
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the nature of the passivation layer enabling reversible calcium plating†
Juan
Forero-Saboya
 a,
Carine
Davoisne
bcd,
Rémi
Dedryvère
cde,
Ibraheem
Yousef
f,
Pieremanuele
Canepa
*g and
Alexandre
Ponrouch
*ac
a,
Carine
Davoisne
bcd,
Rémi
Dedryvère
cde,
Ibraheem
Yousef
f,
Pieremanuele
Canepa
*g and
Alexandre
Ponrouch
*ac
aInstitut de Ciència de Materials de Barcelona (ICMBA-CSIC), Campus UAB, 08193 Bellaterra, Spain. E-mail: aponrouch@icmab.es
bLaboratoire de Réactivité et Chimie des Solides, Université de Picardie Jules Verne, CNRS UMR7314, 33 rue Saint Leu, 80039 Amiens, France
cALISTORE – European Research Institute, CNRS FR3104, Hub de l’Energie, 15 Rue Baudelocque, 80039 Amiens, France
dRéseau sur le Stockage Electrochimique de l’Energie (RS2E) – FR CNRS 3459, 80039 Amiens Cedex, France
eIPREM, E2S-UPPA/CNRS/Université Pau & Pays Adour, 64000 Pau, France
fMIRAS Beamline, ALBA Synchrotron Light Source, Carrer de la Llum 2-26, 08290 Cerdanyola del Vallès, Spain
gDepartment of Materials Science and Engineering, The National University of Singapore, 117576, Singapore. E-mail: pcanepa@nus.edu.sg
First published on 26th August 2020
Abstract
As for other multivalent systems, the interface between the calcium (Ca) metal anode and the electrolyte is of paramount importance for reversible plating/stripping. Here, we combined experimental and theoretical approaches to unveil the potential solid electrolyte interphase (SEI) components enabling facile Ca plating. Borates compounds, in the form of cross-linked polymers are suggested as divalent conducting component. A pre-passivation protocol with such SEI is demonstrated and allows to broaden the possibility for electrolyte formulation. We also demonstrated a 10-fold increase in Ca plating kinetics by tuning the cation solvation structure in the electrolyte limiting the degree of contact ion pair.
Broader contextIn the 1990s, the need of a light, high capacity battery concept to power the emerging market of portable electronics fuelled the development of modern Li-ion batteries (LIBs). Since then, LIBs have allowed the proliferation of diverse devices which are now part of our daily lives. We are currently facing a similar challenge: to develop a trustable energy storage solution for the growing energetic demands of electric vehicles and grid electricity, for which LIBs might be insufficient. Among the various next-generation batteries, technology based on calcium-metal has recently gained much attention given its high theoretical capacity and low redox potential (potentially resulting in high energy density at the cell level). However, given its low redox potential, a surface layer would naturally form over the metallic electrode. In most cases, this surface film blocks any further electrochemical response, in other few reported cases, it allows stable migration of cations and so, an efficient solid–electrolyte interphase (SEI) is formed. Understanding the chemical nature of such passivation films and being able to engineer them will be crucial for the future development of calcium-metal and other divalent metal batteries. |
Introduction
Post-lithium battery technologies are currently attracting significant attention fuelled by the controversial debate on the sustainability of lithium technologies and the supply risk of future raw-materials.1 While Na-ion and redox flow batteries are very attractive, having potential for lower cost (ideally below 100$ per kW h) than Li-ion analogues,2 the energy densities attainable by such systems are inherently lower, or at best equivalent. Divalent batteries based on magnesium (Mg) and calcium (Ca) set the prospect to outperform commercial Li-ion batteries in terms of energy density, since they rely on potentially safer metal anodes (Ca or Mg) with high theoretical specific capacity (∼1340 and ∼2205 mA h g−1, respectively) and high volumetric capacity (∼3833 and ∼2073 mA h ml−1, respectively).3While the Mg-battery technology is significantly more mature, with continuous development of this field since the early 90's,4 Ca-based technologies have only recently gained much interest given a number of advantages of a Ca chemistry over Mg. Calcium has a lower standard redox potential (−2.87 V vs. NHE and −2.37 V for Mg2+/Mg vs. NHE) and Ca2+ is larger than Mg2+ ensuring a lower polarizing power.5 The lower polarizability of Ca2+ implies a milder electrostatic interaction (compared to Mg) with both the anion framework of the cathode material host structure, as well as the electrolyte components, resulting in higher cation mobilities.5
Since, the standard redox potential of Ca is more than ∼500 mV lower than Mg, most conventional electrolyte formulations are not thermodynamically stable against Ca metal and the formation of passivating layers or continuous electrolyte decomposition occurs.
The group of Prof. Aurbach6 was the first to explore the formation of passivation layers on calcium metal electrodes. When studying the calcium plating from an organic solution of Ca(BF4)2 or Ca(ClO4)2 dissolved in propylene carbonate (PC), gamma-butyrolactone, tetrahydrofuran (THF) and acetonitrile, it was determined that the electrochemical deposition of calcium was surface limited, as the electrolyte decomposes immediately to form an ionically insulating passivation layer. The chemical composition of the passivation layer was identified via infrared (IR) spectroscopy and was mostly composed by calcium carbonate and hydroxide.
In 2016, the first report on Ca plating using Ca(BF4)2 in mixtures of ethylene carbonate (EC) and propylene carbonate (PC) with operation temperatures >75 °C was reported.7 Recent studies reported Ca plating and stripping at ambient temperature in different organic electrolytes, including Ca(BH4)2 in THF8 and fluorinated alkoxyborate calcium salts (calcium tetrakis(hexafluoroisopropyloxy)borate:Ca[B(hfip)4]2) in dimethoxy ethane (DME).9,10
All these studies report the presence of a passivation layer at the interface between the Ca-metal anode and the electrolyte solution. For Ca(BH4)2/THF electrolyte, the passivation layer was found entirely composed by CaH2, which forms by the reaction of Ca metal and THF or BH4−.8,11 On the other hand, CaF2 was reported as an electrolyte decomposition product when employing solutions of Ca[B(hfip)4]2/DME.
In the case of Ca(BF4)2/EC:PC, where reversible plating and stripping occurs, the IR spectroscopy study of the Ca metal interface revealed several bands attributed to the decomposition products resulting from the solvent breakdown. In contrast, when calcium bis(trifluoromethansulfonil)imide (Ca(TFSI)2) is used, no electrochemical response is observed, which suggests that the BF4− anion and/or its decomposition product(s) play a crucial role in the process of Ca plating and stripping. None of the aforementioned studies provided a full characterization of the passivation layer of the Ca-metal electrodes. Understanding the composition of the passivation layer, its thickness and the coverage of the Ca electrode is still largely lacking, and requires additional work to identify the passivation layer's components that enable for divalent cation migration and reversible plating and stripping of Ca.
Here, we report a detailed comparative study of the passivation films formed in Ca(BF4)2/EC:PC and Ca(TFSI)2/EC:PC electrolytes, only the former allowing calcium plating and stripping. This study evidences the crucial role of the anion chemistry at the Ca-metal/electrolyte interface, impeding or allowing the Ca deposition/stripping. A potential passivation film component (comprising of borate species), that enables for Ca plating, is thus unravelled for the first time and other potential candidates are explored combining the nudged elastic band (NEB) method with density functional theory (DFT). A parallel is drawn with the Mg(TFSI)2/EC:PC system, allowing for a better understanding of the strategies to optimize divalent cation mobility in passivation layer. The comparison of our results on the Ca-metal/electrolyte interface with the Mg analogues, suggest remarkable differences of these two chemistries, while they are often gathered under the hood of multivalent batteries. Finally, we demonstrated that the borate-rich passivation layer effectively works as a solid electrolyte interphase (SEI) enabling calcium migration and subsequent calcium plating in Ca(TFSI)2 based electrolyte.
Results
Morphology of the deposited passivation layers
The morphology of the passivation layers formed electrochemically from the two model electrolytes, i.e. Ca(TFSI)2 or Ca(BF4)2 in a mixture of EC:PC solvents, was studied by transmission electron microscopy (TEM). For the deposition of the films, nickel powder particles were used as a working electrode without the addition of any binder (Fig. 1a). Although the exact composition and nature of the passivation layer formed on Ni particles may differ from that formed on Ca particles, the differences observed between the two electrolyte formulations should be maintained for both metals. In addition, the chemical modifications and microstructural investigations are coherent with the findings in the two systems with the different techniques, as it will be highlighted in the following paragraphs. The formation of a passivation layer on the Ni particles was ensured by potentiostatic deposition (−1.4 V vs. Ca/Ca2+ for 48 hours) and TEM images are shown in Fig. 1. | ||
| Fig. 1 Bright field TEM images of Ni particles (a) before and after formation of a surface layer in (b) Ca(TFSI)2 or (c) and (d) Ca(BF4)2 based electrolytes, respectively. Red and blue dotted lines are as a guide for the eye to indicate the thickness of each passivation layer. The inset in panel (d) corresponds to the diffraction pattern associated to the deposit. | ||
In the case of Ca(TFSI)2-based electrolyte, a 12–20 nm thick amorphous layer covers homogeneously the nickel surface (Fig. 1b), while some protruding areas were observed in a few spots (Fig. S1, ESI†). In the case of the Ca(BF4)2 electrolyte, the deposit is also fully conformal but it is thicker ∼70–80 nm (named SEI2) and presents a polycrystalline morphology, as observed by the presence of well-defined rings in the selected area electron diffraction (SAED) in the inset of Fig. 1d. The layer is composed of small crystallites of ∼10 nm in size and the indexing of the diffraction pattern is in good agreement with that of CaF2. In some areas, additional amorphous sheet-like features (named SEI1) are observed. The chemical composition of such passivation layers was then investigated by X-ray Photoelectron Spectroscopy (XPS), Fourier-transform Infrared Spectroscopy (FTIR), and Electron Energy Loss Spectroscopy (EELS).
Chemical composition of the passivation layers
The passivation layers in BF4− or TFSI electrolytes were obtained by polarizing a Ca disks at −1.4 V vs. a Ca reference electrode for 48 hours, thus forcing the reduction of the electrolyte as well as the formation of a passivation layer (current vs. time plots are shown in Fig. S2, ESI†). The passivated Ca disks appeared visually different depending on the anion used. Indeed, the Ca disks prepared in BF4− electrolyte seems completely covered by a dark deposit, while the disk passivated in the TFSI electrolyte remained shinny and visually unaltered, as shown in the inset of Fig. 2.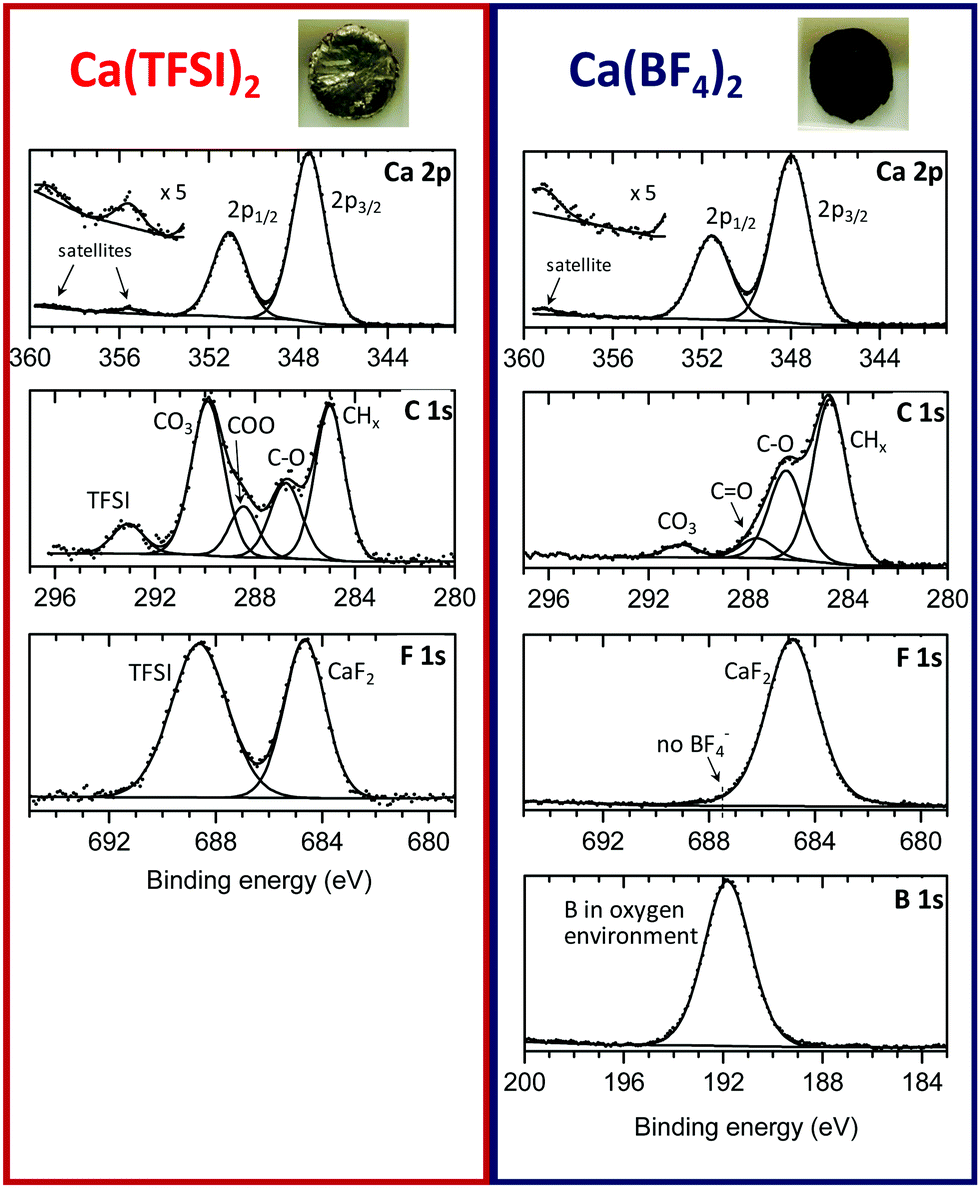 | ||
| Fig. 2 XPS spectra of the passivation layers formed on a calcium disk in (left-panel) Ca(TFSI)2 electrolyte, and (right-panel) Ca(BF4)2 electrolyte, respectively. Insets pictured the appearance of each calcium disk after negative polarization for 48 hours. | ||
XPS measurements were carried out to elucidate the chemical composition of the deposited films. Fig. 2 shows the C 1s, Ca 2p, F 1s and B 1s of the surface layers obtained from the two electrolytes. Quantitative data for each element are given in the ESI† (Table S1). For the two samples, the signal of the metallic calcium was not detectable, meaning that in each case the calcium surface is covered by a thicker layer than the maximum depth that XPS can probe (>5 nm). The sample with Ca(TFSI)2 electrolyte could be recorded without any charge compensation, whereas the sample with Ca(BF4)2 electrolyte required charge compensation. These observations indicate the formation of a very thin SEI for TFSI and a much thicker one in the case of BF4−, in agreement with TEM observations of Fig. 1. The Ca(TFSI)2 sample was characterised by a substantial quantity of carbonate, accompanied by mono- and di-oxygenated carbon environments from undefined species. Moreover, in the Ca 2p spectrum, the two satellite (plasmon) features at +8 eV from 2p3/2 and 2p1/2 main peaks (355.5 and 359 eV) correspond with previous reports of CaCO3 and CaO.12,13 Assuming that CaCO3 is the only carbonate species present in the deposit, the quantitative analysis leads to ∼34% CaCO3, 2% CaF2 and 7% Ca(TFSI)2 at the surface. While the Mg system is discussed in more details latter on, it is worth noting that the passivation layer formed onto Mg (in Mg(TFSI)2 in EC:PC) contains a significantly higher proportion of MgO (∼40%) and lower amounts of MgCO3 (∼15%) (see Fig. S3 and Table S2, ESI†). The passivation layer is also thinner than those formed on calcium electrodes, as the Mg 2p core peak of metallic Mg could be very well detected by XPS.
The chemical composition of the Ca(BF4)2 sample was very different. At the surface of this thicker SEI, the amount observed of carbonate was much lower (∼8% CaCO3), as clearly shown by the C 1s spectrum. The cumulated contribution of carbon in C–C, C–H and oxygenated environments (other than carbonate) corresponds to ∼35 at% of the surface for BF4−, instead of ∼16 at% for TFSI (Table S1, ESI†). Thus, the BF4−-derived SEI is much richer in organic species than the TFSI-derived one.
The B 1s spectrum revealed the great amount of boron (∼14%) in oxygenated environment,14 with no evidence of boron–fluorine coordination in the passivation layer compounds. The absence of B–F coordination points towards a complete displacement of the fluoride ligands by oxygen atoms, in agreement with previous reports on the labile character of the tetrafluoroborate anion.15 Unfortunately, XPS is unable to clearly identify the resulting boron species. Besides, as aforementioned, the amount of carbon in different environments (other than carbonate) is much more prominent than in the Ca(TFSI)2 sample: 35% instead of 16%, revealing a much greater proportion of organic species at the surface of the Ca(BF4)2 sample. On the basis of this observation, we envisage the formation of organic (polymeric) species potentially including boron element.
Finally, in the Ca 2p spectrum, the satellite was shifted to +11 eV with respect to the 2p3/2 main peak (359 eV), which is in good agreement with CaF2.16 The presence of calcium fluoride also agrees with the higher fluorine content detected, compared to the TFSI-derived SEI. The quantitative analysis leads to ∼15% CaF2 at the surface.
Fig. 3a shows the IR spectra (acquired in diffuse reflectance mode, DRIFTS) of the films deposited in both electrolytes under study (Ca(TFSI)2 and Ca(BF4)2 in EC:PC) and the assignment of some relevant bands, a comparison with the IR signatures of some relevant compounds is shown in Fig. S4 of the ESI.† The spectrum of the film formed in the TFSI-based electrolyte showed no distinguishable bands, because the SEI was too thin and the cumulated mass of deposited species was too low to be clearly detected. In contrast, in the IR spectrum of the film deposited from the BF4− electrolyte several adsorption peaks can be assigned to a number of functional groups, including ∼3500 cm−1 the O–H stretching, ∼2900 cm−1 the C–H stretching, 1850–1700 cm−1 the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, ∼1336 cm−1 the B–O stretching, ∼1073 cm−1 the C(O)–O stretching, and ∼780 cm−1 the CH2 rocking (see Fig. S4 of ESI†).
O stretching, ∼1336 cm−1 the B–O stretching, ∼1073 cm−1 the C(O)–O stretching, and ∼780 cm−1 the CH2 rocking (see Fig. S4 of ESI†).
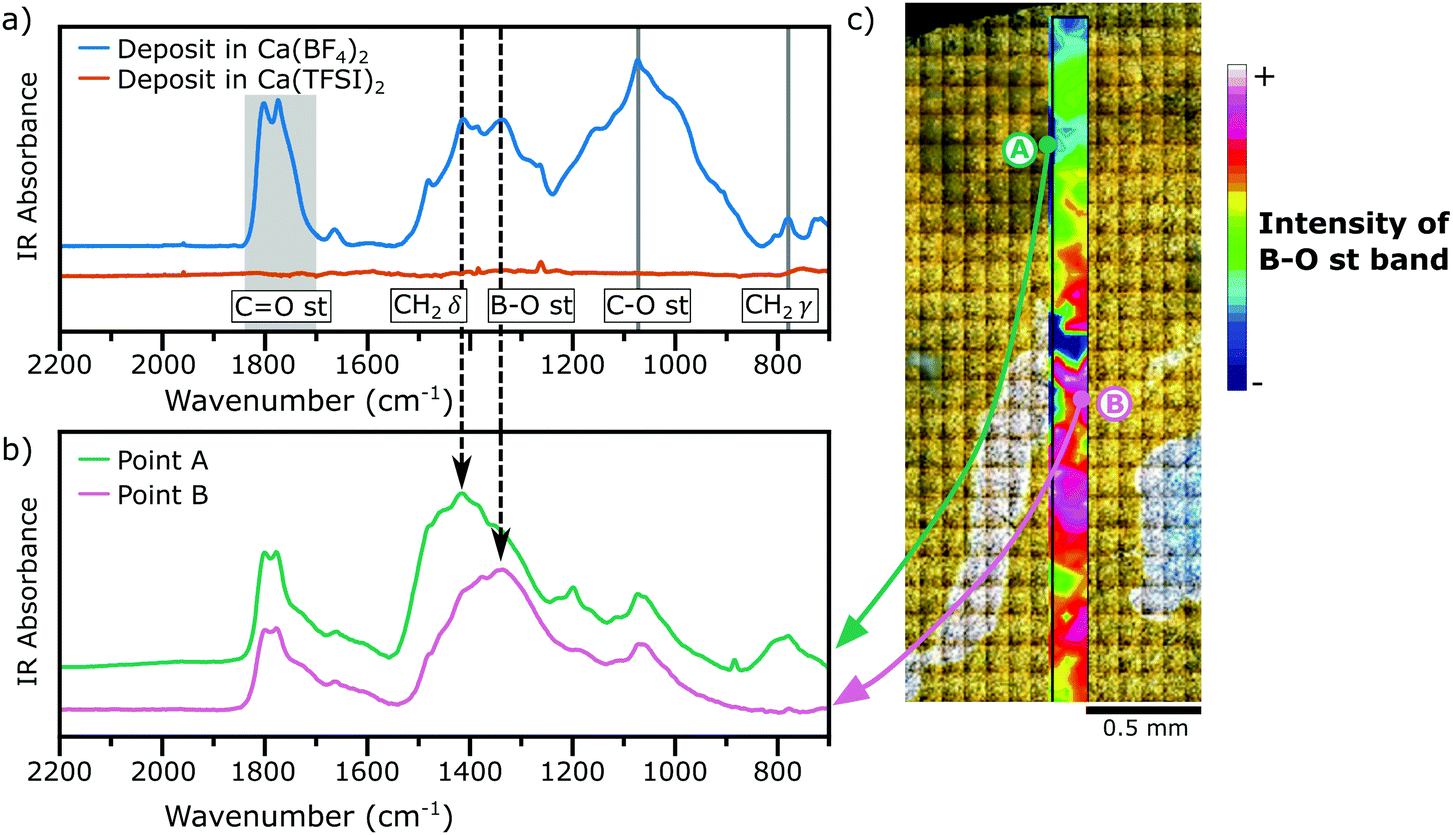 | ||
| Fig. 3 IR analysis of the SEI layers. (a) Diffuse reflectance FTIR spectra of the deposit obtained in the BF4− electrolyte and compared to the TFSI− electrolyte, (b) FTIR-microspectroscopy spectra of regions A and B marked in panel (c). (c) Shows an image of the surface of the deposit formed in Ca(BF4)2 electrolyte. | ||
The presence of a weak O–H stretching band is linked to a small amount of Ca(OH)2 formed by residual water in the electrolyte,7,17 whereas the B–O stretching is a characteristic signature of borates groups. The weak bands at 1387 and 707 cm−1 were assigned to CaCO3, which is present at low concentration in the SEI, as observed by XPS. Other vibrational modes observed here have been previously assigned to carbonate-like structures formed by solvent decomposition.18 For example, the strong absorption bands observed between 1750–1800 cm−1 may arise from ring-opening decomposition of EC or PC, in which the solid product deposited on the surface keeps the CO or C(O)–O moieties. It is also possible, however, that some solvent molecules are trapped inside the SEI structure and are not removed by the rinsing of the electrodes with DMC.19
Note that the probe depth of DRIFTS measurements is in the order of some hundreds of μm, much greater than XPS's probe depth, and thus it allows to detect species located in the buried layers of the SEI. However, the results obtained by the two techniques remain in good agreement, suggesting that the composition of the SEI is homogeneous in depth.
While FTIR spectra obtained by diffuse reflectance provide an average contribution of different compounds present in a relatively large area of analysis (typically few mm2), synchrotron FTIR microspectroscopy can resolve the different contributions from different regions of the deposit with a spatial resolution of ∼50 μm × ∼50 μm. Fig. 3b shows the IR spectra for a passivation layer formed in a BF4−-based electrolyte and for two different regions, Region A and Region B, which are marked in Fig. 3c. Region A in Fig. 3b shows a high concentration of carbonaceous species, inferred by the presence of intense bands of the CH2 scissoring (∼1414 cm−1) and rocking (∼780 cm−1) modes. Instead, Region B shows a strong contribution of the B–O stretching mode (∼1336 cm−1). Additionally, the absence of any vibrational mode in the range 800–1000 cm−1 is a signature that each boron species is coordinated by three oxygen atoms as in BO3 trigonal planar geometries instead of BO4 tetrahedra moieties.20,21 Using the intensity of the B–O stretching band, a chemical map was constructed (Fig. 3c) evidencing the heterogeneity of the deposit in the μm range.
The deposits were also studied by EELS and the normalized spectra of B K-edge, Ca L2,3 edge, O K-edge and low-loss region are presented in Fig. 4 and Fig. S5, S6 (ESI†) together with the spectra of some reference compounds.
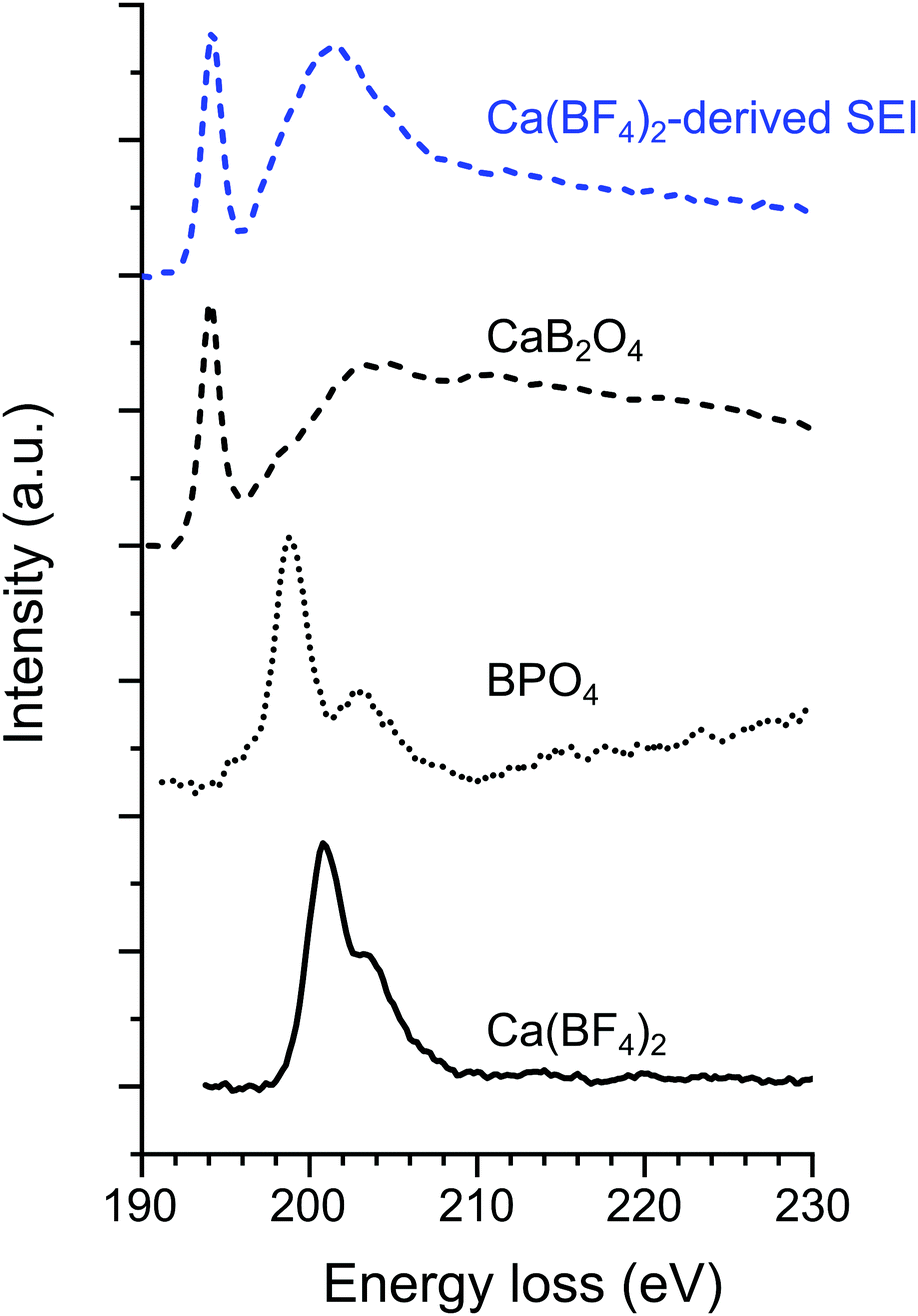 | ||
| Fig. 4 Normalized EELS spectra on the B K-edge for the Ca(BF4)2-derived passivation layer (“SEI1” deposit shown in Fig. 1c) together with three boron-containing compounds serving as references i.e., CaB2O4, BPO4, and Ca(BF4)2. | ||
In the case of the Ca(TFSI)2-derived deposit, the presence of calcium and oxygen is clearly visible in the spectrum of the deposit. The position and shape of the Ca L2,3 edge is similar to the other calcium compounds (e.g. CaB2O4, CaF2). Yet, the Ca-edge low sensitivity to the chemical environment prevents us to fingerprint the compounds formed. The O K-edge itself is not enough to make a clear identification of the compound(s). In addition, CaCO3, which is one of the SEI compounds determined by XPS and FTIR, is highly reactive under the electron beam and readily decomposes to CaO modifying the O K-edge.22
In the case of the Ca(BF4)2 electrolyte, the EELS measurements were performed in the two different areas observed by TEM (SEI1 and SEI2 in Fig. 1). For SEI2, the EELS analysis in the core-loss region highlights the presence of F (not shown here) and Ca (Ca L2,3 in Fig. S5, ESI†). The shape of the spectrum in the low-loss region (energy loss <50 eV) has previously been used as fingerprint,23 and allows us to confirm the presence of CaF2.
In the case of SEI1, the presence of B, Ca and O is confirmed due to the contribution observed in EELs spectra (Fig. 4 and Fig. S5, S6, ESI†). Fig. 4 shows the EELS spectra on the B K-edge for the “SEI1” structure, together with the spectra of BPO4 and Ca(BF4)2 as references for tetra-coordinated boron and CaB2O4 as reference for tri-coordinated boron. In the case of Ca(BF4)2, in the B K-edge the main peak σ* appears at 200.8 eV, in agreement with previous studies on metal-BF4 compounds.24 Similar peak positions were observed for BPO4 with the most prominent contribution σ* at 198.9 eV and a second peak at 203.2 eV. In contrast with CaB2O4, the main component π* and a broader σ* appeared at 194.2 and 203 eV, respectively. The peaks position and shape of these two references are in agreement with previous EELS studies on minerals with boron in trigonal and tetrahedral coordination, respectively.25,26 The B K-edge obtained on “SEI1” is composed of a primary π* peak at 194.3 eV and followed by a broader σ* peak at 201.3 eV. These signatures correspond in position and shape to the signal of a boron in a BO3 coordination environment confirming the FTIR analyses. However, the general shape of the plasmon related to SEI1 (Fig. S6, ESI†) is different from the CaB2O4, used here as reference, indicating that the boron-containing phase is most probably not this specific calcium borate.
Migration of divalent cations in the passivation layer
Following the experimental assessment of the previous section, the migration of Ca2+ (and Mg2+) was modelled in a number of materials, including borates, carbonates, hydrides, and fluorides that may form and/or “precipitate” at the Ca-metal (Mg-metal) electrode and during the early stage of electrolyte decomposition. The rationale behind the selection of these phases is driven both by experimental evidences of the organic electrolytes decomposing at the electrode, or computationally whenever the experimental data is not sufficient to justify the compound selection. For example, while XPS, FTIR and EELS experiments demonstrate the presence of borates (with BO3 moieties), the B speciation can result extremely complex, hence we invoked stable compounds of the computed Ca–O–B phase diagram (Fig. S7, ESI†). These include: CaO, CaB6, as well as ternary compounds, such as Ca2B2O5 and Ca3(BO3)2. Clear signatures of CaF2 and CaCO3 were also evidenced by XPS and FTIR. CaH2 has been also considered here as it was previously suggested as a potential “SEI” component.8 Therefore, the following materials were considered in the computational assessment: CaH2 (space group: Pnma and ICSD: 260873), CaB6 (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, 196516) CaO (Fmm, 51409), CaF2 (Fmm, 60368), Ca3(BO3)2 (Fmm, 60368), Ca2B2O5 (P121/c1, 190680), CaCO3 (Rc, 18166), rutile-MgH2 (P42/mnm, 1155807), rutile-MgF2 (P42/mnm, 394), MgO (Fmm, 9863), Mg2B2O5 (P121/c1, 81229) and MgCO3 (Rc, 10264). We extend this analysis to the Mg parent compounds, thus informing the feasibility of Mg migration if these phases were to be found at the anode electrode. Although the experiments of this study concern mostly with the interface of Ca-electrode/electrolyte, the comparison between potential Ca and Mg compounds forming at the interface is important to highlight the substantial differences of these two chemistries, which are otherwise considered very similar.
m, 196516) CaO (Fmm, 51409), CaF2 (Fmm, 60368), Ca3(BO3)2 (Fmm, 60368), Ca2B2O5 (P121/c1, 190680), CaCO3 (Rc, 18166), rutile-MgH2 (P42/mnm, 1155807), rutile-MgF2 (P42/mnm, 394), MgO (Fmm, 9863), Mg2B2O5 (P121/c1, 81229) and MgCO3 (Rc, 10264). We extend this analysis to the Mg parent compounds, thus informing the feasibility of Mg migration if these phases were to be found at the anode electrode. Although the experiments of this study concern mostly with the interface of Ca-electrode/electrolyte, the comparison between potential Ca and Mg compounds forming at the interface is important to highlight the substantial differences of these two chemistries, which are otherwise considered very similar.
Fig. 5a shows the maximum attainable migration barriers in selected materials specified by the experimental conditions of the electrochemical measurements. The input parameters of our model are:4,27 (i) the temperature, here set at 25 °C and 120 °C. High temperature is adopted experimentally to facilitate the sluggish kinetics of multivalent ions. (ii) The C-rate which controls the rate of charge/discharge of a battery and in this case the rate of Ca-deposition, and here is set to C/10 to C/100 (10 and 100 hours respectively). (iii) The thickness of the passivation layer, ranging from 10 nm to 100 μm. Within these boundary conditions, one can see, for example, that candidate products of decomposition displaying large Ca/Mg migration barriers (>500 meV) become only viable @25 °C and C/10 only if the thickness of the deposited layer falls below ∼100 nm. Unsurprisingly, materials displaying large Ca/Mg migration barriers will be able to contribute to ion transport only if the operational temperature is large (120 °C), extremely low C-rates (e.g. C/100) and/or very thin (nm-size) cross-section of the deposit. While extremely thin-size (nm-like) passivation layer are common, such temperature and C-rates are quite unrealistic for battery operations. Fig. 5a serves to guide the identification of possible materials candidates that may form at the interface Ca-metal/electrolyte.
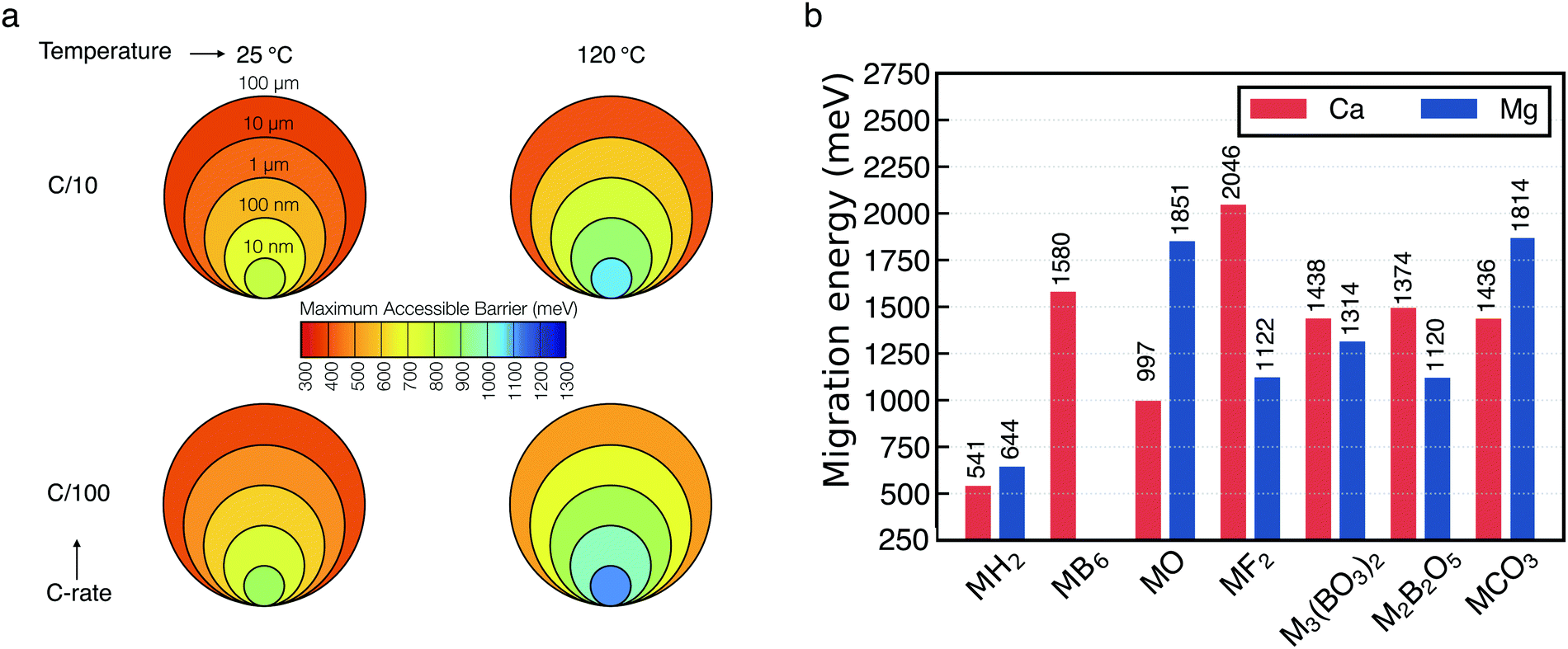 | ||
| Fig. 5 (a) Maximum particle size tolerating reasonable diffusivity as function of M2+ migration energy in the context of battery performance at various charging rates and temperatures (b) energies of Ca2+ (red) and Mg2+ (blue) migration (y-axis in meV) in a number of materials as computed from first-principles calculations. M indicates either Ca2+ or Mg2+. Values on top of each bar provide the magnitude of the barriers. The migration energy path of each material are reported in Fig. S8 and S9 (ESI†). | ||
Fig. 5b shows the computed barriers for either Mg2+ (blue bar) or Ca2+ (red bar) migration in the materials considered. In particular, the magnitude of the computed barriers of Fig. 5b are used together with the macroscopic model of Fig. 5a to eliminate poor Ca2+ (and Mg2+)-ion conductors that cannot be responsible for Ca percolation during electrochemical stripping and deposition. From Fig. 5b, one can observe that all the computed migration barriers are above 1000 meV, with exception of the hydrides (MgH2 and CaH2), which are just above 500 meV. For binary compounds the trend of the barriers’ magnitude can be rationalized with respect to the relative position of the anion groups in the periodic table.27 Except from MgF2, unsurprisingly, our data shows that more electronegative anions will tend to increase the barrier for multi-valent migration. The largest barriers in Mg compounds is computed for MgO (∼1851 meV), while for Ca the largest barrier is given by CaF2 (∼2046 meV). Also, in stark contrast with MgO, CaO is one of the Ca compounds with the lowest migration barrier (∼997 meV), lower than borates and carbonates, which emphasizes the remarkable difference between Ca-based batteries and their Mg analogue.
Notably, the migration barriers reported here were computed for pristine materials, and provide a lower bound of Ca2+ migration in these chemistries. The occurrence of defects such as dislocation and grain-boundaries may increase further the migration barriers, with direct effect on the kinetics of Ca deposition.28
Influence of the passivation layer on the calcium plating/stripping
The passivation layers formed in TFSI or BF4−-based electrolytes were found to differ significantly on composition, the former contains mostly carbonates, whereas the latter contains mostly borates, CaF2 and organic (polymeric) species. As aforementioned, TFSI-based electrolytes does not allow for Ca plating and stripping which can only be achieved when Ca(BF4)2 salt is used.7In order to ascertain the role of the passivation layer, a pre-passivation protocol was established (see experimental section in ESI†) ensuring that the stainless steel working electrode is fully covered by a borate containing passivation layer (formed in Ca(BF4)2 in EC:PC). Fig. 6 shows the electrochemical plating/stripping curves using such pre-passivated substrates, which were transferred to new cells and cycled in Ca(BF4)2 or Ca(TFSI)2 in EC:PC. After pre-passivation, not only Ca plating and stripping can be observed in TFSI-based electrolyte but a remarkable increase in the charge associated with Ca stripping (220 vs. 28 mC cm−2) and decrease of the polarization (ipa − ipc = 250 vs. 670 mV) are observed in Ca(TFSI)2 in EC:PC (red curve) when compared to the BF4− case (blue curve). While this experiment confirms the key role of the passivation layer in order to realize Ca plating and stripping, it also highlights another parameter affecting the overall kinetics of the process – the electrolyte formulation.
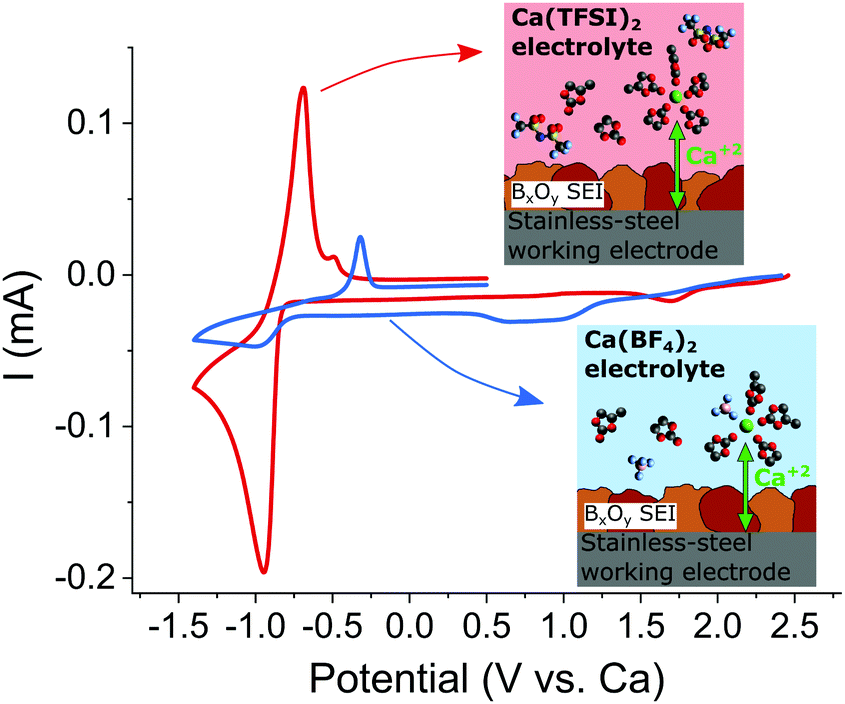 | ||
| Fig. 6 Cyclic voltammograms (0.1 mV s−1) of pre-passivated stainless-steel electrodes (covered with borate films) using either Ca(TFSI)2 (red curve) or Ca(BF4)2 (blue curve) electrolytes. Insets show the proposed cation solvation structures in each electrolyte, which are expected to be responsible for the difference in electrochemical behaviour between the two electrolytes. | ||
Discussion
We now discuss the formation and influence of the passivation layer with respect to the cation solvation structure in different electrolyte formulations.First, we have shown that the electrochemically formed passivation layer, rich in borate and CaF2, acts as an SEI. Indeed, the latter allows facile Ca2+ migration (and plating/stripping) and, even though a thicker layer is observed, meaning an initially more important electrolyte decomposition, a stable layer is eventually formed preventing further electrolyte reduction, as previously demonstrated.7 This signifies that the SEI is electronically insulating, but also an ionic conductor. We have observed that Ca plating/stripping kinetics differ greatly between electrolyte formulations, which underlines the important role played by the cation mobility in solution or by interfacial processes, such as desolvation at the electrolyte interface. Previous reports have shown that the desolvation process at the electrode surface is no-trivial for Mg electrolytes and can be the rate limiting step for Li+ insertion into graphite anode.29–32
The physicochemical properties of 0.45 M Ca(BF4)2 or Ca(TFSI)2 in EC:PC, were previously investigated and significant differences in terms of Ca2+ solvation structure were demonstrated with notable degree of ion-pair formation observed in BF4− electrolyte,33 which was attributed to the highly delocalized charge of the TFSI anion.34 The presence of contact ion-pairs in solution can have two detrimental effects on the plating kinetics. They can affect (i) the migration and overall cation complex mobility (transference number) due to the change of solvation structure and charge, and (ii) the existence of tight ion couples will affect the desolvation energy required in order to strip-off the molecules complexing Ca2+ before it can “enter” the SEI layer. It is worth noting that binding energies (related to desolvation energies) calculated by means of DFT are approximately four times higher for anion–Ca2+ than for EC–Ca2+ complexes (see Table S6 in ref. 33). The higher plating/stripping kinetics observed in Fig. 6 (nearly an order-of-magnitude difference) in TFSI electrolyte can be speculatively attributed to the lower presence of ion pairs in the Ca(TFSI)2 electrolyte.
The cation solvation shell in the electrolyte impacts, also, the chemical composition and properties of the SEI layer. Such a relation has been broadly studied in Li systems35,36 and recently demonstrated by real-time mass spectroscopy.37 The lithium ions accumulate at the surface of the negatively polarized electrode and carry their solvation shells with them. Thus, the main building blocks of the SEI formation are the decomposition products of the cation solvation shell. Similarly, we have observed that TFSI-based electrolyte mostly leads to solvent decomposition products, such as calcium carbonate and other carbonaceous species, while in BF4− electrolyte a substantial amount of borates and CaF2 are formed from the anion decomposition. In agreement with the higher tendency for ion pair formation with BF4− anion compared to TFSI anion, at the concentrations used in this study.33
The thin layer (∼20 nm) of CaCO3 formed in the TFSI-based electrolyte is sufficient to block any continuous migration of calcium ions as its computed migration barrier is ∼1436 meV, much higher than the maximum tolerable migration barrier for electrodeposition even at C/100 (Fig. 5a). On the contrary, in BF4− electrolytes, where the electrochemical plating/striping of calcium is observed, there should be one (or several) chemical compounds in the SEI which allow the cation migration towards the anode. However, even when the chemical analysis shows a high content of borates, none of the two structures computed with DFT appears to be responsible for any ionic conductivity, neither does the calcium fluoride nor the hexaboride, as these compounds have migration barriers higher than 1300 meV.
According to the computed barriers of Fig. 5b, CaO layers with thickness in the tens of nanometers might sustain cation migration (∼997 meV, Fig. 5a). We speculate that a layer of calcium oxide might be present in the inner stratum of the BF4−-derived SEI as the computed Ca–O–B phase diagram (Fig. S7, ESI†) shows that none of the calcium borates can be in direct equilibrium with Ca metal, which remains always separated by CaO. The evidence of borate presence at the interface might be linked to (i) some kinetic stabilization preventing further decomposition (e.g. the presence of an intermediate CaO phase) and/or (ii) the presence of a different (potentially non-crystalline) borate phase.
An interesting comparison arises when performing similar experiments using metallic Mg electrode. In this case only the TFSI electrolyte could be prepared since Mg(BF4)2 has not been reported yet. As observed in the case of calcium, no electrochemical plating of magnesium is observed, but as mentioned before, the passivation layer formed is thinner than the one obtain with Ca, richer in MgO (∼40%) and contains less MgCO3 (∼15%). This suggest a stronger blocking nature (ionically and electronically) of the Mg based passivation layer when compared with its Ca analogue in agreement with the high Mg2+ migration barriers from our computed data for MgO and MgCO3, respectively 1851 and 1814 meV (Fig. 5b).
The sizeable barrier for Mg migration in MgO is in stark contrast with the calcium system for which, nano-sized CaO is the only crystalline compound investigated which might allow Ca-metal plating and stripping with a migration barrier of 997 meV (Fig. 5b). We speculate that the significant difference in the migration barrier between MgO and CaO plays a major role in the reactivity of the metal anodes. While it is virtually impossible to fully remove a native oxide surface layer on a metal anode, its presence on a Mg electrode will fully hamper any electrochemical response, while some degree of divalent conduction can be expected for Ca depending on the experimental conditions and the oxide layer thickness. This could also help rationalizing the fact that, in contrast to Mg, Ca metal does not require cumbersome surface treatment or the use of corrosive electrolytes since the presence of a thin oxide layer would not preclude plating/stripping.
Altogether, except for CaO, none of the other crystalline compounds suggested by computation are likely to enable Ca2+ migration through the 80 nm thick SEI formed in Ca(BF4)2 electrolytes. However, a comparison with previously reported Li system can shed light on the borate chemistry resulting from BF4− and alkyl carbonate based electrolytes. In Li cells the reduction of LiBF4 coupled with a ring-opening of EC has been found to form Li(BF2O)n linear polymeric structures along with the formation of CO2, ethylene, and LiF.38,39 Other borate polymers were also reported when other anions are used, such as bis(oxalate)borate (BOB) and difluoro(oxalate)borate, with excellent Li-ion conductivity.40–42 The electrochemical reduction of BOB anions involved a change in the oxygen coordination around the B atom from tetrahedral BO4 to trigonal BO3, with the end products possibly including oligomeric borate carbonyl moieties.43,44 These polymeric structures can potentially present a sizable degree of cross-linking and may be the origin of good mechanical stability and high temperature behaviour. In this work, we confirmed that all boron is surrounded by oxygen ligands, which may indicate the existence of these polymeric units.
While carbonate solvent reduction can be rather complex and involve several possible reaction pathways,45,46 most intermediate products (e.g., alkoxides) lead to charged-oxygen species, whose nucleophilic nature is sufficient to substitute the fluorine ligands of the BF4− anion (Fig. 7). Upon further reduction a cross-linked boron polymer can be produced.
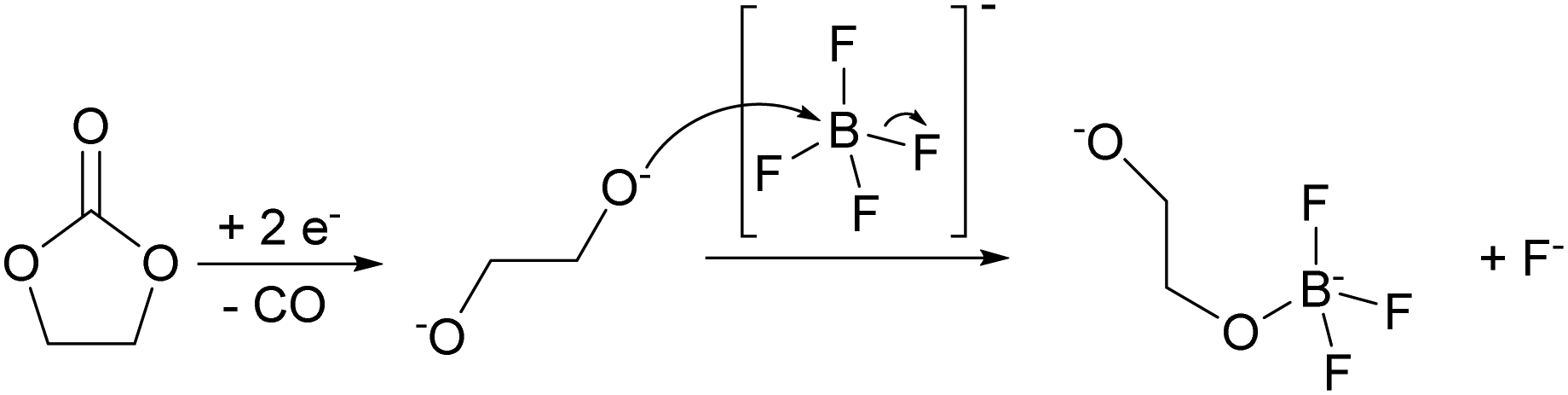 | ||
| Fig. 7 Proposed mechanism for the anion decomposition upon reduction in Ca(BF4)2/EC:PC electrolytes. Further substitution of F by O can lead to cross-linked boron polymers. | ||
Another point of discussion is the presence of CaF2 nanocrystalline domains, observed by TEM. While computation predicts a prohibitive migration barrier for Ca2+ in crystalline CaF2 (>2 eV, Fig. 5), its presence as nanocrystals embedded in a cation conducting compound may promote space charge accumulation and an increase of ionic conductivity.47,48
Finally, the high inhomogeneity of the passivation layer evidenced by synchrotron-based FTIR microspectroscopy (Fig. 3c), suggests a number of possible strategies to improve Ca plating kinetics. While, the optimization of the polymeric borate content and the homogeneity of its electrode surface coverage through careful electrolyte formulation appears as the most obvious strategy in order to engineer the passivation layer, surface pre-treatment and thin film deposition could also be foreseen as a viable strategy.
Conclusions
Understanding the nature and composition of Ca-metal/electrolyte interfaces represents a crucial task in the development of electrolytes that can enable facile Ca plating/stripping. Here we have used a multi-technique approach encompassing experimental measurements and theoretical calculations to rationalize the complex composition of the passivation layer formed on the Ca metal electrode in different electrolyte formulations. DFT predictions are used to aid the elimination of inorganic compounds that display low propensity for Ca migration and thus cannot participate actively towards Ca deposition.We have demonstrated the effectiveness of a borate-based passivation layer to allow reversible calcium plating and stripping. This layer, derived from the BF4− decomposition, acts as an SEI by allowing Ca2+ percolation, while limiting further the electrolyte reduction. Once this protective SEI layer is formed on the working electrode, it is possible to transfer the pre-passivated electrode to new electrochemical cells with other electrolytes that would not allow reversible Ca plating otherwise. The effectiveness of this protocol was evidenced by using a Ca(TFSI)2 based electrolyte. A ten-fold increase in Ca plating kinetics was then observed and attributed to the difference in cation-solvation structure between a TFSI and BF4− electrolytes. Thus, borates are regarded as key component in the SEI layer and tricoordinated boron (BO3) was identified. While the exact structure/stoichiometry of this borate species has not yet been fully elucidated, the formation of cross-linked boron polymer can be hypothesized. This work paves the way for strategic design of (artificial) SEI layers and electrolyte formulations with enhanced Ca plating kinetics. This entails a better understanding of the reduction process of boron containing species leading to the formation of the passivation layer and a better control of it through systematic electrolyte formulation optimization and considering new boron containing anions, solvents and/or additives.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the European Union's Horizon 2020 research and innovation program H2020 is acknowledged: European Research Council (ERC-2016-STG, CAMBAT grant agreement no. 715087) and H2020-MSCA-COFUND-2016 (DOC-FAM, grant agreement no. 754397). A. Ponrouch is grateful to the Spanish Ministry for Economy, Industry and Competitiveness Severo Ochoa Programme for Centres of Excellence in R&D (SEV-2015-0496) and to Prof. D. Lemordant (Tours University, France) for stimulating discussions. P. Canepa acknowledges funding from the National Research Foundation under his NRFF NRFF12-2020-0012 and the ANR-NRF NRF2019-NRF-ANR073 Na-MASTER. P. Canepa acknowledges the National Supercomputing Centre, Singapore (https://www.nscc.sg). This work has been done in the frame of the Doctoral Degree Program in Materials Science by the Universitat Autònoma de Barcelona. The FTIR experiments were performed at MIRAS beamline at ALBA Synchrotron with the collaboration of ALBA staff.Notes and references
- A. El Kharbachi, O. Zavorotynska, M. Latroche, F. Cuevas, V. Yartys and M. Fichtner, J. Alloys Compd., 2020, 817, 153261 CrossRef CAS
.
- C. P. Grey and J. M. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS
- M. E. Arroyo-de Dompablo, A. Ponrouch, P. Johansson and M. R. Palacín, Chem. Rev., 2020, 120(14), 6331–6357, DOI:10.1021/acs.chemrev.9b00339
- Z. Lu, A. Schechter, M. Moshkovich and D. Aurbach, J. Electroanal. Chem., 1999, 466, 203–217 CrossRef CAS
- A. Ponrouch, C. Frontera, F. Bardé and M. R. Palacín, Nat. Mater., 2016, 15, 169–172 CrossRef CAS
- D. Wang, X. Gao, Y. Chen, L. Jin, C. Kuss and P. G. Bruce, Nat. Mater., 2017, 17, 16–20 CrossRef
- Z. Li, O. Fuhr, M. Fichtner and Z. Zhao-Karger, Energy Environ. Sci., 2019, 12, 3496–3501 RSC
- A. Shyamsunder, L. E. Blanc, A. Assoud and L. F. Nazar, ACS Energy Lett., 2019, 4, 2271–2276 CrossRef CAS
- K. Ta, R. Zhang, M. Shin, R. T. Rooney, E. K. Neumann and A. A. Gewirth, ACS Appl. Mater. Interfaces, 2019, 11, 21536–21542 CrossRef CAS
- S. L. Stipp and M. F. Hochella, Geochim. Cosmochim. Acta, 1991, 55, 1723–1736 CrossRef CAS
- L. Järvinen, J. Leiro and M. Heinonen, Surf. Interface Anal., 2014, 46, 399–406 CrossRef
- W. E. Moddeman, A. R. Burke, W. C. Bowling and D. S. Foose, Surf. Interface Anal., 1989, 14, 224–232 CrossRef CAS
- M. G. Freire, C. M. S. S. Neves, I. M. Marrucho, J. A. P. Coutinho and A. M. Fernandes, J. Phys. Chem. A, 2010, 114, 3744–3749 CrossRef CAS
- B. W. Veal and A. P. Paulikas, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 5399–5416 CrossRef CAS
- D. Aurbach, R. Skaletsky and Y. Gofer, J. Electrochem. Soc., 1991, 138, 3536 CrossRef CAS
- L. Gireaud, S. Grugeon, S. Laruelle, S. Pilard and J.-M. Tarascon, J. Electrochem. Soc., 2005, 152, A850 CrossRef CAS
- D. Aurbach, B. Markovsky, A. Shechter, Y. Ein-Eli and H. Cohen, J. Electrochem. Soc., 1996, 143, 3809 CrossRef CAS
- C. E. Weir and R. A. Schroeder, J. Res. Natl. Bur. Stand., Sect. A, 1964, 68A, 465 CrossRef CAS
- A. C. Wright, Int. J. Appl. Glass Sci., 2015, 6, 45–63 CrossRef
- U. Golla-Schindler, G. Benner, A. Orchowski and U. Kaiser, Microsc. Microanal., 2014, 20, 715–722 CrossRef CAS
- M. Boniface, L. Quazuguel, J. Danet, D. Guyomard, P. Moreau and P. Bayle-Guillemaud, Nano Lett., 2016, 16, 7381–7388 CrossRef CAS
- W. H. E. Schwarz, L. Mensching, K. H. Hallmeier and R. Szargan, Chem. Phys., 1983, 82, 57–65 CrossRef CAS
- L. A. J. Garvie, A. J. Craven and R. Brydson, Am. Mineral., 1995, 80, 1132–1144 CAS
- H. Sauer, R. Brydson, P. N. Rowley, W. Engel and J. M. Thomas, Ultramicroscopy, 1993, 49, 198–209 CrossRef CAS
- T. Chen, G. Sai Gautam and P. Canepa, Chem. Mater., 2019, 31, 8087–8099 CrossRef CAS
- J. A. Dawson, P. Canepa, T. Famprikis, C. Masquelier and M. S. Islam, J. Am. Chem. Soc., 2018, 140, 362–368 CrossRef CAS
- P. Canepa, G. S. Gautam, R. Malik, S. Jayaraman, Z. Rong, K. R. Zavadil, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 3317–3325 CrossRef CAS
- K. A. See, K. W. Chapman, L. Zhu, K. M. Wiaderek, O. J. Borkiewicz, C. J. Barile, P. J. Chupas and A. A. Gewirth, J. Am. Chem. Soc., 2016, 138, 328–337 CrossRef CAS
- V. Vasudevan, M. Wang, J. A. Yuwono, J. Jasieniak, N. Birbilis and N. V. Medhekar, J. Phys. Chem. Lett., 2019, 10, 7856–7862 CrossRef CAS
- K. Xu, A. von Cresce and U. Lee, Langmuir, 2010, 26, 11538–11543 CrossRef CAS
- J. D. Forero-Saboya, E. Marchante, R. B. Araujo, D. Monti, P. Johansson and A. Ponrouch, J. Phys. Chem. C, 2019, 123, 29524–29532 CrossRef CAS
- N. Chapman, O. Borodin, T. Yoon, C. C. Nguyen and B. L. Lucht, J. Phys. Chem. C, 2017, 121, 2135–2148 CrossRef CAS
- T. Yoon, N. Chapman, D. M. Seo and B. L. Lucht, J. Electrochem. Soc., 2017, 164, A2082–A2088 CrossRef CAS
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H.-H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS
- Y. Zhou, M. Su, X. Yu, Y. Zhang, J.-G. Wang, X. Ren, R. Cao, W. Xu, D. R. Baer, Y. Du, O. Borodin, Y. Wang, X.-L. Wang, K. Xu, Z. Xu, C. Wang and Z. Zhu, Nat. Nanotechnol., 2020, 15, 224–230 CrossRef CAS
- A. Xiao, L. Yang, B. L. Lucht, S.-H. Kang and D. P. Abraham, J. Electrochem. Soc., 2009, 156, A318 CrossRef CAS
- B. S. Parimalam and B. L. Lucht, J. Electrochem. Soc., 2018, 165, A251–A255 CrossRef CAS
- S. Shui Zhang, Electrochem. Commun., 2006, 8, 1423–1428 CrossRef
-
C. Charton, PhD thesis, Université François-Rabelais de Tours, 2017
- Z. Liu, J. Chai, G. Xu, Q. Wang and G. Cui, Coord. Chem. Rev., 2015, 292, 56–73 CrossRef CAS
- G. V. Zhuang, K. Xu, T. R. Jow and P. N. Ross, Electrochem. Solid-State Lett., 2004, 7, A224 CrossRef CAS
- K. Xu, U. Lee, S. Zhang, M. Wood and T. R. Jow, Electrochem. Solid-State Lett., 2003, 6, A144 CrossRef CAS
- J. Henschel, C. Peschel, S. Klein, F. Horsthemke, M. Winter and S. Nowak, Angew. Chem., Int. Ed., 2020, 59, 6128–6137 CrossRef CAS
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS
- N. Sata, K. Eberman, K. Eberl and J. Maier, Nature, 2000, 408, 946–949 CrossRef CAS
- W. Puin, S. Rodewald, R. Ramlau, P. Heitjans and J. Maier, Solid State Ionics, 2000, 131, 159–164 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization techniques and computational methods are provided, together with supporting tables and figures. See DOI: 10.1039/d0ee02347g |
| This journal is © The Royal Society of Chemistry 2020 |