DOI:
10.1039/D0MA00476F
(Paper)
Mater. Adv., 2020,
1, 3358-3368
Cyclic siloxanes conjugated with fluorescent aromatic compounds as fluoride sensors†
Received
2nd July 2020
, Accepted 13th October 2020
First published on 11th November 2020
Abstract
Cyclic siloxanes conjugated with anthracene (D4An) and pyrene (D4Py) were prepared via Heck coupling reactions of 2,4,6,8-tetramethyl-2,4,6,8-tetravinylcyclotetrasiloxane with 9-bromoanthracene and 1-bromopyrene, respectively. These materials exhibited solvent-dependent fluorescence with the largest Stokes shift of up to ∼146 nm, confirming the presence of intramolecular exciplex formation among the fluorophores within a cyclic siloxane. After reacting with fluoride ions, blue-shifted emissions of D4An and D4Py in organic solvents were observed; it suggests an increase in monomer emission as a result of exciplex preclusion via Si–F bond formation. Moreover, the colorless solutions of D4An and D4Py in THF turned pink and orange, respectively, in the presence of excessive F−, which also suggested the formation of charge-transfer complexes. The interactions between cyclic siloxanes and fluoride ions were also studied via FTIR, ESI-MS and NMR experiments. Computational modeling studies revealed that fluoride played an important role in distorting the geometry of cyclic siloxanes by interrupting excimers of fluorescent aromatic rings in the side-chain.
Introduction
Polysiloxane (R2SiO)n can be found as major components in commercial products in our daily life such as scented hydrogels, silicon ware, household products, personal care products, electrical devices and cookware.1,2 They are described as organosilicon compounds that include silicon–oxygen backbones with organic side chains attached to each silicon atom.3–5 Flexible siloxane polymers or silicones have been extensively used due to its high thermal stability, chemical resistance, low tension, smooth texture, and non-toxicity and biocompatibility of compounds depending on functionalization.6–9 Siloxanes have two general forms: cyclic and linear.10 For cyclic siloxanes, silicon and oxygen atoms bond and connect at each end to form a ring structure.4,11 Cyclic siloxanes are used in a wide range of applications due to the hydrocarbon side chain of siloxane, which is beneficial for side-chain functionalization. In terms of synthesis, cyclic siloxanes are frequently used as precursors for the synthesis of high performance polysiloxane elastomers,12,13 and as cross-linking agents to strengthen materials.14–16 Recently, polysiloxanes and cyclic siloxanes functionalized with fluorophores have exhibited excellent sensing ability to detect specific chemical species. For example, Ren et al. reported tetraphenylethene units modified into polysiloxane chains possessing fluorescence ability and capable of detecting the vapor-phase of explosive compounds.17 Lang et al. successfully synthesized a siloxane polymer containing fluoranthene groups that showed high thermal stability and strong fluorescence emission that can be quenched by picric acid detection.18 Polydimethylsiloxane bearing spirobenzopyran, which acted as a colorimetric probe for pH determination and Ag+ and Fe3+ ion detection, was reported by Li and co-workers.19 Recently, Lin et al. studied pyrenyl-functionalized cyclic siloxane and polysiloxane for nitroaromatic compound detection.20 Along this line, the development of anion sensors from cyclic siloxanes would be very intriguing and challenging. In the meantime, a lot of molecular interactions were proposed for designing probes, including hydrogen bonding,21,22 electrostatic interaction,23 deprotonation,24,25 nucleophilic substitution26 and Si–O bond cleavage.27,28
In the last two decades, the usage of organosilica-based cage materials has expanded to cover many fields of applications such as catalytic supports, polymers, biocompatible materials and sensors.29 Previously, we have reported fluorescent organosilica-based cage materials for both efficient fluoride detection and adsorption.30–32 The principle of electrostatic interaction between fluoride ions and the potential surface of organosilica cages, which plays a role in the attraction of positive and negative parts to each other, in those materials has been proposed.33 Those responsive changes in fluorescence emission involve the conversion of an exciplex signal to a monomer signal due to F− induced conformational changes of the cage.34 Intriguingly, the less rigid cyclic siloxanes, in comparison with the cage siloxanes, are worth exploring as they are more common starting materials for silicone and gel materials. Herein, two cyclic siloxanes conjugated with anthracene (D4An) and pyrene (D4Py) were successfully synthesized with high fluorescence emission. UV-vis absorption and fluorescence spectroscopy were used to investigate their absorption and emission. Furthermore, the interactions of fluoride and cyclic siloxanes were supported by computations as well as FTIR and 19F NMR spectroscopy. Quantum calculations were performed to propose the optimized geometry as well as changes in the conformation of the fluorophores. This work aims to demonstrate the application of cyclic siloxanes as an anchoring platform for anion sensors.
Results and discussion
Synthesis and solvent effects of cyclic siloxanes (D4An and D4Py)
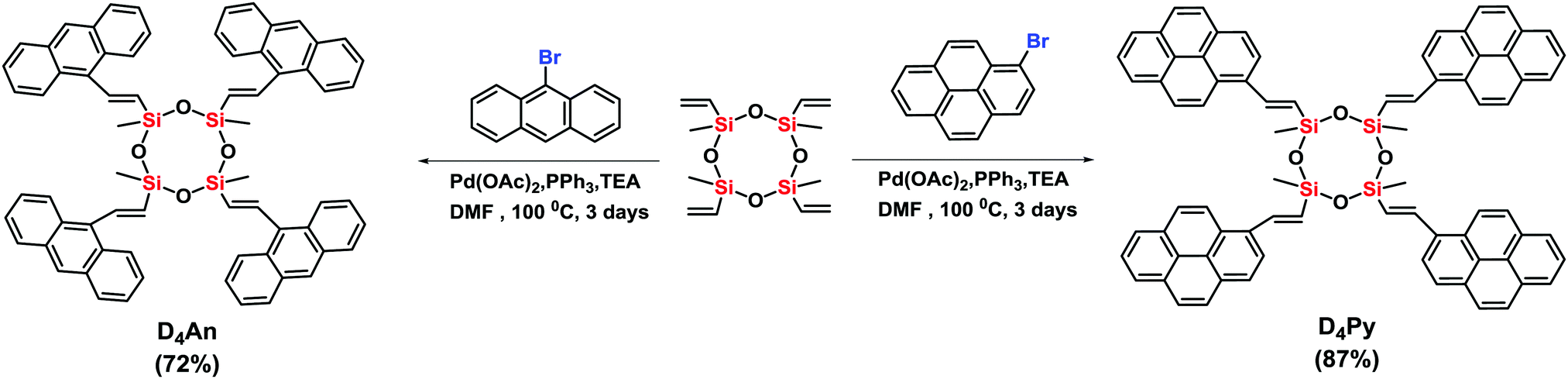
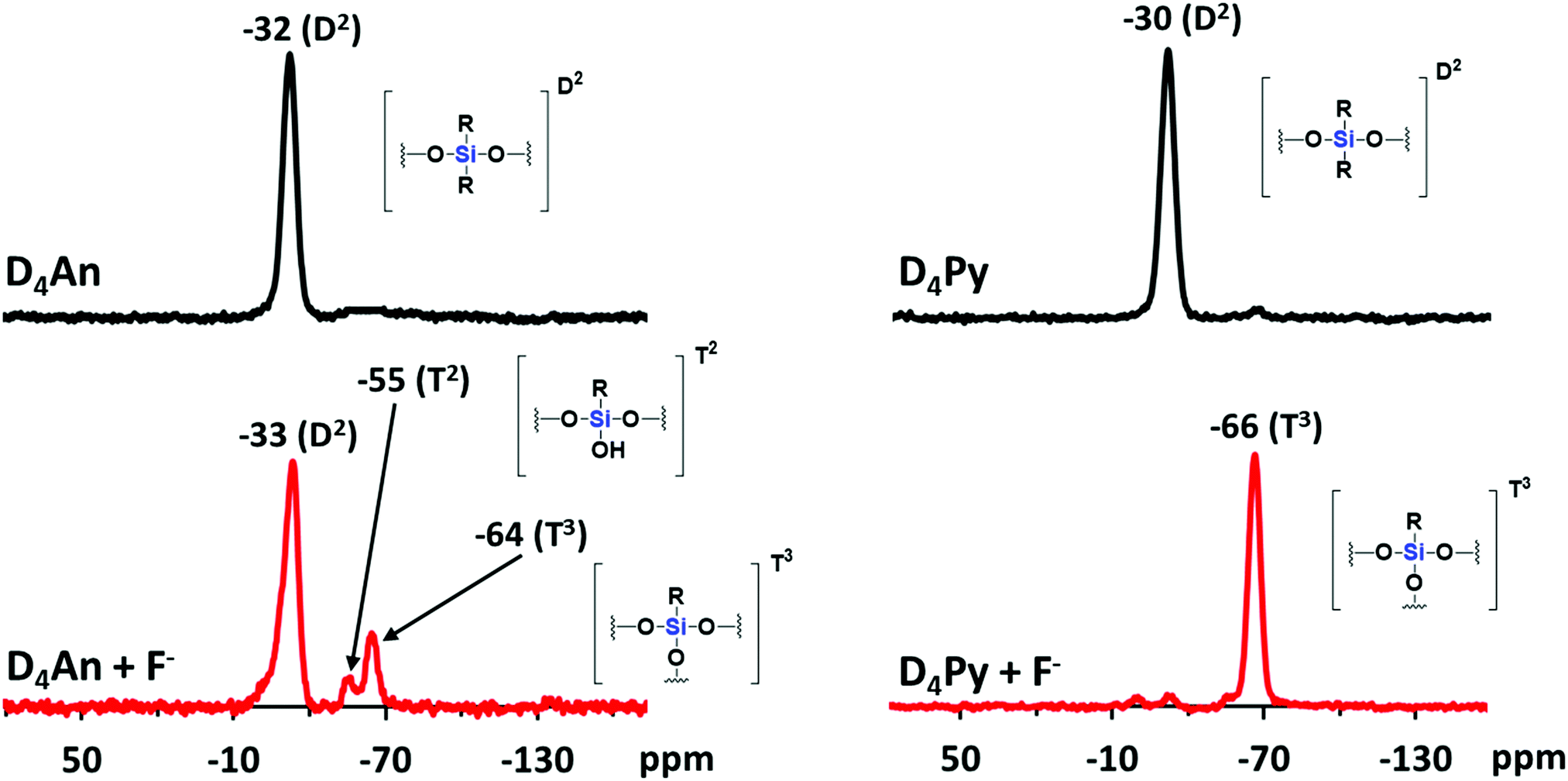
Compounds of D4An and D4Py were synthesized via Heck coupling reaction from 2,4,6,8-tetramethyl-2,4,6,8-tetravinylcyclotetrasiloxane and 9-bromoanthracene or 1-bromopyrene as precursors (Scheme 1). A dark-orange powder of D4An and yellow powder of D4Py were obtained in 72% and 87% yields, respectively (Fig. S1, ESI†). The solid state 1H MAS NMR spectrum of D4An shows a broad peak at 6.28 ppm and sharp peaks in the range of 0.02–1.29 ppm, which indicate the presence of aromatic and methyl groups, respectively. For D4Py, a broad peak of pyrene appeared at 5.97 ppm while a sharp peak of the methyl group was found at 0.03 ppm (Fig. S2, ESI†). The solid state 29Si MAS NMR spectra of D4An and D4Py only showed a singlet peak at −32 and −30 ppm, respectively (Fig. 5), confirming the formation of the siloxane (D type) network. These solid products could be a mixture of 2 and 4 fluorophore units on cyclic siloxanes, as shown in the MALDI-TOF MS spectra, confirming the coupling of the cyclic products with 4 anthracene units with the found m/z at 1049.429 (calcd [C68H56O4Si4 + H+]: m/z 1049.333) as well as 4 pyrene units with the found m/z at 1145.778 (calcd [C76H56O4Si4 + H+]: m/z 1145.333) (Fig. S3, ESI†).
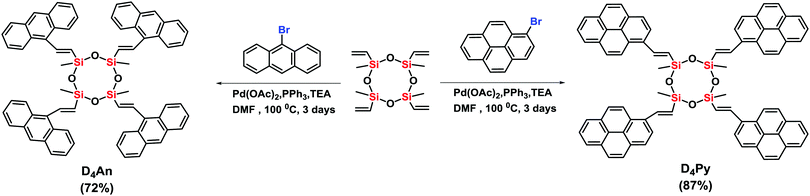 |
| | Scheme 1 Synthesis of cyclic siloxanes conjugated with anthracene (D4An) and pyrene (D4Py). | |
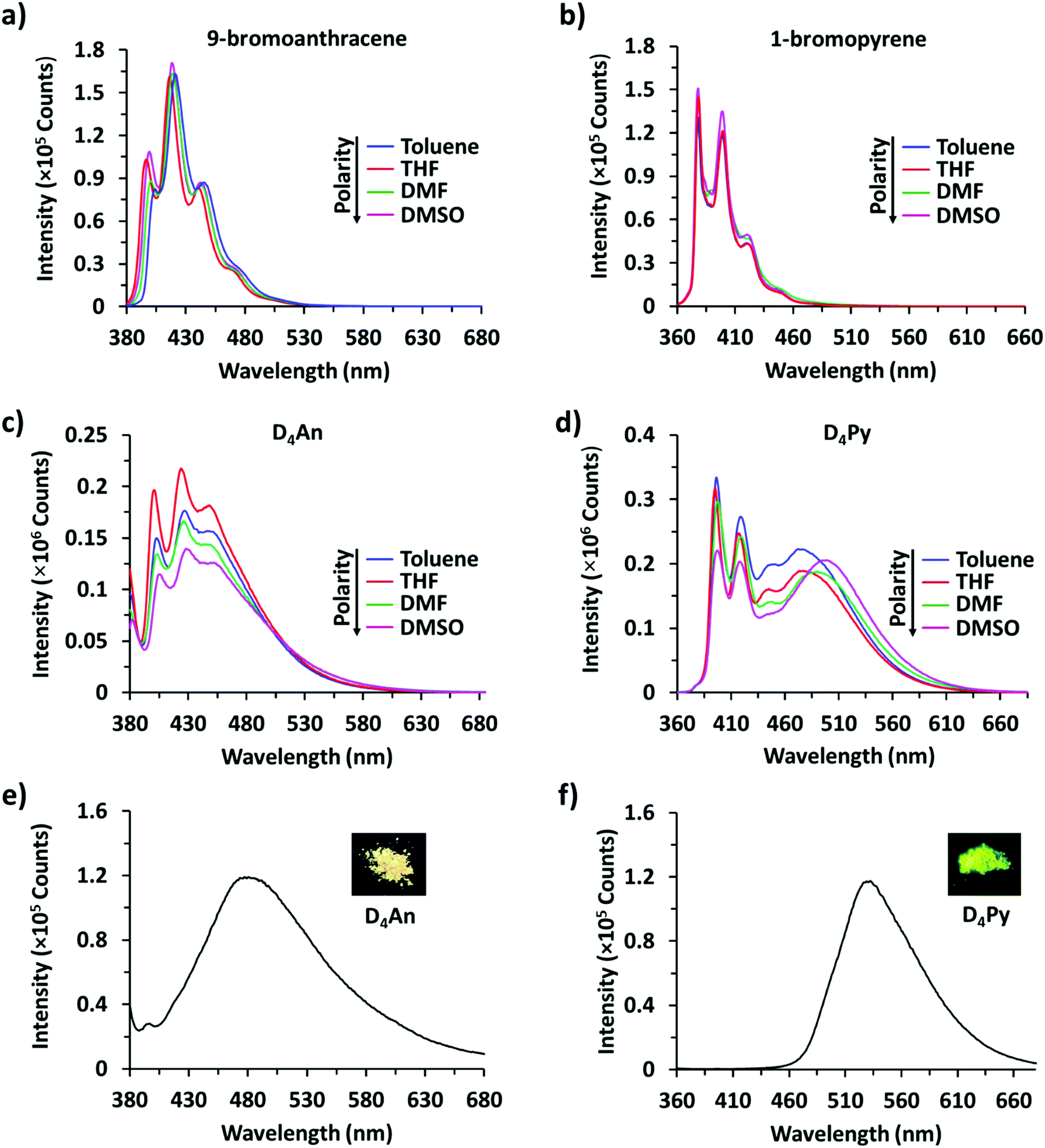
It is well-known that anthracene and pyrene monomers possess solvent-dependent properties where solvent polarity could induce different ratios of monomer and excimer emissions.35–38 The fluorescence intensity of both 9-bromoanthracene and 1-bromopyrene slightly increased with the increase of solvent polarity while their fluorescence intensity showed two or three sharp peaks (Fig. 1a and b), confirming the presence of monomer emission. However, small broad peaks at longer wavelengths were observed, attributed to intermolecular excimer formation.39,40 Confirming the successful synthesis of cyclic siloxanes conjugated with fluorescent aromatic compounds, the spectra of both cyclic siloxanes in organic solvents appeared broader and showed a red-shift of fluorescence emissions at longer wavelengths (450–600 nm) (Fig. 1c and d). This result indicated significant intramolecular exciplex formation among the fluorophore units within the cyclic siloxanes;41 at the same time, the largest Stokes shift (Δλ = 146 nm) from the absorption band at 350 nm to the excimer emission band at 496 nm of D4Py was observed in DMSO as a representative of highly polar solvents (Fig. 1d). In slightly polar solvents, the exciplex emissions of D4Py showed a shorter Stokes shift (126 nm for toluene and THF). Therefore, intramolecular exciplex formation within cyclic siloxanes of both D4An and D4Py is favourable in polar organic solvents. In comparison with D4Py, all D4An solutions gave bright blue emission and a shorter Stokes shift of 68–73 nm, measured using the differences between an absorption peak at 376–379 nm and a peak of excimer emission at 446–452 nm (Fig. 1c). It was suggested that π–π interactions in anthracene units within cyclic siloxanes are weaker than those within pyrenyl siloxanes. This suggestion can also be confirmed by the solid-state fluorescence spectra of D4An and D4Py that showed broad spectra with only single peaks at 477 and 532 nm, respectively (Fig. 1e and f).
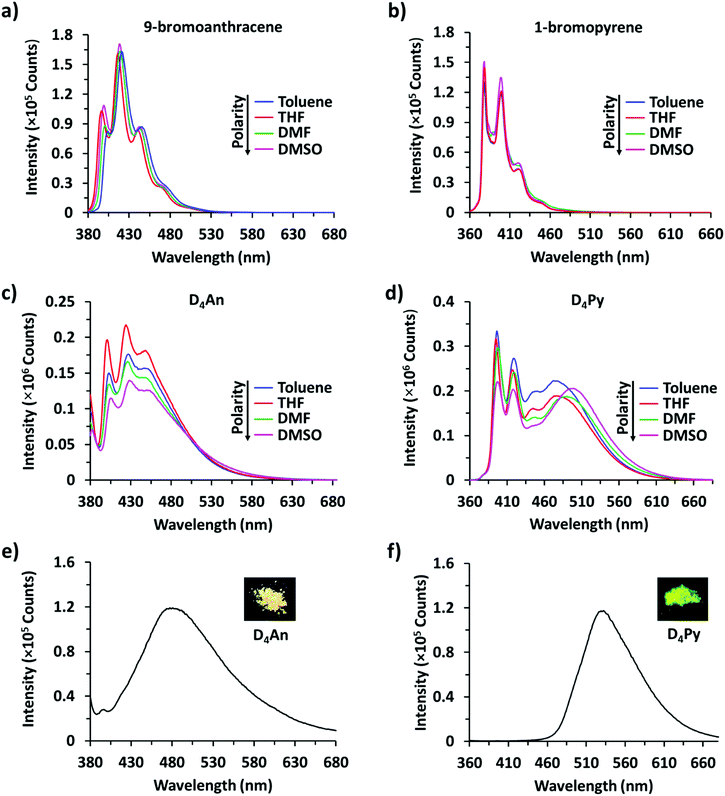 |
| | Fig. 1 Fluorescence spectra of (a) 9-bromoanthracene (1 × 10−4 M) at λex = 370 nm, (b) 1-bromopyrene (1 × 10−4 M) at λex = 346 nm, (c) D4An (2 × 10−5 M) at λex = 378 nm, and (d) D4Py (1 × 10−5 M) at λex = 350 nm; solid-state fluorescence spectra of (e) D4An at λex = 378 nm and (f) D4Py at λex = 350 nm. | |
Selectivity test
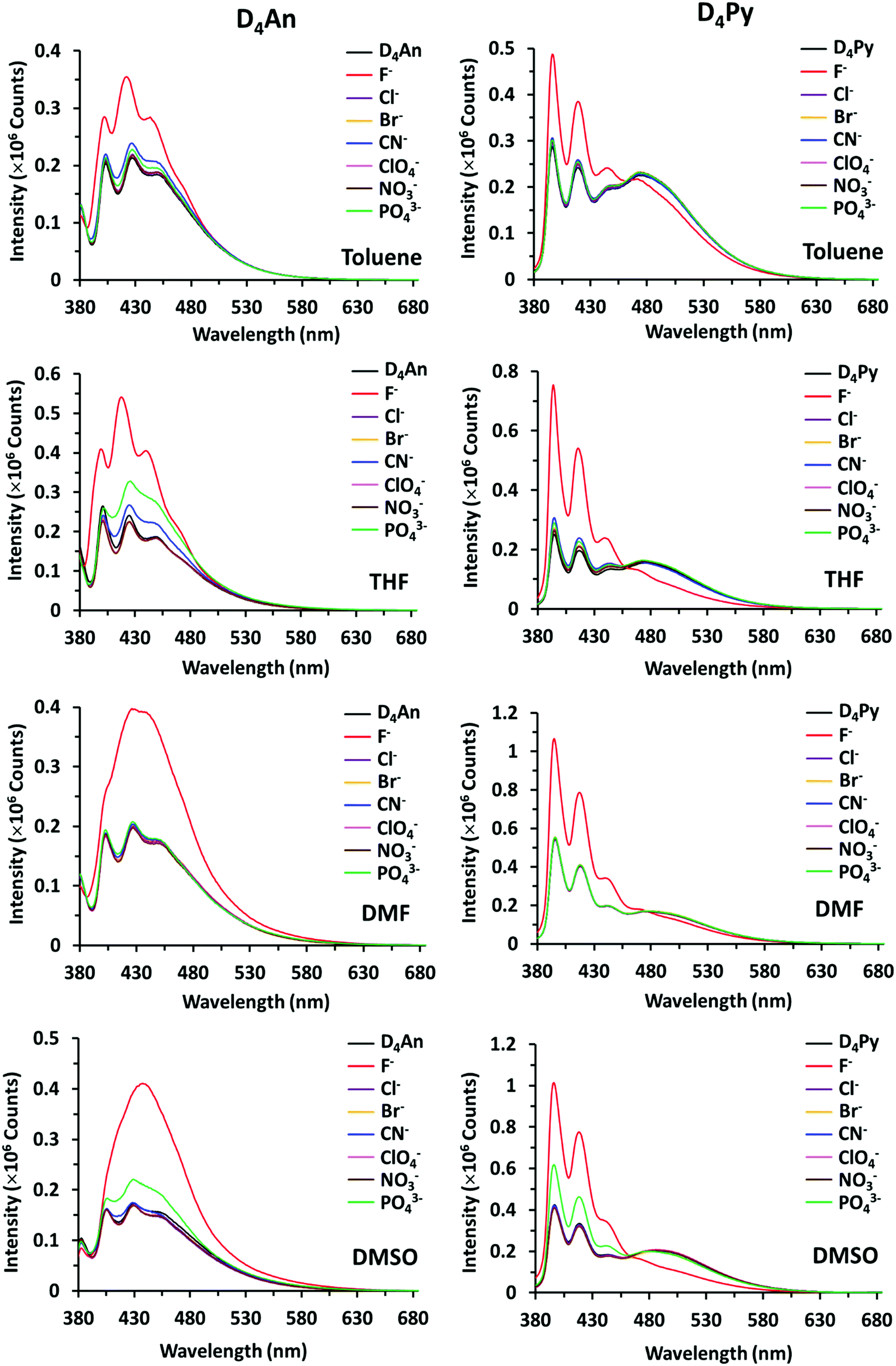
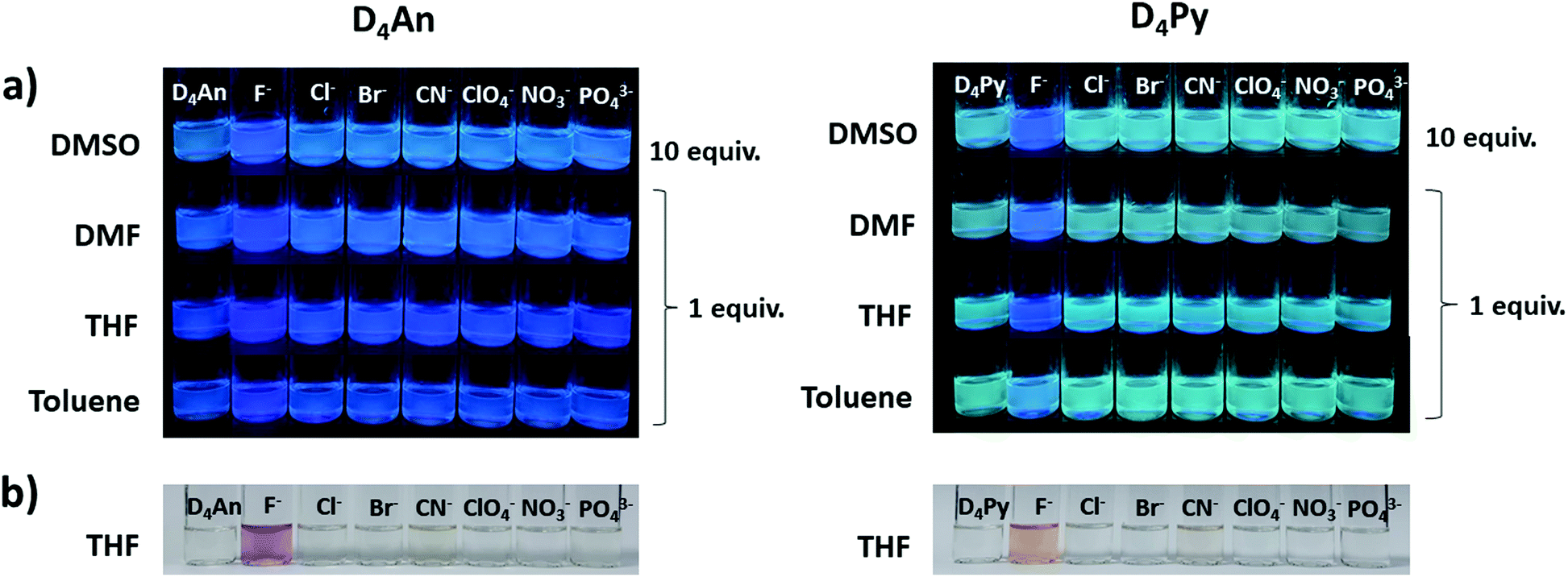
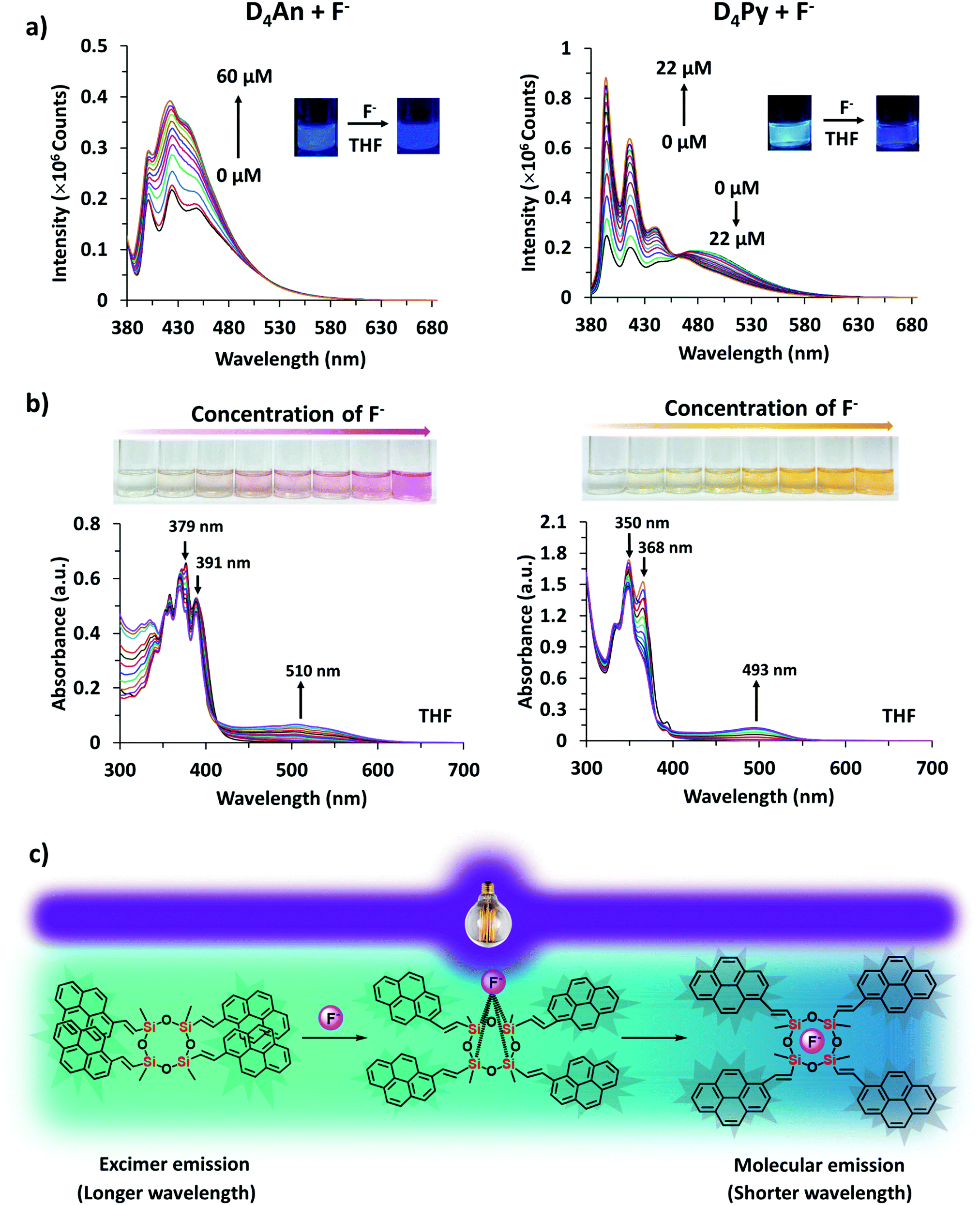
The fluorescence emission patterns of D4An and D4Py before and after the addition of anions were investigated in four solvents (e.g. toluene, THF, DMF and DMSO). When 1 equiv. of tetrabutylammonium salts (TBA-X: X = F−, Cl−, Br−, CN−, ClO4−, NO3− and PO43−) was added into both D4An and D4Py solutions except in DMSO where 10 equiv. was added, only F− can significantly enhance the fluorescence intensity of the monomer emission (Fig. 2) with a deep blue color in all solvents (Fig. 3a). For example, upon addition of F− into D4Py solutions, the monomer emissions (λmax ∼ 395 nm) increased while the intramolecular exciplex emissions (λmax ∼ 480 nm) decreased (Fig. 2). This result confirmed that F− was involved in the reorganization and orientation of the pyrene units. The green emission of the D4Py solution changed to deep blue emission after adding fluoride, as shown in Fig. 3a, which also supported the separation of the intramolecular pyrene–pyrene excimer into a single monomer. Moreover, after the addition of excess anions (200 equiv.) into both cyclic siloxanes in all solvents, these compounds additionally responded to CN− and PO43− (Fig. S4 and S5, ESI†). The fluorescence emissions of D4An and D4Py could also be intensified in highly polar solvents (DMF and DMSO) after the addition of F−, CN− and PO43−. Nonetheless, the fluorescence emissions of D4Py in THF with a large excess of F− were completely quenched (Fig. S5, ESI†). Furthermore, only F− (200 equiv.) could induce naked-eye color changes of D4An and D4Py solutions in THF from colorless to pink and orange, respectively (Fig. 3b). These results suggested charge-transfer (CT) complexation between F− and the fluorophores via anion–π interaction.31 Notably, single fluorophore compounds such as 1-bromopyrene and 9-bromoanthracene did not show any color change suggesting that the anchoring of multiple fluorophores within the same molecule was necessary to promote this interaction.
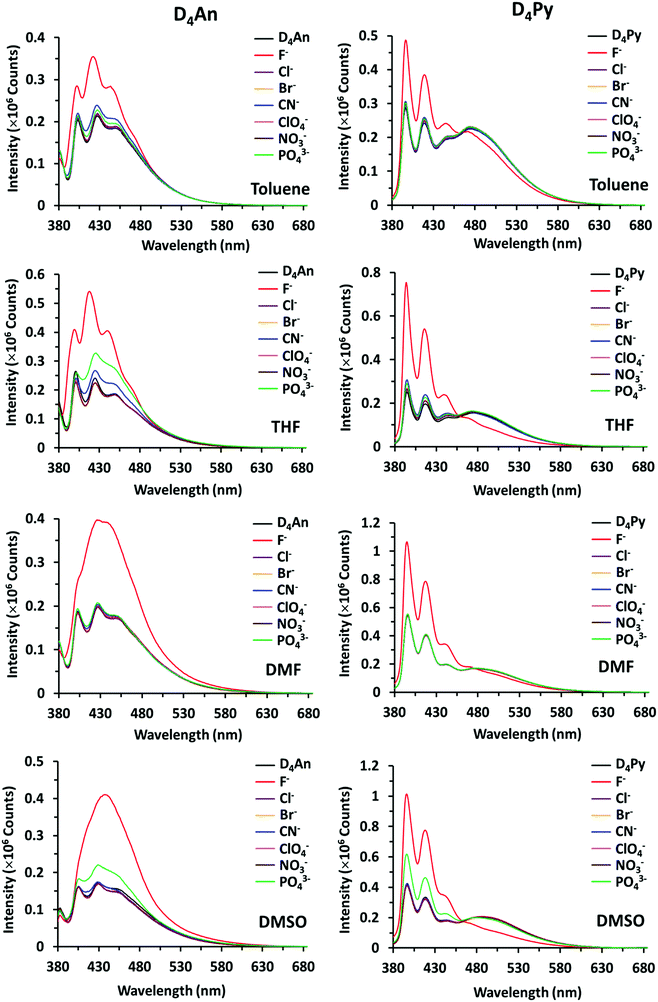 |
| | Fig. 2 Fluorescence spectra of D4An (2 × 10−5 M) at λex = 378 nm (left) and D4Py (1 × 10−5 M) at λex = 350 nm (right) in various solvents before and after the addition of 1 equiv. of anions. | |
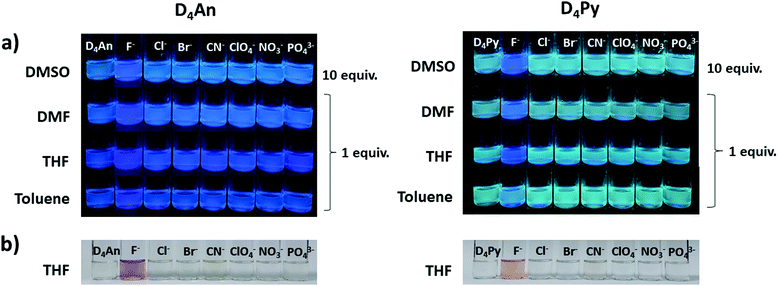 |
| | Fig. 3 Selective images of (a) D4An (2 × 10−5 M) (left) and D4Py (1 × 10−5 M) (right) after the addition of anions (1 and 10 equiv.) in various solvents under a UV lamp; (b) D4An (4 × 10−5 M) (left) and D4Py (2 × 10−5 M) (right) in THF upon the addition of anions (200 equiv.) under daylight. | |
Kinetic study
The fluorescence responses of D4An and D4Py after adding 200 equiv. of F−, CN−, and PO43− were measured at a fixed excitation wavelength for 30 minutes. For F−, the change in fluorescence signal occurred and completed almost instantly in DMF. In contrast, the response was slower in slightly polar solvents like toluene, as shown in Fig. S6 (ESI†). For CN−, D4An responses were slower in all solvents, but a faster response was seen in slightly polar solvents for D4Py. In addition, PO43− caused a fast kinetic response of D4Py in highly polar solvents (DMSO and DMF), but a similar rate in all solvents for D4An. The pseudo-first order kinetic rate constants of the three ions were calculated and displayed in Table S1 (ESI†).
Quantitative analyses of fluoride
An optimized concentration of D4An at 2 × 10−5 M was used in fluorescence titration for quantitative analysis of F−. The fluorescence emission of D4An in THF displayed a peak maximum at 427 nm; after the addition of F− into the D4An solution, the peak shifted to 420 nm, and it then increased when the concentration of F− was increased (Fig. 4a). The same trend of emission spectra was observed in toluene, DMF and DMSO, as shown in Fig. S7 (ESI†). The association constant (Ka) was calculated based on Benesi–Hildebrand plots from titration spectra. The Ka values were 4.9, 11.8, 2.5 and 3.1 (×104 M−1) in toluene, THF, DMF and DMSO, respectively. The detection limits of F− in D4An in toluene, THF, DMF and DMSO were found to be 0.2, 0.3, 0.8 and 1.7 μM, respectively. However, the optimized concentration of D4Py of 1 × 10−5 M was used for performing fluoride titration. The monomeric emission spectra in the range of 395–397 nm increased, but the peak intensities in the range of 475–482 nm, which represented intermolecular exciplex formation dropped and generated isosbestic points at 470, 462, 474 and 476 nm in toluene, THF, DMF and DMSO, respectively (Fig. 4a and Fig. S7, ESI†). The association constant values of D4Py were calculated as 6.3, 3.4, 4.5 and 3.3 (×105 M−1), which were higher than those of D4An, indicating that D4Py could bind more strongly with fluoride ions than D4An. Notably, the detection limits of D4Py in toluene, THF, DMF and DMSO were 0.3, 0.4, 1.0 and 1.0 μM, respectively. These results indicate that D4An and D4Py exhibit good selectivity and sensitivity for detecting fluoride ions. The association constant (Ka), LOD and LOQ of D4An and D4Py with fluoride in various solvents are summarized in Table S2 (ESI†).
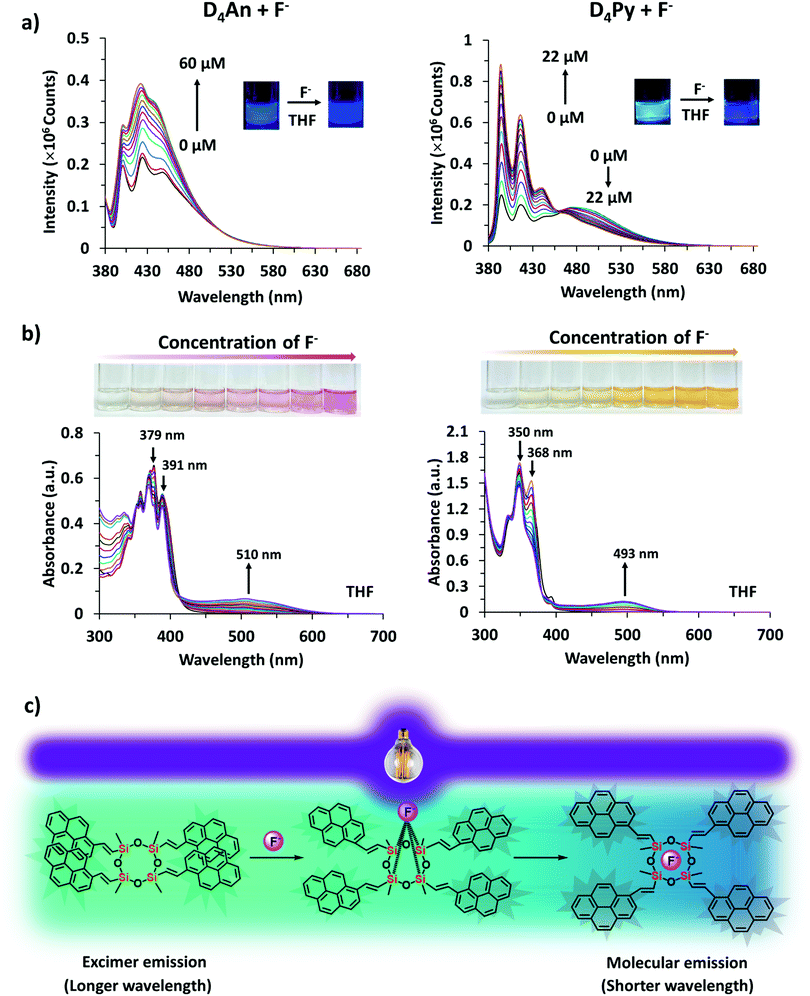 |
| | Fig. 4 (a) Fluorescence titration spectra of D4An (2 × 10−5 M) at λex = 378 nm (left) and D4Py (1 × 10−5 M) at λex = 350 nm (right) in THF at different concentrations of fluoride; (b) UV-vis adsorption spectra of D4An (4 × 10−5 M) (left) and D4Py (2 × 10−5 M) (right) in THF at different concentrations of fluoride; and (c) proposed interactions between D4Py and fluoride according to their fluorescence characters. | |
Proposed mechanism of fluoride interaction
Since D4An and D4Py are structures of cyclic siloxanes conjugated with a polyaromatic group, both compounds exhibited a positive contour within the cyclic siloxanes, which provides good possibilities for attracting F−.33 After the addition of F− in the system, the Si–F bond can change the conformation of fluorophores on cyclic siloxanes, the destruction of excimers was observed from the fluorescence emission spectra (Fig. 4a and c) and then the cyclic siloxanes were cleaved. The FTIR results showed that D4An and D4Py exhibited the asymmetric stretching vibration of Si–O–Si at 1025 cm−1. Nevertheless, a broad peak at around 3378 cm−1 (–OH) and a sharp peak at 882 cm−1 (Si–OH) appeared after the addition of fluoride (1 equiv.) into both D4An and D4Py (Fig. S8, ESI†). These results confirmed that the cyclic siloxane was cleaved by fluoride ions. Moreover, the 19F NMR titration experiment in CDCl3 showed free TBAF at −112 ppm. The signal appeared at −124 ppm represented the free F− on a glass tube (SiF62−).42,43 Upon the addition of fluoride into D4An and D4Py solutions, new peaks appeared at −129 ppm, indicating the interaction between the cyclic siloxane and F− (Fig. S9 and S10, ESI†). Interestingly, their solid state 29Si MAS NMR spectra after the addition of F− showed new smaller peaks at −55 and −64 ppm for D4An, while the peak of D4Py almost completely shifted from −30 to −66 ppm. These NMR results suggested that the Si–O linkages of cyclic siloxanes were not only cleaved using F−, but some Si–C bonds were also broken and rearranged to form silsesquioxanes (T unit) of silicon (Fig. 5).44,45 Furthermore, the ESI-MS results of D4An and D4Py with fluoride also showed small fractions of cyclic siloxane caused by fluoride ions (Fig. S11 and S12, ESI†). These results also validated the fact that the interaction between fluoride and D4An or D4Py cleaved the cyclic siloxane.
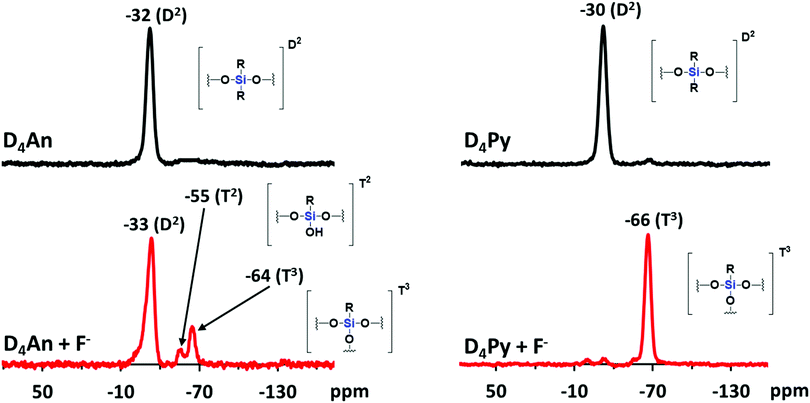 |
| | Fig. 5
29Si MAS NMR spectra of D4An (left) and D4Py (right) before and after the addition of fluoride (1 equiv.). | |
UV-vis spectroscopy
In the UV-vis spectra (Fig. S13, ESI†), the absorption spectrum of D4An showed three major peaks at 360, 379 and 391 nm while that of D4Py also showed three peaks at 337, 350 and 370 nm. The absorption spectra of D4An and D4Py in the presence of various anions were studied in an excessive amount (200 equiv.) in THF solvent, as shown in Fig. S14 (ESI†). The result suggested that D4An exhibited a new absorption peak at 510 nm while peaks at 379 and 391 nm dropped in the presence of F−, and a clear isosbestic point at 415 nm was observed. The absorption spectra of D4Py at 350 and 368 nm plummeted whereas a new peak at 493 nm increased with the isosbestic point at 382 nm after the addition of F− (Fig. 4b); however, the other anions did not significantly cause a change to the spectral pattern (Fig. S14, ESI†). The new red-shift band along with the isosbestic point indicated a conversion of the fluorophore to its CT complex with F−. Moreover, the association constant (Ka) values for the CT complex formation of D4Py and D4An were 1.2 × 103 and 394 M−1, respectively. The detection limits of F− obtained via UV-vis absorption titration for D4An and D4Py were 3.6 × 10−5 and 1.6 × 10−5 M, respectively. The association constant (Ka), LOD and LOQ of D4An and D4Py with fluoride in THF are summarized in Table S2 (ESI†). These results indicated that D4Py is more sensitive for F− detection in comparison with D4An, which is consistent with the fluorescence titration data described earlier.
Computational study
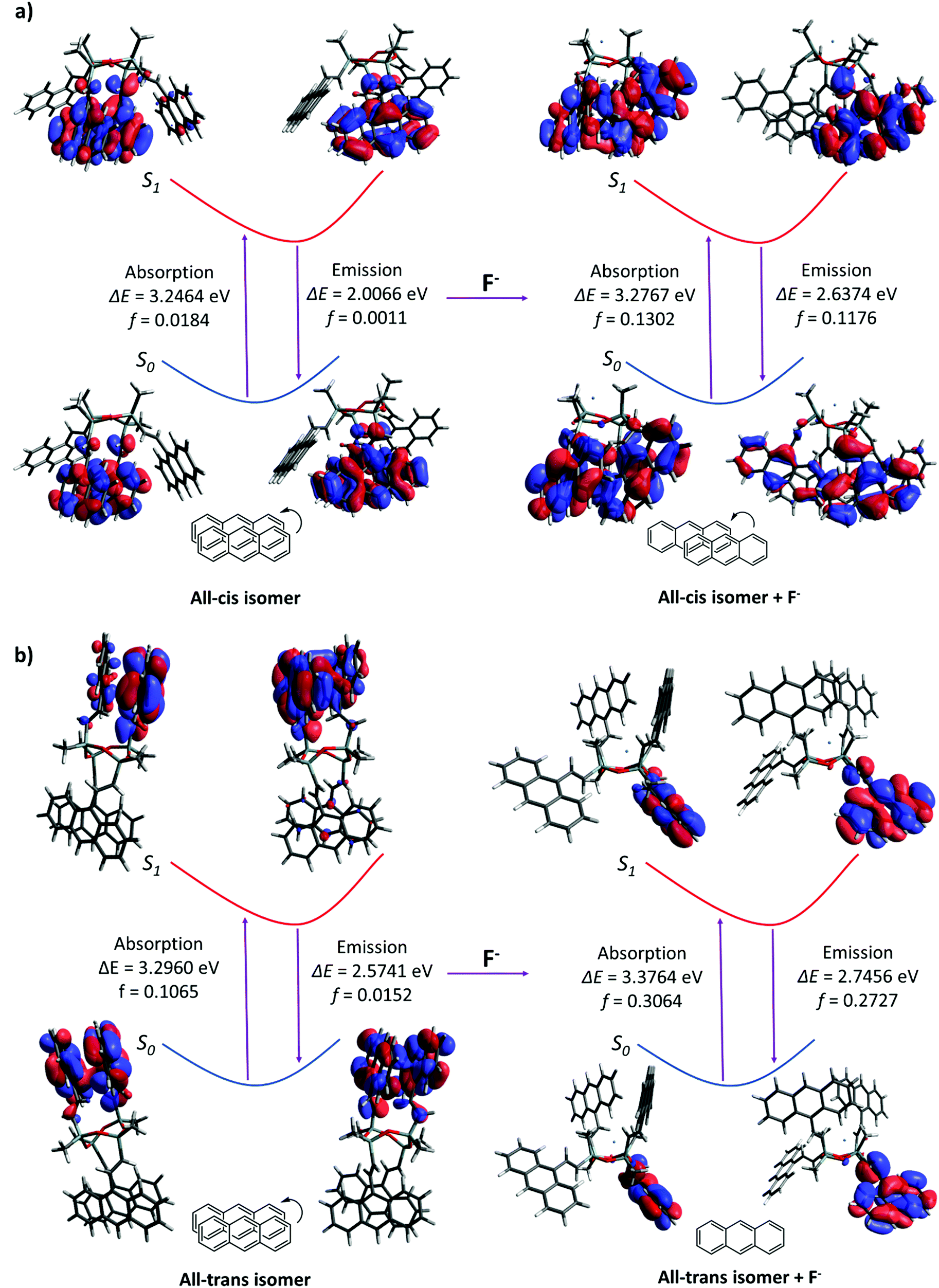
Quantum calculations were performed to investigate the changes in fluorescence emission upon the formation of the D4An-F− model, which has been speculated as an intermediate before the cleavage of the D4 ring. D4 isomerism basically has four major isomers, which are all-cis, all-trans, cis–trans–cis and cis–cis–trans (Fig. S21, ESI†).46 After geometrical optimization, all-cis and all-trans isomers were the most stable species due to their lower energy compared to the rest (Table S3, ESI†); therefore, all-cis and all-trans isomers of D4An were chosen for further study. The photon absorption and fluorescence of all-cis and all-transD4An are shown in Fig. 6, and the obtained results were in agreement with our previous report.31 A blue shift of fluorescence wavelength was found in both structures evidently from the different conformations of fluorophores caused by F−.47 The pristine formation of anthracene was dominated by graphite-like stacking. When D4 interacted with fluoride, the formation of stacking anthracene changed to slipped-parallel and single monomers in the all-cis and all-trans isomers of D4An, respectively. Therefore, the quantum calculations can well explain the fluorescence wavelength changes after fluoride addition.
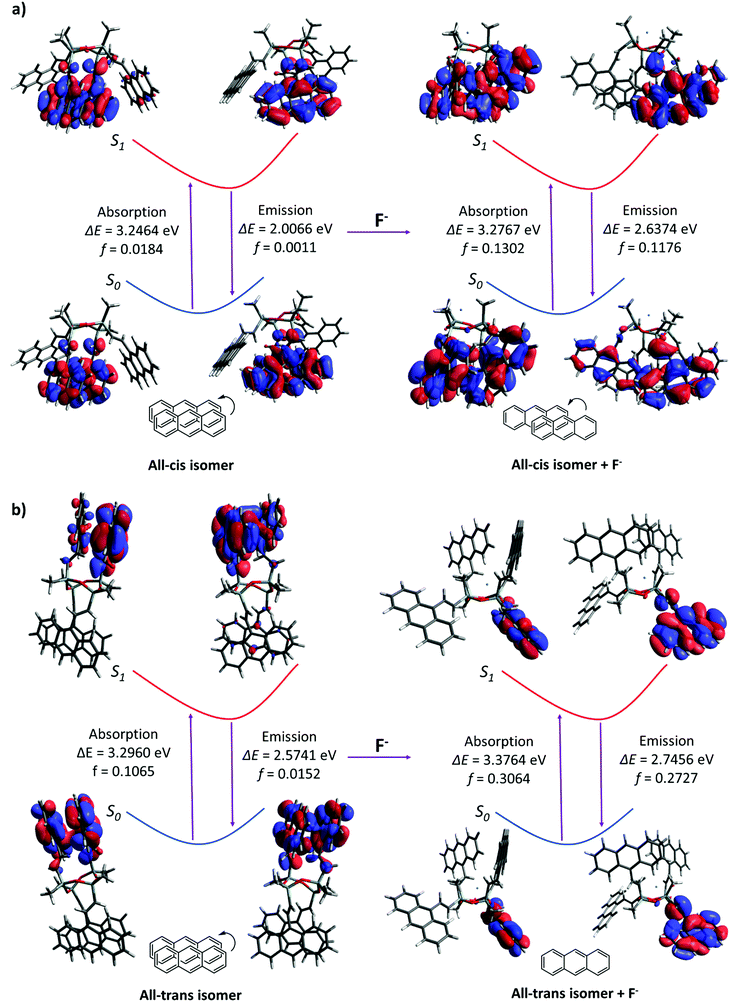 |
| | Fig. 6 Photoexcitation and emission including oscillator strength (f) in Frank-Condo state of (a) all-cisD4An and (b) all-transD4Anin vacuo. | |
Conclusions
Cyclic siloxane-based anthracene (D4An) and pyrene (D4Py) were successfully synthesized via Heck coupling reaction and showed solvent-dependent fluorescence with a largest Stokes shift (Δλ = 146 nm). These functionalized cyclic siloxanes in organic solvents exhibited strong fluorescence of intramolecular excimers, which showed excellent selectivity towards F− over other anions. In the presence of fluoride ions, their fluorescence emissions at longer wavelengths, mainly from the radiative relaxation of exciplex species of fluorophores, decreased, while those at shorter wavelengths, caused by the emission of separated fluorophores, increased. The synthesized molecules provided low detection limits in the ranges of 0.2–1.7 and 0.3–1.0 μM for D4An and D4Py, respectively. Moreover, the naked eye detection of fluoride ions was achieved in THF solvent as the colour changed to pink in D4An and to orange in D4Py. Interestingly, it was found that not only the Si–O linkages of cyclic siloxanes were cleaved using F−, but some Si–C bonds could also be broken and rearranged to form the silsesquioxane (T unit) network of silicon. The host–guest interaction was also studied through computational modelling; intramolecular excimer formation within cyclic siloxanes can be interrupted by fluoride ions leading to monomer formation causing a blue-shift of fluorescence emissions. Therefore, cyclic siloxanes can be one of the promising material candidates for F− sensors in organic solvents.
Experiments
Materials and methods
2,4,6,8-Tetramethyl-2,4,6,8-tetravinylcyclotetrasiloxane, 1-bromopyrene, 9-bromoanthracene, palladium(II) acetate, triphenylphosphine and triethylamine were purchased from TCI Chemicals. Anhydrous dimethylformamide was purchased from Sigma-Aldrich. Commercial solvents such as dichloromethane were further distilled. UV-vis spectra were recorded on a UV-vis spectrometer (Shimadzu UV-2600). All fluorescence emission spectra were recorded using a spectrometer (Horiba FluoroMax4+, slit width: 2 nm). The FTIR spectra were recorded using the attenuated total reflectance (ATR) technique on a Bruker Alpha spectrometer. High resolution ESI-MS spectra (positive mode) were measured on a micrOTOF instrument. MALDI-TOF MS was performed on a Bruker Autoflex instrument with trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile as a matrix.
Synthetic procedures
Synthesis of 2,4,6,8-tetrakis((E)-2-(anthracen-9-yl)vinyl)-2,4,6,8-tetramethyl-1,3,5,7,2,4,6,8-tetraoxatetrasilocane (D4An).
First, 9-bromoanthracene (1.54 g; 6 mmol) and triphenylphosphine (0.1311 g; 0.5 mmol) were added into a 200 mL heavy wall pressure vessel tube. Then, anhydrous dimethylformamide (15 mL), triethylamine (3.5 mL) and 2,4,6,8-tetramethyl-2,4,6,8-tetravinylcyclotetrasiloxane (0.35 mL; 1 mmol) were added into the tube, respectively. The reaction mixture was exposed to a flow of argon gas for 5 minutes, and then palladium(II) acetate (0.0229 g; 0.1 mmol) was added into the solution. The reaction mixture was stirred at 100 °C for 3 days. After cooling to room temperature, the solution was filtered for removing the palladium catalyst using Celite. The filtrate was precipitated with ice and a dark crude product was obtained, which was filtered and subsequently washed with deionized water. Then, the crude product was eluted via flash column chromatography using dichloromethane as an eluent. A dark orange powder was obtained, which was further purified via Soxhlet extraction using methanol and hexane (one day per extraction). A dark orange product was obtained (0.7634 g, 73% yield). Solid state 1H MAS NMR (400 MHz, δ): 0.02–1.29 ppm (–CH3) and 6.28 ppm (Ar-H), as shown in Fig. S2 (ESI†). Solid state 29Si MAS NMR (400 MHz, δ): −32 ppm, as depicted in Fig. 5. MALDI-TOF MS for 4 anthracene subunits (n = 4), calcd C68H56O4Si4 + H+: m/z 1049.333 [M + H+]; found: m/z 1049.429 (Fig. S3, ESI†). The FTIR spectrum showed a vibration band in the range of 2873–3048 cm−1 (–CH stretching) and at 1049 cm−1 (Si–O–Si stretching), as shown in Fig. S8 (ESI†).
Synthesis of 2,4,6,8-tetramethyl-2,4,6,8-tetrakis((E)-2-(pyren-1-yl)vinyl)-1,3,5,7,2,4,6,8-tetraoxatetrasilocane (D4Py).
1-Bromopyrene (1.69 g; 6 mmol) and triphenylphosphine (0.1311 g; 0.5 mmol) were added into a 200 mL heavy wall pressure vessel tube. Then, 15 mL of anhydrous dimethylformamide, 3.5 mL of triethylamine and 2,4,6,8-tetramethyl-2,4,6,8-tetravinylcyclotetrasiloxane (0.35 mL; 1 mmol) were added into the vessel tube. Argon flow was introduced into the solution for 5 minutes, and then palladium(II) acetate (0.0229 g; 0.1 mmol) was added into the solution. The reaction mixture was stirred and heated at 100 °C for 3 days. Then, the solution was cooled to room temperature and filtered using a Buchner funnel to remove the insoluble catalyst. The solution was precipitated with crushed ice cubes. The crude product was filtered and washed with deionized water. The residue was eluted via flash column chromatography on silica gel using dichloromethane as an eluent and then further purified by using Soxhlet extraction in methanol and hexane (one day per extraction). A yellow powder was obtained (0.9968 g, 87% yield). Solid state 1H MAS NMR (400 MHz, δ): 0.03 ppm (–CH3) and 5.97 ppm (Ar-H), as shown in Fig. S2 (ESI†). Solid state 29Si MAS NMR (400 MHz, δ): −30 ppm, as depicted in Fig. 5. MALDI-TOF MS for 4 substitutions of pyrene (n = 4), calcd C76H56O4Si4 + H+: m/z 1145.333 [M + H+]; found: m/z 1145.778 (Fig. S3, ESI†). The FTIR spectrum showed a vibration peak in the range of 2872–3082 cm−1 (–CH stretching) and at 1039 cm−1 (Si–O–Si stretching), as shown in Fig. S8 (ESI†).
Fluorescence and absorption studies of D4An and D4Py with anions
The stock solutions of anions (e.g. F−, Cl−, Br−, CN−, ClO4−, NO3− and PO43−) in the form of tetrabutylammonium salts in THF were each prepared at 0.1 M. Then, solutions of D4An (2 × 10−5 M) and D4Py (1 × 10−5 M) in all solvents (e.g. toluene, THF, DMF and DMSO) were prepared. The fluorescence spectra were measured with the addition of anions at 1 and 200 equiv. into the solution of D4An or D4Py (2 mL). On the other side, D4An (4 × 10−5 M) and D4Py (2 × 10−5 M) in THF were used for the absorption study. The UV-vis adsorption spectra were recorded in the solution of D4An or D4Py (2 mL) after the addition of anions (200 equiv.). All UV-vis and fluorescence spectra were recorded at room temperature.
Fluorescence and absorption titration of D4An and D4Py with fluoride
All tests were carried out at room temperature. The concentrations of D4An (2 × 10−5 M) and D4Py (1 × 10−5 M) in all solvents were used for the fluorescence experiment. The solutions of D4An and D4Py (2 mL) were added into a quartz cuvette. Then, 5–140 μL of the fluoride solution (10−3 M) was added into the cuvette. After stirring the solution for 3 minutes, the fluorescence spectra were recorded. For the absorption experiment, D4An (4 × 10−5 M) and D4Py (2 × 10−5 M) in THF (2 mL) were placed in the cuvette, followed by the addition of the fluoride solution (0.06 M and 0.03 M) in the range of 5–60 μL into D4An and D4Py solutions, respectively. The absorption spectra were recorded after stirring the solution for 3 minutes.
Computational study
The quantum calculations were performed using Gaussian 16 using density functional theory (DFT) and time-dependent density functional theory (TD-DFT); the M062X functional and Def2SVP basis set were used for all calculations in vacuo.48 The positive frequency of each geometry in its ground state was checked to validate the optimized structure. The molecular orbitals were visualized using the Avogadro software.49
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research project was supported by the Thailand Research Fund (RSA6280049 and RTA6180007), the Center of Excellence for Innovation in Chemistry (PERCH-CIC), the Ministry of Higher Education, Science, Research and Innovation (MHESI), and the Faculty of Science, Mahidol University through a CIF grant.
Notes and references
- Y. Horii and K. Kannan, Arch. Environ. Contam. Toxicol., 2008, 55, 701 CrossRef CAS.
- C. Racles, M. Silion and L. Sacarescu, Colloids Surf., A, 2018, 547, 102–110 CrossRef CAS.
-
Z. Han, A. Fina and G. Camino, in Polymer Green Flame Retardants, ed. C. D. Papaspyrides and P. Kiliaris, Elsevier, Amsterdam, 2014, pp. 389–418, DOI:10.1016/B978-0-444-53808-6.00012-3.
- H. Brothers, T. Boehmer, R. Campbell, S. Dorn, J. Kerbleski, S. Lewis, C. Mund, D. Pero, K. Saito, M. Wieser and W. Zoller, Int. J. Cosmet. Sci., 2017, 39, 580–588 CrossRef CAS.
- E. Yilgör and I. Yilgör, Prog. Polym. Sci., 2014, 39, 1165–1195 CrossRef.
- T. M. Tran, H. T. Le, N. D. Vu, G. H. Minh Dang, T. B. Minh and K. Kannan, Chemosphere, 2017, 184, 1117–1124 CrossRef CAS.
- T. B. Longenberger, K. M. Ryan, W. Y. Bender, A.-K. Krumpfer and J. W. Krumpfer, J. Chem. Educ., 2017, 94, 1682–1690 CrossRef CAS.
- Y. Song, M. Liu, L. Zhang, C. Mu and X. Hu, Chem. Eng. J., 2017, 328, 274–279 CrossRef CAS.
- T. Zhao, R. Yu, S. Li, X. Li, Y. Zhang, X. Yang, X. Zhao, C. Wang, Z. Liu, R. Dou and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 14391–14398 CrossRef CAS.
-
H. Fromme, in Encyclopedia of Environmental Health, ed. J. Nriagu, Elsevier, Oxford, 2nd edn, 2019, pp. 805–812, DOI:10.1016/B978-0-12-409548-9.11241-2.
- N. Espinosa, J. López, F. Flores Gracia, A. D. de la Luz and J. Martínez-Juárez, Adv. Mater. Lett., 2016, 7, 480–484 CrossRef.
- C. Racles, M. Cazacu, B. Fischer and D. Opris, Smart Mater. Struct., 2013, 22, 104004 CrossRef.
- Y. Meng, J. Chu, J. Xue, C. Liu, Z. Wang and L. Zhang, RSC Adv., 2014, 4, 31249–31260 RSC.
- X. Zhan, X. Cai and J. Zhang, RSC Adv., 2018, 8, 12517–12525 RSC.
- Y. Looi, M. Riduwan, M. B. H. Othman, R. Ramli, Z. Ishak and Z. Ahmad, Polym. Int., 2013, 62, 382–389 CrossRef.
- L. Y. Tyng, M. R. Ramli, M. B. H. Othman, R. Ramli, Z. A. M. Ishak and Z. Ahmad, Polym. Int., 2013, 62, 382–389 CrossRef CAS.
- Q. Li, Z. Yang, Z. Ren and S. Yan, Macromol. Rapid Commun., 2016, 37, 1772–1779 CrossRef CAS.
- Y. Liang, L. Xu, F. Qu, K. Tang, H. Wang and W. W. Yu, Polym. Chem., 2019, 10, 4818–4824 RSC.
- T. Zheng, Z. Xu, Y. Zhao, H. Li, R. Jian and C. Lu, Sens. Actuators, B, 2018, 255, 3305–3315 CrossRef CAS.
- Z. Gou, X. Zhang, Y. Zuo, M. Tian, B. Dong and W. Lin, ACS Appl. Mater. Interfaces, 2019, 11, 30218–30227 CrossRef CAS.
- Y. Li, J. Chen and T.-S. Chu, J. Comput. Chem., 2018, 39, 1639–1647 CrossRef CAS.
- N. Borah, B. Nayak, A. Gogoi and G. Das, New J. Chem., 2019, 43, 16497–16505 RSC.
- W. Ding, J. Xu, Y. Wen, J. Zhang, H. Liu and Z. Zhang, Anal. Chim. Acta, 2017, 967, 78–84 CrossRef CAS.
- D. Liu, L. Shi, S.-H. Gao, Y.-H. Wu, G.-Y. Li and C.-H. Zhou, Chem. Phys. Lett., 2020, 738, 136894 CrossRef CAS.
- Z. Li, S. Wang, L. Xiao, X. Li, X. Shao, X. Jing, X. Peng and L. Ren, Inorg. Chim. Acta, 2018, 476, 7–11 CrossRef CAS.
- L. Wang, W. Li, J. Lu, J.-P. Zhang and H. Wang, Tetrahedron, 2014, 70, 3172–3177 CrossRef CAS.
- P. Hou, S. Chen, H. Wang, J. Wang, K. Voitchovsky and X. Song, Chem. Commun., 2014, 50, 320–322 RSC.
- B. Qiu, Y. Zeng, L. Cao, R. Hu, X. Zhang, T. Yu, J. Chen, G. Yang and Y. Li, RSC Adv., 2016, 6, 49158–49163 RSC.
- N. Prigyai, S. Chanmungkalakul, V. Ervithayasuporn, N. Yodsin, S. Jungsuttiwong, N. Takeda, M. Unno, J. Boonmak and S. Kiatkamjornwong, Inorg. Chem., 2019, 58, 15110–15117 CrossRef CAS.
- S. Chanmungkalakul, V. Ervithayasuporn, S. Hanprasit, M. Masik, N. Prigyai and S. Kiatkamjornwong, Chem. Commun., 2017, 53, 12108–12111 RSC.
- S. Chanmungkalakul, V. Ervithayasuporn, P. Boonkitti, A. Phuekphong, N. Prigyai, S. Kladsomboon and S. Kiatkamjornwong, Chem. Sci., 2018, 9, 7753–7765 RSC.
- C. Wannasiri, S. Chanmungkalakul, T. Bunchuay, L. Chuenchom, K. Uraisin, V. Ervithayasuporn and S. Kiatkamjornwong, ACS Appl. Polym. Mater., 2020, 2, 1244–1255 CrossRef CAS.
- S. E. Anderson, D. J. Bodzin, T. S. Haddad, J. A. Boatz, J. M. Mabry, C. Mitchell and M. T. Bowers, Chem. Mater., 2008, 20, 4299–4309 CrossRef CAS.
- P. K. Lekha and E. Prasad, Chem. – Eur. J., 2010, 16, 3699–3706 CrossRef CAS.
- H. Liu, X. Yan, L. Shen, Z. Tang, S. Liu and X. Li, Mater. Horiz., 2019, 6, 990–995 RSC.
- E. V. Bichenkova, A. R. Sardarian, A. N. Wilton, P. Bonnet, R. A. Bryce and K. T. Douglas, Org. Biomol. Chem., 2006, 4, 367–378 RSC.
- M. Kanai, T. Hirano, I. Azumaya, I. Okamoto, H. Kagechika and A. Tanatani, Tetrahedron, 2012, 68, 2778–2783 CrossRef CAS.
- Z. Guo, S. Jin and B. Liu, Spectrochim. Acta, Part A, 2007, 66, 672–675 CrossRef.
- M. M. Islam, Z. Hu, Q. Wang, C. Redshaw and X. Feng, Mater. Chem. Front., 2019, 3, 762–781 RSC.
- J. Kim, L. J. Cote, F. Kim and J. Huang, J. Am. Chem. Soc., 2010, 132, 260–267 CrossRef CAS.
- M. Danko, P. Kasák and P. Hrdlovič, J. Photochem. Photobiol., A, 2015, 307-308, 79–87 CrossRef CAS.
- Å. Östlund, D. Lundberg, L. Nordstierna, K. Holmberg and M. Nydén, Biomacromolecules, 2009, 10, 2401–2407 CrossRef.
- K. O. Christe and W. W. Wilson, J. Fluorine Chem., 1990, 46, 339–342 CrossRef CAS.
-
R. M. Laine and M. Z. Asuncion, US20140120243A1, 2014.
- M. Ge and H. Liu, Chem. – Eur. J., 2018, 24, 2224–2231 CrossRef CAS.
- I. Ryuichi, K. Yuriko and K. Yusuke, Chem. Lett., 2009, 38, 364–365 CrossRef.
- C. Zhao, X. Cai, Z. Ma, J. Shi, L. Xu and H. Wang, J. Photochem. Photobiol., A, 2018, 355, 318–325 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00476f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a