thiolated ssDNA monolayers (SAMs) on gold has been extensively used to functionalize gold electrodes with well-controlled packing densities.30 However, the interaction between gold and thiolated ssDNA limits the operating potential window within ±0.6 V with reference to Ag/AgCl at pH 7, in order to prevent the reductive desorption of thiolated ssDNA and oxidation of guanine nucleobase and thiol groups.17,31 Alternatively, a robust way of immobilizing ssDNA on an electrode is to take advantage of the rapid and strong biotin–avidin interaction by attaching biotinylated ssDNA to a streptavidin-covered electrode surface.32 While the conformation of ssDNA strands exhibits mechanical flexibility, double-stranded (ds) DNA possesses a rigid rod-like behaviour, capable of bending towards the electrode surface. dsDNA also mediate electron transfer through its structure,33 effectively acting as an extended nanoelectrode.34,35 Actually, at the current time, the mechanisms behind the DNA-mediated charge transport remain controversial.36,37 For example, a number of theories have been proposed based on the long-range charge transport through the π–π stacking of nucleotide orbitals mediated by the oxidation of guanine.35 Conversely, Plaxco and co-workers have suggested that electrons are not transferred through the DNA strands themselves but through a contact-mediated mechanism, with the DNA duplexes bent towards the electrode surface. These claims have been supported by experiments in which guanine content, base mismatches and dsDNA probe lengths are varied.373.3 Thermodynamic and diffusion limitations
Since targets are often at ultra-low concentrations (down to the single molecule level), the thermodynamics of binding affinity must also be considered; specifically in regard to the kon and koff, of the biomolecular interaction. Indeed, the sensor response will not reflect the measurement of an ensemble of bound molecules but is triggered by individual binding events.38,39 Accordingly, the measurement time plays a crucial role when probing ultra-low concentrations, as this will be strongly influenced by the surface coverage of analytes and the capturing elements on the surfaces. Calculations by Sheehan and Whitman40 suggest that concentration detection limits within the femtomolar range are limited by mass transport, which may require hours or even days for the analyte molecules to diffuse to the electrode surface.41 However, this suggestion has been challenged by recent reports claiming sub-femtomolar detection limits,42 and is most likely due to inconsistent definitions of detection time scales in the theoretical and experimental frameworks.43 Specifically, it has been suggested that theoretical approaches actually model the mean diffusion time for an analyte to the electrode, whereas in practice, the sensor response corresponds to the minimum diffusion time that generates a detectable signal (from the tail of the statistical distribution of Brownian motion).43 Furthermore, it should be remembered that a number of experimental techniques can overcome mass transport limitations, including sample pre-concentration using magnetic beads or flow-based assays that will be discussed later. Finally, reaction volumes must also be taken into account, since reductions in volume will decrease diffusion times but will also lower the mass of analyte. In this regard, one may quantitatively analyse such an issue through estimation of the Damköhler number, which relate reaction and mass transport time scales.443.4 Electrochemical detection of DNA
Early electrochemical approaches for the detection of nucleic acids involved the inherent electroactivity of nucleotide bases via either reduction or oxidation pathways.45,46 For example, the use of reduced graphene oxide (rGO) as an electrode material enables the detection of the four specific nucleobases within a potential window of 0.5–1.5 V vs. Ag/AgCl under physiological conditions, and lower than their traditional oxidative potentials.47 It should be noted that the redox potentials strongly depend on the properties of the electrode surface and history of pre-treatment. Nevertheless, nucleobase oxidation peaks are relatively weak and overlap with those of water oxidation, thereby limiting the scope of such approaches for biosensors.Hybridization-based approaches later emerged for the detection of specific NA sequences by utilizing specific physical and electrical properties of ss- and dsDNA.17 In these systems, redox indicators capable of reporting the presence of hybridized probes on the electrode have attracted considerable attention.48 In particular, the ferro-/ferricyanide anion couple ([Fe(CN)6]3−/4−) has become one of the most widely used redox indicators due to its sensitivity to surface coverage.48 The presence of negatively charged duplex DNAs on the surface reduces the voltammetric redox signal, which enables the quantification of hybridized targets.48 As an alternative redox marker, the hexaamineruthenium(III) ion (RuHex, [Ru(NH3)6]3+/2+) binds to the phosphate backbone of nucleic acids, forming conductive wires that generates electrochemical signals proportional to the amount of nucleic acids adjacent to the electrode surface.17,48 The formation of molecular wires can be used in combination with other redox labels, which increases the signal by up to 5-fold,49via electron transfer from the DNA layer to the electrode through the inner Helmholtz plane (IHP).26 There are two main types of electroactive species; inner sphere redox probes (e.g. ferro-/ferricyanide and dopamine) that require direct contact with the electrode surface, and outer sphere redox probes (e.g. RuHex) that transfer electrons over short distances (within 2–3 Å) to the electrode via electron tunneling.26 Inner sphere redox probes are much more sensitive to the electrode surface chemistry than outer sphere probes.26 Alternatively, duplex-specific redox indicators, such as methylene blue (MB), can intercalate into dsDNA strands to allow the specific detection of hybridized probes.17 MB is also known to covalently link to the end of hairpin probes that are tethered on the electrode, building switchable DNA architectures labelled with a redox indicator.50
Due to the fact that clinical samples are highly heterogeneous, molecular diagnostic methods strongly benefit from molecular signal amplification methods. Inspired by the standard enzyme-linked immunosorbent assay (ELISA), electrochemical equivalents, namely, electrochemical ELISA or E-ELISA, hold promise towards ultrasensitive detection of nucleic acids.51 Such an approach uses an enzyme label in a sandwich assay to amplify reporter molecules, in which the target NA is immobilized on a transducer substrate by a capture probe, followed by tagging with a label. Together with the rise of nanotechnology, a range of novel strategies have emerged to amplify electrochemical signals.7 We now review the development of nanomaterial-based signal amplification techniques.
4. Nanomaterials for molecular signal amplification
An important feature of a nanomaterials-based biosensing platform is that it is able to amplify electrochemical signals under mild conditions, i.e. at lower oxidation potentials when compared to the direct oxidation of nucleobases, by generating electroactive species that subsequently produce detectable redox events on the electrode surface. Nanoparticles normally act as labels for captured nucleic acids, either through specific interactions, such as DNA hybridization or aptamer recognition, or non-specifically, such as electrostatic or π–π interactions.7 Alternatively, “triggerable” amplification approaches, in which the presence of target NAs initiates the amplification activity of a freely-diffusing nanomaterial, have also been reported. In the current discussion, and as illustrated in Fig. 2, amplification strategies are categorized on the basis of the role of the nanomaterial, namely, nanocatalysts (where nanomaterials act as catalysts that facilitate the production of electroactive species), nanoreporters (where the nanomaterials themselves act as redox active species) and nanocarriers (where the nanomaterials act as cargos loaded with reporter molecules).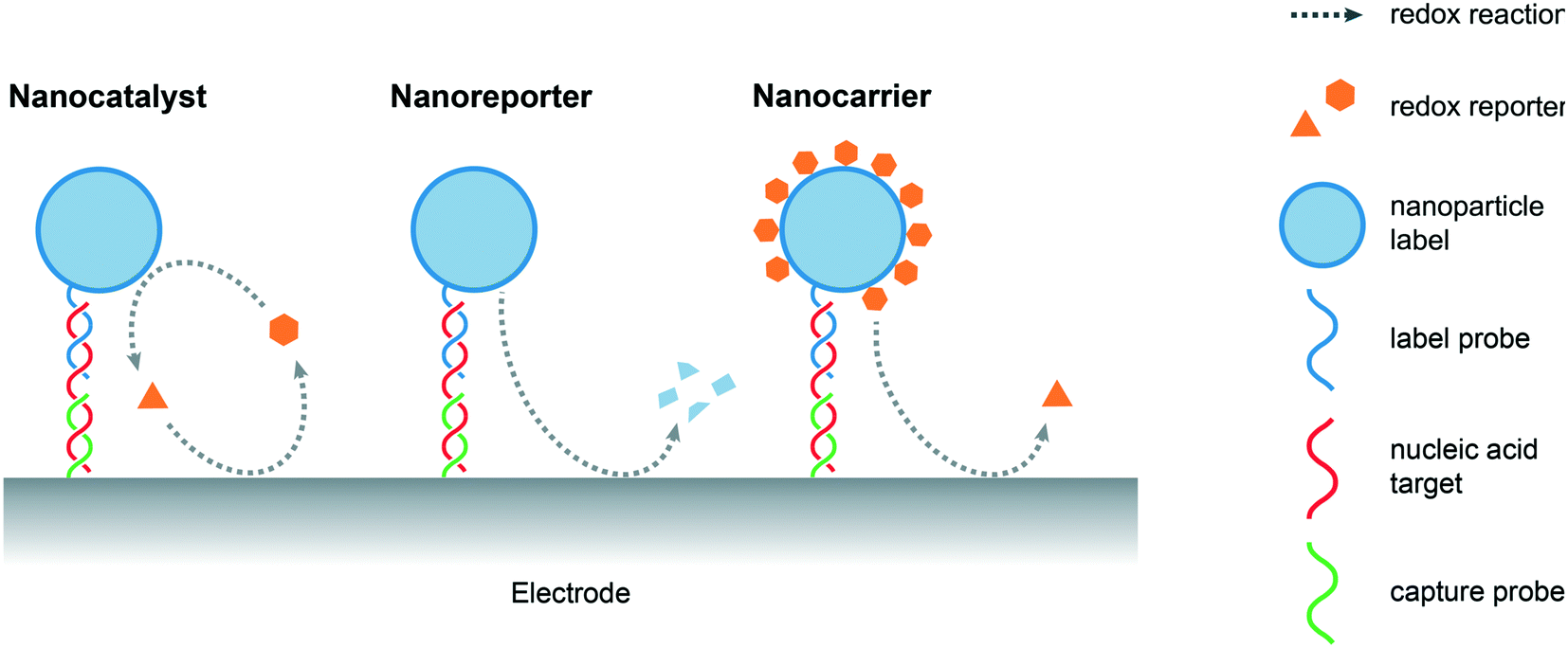 | ||
| Fig. 2 Schematic overview of the role of nanomaterials in electrochemical signal amplification (based on a simplified sandwich assay) for the detection of nucleic acids. Nanomaterials can act as (i) nanocatalysts that promote the generation of electrochemically active species, (ii) redox-active nanoreporters and (iii) nanocarriers loaded with redox reporter molecules. | ||
4.1 Nanocatalysts
Catalytic signal amplification has become an essential tool in biology, where methods such as ELISA incorporating colorimetric, fluorescent or electrochemical detection are well-established.52 Despite their high activity and specificity, enzyme-based assays suffer from a number of intrinsic limitations, such as poor stability, high production costs, and difficulties associated with storage. All of these prevent them from applications in harsh or remote environments.53 Nanomaterials have emerged as an attractive alternative to enzyme-based assays, not only due to their enzyme-mimicking characteristics (which gives rise to the term ‘nanozymes’54) but also due to their multi-functionality. In a seminal report, Gao et al. reported the use of magnetic Fe3O4 nanoparticles as peroxidase-mimicking nanomaterials and demonstrated their multifunctional nature. In such a system, nanoparticles are able to capture target analytes, extract them from the sample matrix and amplify the signal for the colorimetric immunoassay.55 In other words, nanomaterials are able carry a large amount of recognition elements, such as antibodies, ssDNA or aptamers, but still preserve their active surface. In this regard, surface engineering of nanomaterials plays a key role in the optimization of biosensor performance, balancing the trade-off between capturing and catalytic capabilities.56 In an optimized assay, nanocatalysts decorated with antibodies can lose up to 50% of their activity compared to their ‘naked’ counterparts in conditions yielding maximal signal intensity.57 Accordingly, catalytic performance will be determined by a number of parameters, including composition, doping, size, surface coverage, experimental conditions, sample type, and detection methods.Electrocatalytic amplification for microRNA detection was reported in 2006 by Gao and co-workers, using OsO2 nanoparticles to catalyze the decomposition of hydrazine.58 Here, an indium tin oxide (ITO) electrode, inactive to hydrazine at low potentials, was used to re-oxidize the catalyst, which enabled a detection limit of 80 fM for the extraction of RNA in a buffer solution, with a 60 minute hybridization step.58 This electrochemical signal can be further amplified by designing a redox cycling system.59 In such a method, a reversible redox probe with a fast kinetic response, produces a detectable signal at the working electrode that is regenerated by a catalyst (chemical–electrochemical redox cycling)60 or by the counter electrode in a microsystem (electrochemical–electrochemical redox cycling),52 effectively forming a cyclic reaction pathway. It should be noted that in order to achieve ultrasensitive detection, redox-cycling systems should be designed to have faster kinetics than any side reactions.59 Design requires careful consideration of the electroactive species involved and the types of electron transfer involved.61
Progress in enzyme-mimicking nanomaterials has been extensively discussed in an excellent recent review.56 In electrochemical systems, an emerging strategy is to employ composite nanomaterials to improve catalytic performance or add new functionalities. Fe3O4 nanoparticles were amongst the first nanocatalysts used for signal amplification in a biological assay,62 due to their ferromagnetic properties that greatly facilitate sample preconcentration and washing steps. Their multifunctional nature has facilitated new strategies in electrochemical biosensing. For example, Yadav et al. developed a composite nanomaterial comprising Fe3O4 core particles decorated with Au nanoclusters that functionalize the particle with nucleic acids via gold–RNA interactions.63 In follow-up studies, the authors further presented a complete assay for miRNA detection from cell culture and patient samples.64,65 As illustrated in Fig. 3, miRNA is purified and extracted from the sample using commercial magnetic particles. This is followed by the conjugation and immobilization of the target on the electrode using the Fe3O4@Au nanoparticles. Subsequently, ruthenium-based redox reporter molecules can be detected by chronocoulometry. In addition, results demonstrated an improvement in the limit of detection (LOD) of three orders of magnitude (to 100 aM) when using ferrocyanide as the reducing agent.64,65 That said, the exact contribution of the nanoparticles to the catalytic cycles with the reducing agent remains unclear. Overall, the method leads to a time-to-result of approximately 2 hours and highlights the importance and challenges associated with the development of a full diagnostic assay from sample collection to result. Finally, other approaches have been proposed to increase the activity of the catalytic cycle using the synergetic effects of Fe3O4 nanoparticles bound to metal complexes, such as Cu(II)-based organometals.66
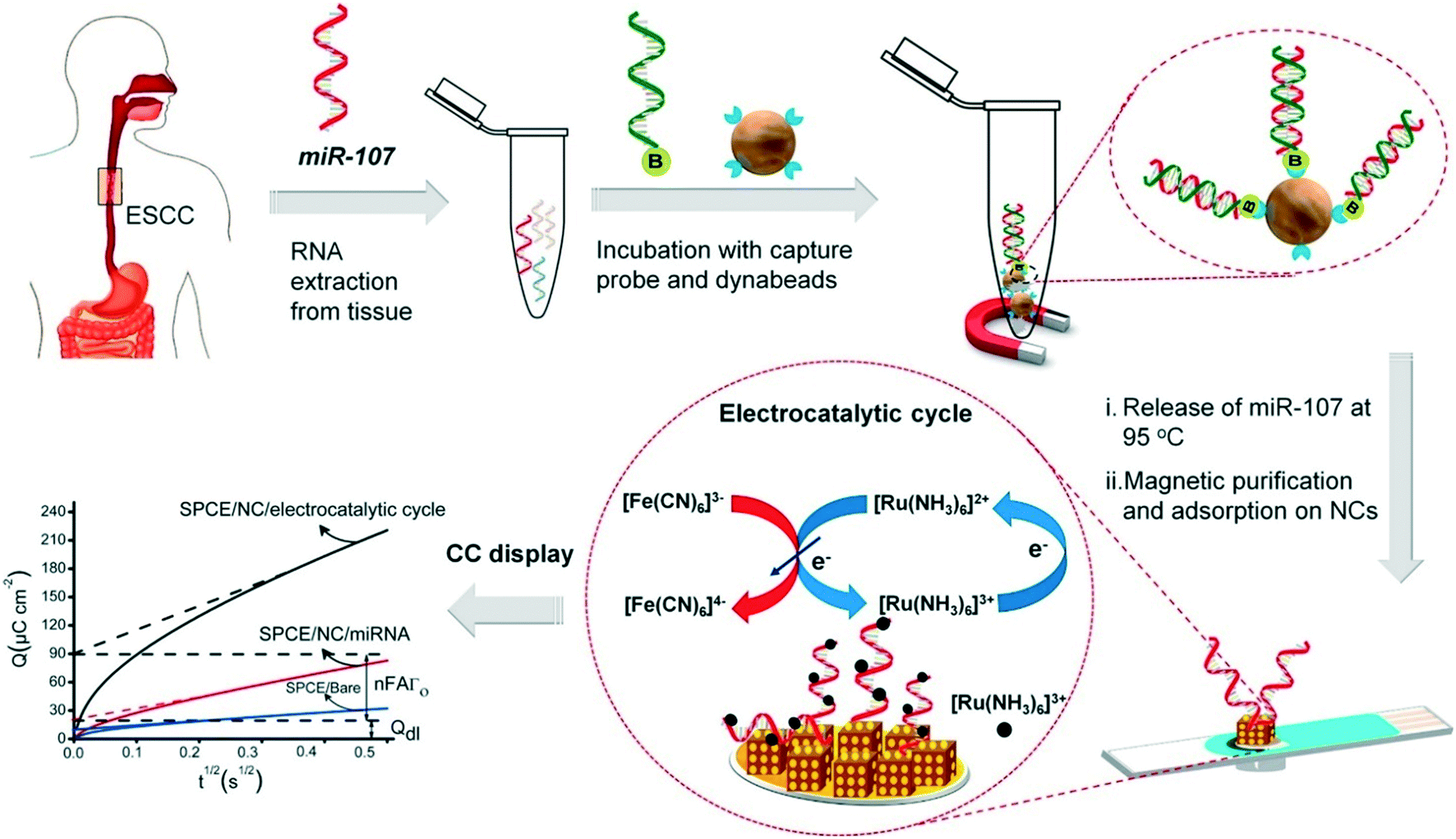 | ||
| Fig. 3 Multifunctional capabilities of iron oxide nanoparticles in electrochemical biosensing, include analyte capture and sample preconcentration and electrocatalysis for signal amplification. Images reproduced with permission from ref. 65. Copyright © (2018) Elsevier. | ||
Catalytically active nanoparticles benefit from a support material that maximizes the number of catalytic sites, whilst minimizing the amount of material used.67 A number of systems have been recently reported in the literature for miRNA and DNA detection using supported catalysts for signal amplification, such as Pt nanoparticles supported by TiO2 nanospheres,68 Pt or Pd nanoparticles supported by metal–organic frameworks (MOFs),69,70 Pt nanoparticles supported by graphene,71 fullerene-CeO2 composites with Pt nanoparticles72 and Fe3O4/CeO2 composites decorated with Au nanoparticles.73 These approaches demonstrate superior performance when compared to associated bare catalyst nanoparticle systems, owing to the increase of active surface area and synergetic interactions with the support materials. Nevertheless, composite nanomaterial platforms often demand multiple synthetic steps, which limit their scalability. Accordingly, MOFs (metal organic frameworks), which can be synthesized in a single step, have been suggested as a promising electrocatalyst for biosensing.74 The metal centers affords catalytic activity to the MOF nanomaterials, with the surface open to functionalization with biomolecules via organic ligands. There have been several reports demonstrating the peroxidase-mimicking properties of MOFs functionalized with streptavidin for DNA detection, either as an indirect label that binds to a streptavidin aptamer when target DNA is absent,75 or as a direct label, binding the biotinylated complementary DNA strand.76,77 Recent advances in catalysis and magnetic separation of MOFs, suggest that such materials hold significant promise for electrochemical signal amplification.78,79
Sandwich type assays, where the target is immobilized between a capture and label probe, represent the most common approach for nanoparticle-based nucleic acid sensors. Taking advantage of direct interactions between nanomaterials and nucleic acids, the affinity reaction of the label may be bypassed. For example, carbon nitride nanosheets have been used as indirect labels in biosensing due to their strong interactions with ssDNA via π–π interactions. When no target NA strands are present, nanosheets bind the unhybridized capture NA probes and subsequently catalyze the oxidation of H2O2, thereby generating an electrochemical response.80 Alternatively, certain metals ions, such as Ag+,81 Cu2+,82 or Pd2+,83 interact with dsDNA and can be metallized in situ to form electrocatalytically active nanoclusters, which will be discussed in detail later. The non-specific labelling the NA targets implies that the detection of nucleic acids does not discriminate based on NA sequence, but can be achieved without the hybridization step. In other words, a fully non-specific approach does not result in a stand-alone diagnostic assay but serves as a test to detect the presence of nucleic acids within a sample. In this regard, Kim et al. reported a sensor based on CeO2 nanoparticles for the detection of PCR products.84 The catalytic activity of CeO2 nanoparticles was found to be inhibited when dsDNA amplicons are adsorbed on the surface, as shown in Fig. 4.84 Such an electrochemical amplification and detection required only 6 minutes, with a proof-of-concept assay being applied to clinical samples. In this case, for the detection of amplified products, an ultralow detection limit is not necessary as the sensors should return a negative response when measuring unamplified DNA. Nevertheless, the assay remained prone to interfering agents present in human serum (due to the non-specific nature of the detection) and exposed the assay to a large number of false positives.
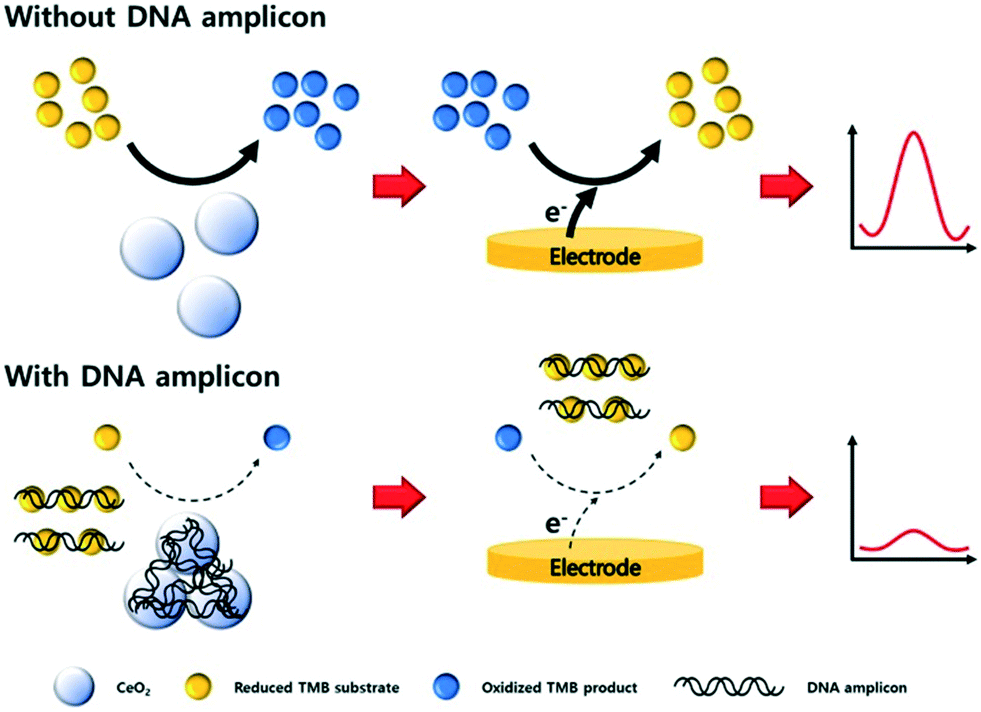 | ||
| Fig. 4 Non-specific detection of PCR products with cerium oxide nanoparticles. The adsorption of amplicons on the surface of the particles block the electrocatalytic activity of the material, thus diminishing the biosensor response proportionally to the concentration of PCR products. Images reproduced with permission from ref. 84. Copyright © (2019) Elsevier. | ||
Due to their rapid and quantitative nature, one can also incorporate electrochemical biosensors with other established nucleic acid amplification strategies. For example, the Merkoçi group reported an assay for the Leishmania DNA detection from blood samples using isothermal amplification with modified primers and electrochemical detection.85 The reverse primer was labelled with a magnetic bead for facile purification and immobilization of the amplicon on the electrode, and the forward primer was bound to a gold nanoparticle to amplify the signal by catalysing the hydrogen evolution reaction (Fig. 5). Importantly, the entire procedure required less than 20 minutes at a constant temperature of 37 °C, with a detection limit of approximately one parasite per milliliter of blood, offering a promising approach for point-of-care testing on clinical sample with minimal sample pretreatment.
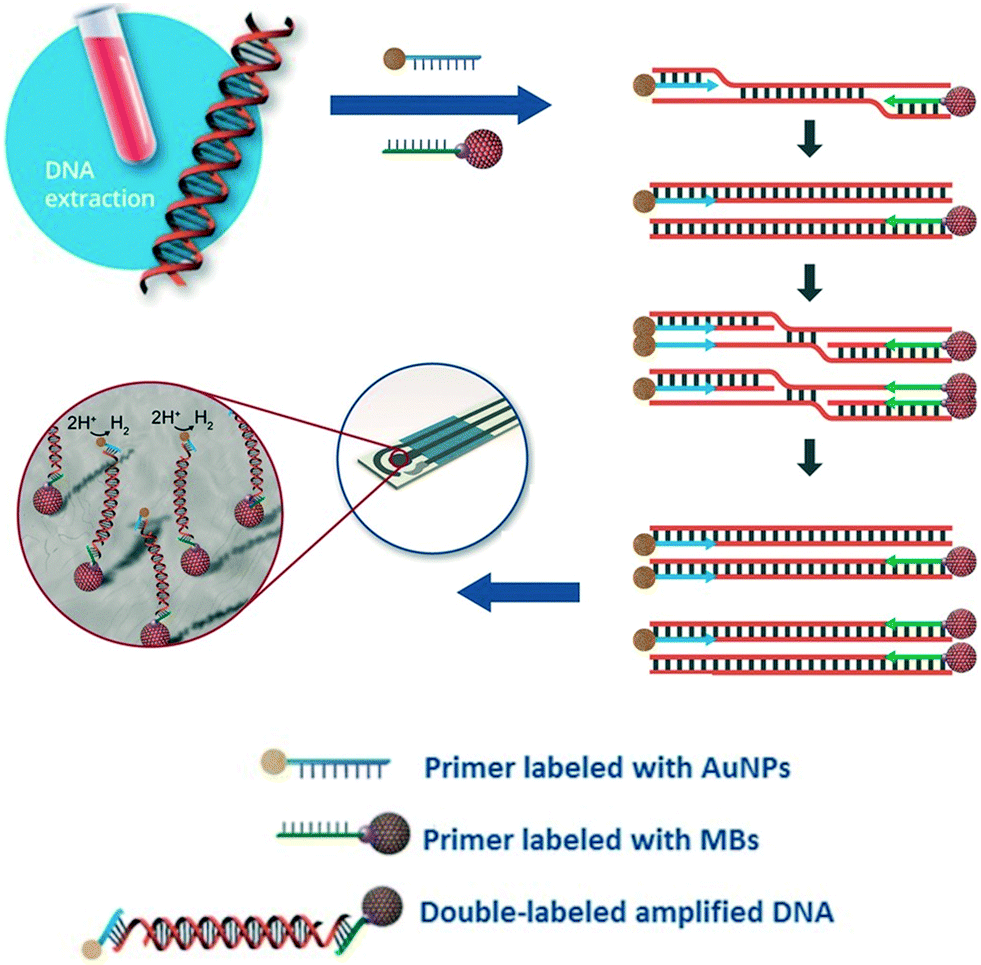 | ||
| Fig. 5 Incorporation of nanoparticles with isothermal nucleic acid amplification. The forward and reverse primers are modified with gold and iron oxide nanoparticles, respectively, enabling simple sample pre-concentration and signal amplification. Images adapted with permission from ref. 85. Copyright © (2016) John Wiley and Sons. | ||
In most of the approaches discussed so far, the target DNA and nanoparticle labels are immobilized on the electrode surface. Conversely, Crooks and co-workers have described a fundamentally different approach, where platinum nanocatalysts freely diffused in solution and generate a detectable signal on colliding with the electrode surface (Fig. 6).86 Specifically, nanoparticles were initially functionalized with ssDNA complementary to the target microRNA. This was followed by incubation with the target probe and a nuclease enzyme. Subsequently, cleavage of the hybridized probes exposes the catalytically-active nanoparticle surface, which in turn activates target recycling. Finally, the particle/DNA complexes were incubated in hydrazine, so that an electrochemical signal was generated from the catalyzed hydrazine decomposition reaction as a result of particle collisions with the gold ultra-microelectrode. The merit of this approach lies in its simplicity and versatility when compared to other methods. However, to date it has only been reported in buffer (and not complex biological media) and concentration detection limits are relatively high (∼5 pM).
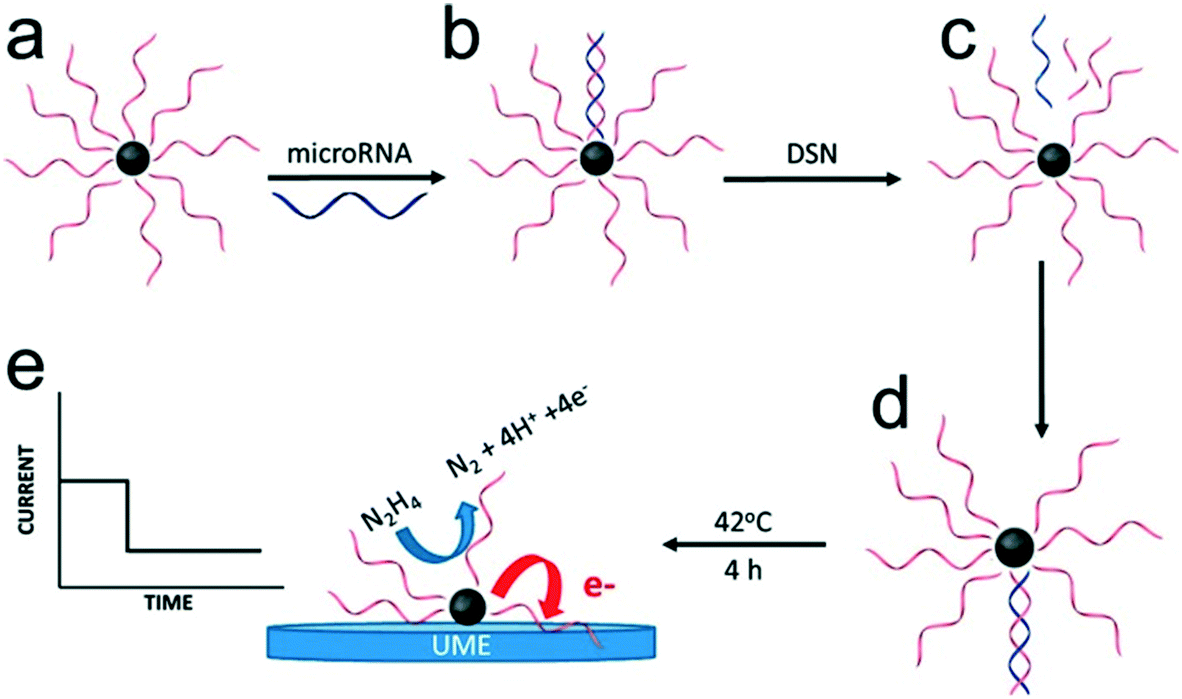 | ||
| Fig. 6 Electrocatalysis of freely diffusing platinum nanoparticles. An enzyme-assisted target recycling mechanism exposes the catalytically-active nanoparticle surface in the presence of target miRNAs, and is followed by the addition of hydrazine. The electrochemical signal generated by the catalytic decomposition of hydrazine is proportional to the exposed surface of Pt nanoparticles. Images reproduced with permission from ref. 86. Copyright © (2017) American Chemical Society. | ||
In electrocatalysis, amperometry and voltammetry are the primary electrochemical detection methods used to quantify electroactive species produced by catalytic amplification. Alternatively, electrochemical impedance spectroscopy (EIS), one of the most well-established electrochemical methods, has also been leveraged to measure the extent of electrocatalytic reactions via the determination of the charge transfer resistance.87 For example, Peng et al. developed a hybridization-based miRNA biosensor using RuO2 nanoparticle labels to catalyze the polymerization of 3,3-dimethoxybenzidine (DB). This reaction produces an insulating layer of poly-DB on the electrode that alters the impedance response.88 That said, the polymerization reaction has be run for at least an hour before detection by EIS. In conclusion, electrocatalysis offers excellent performance with respect to signal amplification, especially when combined with redox cycling. However, careful consideration of nanomaterial surface engineering is required to ensure optimal performance.
4.2 Nanoreporters
In regard to the use of electroactive nanomaterials as reporters in biosensor applications, we now discuss redox-active nanomaterials, particularly those containing transition metals, which can be detected electrochemically. It should be noted that electrochemical signals scale with the number of detected atoms or ions, and thus are proportional to the volume of reporter. A simple approach, for example, involves labelling the target nucleic acid with a single nanoparticle. In this regard, Crooks and co-workers reported a method for the detection of hepatitis B DNA by hybridizing the target probe between a magnetic bead and a silver nanoparticle (AgNP).89 After hybridization and magnetic extraction, AgNPs were mixed with an oxidizing agent, to allow the detection of silver ions by anodic stripping voltammetry. Significantly, the authors reported a 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold signal amplification using 20 nm AgNPs (when compared to methylene blue-labelled DNA) and a concentration detection limit of 85 pM on a paper-based device containing screen-printed electrodes. The ability to perform signal amplification within a paper device demonstrates the strength and simplicity of the approach, with only mild reagents and short incubation time being used. Several methods have also been used to further amplify signal, primarily by increasing the number of AgNPs per target strand. For example, a rolling circle amplification step was implemented followed by hybridization of AgNPs periodically along the elongated DNA strand (Fig. 7).90 Alternatively, the use of a support material,91 dendritic AgNPs,92 and AgNPs covalent aggregates93 have also examined.
000-fold signal amplification using 20 nm AgNPs (when compared to methylene blue-labelled DNA) and a concentration detection limit of 85 pM on a paper-based device containing screen-printed electrodes. The ability to perform signal amplification within a paper device demonstrates the strength and simplicity of the approach, with only mild reagents and short incubation time being used. Several methods have also been used to further amplify signal, primarily by increasing the number of AgNPs per target strand. For example, a rolling circle amplification step was implemented followed by hybridization of AgNPs periodically along the elongated DNA strand (Fig. 7).90 Alternatively, the use of a support material,91 dendritic AgNPs,92 and AgNPs covalent aggregates93 have also examined.
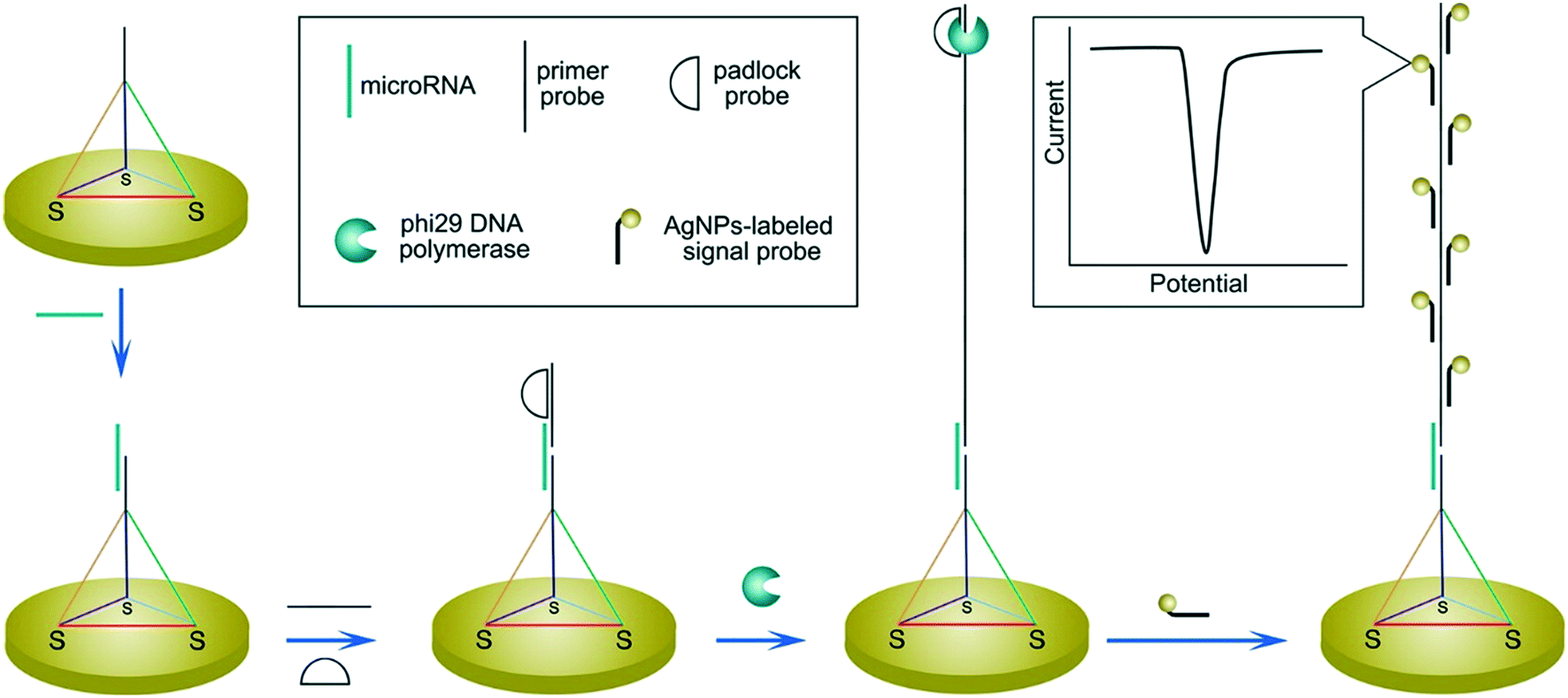 | ||
| Fig. 7 Increasing the number of binding sites for electroactive reporters via nucleic acid elongation. Rolling circle amplification creates periodical hybridization binding sites for DNA-labelled silver nanoparticles along the elongated strand. The increased number of silver nanoparticle labels are subsequently detected by stripping voltammetry. Images reproduced with permission from ref. 90. Copyright © (2015) American Chemical Society. | ||
To amplify electrochemical signals without hybridizing nanoparticles labels to the target strand, in situ metallization of metal nanoclusters on nucleic acid strands has been demonstrated. For example, experimental studies have demonstrated the interaction of silver(I) ions with cytosine–cytosine (C–C) base pairs94 and copper(II) ions with dsDNA.95 Here, a typical assay involves the capture of target NAs on the electrode, which is then contacted with a solution containing metal ions to form metal nanoclusters on the nucleic acid strands, either through redox reactions with a reducing agent or via electrochemical/catalytic pathways.96–100 Subsequently, the metal nanoclusters are dissolved using a strong acid (e.g. for Cu nanoclusters) or electrochemically (e.g. for Ag nanoclusters), and quantified by stripping voltammetry.96,97 Recent studies have also reported the combination of metallization and hybridization chain reaction (HCR) to increase the number of binding sites for metal ions. Elongation probes are designed to form C-rich loops as seed sites for Ag nanoclusters,98 or Y-shaped dsDNA shapes for Cu nanoclusters.99 Gold nanoparticles have also been decorated with C-rich DNA strands to offer an increased number of seed sites for silver metallization.101 Alternatively, graphene oxides can be used as a catalytic platform to assist in the in situ formation of Ag or Prussian blue nanoparticles.102,103 Here, graphene oxide interacts with the unhybridized capture ssDNA via π–π interactions and amplifies the signal when no target analytes are present in the sample, i.e. a ‘signal-off’ sensor.102,103
Direct detection of the nanoparticle labels also offers the possibility of multiplexing signals by incorporating nanomaterials with distinguishable redox peaks. For example, Merkoçi et al. have pioneered the use of quantum dots for multiplexed electrochemical biosensing.104 This approach enabled the simultaneous detection of three DNA targets with PbS, ZnS and CdS quantum dot (QD) labels, with detection limits down to 270 pM for single QD labels and 2.7 pM for QDs loaded on latex beads. Building on this approach, recent advances include the integration of QDs labels with other amplification methods, such as nucleic acid amplification,105 target recycling106 or DNA elongation to increase the number of labels per target.107 Despite the excellent detection limits, down to femtomolar levels, the use of toxic heavy metals and the requirement of time-consuming dissolution steps remain a concern.105,106
4.3 Nanocarriers
Nanomaterials acting as cargos for redox reporters represent one of the most straightforward approaches for electrochemical signal amplification and have been widely used for nucleic acid detection. As one of the first strategies for amplifying biosensing signals, nanocarrier methods have seen relatively limited development in the past five years. That said, Kaur et al. reported a DNA biosensor based on a cobalt-porphyrin redox marker.108 They observed a 1000-fold enhancement in the detection limit (3.8 aM) of short DNA sequences in buffer when loading the organometallic compounds on gold nanoparticles.108,109 A variety of nanoparticle-marker systems have been reported in recent years, including gold nanoparticles with methylene blue110 or ferrocene,111 Fe3O4 nanoparticles with thionine or ferrocene,112 multi-walled carbon nanotubes with thionine,113 polymer nanoparticles with Cu2+ ions,114 and titanium phosphate nanoparticles with Cd2+ ions.49 More recently, versatile nanoporous MOFs have demonstrated high loading capacities for a variety of redox reporters, including ions (e.g. Cd2+, Pb2+),115 organometals (e.g. ferrocene),116 and small molecules (e.g. methylene blue, 3,3′,5,5′-tetramethylbenzidine).116,117Interactions between redox markers and nanoparticles include covalent bonds,112,113 electrostatic interactions,49 nanopore intercalation,114,115 physisorption,116,117 and gold–thiol interactions,110,111 which control the loading process. The ability to load nanoparticle labels with several signal probes, such as Cd2+/Pb2+ or methylene blue/ferrocene, enables a ratiometric signal analysis, using a dual-probe system to detect the ratio of the probe signal with respect to a reference. By using a ferrocene labelled hairpin probe on an electrode surface, and methylene blue on the nanoparticle tag, detection of the target analyte is more robust, since non-specific adsorptions events of the nanoparticle labels on the electrode can be accounted for from the signal ratio.110
More generally, recent research efforts have been focused on surface engineering of nanoparticle labels, with a view to increasing the loading capacity of redox indicators. For example, one can functionalize gold nanoparticles with dsDNA or ssDNA to increase the number of possible binding sites for RuHex118 or methylene blue labels,119 respectively. Alternatively, gold nanoparticles can form hybridization-induced aggregates upon target recognition, which offer more binding sites for RuHex. In this manner, a 32-fold enhancement in detection limits has been reported when compared to a single gold nanoparticle label.120,121
On the other hand, a unique approach for miRNA detection was recently described by Gooding and co-workers, as illustrated in Fig. 8.122 Here, gold-coated magnetic nanoparticles were conjugated with complementary ssDNA labelled with methylene blue, and mixed with the sample solution containing target miRNAs. Nanoparticles were then brought in contact with the electrode using a magnetic field, generating a superlattice whose structure depends on the presence of dsDNA, and thus indicative of hybridized targets. Gold nanoparticles acted as the electrode material and the recorded signal was minimized as the presence of rigid hybridized probes (dsDNA) diminishes structural compactness. In this instance, the nanoparticles serve a dual purpose, not only acting as a nanocarrier for redox reporters, but also improving charge transport properties and thus signal transduction. It was found that the method enables the detection of targets down to 10 aM in whole blood, with a remarkably broad linear range of over 7 orders of magnitude. The use of magnetic core–shell nanoparticles enables the extraction of target miRNAs from blood samples after a 30 minute hybridization step, followed by a 5 minute step for electrochemical quantification.
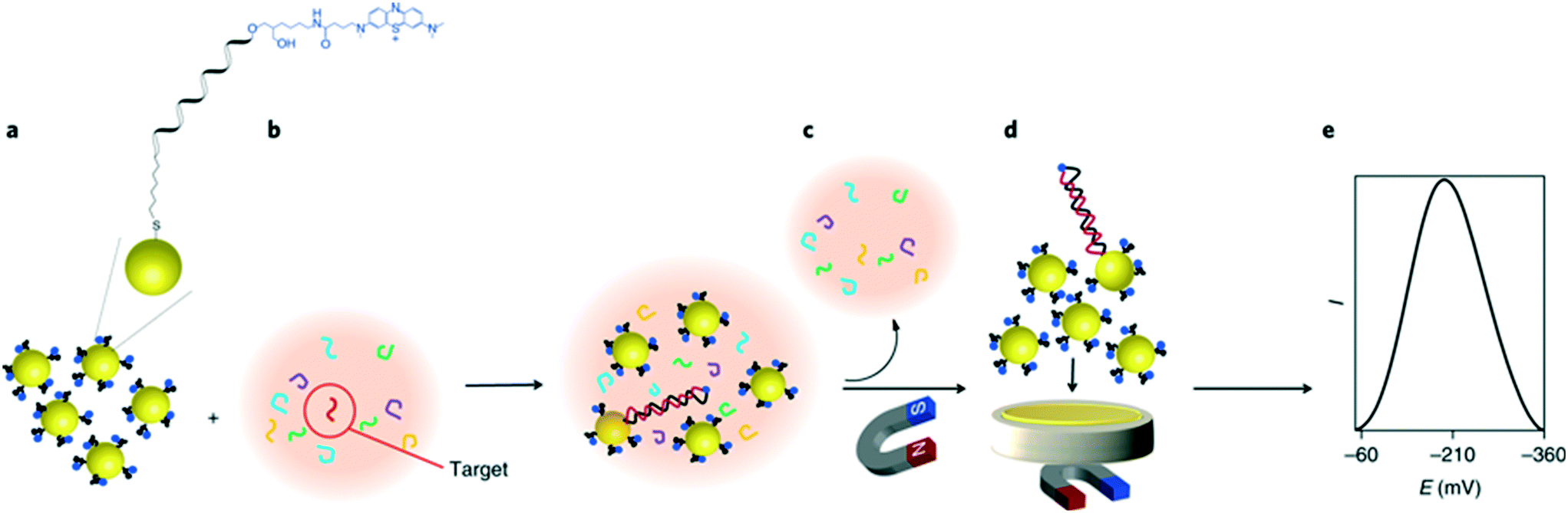 | ||
| Fig. 8 Gold nanoparticle networks as a DNA sensitive electrode. Iron oxide core/gold shell nanoparticles, functionalized with redox labelled single stranded DNA form a compact superlattice on the electrode when a magnetic field is applied. The compactness of this network is sensitive to the presence of hybridized double stranded DNA on the particle surface, therefore strongly impacting the electrochemical signal when a target sequence is present in the sample. Images reproduced with permission from ref. 122. Copyright © (2018) Springer Nature. | ||
4.4 Materials considerations
Table 1 summarizes the aforementioned roles of nanomaterials for amplifying electrochemical signals. The limits of detection and sample matrices are also indicated. We now discuss the application of noble metals,123 metal oxides,124 quantum dots,125 2D nanomaterials, and MOFs126 for electrochemical signal amplification.| Type | Nanomaterials | Signal amplification strategy | Redox indicator | Nucleic acid amplification | Sample | LOD | Ref. |
|---|---|---|---|---|---|---|---|
| Abbreviations: CHA: catalytic hairpin assembly, EATR: enzyme-assisted target recycling, HCR: hybridization chain reaction, LOD: limit of detection, MOF: metal–organic framework, NP: nanoparticle, PCR: polymerase chain reaction, RCA: rolling circle amplification, RPA: recombinase polymerase reaction, RuHex: hexaamineruthenium(III), TMB: 3,3′,5,5′-tetramethylbenzidine. | |||||||
| Noble metals | Au NPs | Nanocatalyst | Hydrogen evolution | RPA | Blood | ∼20 aM | 85 |
| Au NPs | Nanocarrier | Co(II) porphyrin | — | Buffer | 3.8 aM | 108 | |
| Au NPs | Nanocarrier | Ferrocene | — | Buffer | 0.37 fM | 111 | |
| Au NPs | Nanocarrier | RuHex | — | Buffer | 1.0 fM | 118 | |
| Au NPs | Nanocarrier | RuHex | EATR + CHA | Buffer | 25 aM | 120 | |
| Au NPs | Nanocarrier | RuHex | HCR | Serum | 0.12 fM | 121 | |
| Au NPs | Nanocarrier | Methylene blue | — | Blood | 10 aM | 122 | |
| Ag NPs | Nanoreporter | Ag+ | — | Buffer | 85 pM | 89 | |
| Ag NPs | Nanoreporter | Ag+ | RCA | Serum | 50 aM | 90 | |
| Ag NP dendrimers | Nanoreporter | Ag+ | — | Buffer | 0.78 pM | 92 | |
| Ag NP aggregates | Nanoreporter | Ag+ | — | Serum | 20 aM | 93 | |
| Metallized Ag nanoclusters | Nanoreporter | Ag+ | EATR + HCR | Serum | 0.64 fM | 98 | |
| Metallized Ag nanoclusters | Nanoreporter | Ag+ | — | Buffer | 0.16 fM | 101 | |
| Metallized Cu nanoclusters | Nanoreporter | Cu2+ | EATR + HCR | Serum | 10 aM | 99 | |
| Metallized Cu nanoclusters | Nanoreporter | Cu2+ | EATR + HCR | Cell lysate | 45 aM | 96 | |
| Pt NPs | Nanocatalyst | Hydrazine | — | Buffer | 45 pM | 86 | |
| Metal oxides | Fe3O4@Au NPs | Nanocatalyst | RuHex | — | Cell lysate | 100 fM | 64 |
| Fe3O4 NPs @ Cu(II) thiosemicarbazide | Nanocatalyst | TMB | HCR | Serum | 33 aM | 66 | |
| Fe3O4 NPs | Nanocarrier | Thionine/ferrocene | HCR | Cell lysate | 0.28 fM | 112 | |
| CeO2 NPs | Nanocatalyst | TMB | PCR | Serum | 33 aM | 84 | |
| CeO2@Pt NPs | Nanocatalyst | H2O2 | — | Serum | 0.33 fM | 72 | |
| CeO2/Fe3O4@Au NPs | Nanocatalyst | Methylene blue | CHA | Serum | 0.33 fM | 73 | |
| Quantum dots | PbS and CdS NPs | Nanoreporter | Pb2+ and Cd2+ | — | Serum | 12 fM | 105 |
| CdS NPs | Nanoreporter | Cd2+ | EATR | Cell lysate | 0.48 fM | 106 | |
| Carbon-based naomaterials | Graphene@Ag NPs | Nanocatalyst/nanoreporter | Ag+ | EATR | Buffer | 57 aM | 71 |
| Graphene oxide@Ag NPs | Nanocatalyst/nanoreporter | Ag+ | — | Serum | 7.6 fM | 102 | |
| Graphene oxide@Prussian blue | Nanocatalyst/nanoreporter | Prussian blue | — | Serum | 1.5 fM | 103 | |
| Multiwalled carbon nanotubes | Nanocarrier | Thionine | — | Buffer | 32 fM | 113 | |
| MOFs | MOF@Pt NPs | Nanocatalyst | H2O2 | — | Serum | 33 aM | 134 |
| MOF@Pd NPs | Nanocatalyst | NaBH4 | — | Buffer | 33.6 fM | 69 | |
| MOF@Fe(III) porphyrin | Nanocatalyst | o-Phenylenediamine | — | Serum | 0.48 fM | 75 | |
| MOF@Fe(III) porphyrin | Nanocatalyst | H2O2 | — | Buffer | 0.69 fM | 76 | |
| Zr(IV)-MOF | Nanocatalyst | O2 | EATR | Serum | 0.29 fM | 77 | |
In conclusion, signal amplification using nanomaterials as cargos has evolved from commonly used single redox marker labels, and provides for an increased number of redox reporters at the electrode. The loading capacity of nanoparticle labels however limits amplification potential. The use of nanomaterials as redox reporters is simple to implement, provides for amplification factors of five to six orders of magnitude and can be easily combined with in situ metallization. That said, performance highly depends on the choice of nanoparticle label, and the general method requires an additional step to convert the nanomaterial to a detectable form. Finally, electrocatalysis represents one of the most powerful amplification tool in electrochemical biosensing and strongly benefit from the high catalytic activity and multifunctional nature of nanomaterials. However, the approach requires an additional incubation period with the substrate solution and does not currently allow multiplexed analysis.
5. Applications for point-of-care diagnostics
In 2003, the World Health Organization (WHO) introduced the ‘ASSURED’ criterion to define the ideal features of point-of-care (POC) diagnostic tests.39 Put simply, such tests should be affordable, sensitive, specific, user-friendly, rapid and robust, equipment-free and deliverable to end-users.39 Recently, Peeling and colleagues135 suggested two additions to these basic requirements. Real-time connectivity for digital healthcare136 and ease of sample collection yield the ‘REASSURED’ criterion.135 Indeed, sample collection and pretreatment remain a major challenge hindering the translation of biosensing strategies from laboratory settings to the field. To address this wish list, analytical instrumentation can be miniaturized and made to handle ultra-small sample volumes with minimal user input using the principles of microfluidics.137–139 Such microscale systems can be easily integrated with external electronic architecture, to enable the performance of multistep assays with minimal user input.140 In particular, microfluidic paper-based analytical devices (μPADs) offer an ultra-low cost platform to perform assays. Here, capillary flow is driven by the wicking properties of paper, owing to its microporous and hydrophilic structure, and thus pumps or other fluid actuation systems are not required.141 μPADs have been extensively applied for the detection of infectious diseases, with several diagnostic tests being approved by the WHO for the detection of malaria and HIV at the POC.141,142 However, colorimetric readout only provides for a semi-quantitative interpretation of the result, with bulky and expensive instrumentation normally being needed for quantification. To address this need, Henry and co-workers pioneered electrochemical detection in μPADs, using screen-printed electrodes for the rapid and quantitative determination of analyte concentrations.143 Since this seminal work, a wide range of techniques have been developed to fabricate and pattern electrodes on μPADs; including screen printing and inkjet printing of conductive inks,144 evaporation or sputtering of metals,145 as well as “reagentless” techniques such as laser scribing.146Very recently, exciting advances have been made in relation to the detection of nucleic acids on μPADs. For example, Crooks and co-workers presented an origami-inspired device comprising multiple sliding layers of paper that allowed for electrochemical detection of short target DNA sequences.147 In a subsequent study, the authors implemented a signal amplification strategy (using silver nanoparticles) for the detection of the hepatitis B virus at picomolar concentrations (see Fig. 9A).148 Briefly, the hybridization of the target was first performed in a vial followed by introduction onto the μPAD. Subsequently, the top layer was slided to dispense pre-dried KMnO4, with silver ion concentration being quantified by the stripping voltammetry. Alternatively, Kokkinos et al. more recently described a paper-based lateral flow assay with quantum dots labels using a sputtered tin working electrode, in which CdSe/ZnS core/shell nanoreporters were dissolved using HCl and Cd2+ ions detected by stripping voltammetry.145 Furthermore, Whitesides and colleagues reported the first fully integrated and portable μPAD that combines nucleic acid isothermal amplification with electrochemical readout for the detection of M. tuberculosis genomic DNA (Fig. 9B).149 Here, the paper layer is sandwiched between the screen-printed carbon electrodes and a heating element. Recombinase polymerization reaction (RPA) reagents were mixed with the target DNA and RuHex redox reporters, and loaded onto paper. The detection of amplicons by square wave voltammetry afforded a detection limit of 11 CFU mL−1 after a 20 minute RPA. This proof-of-concept study demonstrated a fully integrated and automated assay for pathogen detection with little demand for equipment. Although μPADs are ideal for simple assays, protocols requiring long incubation times and high temperatures are often compromised due to fluid vaporation. To this end, polymeric or glass microfluidic devices are better suited to fully-integrated and complex assays. For example, Plaxco et al. integrated a gold microelectrode and a heating block module in a polydimethylsiloxane/glass microchip to perform loop-mediated isothermal amplification (LAMP) at 65 °C over 60 minutes, and achieved a detection limit of 16 copies of Salmonella genomic DNA.150 However, these microsystems require the use of external pumps, which hinders their use in resource-limited settings. It is clear that even though microfluidic systems greatly facilitate sample handling and integration of functional components,137 they come with new challenges associated with electrochemical detection. These include robust electrode modification and the ability to measure ultra-small currents from microelectrodes.140
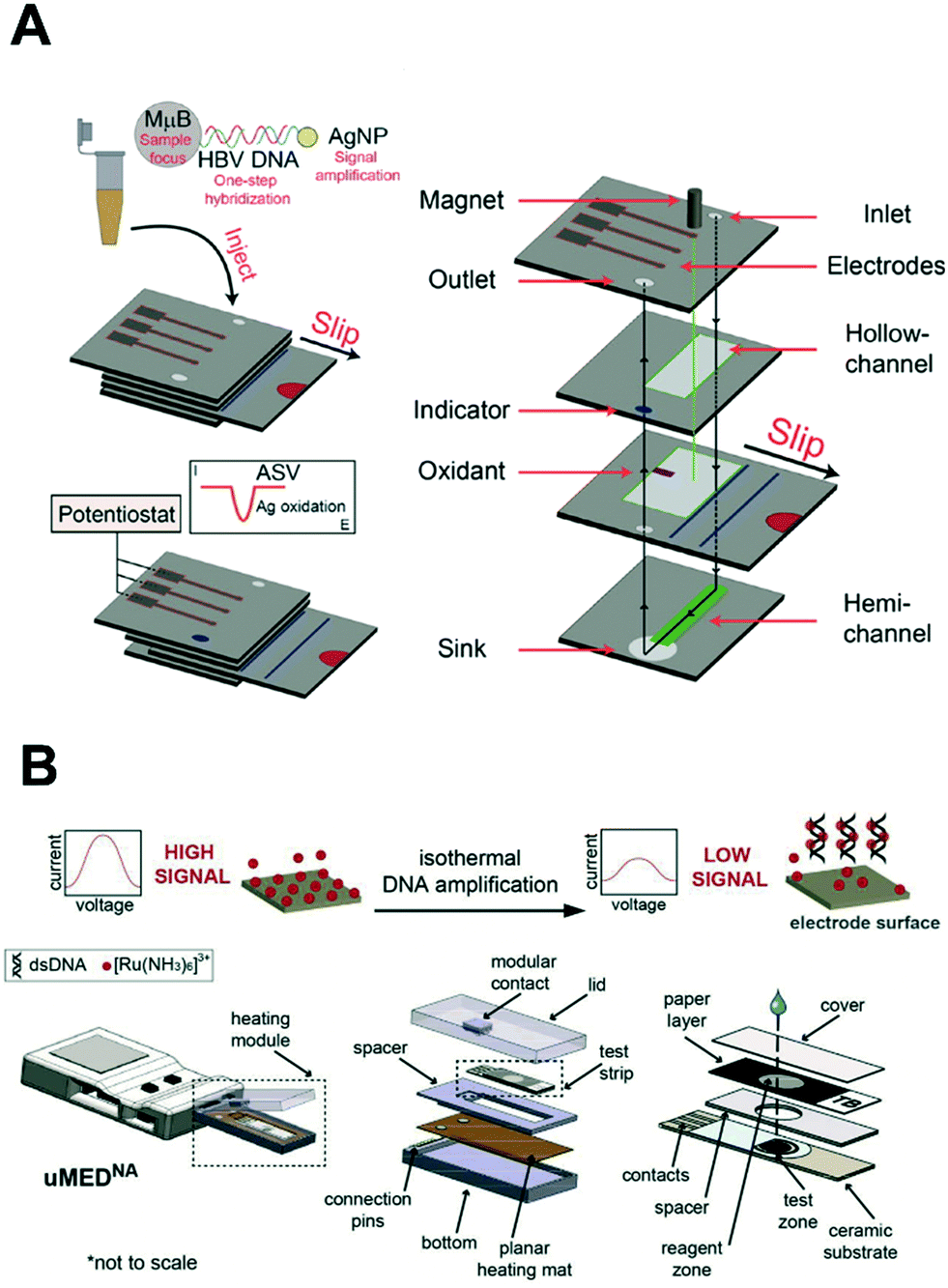 | ||
| Fig. 9 Integration of nucleic acid electrochemical detection in self-contained device for point-of-care application. (A) Multilayer paper-based device with screen-printed electrodes capable of performing a multi-step assay with electrochemical detection for the detection of hepatitis B virus. The sliding mechanism enables the control of the incubation and dispensing of the required reagents for the labeling of the target DNA with silver nanoparticles. Images adapted with permission from ref. 89. Copyright © (2015) American Chemical Society. (B) Fully automated and connected device for the detection of M. tuberculosis. The genomic DNA is amplified by isothermal amplification and detected electrochemically with limited user input. Images adapted with permission from ref. 147. Copyright © (2018) Elsevier. | ||
Another issue relating to electrochemical biosensing is powering. Self-powered biosensors have already been demonstrated. For example, enzymatic biofuel cells at the counter electrode can provide power to the device whilst also setting the reference potential. This could be an interesting opportunity to drastically simplify device hardware.151 Indeed, it is well-recognized that one of main obstacles for the implementation of electrochemical sensors, compared to colorimetric assays, at the POC is the requirement of a potentiostat.26 Although portable potentiostats are now commercially available, devices with sufficient accuracy for ultrasensitive detection remain prohibitively expensive.152–154 To this end, Whitesides and colleagues has developed an open-source potentiostat built using off-the-shelf components and capable of wireless communication via Wi-Fi or Bluetooth,155,156 greatly reducing the equipment cost by more than one order of magnitude. Additionally, the connectivity of potentiostats to smartphones opens the possibility of real-time data analysis, storage and transmission of patient data, and connectivity to cloud services. With the widespread development of cellular network technologies in countries with poor access to healthcare facilities, connected diagnostic devices are expected to transform patients' access to advice and treatment.136 We anticipate that the development of low-cost connected sensors will be a crucial step towards next-generation digital diagnostics for mobile health.
6. Outlook
6.1 Nanomaterial engineering challenges
As previously illustrated, nanomaterials have many advantageous features for the amplification of electrochemical signals. That said, the use of nanoparticle labels is often constrained by their high surface-to-volume ratios, which means that nanoparticles aggregate in the high ionic strength solutions commonly used for electrochemical detection. To this end, surface engineering of nanoparticles is key in generating colloidally stable nanoparticles in high saline solutions, without compromising other functions, such as the affinity of biorecognition elements, the active surface for electrocatalysis and the loading capacity of redox reporter. In addition, non-specific binding of nanoparticles to proteins and device substrates can compromise detection specificity and sensitivity, and remains one of the most common factors limiting the performance of nanoparticle-based biosensors at ultra-low concentrations. This issue can be addressed by blocking either the electrode or nanomaterial surface, but to date these approaches remain empirical and are seldom systematically optimized. Rational approaches are emerging, with progress in surface engineering of nanomaterials and fundamental interaction of materials-biomolecules interfaces.We also note that, despite recent advances in the use of nanomaterials in electrochemical biosensing, there are no widely-accepted benchmark method, such as gold nanoparticle labels in the case of colorimetric diagnostic tests. In this regard, there is an urgent need to establish methods that quantitatively compare novel amplification strategies. For example, the intrinsic amplification capability of a given nanoparticle label could be evaluated by directly capturing the labels on the electrode via biotin/avidin interaction. Put simply, standardized approaches is seldomly presented, but must be adopted to accelerate advancements in the field.
Extensive characterization of novel nanomaterials represent a crucial step for the progress of nanotechnology. In particular, electrocatalysis involves complex interfacial processes that have been examined in systems such as noble metals, metal oxides, MOFs, and carbon nanomaterials. Nevertheless, each material will catalyze a reaction via different mechanisms, which are poorly understood.157 This issue limits the optimization and rational design of novel materials for electrocatalytic sensing. Recently, considerable efforts have been made to better characterize electrocatalytic materials, in particular with regard to stability, morphology and reaction mechanisms, using analytical tools such as mass spectrometry, electrochemical atomic force microscopy and photoelectrospectroscopy.158–160
6.2 Next-generation nucleic acid detection approaches
Nucleic acid detection technologies have advanced significantly in recent years. In particular, the CRISPR/Cas technologies, widely studied for their genome editing capabilities, have attracted considerable attention as a tool for nucleic acid detection.161 For example, CRISPR/Cas13 and Cas12a effectors exhibit ribonuclease activity upon target recognition, enabling the detection of DNA and RNA with remarkable sensitivity.162,163 This system, when combined with an isothermal amplification step, has been shown allow detection of a single target copy per microliter of biofluid within 2 hours, using gold nanoparticle labels for colorimetric readout.162,164 The assay can be adapted to other target analytes through design of an appropriate single guide RNA (sgRNA) and possesses multiplexing capabilities by combining orthogonal CRISPR/Cas systems.163 Alternatively, a proof-of-concept strategy called CRISPR-Chip has described the immobilization of CRISPR/Cas9 on graphene field effect transistors (gFET) for the label-free (electrochemical) detection of unamplified DNA.165 It is only a matter of time before this technology is applied to electrochemical detection with electroactive nanomaterials, representing exciting new avenues for double-signal amplification.Next, it should be remembered that multiplexed detection of several analytes by a single biosensor remains a difficult task.166 Current electrochemical strategies can detect up to three or four analytes on a single sensor; a number limited by the potential window and the intrinsic bandwidth of the redox reporter peaks.26 Parallelization of biosensors detection offers a robust approach to increasing the multiplexing capabilities of a device using standard microfabrication technologies. Systems with up to several thousand parallel electrodes have been reported for oligonucleotide detection, using enzymatic labels for signal amplification.167 However, most of these approaches require bulky instrumentation or robots to pipette the reagents onto microelectrodes. Microfluidics provide a promising platform to miniaturize these highly-parallelized systems with integrated signal amplification and detection strategies.
Progress in nucleic acid sequencing is transforming both epidemiological studies and personalized medicine. Next-generation sequencing methods commonly relies on solid-state membrane materials containing nanopores, with the passage of biomolecules through the pores being detected by the a change in current.168 Whilst assays based on biomarker recognition, as discussed in this review, are essentially hypothesis-driven, sequencing pathogen genomes may simultaneously yield information on the genes of interest, e.g. genes from well-conserved regions or those encoding antibiotic resistance, but also on mutations, creating valuable epidemiological information for disease surveillance. The commercialization and miniaturization of next-generation sequencing technology is likely to make it an interesting addition to the molecular diagnostics toolkit. Indeed, portable devices, such as the MinION by Oxford Nanopore,169 are now commercially available and capable of sequencing DNAs or RNAs when simply connected via USB to a laptop; highlighting their potential for rapid clinical response in resource-limited settings.169
6.3 Alternative biomarkers
Much activity has been devoted to the search for alternative biomarkers for disease detection via non-invasive procedures. In particular, circulating biomarkers, such as tumour cells, miRNAs and exosomes, in body fluids has emerged as robust tools for the detection of cancers and bacterial diseases, and additionally show great promise for electrochemical detection.170–172 With the ever-growing number of molecular and physiological biomarkers, precision medicine is moving towards a scenario where data analysis is carried out via machine learning techniques.173 Indeed, the development of biosensors capable of measuring a variety of analytes will require labels and amplification methods capable of high versatility, which could in principle be fulfilled by bespoke multifunctional nanomaterials.6.4 Towards decentralized healthcare
Decentralization of healthcare systems involves moving medical procedures away from hospitals and surgeries and into the home. Such a transition necessitates the development of diagnostic tests that can be reliably used at the point-of-care by untrained users. In this respect, the development of POC tests must consider not only the assay itself, but also every step between sample collection and outcome interpretation. As detection limits and sensitivities improve, key challenges will include standardizing sensor preparation (robustness, scalability, electrode and nanomaterial synthesis), sample collection (pre-treatment and pre-concentration), assay times and quantitative performance (through positive and negative controls).174 In this respect the Foundation for Innovative New Diagnostics (FIND) have compiled a list of guidelines for target product profiles, which details the minimum requirements of priority diagnostic tests to ensure that the needs of end-users are taken into account.175 For example, in the detection of gonorrhoea resistance to antibiotics, an ideal test should require less than 20 minutes (excluding sample preparation), and cost less than 15 USD at volume production.175 These recommendations set valuable guidelines for the development of nanomaterials-based electrochemical amplification for specific target diseases.Ultimately, the interpretation of complex diagnostic data may always require medical specialists, with the decentralization of healthcare relying on the connection of end-users to the specialists via digital technology.136 As such, electrochemical biosensors may become an ideal platform for mobile and digital health as they can be easily integrated with wireless communication and connected to a smartphone. With the development of mobile applications and online tools guiding patients for self-testing and follow-up, sensitive POC diagnostic tests can act as a first-round analysis before conducting more thorough laboratory tests, benefiting patients and the greater community.
7. Conclusion
Recent progress in nucleic acid detection offers multiple options for sensitive diagnostics with minimal equipment. Signal amplification strategies enable the development of electrochemical biosensors that access detection limits close to clinically relevant biomarker concentrations, without the need for a nucleic acid amplification step. In this review, we have attempted to highlight the multifunctional nature of nanomaterials, as electrocatalytic labels, cargos for redox indicators or reporters. Simultaneously, nanotechnology opens exciting opportunities for analyte capture and sample pre-concentration. The integration of these methods with microfluidic tools represents new pathways for the development of self-contained diagnostic tests that minimize sample handling and user input for practical clinical use. Clearly, the translation of research technologies into commercial products is an immense challenge, with few proposed methods being robust enough for commercial applications. Finally, we stress the urgent need for standard characterization procedures to understand, optimize, compare and ultimately improve the activity of nanomaterials for signal amplification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Eugenia Quach for proofreading. L. B. and C. J. S. are grateful for the financial support from the Swiss National Science Foundation (Project Number: 200021-178944). A. S. N. would like to thank the State Secretariat for Education, Research and Innovation (SERI) in Switzerland for a Swiss Government Excellence Scholarship (ESKAS No. 2016.0728).References
- D. P. Nikolelis and G. P. Nikoleli, Nanotechnology and biosensors, Elsevier, 2018, vol. 22 Search PubMed.
- H. Aldewachi, T. Chalati, M. N. Woodroofe, N. Bricklebank, B. Sharrack and P. Gardiner, Nanoscale, 2018, 10, 18–33 RSC.
- World malaria report, World Health Organization, Geneva, 2018 Search PubMed.
- E. H. Yoo and S. Y. Lee, Sensors, 2010, 10, 4558–4576 CrossRef.
- Z. Huang, S. Hu, Y. Xiong, H. Wei, H. Xu, H. Duan and W. Lai, TrAC, Trends Anal. Chem., 2019, 114, 151–170 CrossRef CAS.
- W. Putzbach and N. J. Ronkainen, Sensors, 2013, 13, 4811–4840 CrossRef CAS.
- J. Lei and H. Ju, Chem. Soc. Rev., 2012, 41, 2122 RSC.
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230–249 CrossRef CAS.
- S. Campuzano, P. Yáñez-Sedeño and J. M. Pingarrón, ChemElectroChem, 2019, 6, 60–72 CrossRef CAS.
- World Health Organization, Model List of Essential In Vitro Diagnostics, WHO Press, 2018, p. 35 Search PubMed.
- T. S. Crofts, A. J. Gasparrini and G. Dantas, Nat. Rev. Microbiol., 2017, 15, 422–434 CrossRef CAS.
- J. Wang, J. Chen and S. Sen, J. Cell. Physiol., 2016, 231, 25–30 CrossRef CAS.
- R. E. Drury, D. O'Connor and A. J. Pollard, Front. Immunol., 2017, 8, 1182 CrossRef.
- Y. Peng and C. M. Croce, Signal Transduction Targeted Ther., 2016, 1, 15004 CrossRef PubMed.
- S. Kelley, E. M. Boon, J. K. Barton, N. M. Jackson and M. G. Hill, Nucleic Acids Res., 1999, 27, 4830–4837 CrossRef CAS PubMed.
- S. Karkare and D. Bhatnagar, Appl. Microbiol. Biotechnol., 2006, 71, 575–586 CrossRef CAS.
- E. E. Ferapontova, Annu. Rev. Anal. Chem., 2018, 11, 197–218 CrossRef CAS PubMed.
- G. L. Igloi, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8562–8567 CrossRef CAS PubMed.
- Y. Seok Kim, N. H. Ahmad Raston and M. Bock Gu, Biosens. Bioelectron., 2016, 76, 2–19 CrossRef CAS PubMed.
- E. N. Brody and L. Gold, Rev. Mol. Biotechnol., 2000, 74, 5–13 CrossRef CAS PubMed.
- Y. Xiao, V. Pavlov, T. Niazov, A. Dishon, M. Kotler and I. Willner, J. Am. Chem. Soc., 2004, 126, 7430–7431 CrossRef CAS PubMed.
- E. Paleček, M. Fojta, M. Tomschik and J. Wang, Biosens. Bioelectron., 1998, 13, 621–628 CrossRef.
- W. Zhou, R. Saran and J. Liu, Chem. Rev., 2017, 117, 8272–8325 CrossRef CAS PubMed.
- Y. Zhao, F. Chen, Q. Li, L. Wang and C. Fan, Chem. Rev., 2015, 115, 12491–12545 CrossRef CAS.
- Y. Du and S. Dong, Anal. Chem., 2017, 89, 189–215 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods : fundamentals and applications, Wiley, 2001 Search PubMed.
- A. Chen and B. Shah, Anal. Methods, 2013, 5, 2158 RSC.
- J. G. Osteryoung and R. A. Osteryoung, Anal. Chem., 1985, 57, 101–110 CrossRef.
- P. Dauphin-Ducharme and K. W. Plaxco, Anal. Chem., 2016, 88, 11654–11662 CrossRef CAS PubMed.
- A. B. Steel, T. M. Herne and M. J. Tarlov, Anal. Chem., 1998, 70, 4670–4677 CrossRef CAS PubMed.
- E. E. Ferapontova and E. Domínguez, Electroanalysis, 2003, 15, 629–634 CrossRef CAS.
- I. Mannelli, M. Minunni, S. Tombelli, R. Wang, M. Michela Spiriti and M. Mascini, Bioelectrochemistry, 2005, 66, 129–138 CrossRef CAS PubMed.
- C. J. Murphy, M. R. Arkin, Y. Jenkins, N. D. Ghatlia, S. H. Bossmann, N. J. Turro and J. K. Barton, Science, 1993, 262, 1025–1029 CrossRef CAS PubMed.
- R. Tevatia, A. Prasad and R. F. Saraf, Anal. Chem., 2019, 91, 10501–10508 CrossRef CAS PubMed.
- L. Kékedy-Nagy, S. Shipovskov and E. E. Ferapontova, Electrochim. Acta, 2019, 318, 703–710 CrossRef.
- A. R. Arnold, M. A. Grodick and J. K. Barton, Cell Chem. Biol., 2016, 23, 183–197 CrossRef CAS PubMed.
- P. Dauphin-Ducharme, N. Arroyo-Currás and K. W. Plaxco, J. Am. Chem. Soc., 2019, 141, 1304–1311 CrossRef CAS PubMed.
- Y. Wu, R. D. Tilley and J. J. Gooding, J. Am. Chem. Soc., 2019, 141, 1162–1170 CrossRef CAS PubMed.
- J. J. Gooding and K. Gaus, Angew. Chem., Int. Ed., 2016, 55, 11354–11366 CrossRef CAS.
- P. E. Sheehan and L. J. Whitman, Nano Lett., 2005, 5, 803–807 CrossRef CAS PubMed.
- P. R. Nair and M. A. Alam, Appl. Phys. Lett., 2006, 88, 233120 CrossRef.
- S. J. Smith, C. R. Nemr and S. O. Kelley, J. Am. Chem. Soc., 2017, 139, 1020–1028 CrossRef CAS PubMed.
- J. Go and M. A. Alam, Appl. Phys. Lett., 2009, 95, 033110 CrossRef.
- T. M. Squires, R. J. Messinger and S. R. Manalis, Nat. Biotechnol., 2008, 26, 417–426 CrossRef CAS.
- F. Jelen, M. Tomschik and E. Paleček, J. Electroanal. Chem., 1997, 423, 141–148 CrossRef CAS.
- J. Wang, X. Cai, J. Wang, C. Jonsson and E. Palecek, Anal. Chem., 1995, 67, 4065–4070 CrossRef CAS.
- M. Zhou, Y. Zhai and S. Dong, Anal. Chem., 2009, 81, 5603–5613 CrossRef CAS.
- E. E. Ferapontova, Curr. Anal. Chem., 2010, 7, 51–62 CrossRef.
- F. F. Cheng, T. T. He, H. T. Miao, J. J. Shi, L. P. Jiang and J. J. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 2979–2985 CrossRef CAS.
- A. Abi and E. E. Ferapontova, Anal. Bioanal. Chem., 2013, 405, 3693–3703 CrossRef CAS.
- G. Liu, Y. Wan, V. Gau, J. Zhang, L. Wang, S. Song and C. Fan, J. Am. Chem. Soc., 2008, 130, 6820–6825 CrossRef CAS.
- D. L. Bates, Trends Biotechnol., 1987, 5, 204–209 CrossRef CAS.
- M. Staiano, A. Pennacchio, A. Varriale, A. Capo, A. Majoli, C. Capacchione and S. D'Auria, Enzymes as Sensors, Elsevier Science, 2017, vol. 589 Search PubMed.
- F. Manea, F. B. Houillon, L. Pasquato and P. Scrimin, Angew. Chem., Int. Ed., 2004, 43, 6165–6169 CrossRef CAS PubMed.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- C. N. Loynachan, M. R. Thomas, E. R. Gray, D. A. Richards, J. Kim, B. S. Miller, J. C. Brookes, S. Agarwal, V. Chudasama, R. A. McKendry and M. M. Stevens, ACS Nano, 2018, 12, 279–288 CrossRef CAS PubMed.
- Z. Gao and Z. Yang, Anal. Chem., 2006, 78, 1470–1477 CrossRef CAS PubMed.
- H. Yang, Curr. Opin. Chem. Biol., 2012, 16, 422–428 CrossRef CAS.
- O. Niwa, Electroanalysis, 1995, 7, 606–613 CrossRef CAS.
- M. R. Akanda, Y. L. Choe and H. Yang, Anal. Chem., 2012, 84, 1049–1055 CrossRef CAS.
- Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS.
- S. Yadav, M. K. Masud, M. N. Islam, V. Gopalan, A. K. Lam, S. Tanaka, N.-T. Nguyen, M. S. Al Hossain, C. Li, M. Y. Yamauchi and M. J. A. Shiddiky, Nanoscale, 2017, 9, 8805–8814 RSC.
- M. Kamal Masud, M. N. Islam, M. H. Haque, S. Tanaka, V. Gopalan, G. Alici, N.-T. Nguyen, A. K. Lam, M. S. A. Hossain, Y. Yamauchi and M. J. A. Shiddiky, Chem. Commun., 2017, 53, 8231–8234 RSC.
- M. N. Islam, M. K. Masud, N. T. Nguyen, V. Gopalan, H. R. Alamri, Z. A. Alothman, M. S. Al Hossain, Y. Yamauchi, A. K. Y. Lam and M. J. A. Shiddiky, Biosens. Bioelectron., 2018, 101, 275–281 CrossRef CAS PubMed.
- L. Tian, J. Qi, O. Oderinde, C. Yao, W. Song and Y. Wang, Biosens. Bioelectron., 2018, 110, 110–117 CrossRef CAS.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef CAS.
- X. Wu, Y. Chai, P. Zhang and R. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 713–720 CrossRef CAS.
- T. Yan, L. Zhu, H. Ju and J. Lei, Anal. Chem., 2018, 90, 14493–14499 CrossRef CAS PubMed.
- J. Chen, C. Yu, Y. Zhao, Y. Niu, L. Zhang, Y. Yu, J. Wu and J. He, Biosens. Bioelectron., 2017, 91, 892–899 CrossRef CAS PubMed.
- W. Wang, T. Bao, X. Zeng, H. Xiong, W. Wen, X. Zhang and S. Wang, Biosens. Bioelectron., 2017, 91, 183–189 CrossRef CAS.
- C. Zhang, J. He, Y. Zhang, J. Chen, Y. Zhao, Y. Niu and C. Yu, Biosens. Bioelectron., 2018, 102, 94–100 CrossRef CAS.
- S. Liu, Z. Yang, Y. Chang, Y. Chai and R. Yuan, Biosens. Bioelectron., 2018, 119, 170–175 CrossRef CAS PubMed.
- P. Kumar, A. Deep and K.-H. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef CAS.
- P. Ling, J. Lei, L. Zhang and H. Ju, Anal. Chem., 2015, 87, 3957–3963 CrossRef CAS PubMed.
- D. Cheng, X. Xiao, X. Li, C. Wang, Y. Liang, Z. Yu, C. Jin, N. Zhou, M. Chen, Y. Dong, Y. Lin, Z. Xie and C. Zhang, J. Electrochem. Soc., 2018, 165, B885–B892 CrossRef CAS.
- P. Ling, J. Lei and H. Ju, Biosens. Bioelectron., 2015, 71, 373–379 CrossRef CAS.
- G. M. Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533 RSC.
- L. Jiao, Y. Wang, H. L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- X. Zhu, F. Kou, H. Xu, Y. Han, G. Yang, X. Huang, W. Chen, Y. Chi and Z. Lin, Sens. Actuators, B, 2018, 270, 263–269 CrossRef CAS.
- L. Chen, L. Sha, Y. Qiu, G. Wang, H. Jiang and X. Zhang, Nanoscale, 2015, 7, 3300–3308 RSC.
- F. Sheng, X. Zhang and G. Wang, J. Mater. Chem. B, 2017, 5, 53–61 RSC.
- F. Zhou, Y. Yao, J. Luo, X. Zhang, Y. Zhang, D. Yin, F. Gao and P. Wang, Anal. Chim. Acta, 2017, 969, 8–17 CrossRef CAS PubMed.
- H. Y. Kim, J. K. Ahn, M. Il Kim, K. S. Park and H. G. Park, Electrochem. Commun., 2019, 99, 5–10 CrossRef CAS.
- A. De La Escosura-Muñiz, L. Baptista-Pires, L. Serrano, L. Altet, O. Francino, A. Sánchez and A. Merkoçi, Small, 2016, 12, 205–213 CrossRef PubMed.
- A. D. Castañeda, N. J. Brenes, A. Kondajji and R. M. Crooks, J. Am. Chem. Soc., 2017, 139, 7657–7664 CrossRef PubMed.
- F. Lisdat and D. Schäfer, Anal. Bioanal. Chem., 2008, 391, 1555–1567 CrossRef CAS PubMed.
- Y. Peng and Z. Gao, Anal. Chem., 2011, 83, 820–827 CrossRef CAS PubMed.
- X. Li, K. Scida and R. M. Crooks, Anal. Chem., 2015, 87, 9009–9015 CrossRef CAS PubMed.
- P. Miao, B. Wang, F. Meng, J. Yin and Y. Tang, Bioconjugate Chem., 2015, 26, 602–607 CrossRef CAS PubMed.
- M. You, S. Yang, W. Tang, F. Zhang and P. He, Biosens. Bioelectron., 2018, 112, 72–78 CrossRef CAS PubMed.
- X. Jin, L. Zhou, B. Zhu, X. Jiang and N. Zhu, Biosens. Bioelectron., 2018, 107, 237–243 CrossRef CAS PubMed.
- L. Liu, Y. Chang, N. Xia, P. Peng, L. Zhang, M. Jiang, J. Zhang and L. Liu, Biosens. Bioelectron., 2017, 94, 235–242 CrossRef CAS PubMed.
- A. Ono, S. Cao, H. Togashi, M. Tashiro, T. Fujimoto, T. MacHinami, S. Oda, Y. Miyake, I. Okamoto and Y. Tanaka, Chem. Commun., 2008, 4825–4827 RSC.
- A. Rotaru, S. Dutta, E. Jentzsch, K. Gothelf and A. Mokhir, Angew. Chem., Int. Ed., 2010, 49, 5665–5667 CrossRef CAS PubMed.
- P. Miao, T. Zhang, J. Xu and Y. Tang, Anal. Chem., 2018, 90, 11154–11160 CrossRef CAS PubMed.
- A. Suea-Ngam, P. D. Howes, C. E. Stanley and A. J. Demello, ACS Sens., 2019, 4, 1560–1568 CrossRef CAS PubMed.
- C. Yang, K. Shi, B. Dou, Y. Xiang, Y. Chai and R. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 1188–1193 CrossRef CAS PubMed.
- Y. Wang, X. X. Zhang, L. Zhao, T. Bao, W. Wen, X. X. Zhang and S. Wang, Biosens. Bioelectron., 2017, 98, 386–391 CrossRef CAS PubMed.
- Q. Hu, W. Hu, J. Kong and X. Zhang, Biosens. Bioelectron., 2015, 63, 269–275 CrossRef CAS PubMed.
- Y. Ye, Y. Liu, S. He, X. Xu, X. Cao, Y. Ye and H. Zheng, Sens. Actuators, B, 2018, 272, 53–59 CrossRef CAS.
- N. Gao, F. Gao, S. He, Q. Zhu, J. Huang, H. Tanaka and Q. Wang, Anal. Chim. Acta, 2017, 951, 58–67 CrossRef CAS PubMed.
- F. Wang, Y. Chu, Y. Ai, L. Chen and F. Gao, Microchim. Acta, 2019, 186, 116 CrossRef PubMed.
- J. Wang, G. Liu and A. Merkoçi, J. Am. Chem. Soc., 2003, 125, 3214–3215 CrossRef CAS PubMed.
- W. Zhu, X. Su, X. Gao, Z. Dai and X. Zou, Biosens. Bioelectron., 2014, 53, 414–419 CrossRef CAS PubMed.
- X. M. Li, L. L. Wang, J. Luo and Q. L. Wei, Biosens. Bioelectron., 2015, 65, 245–250 CrossRef CAS PubMed.
- C. C. Li, J. Hu, M. Lu and C. Y. Zhang, Biosens. Bioelectron., 2018, 122, 51–57 CrossRef CAS PubMed.
- B. Kaur, K. Malecka, D. A. Cristaldi, C. S. Chay, I. Mames, H. Radecka, J. Radecki and E. Stulz, Chem. Commun., 2018, 54, 11108–11111 RSC.
- I. Grabowska, D. G. Singleton, A. Stachyra, A. Góra-Sochacka, A. Sirko, W. Zagórski-Ostoja, H. Radecka, E. Stulz and J. Radecki, Chem. Commun., 2014, 50, 4196–4199 RSC.
- L. Wang, R. Ma, L. Jiang, L. Jia, W. Jia and H. Wang, Biosens. Bioelectron., 2017, 92, 390–395 CrossRef CAS PubMed.
- P. Jolly, M. R. Batistuti, A. Miodek, P. Zhurauski, M. Mulato, M. A. Lindsay and P. Estrela, Sci. Rep., 2016, 6, 36719 CrossRef CAS PubMed.
- Y. H. Yuan, Y. Di Wu, B. Z. Chi, S. H. Wen, R. P. Liang and J. D. Qiu, Biosens. Bioelectron., 2017, 97, 325–331 CrossRef CAS PubMed.
- K. Deng, X. Liu, C. Li and H. Huang, Biosens. Bioelectron., 2018, 117, 168–174 CrossRef CAS PubMed.
- D. Gong, X. Hui, Z. Guo and X. Zheng, Talanta, 2019, 198, 534–541 CrossRef CAS PubMed.
- M. Chen, N. Gan, T. Li, Y. Wang, Q. Xu and Y. Chen, Anal. Chim. Acta, 2017, 968, 30–39 CrossRef CAS PubMed.
- S. Huang, N. Gan, T. Li, Y. Zhou, Y. Cao and Y. Dong, Talanta, 2018, 179, 28–36 CrossRef CAS PubMed.
- J. Chang, X. Wang, J. Wang, H. Li and F. Li, Anal. Chem., 2019, 91, 3604–3610 CrossRef CAS PubMed.
- H. F. Cui, T. Bin Xu, Y. L. Sun, A. W. Zhou, Y. H. Cui, W. Liu and J. H. T. Luong, Anal. Chem., 2015, 87, 1358–1365 CrossRef CAS PubMed.
- X. Miao, Z. Li, A. Zhu, Z. Feng, J. Tian and X. Peng, Biosens. Bioelectron., 2016, 83, 39–44 CrossRef CAS PubMed.
- S. Yu, Y. Wang, L. P. Jiang, S. Bi and J. J. Zhu, Anal. Chem., 2018, 90, 4544–4551 CrossRef CAS PubMed.
- D. Zhu, W. Liu, W. Cao, J. Chao, S. Su, L. Wang and C. Fan, Electroanalysis, 2018, 30, 1349–1356 CrossRef CAS.
- R. Tavallaie, J. McCarroll, M. Le Grand, N. Ariotti, W. Schuhmann, E. Bakker, R. D. Tilley, D. B. Hibbert, M. Kavallaris and J. J. Gooding, Nat. Nanotechnol., 2018, 13, 1066–1071 CrossRef CAS PubMed.
- J. Tang and D. Tang, Microchim. Acta, 2015, 182, 2077–2089 CrossRef CAS.
- J. M. George, A. Antony and B. Mathew, Microchim. Acta, 2018, 185, 358 CrossRef PubMed.
- H. Huang and J.-J. Zhu, Analyst, 2013, 138, 5855 RSC.
- F. Zhao, T. Sun, F. Geng, P. Chenand and Y. Gao, Int. J. Electrochem. Sci., 2019, 14, 5287–5304 CrossRef CAS.
- J. Tang and D. Tang, Microchim. Acta, 2015, 182, 2077–2089 CrossRef CAS.
- L. D. Burke and P. F. Nugent, Gold Bull., 1997, 30, 43–53 CrossRef CAS.
- H. Cai, Y. Xu, N. Zhu, P. He and Y. Fang, Analyst, 2002, 127, 803–808 RSC.
- M. Hasanzadeh, N. Shadjou and M. de la Guardia, TrAC, Trends Anal. Chem., 2015, 72, 1–9 CrossRef CAS.
- C. Xu and X. Qu, NPG Asia Mater., 2014, 6, e90 CrossRef CAS.
- G. Yang, C. Zhu, D. Du, J. Zhu and Y. Lin, Nanoscale, 2015, 7, 14217–14231 RSC.
- C.-J. Shih, Chimia, 2016, 70, 800–804 CrossRef CAS PubMed.
- J. Chen, C. Yu, Y. Zhao, Y. Niu, L. Zhang, Y. Yu, J. Wu and J. He, Biosens. Bioelectron., 2017, 91, 892–899 CrossRef CAS PubMed.
- K. J. Land, D. I. Boeras, X. S. Chen, A. R. Ramsay and R. W. Peeling, Nat. Microbiol., 2019, 4, 46–54 CrossRef CAS PubMed.
- C. S. Wood, M. R. Thomas, J. Budd, T. P. Mashamba-Thompson, K. Herbst, D. Pillay, R. W. Peeling, A. M. Johnson, R. A. McKendry and M. M. Stevens, Nature, 2019, 566, 467–474 CrossRef CAS PubMed.
- D. T. Chiu, A. J. deMello, D. Di Carlo, P. S. Doyle, C. Hansen, R. M. Maceiczyk and R. C. R. Wootton, Chem, 2017, 2, 201–223 CAS.
- P. A. Auroux, Y. Koc, A. DeMello, A. Manz and P. J. R. Day, Lab Chip, 2004, 4, 534–546 RSC.
- A. J. De Mello, Lab Chip, 2001, 1, 24N RSC.
- D. G. Rackus, M. H. Shamsi and A. R. Wheeler, Chem. Soc. Rev., 2015, 5320, 5320 RSC.
- K. Yamada, H. Shibata, K. Suzuki and D. Citterio, Lab Chip, 2017, 17, 1206–1249 RSC.
- World Health Organization, WHO list of prequalified diagnostic products, Geneva, 2019 Search PubMed.
- W. Dungchai, O. Chailapakul and C. S. Henry, Anal. Chem., 2009, 81, 5821–5826 CrossRef CAS PubMed.
- K. Yamanaka, M. C. Vestergaard and E. Tamiya, Sensors, 2016, 16, 1761 CrossRef PubMed.
- C. T. Kokkinos, D. L. Giokas, A. S. Economou, P. S. Petrou and S. E. Kakabakos, Anal. Chem., 2018, 90, 13 CrossRef PubMed.
- W. R. de Araujo, C. M. R. Frasson, W. A. Ameku, J. R. Silva, L. Angnes and T. R. L. C. Paixão, Angew. Chem., Int. Ed., 2017, 56, 15113–15117 CrossRef CAS PubMed.
- J. C. Cunningham, N. J. Brenes and R. M. Crooks, Anal. Chem., 2014, 86, 6166–6170 CrossRef CAS PubMed.
- K. Scida, J. C. Cunningham, C. Renault, I. Richards and R. M. Crooks, Anal. Chem., 2014, 86, 6501–6507 CrossRef CAS PubMed.
- M.-N. Tsaloglou, A. Nemiroski, G. Camci-Unal, D. C. Christodouleas, L. P. Murray, J. T. Connelly and G. M. Whitesides, Anal. Biochem., 2018, 543, 116–121 CrossRef CAS PubMed.
- K. Hsieh, A. S. Patterson, B. S. Ferguson, K. W. Plaxco and H. T. Soh, Angew. Chem., Int. Ed., 2012, 51, 4896–4900 CrossRef CAS PubMed.
- M. Zhou and S. Dong, Acc. Chem. Res., 2011, 44, 1232–1243 CrossRef CAS PubMed.
- Metrohm, Portable potentiostats, http://www.metrohm-autolab.com/Products/Echem/PortablePot/PortablePotentiostats.html, (accessed 11 August 2019).
- BioLogic Science Instruments, Portable potentiostat, http://www.bio-logic.net/en/products/potentiostat-galvanostat-eis/pg581-portable-potentiostatgalvanostat/, (accessed 11 August 2019).
- PalmSens Compact Electrochemical Interfaces, EmStat, https://www.palmsens.com/product/emstat/, (accessed 11 August 2019).
- A. Nemiroski, D. C. Christodouleas, J. W. Hennek, A. A. Kumar, E. J. Maxwell, M. T. Fernández-Abedul, G. M. Whitesides, M. T. Fernandez-Abedul and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11984–11989 CrossRef CAS PubMed.
- A. Ainla, M. P. S. Mousavi, M. N. Tsaloglou, J. Redston, J. G. Bell, M. T. Fernández-Abedul and G. M. Whitesides, Anal. Chem., 2018, 90, 6240–6246 CrossRef CAS PubMed.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- E. Pastor, F. Le Formal, M. T. Mayer, S. D. Tilley, L. Francàs, C. A. Mesa, M. Grätzel and J. R. Durrant, Nat. Commun., 2017, 8, 14280 CrossRef CAS PubMed.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, Nat. Catal., 2018, 1, 508–515 CrossRef CAS.
- S. Fatayer, F. Albrecht, Y. Zhang, D. Urbonas, D. Peña, N. Moll and L. Gross, Science, 2019, 365, 142–145 CAS.
- Y. Li, S. Li, J. Wang and G. Liu, Trends Biotechnol., 2019, 37, 730–743 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, M. J. Kellner, J. Joung, J. J. Collins and F. Zhang, Science, 2018, 360, 439–444 CrossRef CAS PubMed.
- C. Myhrvold, C. A. Freije, J. S. Gootenberg, O. O. Abudayyeh, H. C. Metsky, A. F. Durbin, M. J. Kellner, A. L. Tan, L. M. Paul, L. A. Parham, K. F. Garcia, K. G. Barnes, B. Chak, A. Mondini, M. L. Nogueira, S. Isern, S. F. Michael, I. Lorenzana, N. L. Yozwiak, B. L. MacInnis, I. Bosch, L. Gehrke, F. Zhang and P. C. Sabeti, Science, 2018, 360, 444–448 CrossRef CAS PubMed.
- R. Hajian, S. Balderston, T. Tran, T. deBoer, J. Etienne, M. Sandhu, N. A. Wauford, J. Y. Chung, J. Nokes, M. Athaiya, J. Paredes, R. Peytavi, B. Goldsmith, N. Murthy, I. M. Conboy and K. Aran, Nat. Biomed. Eng., 2019, 3, 427–437 CrossRef CAS PubMed.
- M. Díaz-González, X. Muñoz-Berbel, C. Jiménez-Jorquera, A. Baldi and C. Fernández-Sánchez, Electroanalysis, 2014, 26, 1154–1170 CrossRef.
- A. Hassibi, A. Manickam, R. Singh, S. Bolouki, R. Sinha, K. B. Jirage, M. W. McDermott, B. Hassibi, H. Vikalo, G. Mazarei, L. Pei, L. Bousse, M. Miller, M. Heshami, M. P. Savage, M. T. Taylor, N. Gamini, N. Wood, P. Mantina, P. Grogan, P. Kuimelis, P. Savalia, S. Conradson, Y. Li, R. B. Meyer, E. Ku, J. Ebert, B. A. Pinsky, G. Dolganov, T. Van, K. A. Johnson, P. Naraghi-Arani, R. G. Kuimelis and G. Schoolnik, Nat. Biotechnol., 2018, 36, 738–745 CrossRef CAS PubMed.
- S. Goodwin, J. D. McPherson and W. R. McCombie, Nat. Rev. Genet., 2016, 17, 333–351 CrossRef CAS PubMed.
- ‘Oxford Nanopore Technologies’, MinION, https://nanoporetech.com/products/minion, (accessed 4 August 2019).
- S. Saini, Cell. Oncol., 2016, 39, 97–106 CrossRef CAS PubMed.
- K. K. Jain, The Handbook of Biomarkers, Springer New York, New York, NY, 2017 Search PubMed.
- S. Wang, L. Zhang, S. Wan, S. Cansiz, C. Cui, Y. Liu, R. Cai, C. Hong, I. T. Teng, M. Shi, Y. Wu, Y. Dong and W. Tan, ACS Nano, 2017, 11, 3943–3949 CrossRef CAS PubMed.
- V. Gligorijević, N. Malod-Dognin and N. Pržulj, Proteomics, 2016, 16, 741–758 CrossRef PubMed.
- R. W. Peeling and D. Mabey, Clin. Microbiol. Infect., 2010, 16, 1062–1069 CrossRef CAS PubMed.
- FIND, Target product profiles, https://www.finddx.org/target-product-profiles/, (accessed 4 August 2019).
| This journal is © The Royal Society of Chemistry 2020 |