
Facile synthesis of a Ru-dispersed N-doped carbon framework catalyst for electrochemical nitrogen reduction†
Ziqi
Zhang
a,
Kaida
Yao
a,
Linchuan
Cong
a,
Zhuochen
Yu
a,
Lina
Qu
a and
Weimin
Huang
 *ab
*ab
aCollege of Chemistry, Jilin University, Changchun 130012, China. E-mail: huangwm@jlu.edu.cn
bKey Laboratory of Physics and Technology for Advanced Batteries of Ministry of Education, Jilin University, Changchun 130012, China
First published on 13th January 2020
Abstract
Ammonia is one of the most important agricultural fertilizers and renewable energy carriers, and is also one of the most highly produced inorganic compounds in the world. The traditional industrial ammonia synthesis mainly uses the Haber–Bosch method, which consumes a large amount of fossil fuels and emits a lot of greenhouse gas. Thus, a more environmentally friendly method is needed for the production of ammonia. In this paper, we present a new electrocatalyst to produce NH3 under ambient conditions, which was obtained via the carbonization of Ru-dispersed ZIF-8. As a result, the catalyst could achieve a maximum NH3 production rate of about 16.68 μgNH3 h−1 mgcat.−1 at −0.4 V (vs. RHE) and a faradaic efficiency of 14.23% at −0.3 V (vs. RHE) at room temperature and ambient pressure in 0.1 M KOH. Also, the loading of Ru is only 16 wt%. Thus, this work provides a feasible method to design highly dispersed noble metal electrocatalysts for the nitrogen reduction reaction (NRR).
1. Introduction
Ammonia is regarded as one of the most important agricultural fertilizers and renewable energy carriers. Also, it is one of the most highly produced inorganic compounds in the world. The total global NH3 production has been more than 146 million tons since the 21st century.1 However, the traditional industrial synthesis of ammonia mainly uses the Haber–Bosch method, which requires a high temperature and high pressure (300–500 °C and 150–350 atm, respectively) to activate the reaction due to the inertia of nitrogen molecules. Furthermore, it has a low yield (only 10% to 15%).2 It consumes a large amount of fossil fuels and emits lots of greenhouse gas, which not only wastes non-renewable resources, but also destroys the ecological environment.3–6 Thus, more environmentally friendly methods to synthesize ammonia are urgently needed.As is known, the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N (225 kcal mol−1) bond in dinitrogen is one of the strongest chemical bonds, which is difficult to be dissociated under ambient conditions.7 Much effort has been focused on finding more efficient and economical processes to synthesize ammonia. To increase the production of ammonia and reduce its energy consumption, alternative routes for fixing N2 to ammonia have been developed, which include photocatalysis,8–11 biological reduction,12 homogeneous catalysis,13–15 and electrocatalysis.16–21 Interestingly, the electrochemical reduction of N2 to ammonia can use H+ in water as the proton source instead of H2, and the reaction can be powered by renewable energy, which is more sustainable compared with the Haber–Bosch process.22 Thus far, a variety of metals,23–25 metal oxides,26–29 metal nitrides30 and other nonmetallic electrocatalysts31,32 have been reported as electrocatalysts for N2 reduction, which can be divided into two categories, noble metal catalysts, such as Ru, Au, and Pt, and non-noble metal catalysts, such as Fe, Co and Ni. Noble metals are often used as commercial electrochemical catalysts because of their stability and selectivity, which lead to high cost and a low atomic utilization ratio. Hence, cost reduction is also a goal of catalyst development and deserves further exploration.
N (225 kcal mol−1) bond in dinitrogen is one of the strongest chemical bonds, which is difficult to be dissociated under ambient conditions.7 Much effort has been focused on finding more efficient and economical processes to synthesize ammonia. To increase the production of ammonia and reduce its energy consumption, alternative routes for fixing N2 to ammonia have been developed, which include photocatalysis,8–11 biological reduction,12 homogeneous catalysis,13–15 and electrocatalysis.16–21 Interestingly, the electrochemical reduction of N2 to ammonia can use H+ in water as the proton source instead of H2, and the reaction can be powered by renewable energy, which is more sustainable compared with the Haber–Bosch process.22 Thus far, a variety of metals,23–25 metal oxides,26–29 metal nitrides30 and other nonmetallic electrocatalysts31,32 have been reported as electrocatalysts for N2 reduction, which can be divided into two categories, noble metal catalysts, such as Ru, Au, and Pt, and non-noble metal catalysts, such as Fe, Co and Ni. Noble metals are often used as commercial electrochemical catalysts because of their stability and selectivity, which lead to high cost and a low atomic utilization ratio. Hence, cost reduction is also a goal of catalyst development and deserves further exploration.
Metal–organic frameworks (MOFs) are composed of organic ligands and tunable metal ion centers, featuring large surface areas and diverse structural topologies, which offer inherent advantages and accessible active sites for use as catalysts.33–35 The zeolitic imidazolate framework series is a type of MOF that can be easily synthesized, which is suitable to serve as a base to load metal atoms in order to enhance electrocatalytic efficiency and decrease cost. More importantly, the direct carbonization of MOFs can result in the formation of nanoporous carbon materials that possess adjustable N and metal atom contents and species.31
From a thermodynamic perspective, the electrocatalytic NRR is possible.22 However, the low yields of NH3, competing hydrogen evolution reaction (HER),36 costly electrolytes and harsh operating conditions37 hinder its wide use. Also, some doubts on the detection method for the electrochemical synthesis of ammonia have been put forward recently.38 Thus, it is necessary to find a new types of electrocatalysts that are inexpensive, with high ammonia yields, high selectivity and can be employed under ambient conditions. In addition, more reliable and precise experimental methods are needed for the production of ammonia.
In this work, we demonstrate a new electrocatalyst to produce NH3 under ambient conditions, which was obtained via the carbonization of Ru-doped ZIF-8. The doped ZIF-8 after carbonization provided a high surface area and satisfactory stability, and thus was durable and effective during electrocatalysis. Meanwhile, the Zn in the ZIF-8 was removed on account of the high temperature, and thus Ru could access the Zn vacancies, which allowed the Ru atoms to be dispersed uniformly and regularly in the N-doped carbon (NC) framework and eventually improve the rate of atomic utilization. Consequently, the catalyst achieved a maximum NH3 production rate of about 16.68 μgNH3 h−1 mgcat.−1 at −0.4 V (vs. the reversible hydrogen electrode, RHE) and a faradaic efficiency of 14.23% at −0.3 V (vs. RHE) under ambient conditions in 0.1 M KOH. Also, inductively coupled plasma spectroscopy (ICP) showed that the loading of Ru was only 16 wt%.
2. Experimental
2.1 Chemicals and materials
Zinc acetate dihydrate (Beijing Chemical Works, C4H6O4Zn·2H2O, analytical reagent), 2-methylimidazole (C4H6N2, Aladdin), ruthenium chloride hydrate (35.0–42.0% Ru basis, Aladdin), Seignette salt (Beijing Chemical Works, NaKC4H4O6·4H2O, analytical reagent), Nessler's reagent (Macklin), p-dimethylaminobenzaldehyde (Aladdin, analytical reagent), methanol (Beijing Chemical Works, CH4O, ≥99.8%), Nafion (5.0 wt%, Dupont), Nafion 117 membrane (Dupont), ethyl alcohol (Beijing Chemical Works, C2H5OH, ≥99.8%), sulfuric acid (Beijing Chemical Works, H2SO4, 98%), hydrochloric acid (Beijing Chemical Works, HCl, 35.0–38.0%), ammonium chloride (Beijing Chemical Works, NH4Cl, analytical reagent), potassium hydroxide (Beijing Chemical Works, KOH, 96%), N2 gas (99.99%), and Ar gas (99.99%) were employed. All chemical reagents were used as received without further purification. All aqueous solutions were prepared with ultrapure water (resistivity of 18.25 Ω cm).2.2 Physical characterization
Transmission electron microscopy (TEM) and elemental mapping were performed on an FEI Tecnai G20/JEM2010 microscope. X-ray photoelectron spectra (XPS) were recorded on a Thermo ESCALAB 250Xi with an excitation source of Al Kα radiation. X-ray diffraction (XRD) was carried out using an Empyrean (PANalytical B.V.) with a Cu Kα radiation source (λ1 = 1.540598 Å, λ2 = 1.544426 Å) operating at 40.0 kV and 40.0 mA, and diffraction data was recorded in the 2θ range of 5–80° with a scan rate of 12 degrees per min. Absorbance data was measured on a Beijing Purkinje General TU-1900 ultraviolet-visible (UV-vis) spectrophotometer. Gas adsorption–desorption (77 K) analyses were conducted using N2 as the adsorbent on a 3H-2000 PM2 (BeiShiDe Instrument, PR China).2.3 Cathode preparation
In a typical procedure, Zn(OAC)2·2H2O (1.243 g) was dissolved in 25 mL of methanol to form a clear solution, which was subsequently injected into 25 mL of methanol containing 1.848 g of 2-methylimidazole (MeIM) under intense stirring for 1 min and then left to stand for 24 h at room temperature. The mixed solution was centrifuged and washed with methanol several times and dried under vacuum at 60 °C overnight to obtain ZIF-8.400 mg of the as-obtained precipitate (ZIF-8) was dissolved in 5 mL of ethanol and 1, 3 or 5 mL of 10−4 M RuCl3 aqueous mixed solution, and stirred for another 24 h in the dark. The solution was centrifuged and washed with ethanol several times and dried under vacuum at 60 °C overnight to obtain Ru-doped ZIF-8 (ZIF-8@Ru). Then ZIF-8@Ru was heated at 900 °C for 4 h at a rate of 3 °C min−1 in a tube furnace under an N2 atmosphere to obtain the N-doped carbon framework/Ru nanoparticle hybrid, named NC@Ru-1, NC@Ru-3, and NC@Ru-5, respectively (the number indicates the volume of Ru3+ solution added). ZIF-8 was treated the same as that for the N-doped carbon (NC) framework.
5 mg of sample was dispersed 250 μL of isopropyl alcohol, 700 μL of ultrapure water and 50 μL 5 wt% Nafion mixed solution and ultrasonicated for 30 min. 50 μL liquid was pipetted onto the surface of carbon paper (1 × 1 cm) (loading = ∼0.25 mg cm−2) and naturally dried, which served as the working electrode.38 For comparison, N-doped carbon (NC) framework loaded and non-loaded carbon paper electrodes were also prepared.
2.4 Electrochemical measurements
Experiments were carried out in an H-type electrochemical cell separated by a Nafion 117 membrane at room temperature. All the ammonia was collected by the solution in the cathodic chamber and no ammonia was measured in the absorber. Before the NRR test, the Nafion membrane was heated in 5% H2O2, 5% H2SO4 aqueous solution and ultrapure water at 80 °C for 1 h. Electrochemical measurements were performed using a CHI 660E electrochemical workstation with a three-electrode system in 0.1 M KOH and N2 atmosphere. A prepared carbon paper served as the working electrode, and Pt foil and Hg/HgO electrode (1 M KOH electrolyte) were used as the counter electrode and reference electrodes, respectively. The experimental potential values were calibrated using the following equation: E vs. RHE = E vs. Hg/HgO + 0.098 + 0.059 pH. Before the test, an N2 flow was continuously fed to the whole cathode through the electrolyte in the cell for 30 min to saturate it with N2. Also, linear sweep voltammetry (LSV) experiments were performed at a scan rate of 10 mV s−1 from 0.3 to −0.7 V (vs. RHE). NRR potentiostatic tests were conducted in 60 mL of 0.1 M KOH solution at −0.2, −0.3, −0.4, −0.5, and −0.6 V. For comparison, potentiostatic tests were also carried out in an Ar-saturated 0.1 M KOH solution at different potentials.212.5 Detection of ammonia and hydrazine
The concentration of produced ammonia was determined by Nessler's reagent spectrophotometry.40 Firstly, 10 mL of solution in the cathode chamber after 2 h electrolysis was taken into colorimetric tubes. Then 1 mL of 0.2 M potassium sodium tartrate was added to chelate interfering soluble metal ions. Next, 1 mL of Nessler's reagent was mixed with the above solution and left to stand for 20 min for the chromogenic reaction. Finally, the absorbance was measured at 420 nm. The blank control was 10 mL of 0.1 M KOH instead of the solution in the cathode chamber. The calibration curve of ammonia was plotted using a series of different concentrations of standard ammonium chloride diluted by 0.1 M KOH.Determination of hydrazine in 0.1 M KOH solution was performed using the Watt and Chrisp method.41 Firstly, 5.99 g of p-dimethylaminobenzaldehyde, 30 mL of concentrated HCl (AR) and 300 mL of ethanol were mixed to obtain a color reagent. Then 10 mL of solution in the cathode chamber after 2 h electrolysis was taken into colorimetric tubes. Next, 5 mL of prepared color reagent was added and left to stand for 10 min. Finally, the absorbance was measured at 460 nm. The blank control was 10 mL of 0.1 M KOH instead of the solution in the cathode chamber. The calibration curve of hydrazine was made using a series of different concentrations of standard hydrazine hydrate diluted by 0.1 M KOH.
The concentration (μg mL−1) of ammonia and hydrazine was calculated using the calibration curves prepared before. The ammonia production rates (r[NH3]) were calculated by using eqn (1):
| r[NH3] = ([NH3] × V)/(t × A) | (1) |
| r[N2H4] = ([N2H4] × V)/(t × A) | (2) |
Assuming three electrons are consumed to produce one NH3 molecule, and four electrons are consumed to produce one N2H4 molecule, the calculation formula can be written as eqn (3)
| FE = 3F × [NH3] × 10−6 × V/(17 × Q) + 4F × [N2H4] × 10−6 × V/(32 × Q) | (3) |
3. Results and discussion
The precursor of NC@Ru was ZIF-8, which was synthesized using the typical method. Then ZIF-8 was totally exposed to a Ru3+ ion solution by stirring intensely, with the expectation that the Ru3+ ions would adhere to the surface of ZIF-8. Then ZIF-8@Ru was calcined in a tube furnace under a nitrogen atmosphere to obtain NC@Ru. Subsequently, the zinc atoms of ZIF-8 were removed partially by the high temperature. Simultaneously, Ru3+ was reduced to Ru and occupied the Zn vacancy to be immobilized. On the other hand, ZIF-8 transformed into a stable N-doped carbon framework after carbonization, increasing its conductivity and endurability, which can serve as a desirable platform for Ru atoms to disperse (Fig. 1).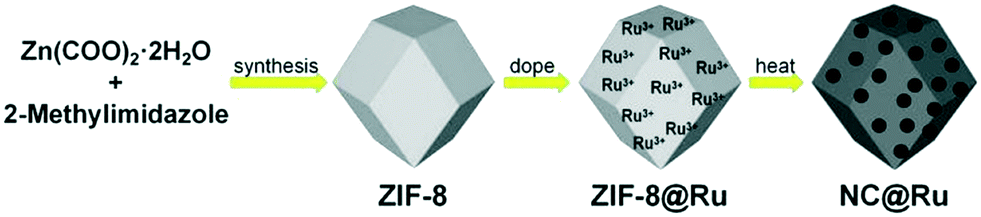 | ||
| Fig. 1 Schematic diagram of preparation process. | ||
To observe the morphology of NC@Ru, high-resolution TEM was used. As shown in Fig. 2a and b, NC@Ru still retained a separate lamellar framework structure, which implies the framework of the precursor did not collapse after calcination and can provide an ideal platform for Ru to be dispersed. Simultaneously, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was used to further observe the surface morphology of NC@Ru. It can be seen clearly that Ru was successfully dispersed and fixed on the surface of the catalyst (Fig. 2c). Also, the corresponding mapping images (Fig. 2d–f) also confirm the dispersion of Ru throughout the N-doped carbon framework, which may consequentially stabilize the Ru atom and prolong the life of the catalyst.
 | ||
| Fig. 2 (a and b) TEM images, (c) HAADF-STEM and (d) C (e) N (f) Ru mapping images of NC@Ru-3. | ||
N2 adsorption–desorption was also carried out to investigate the specific surface area and pore structure of NC@Ru, which are strongly related to the amount of catalytic active sites. As shown in Fig. 3a, the typical shape of the isotherm is a type IV curve with an H4-type hysteresis loop, suggesting the micro/meso-pores in NC@Ru were generated from sheet-like aggregation, corresponding to the TEM result (Fig. 2a). Besides, the calculated Brunauer–Emmett–Teller (BET) surface area of NC@Ru was about 822.05 m2 g−1 (Fig. 3a) with a large BJH average pore diameter of 4.30 nm (Fig. 3b). The sheet-like aggregation pores can not only promote the adsorption and transport of N2 molecules for further reaction, but also provide a considerable amount of Ru active sites for N2 and H+ to interact. On the other hand, the N-doped carbon framework can provide a 3D conductive network to enhance the electron transfer and immobilize the Ru atoms.
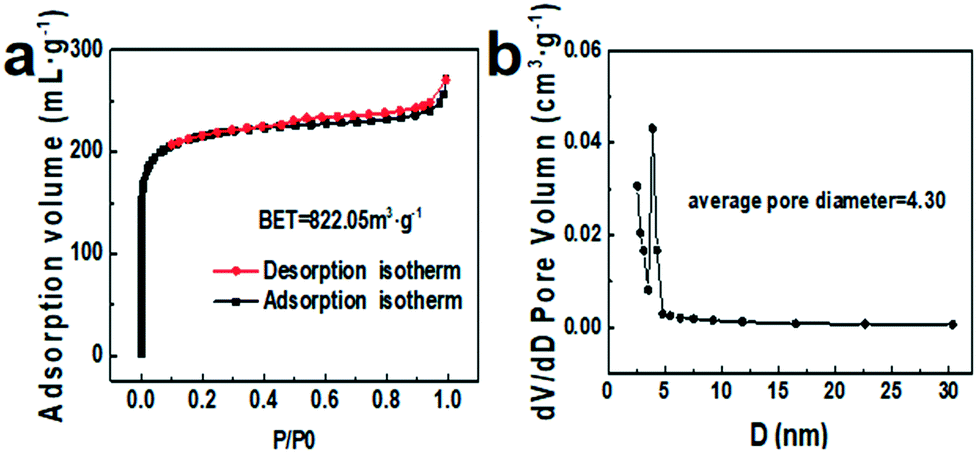 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms (a) and corresponding pore size distribution (b) of NC@Ru-3. | ||
A comparation of the XRD patterns if ZIF-8 synthesized and ZIF-8 simulated is shown in Fig. 4a, which indicates that ZIF-8 was synthesized successfully. The XRD pattern of NC@Ru is shown in Fig. 4b, where the typical peak at ≈25° indicates the (002) face of graphite carbon, which is a characteristic of the carbon framework. The diffraction peaks at 38°, 42°, 44°, 58°, and 69° correspond to the indexed planes of hexagonal closest packed (hcp) crystals of Ru (100), (002), (101), (102) and (110), respectively (JCPDS No: 6-0663).42
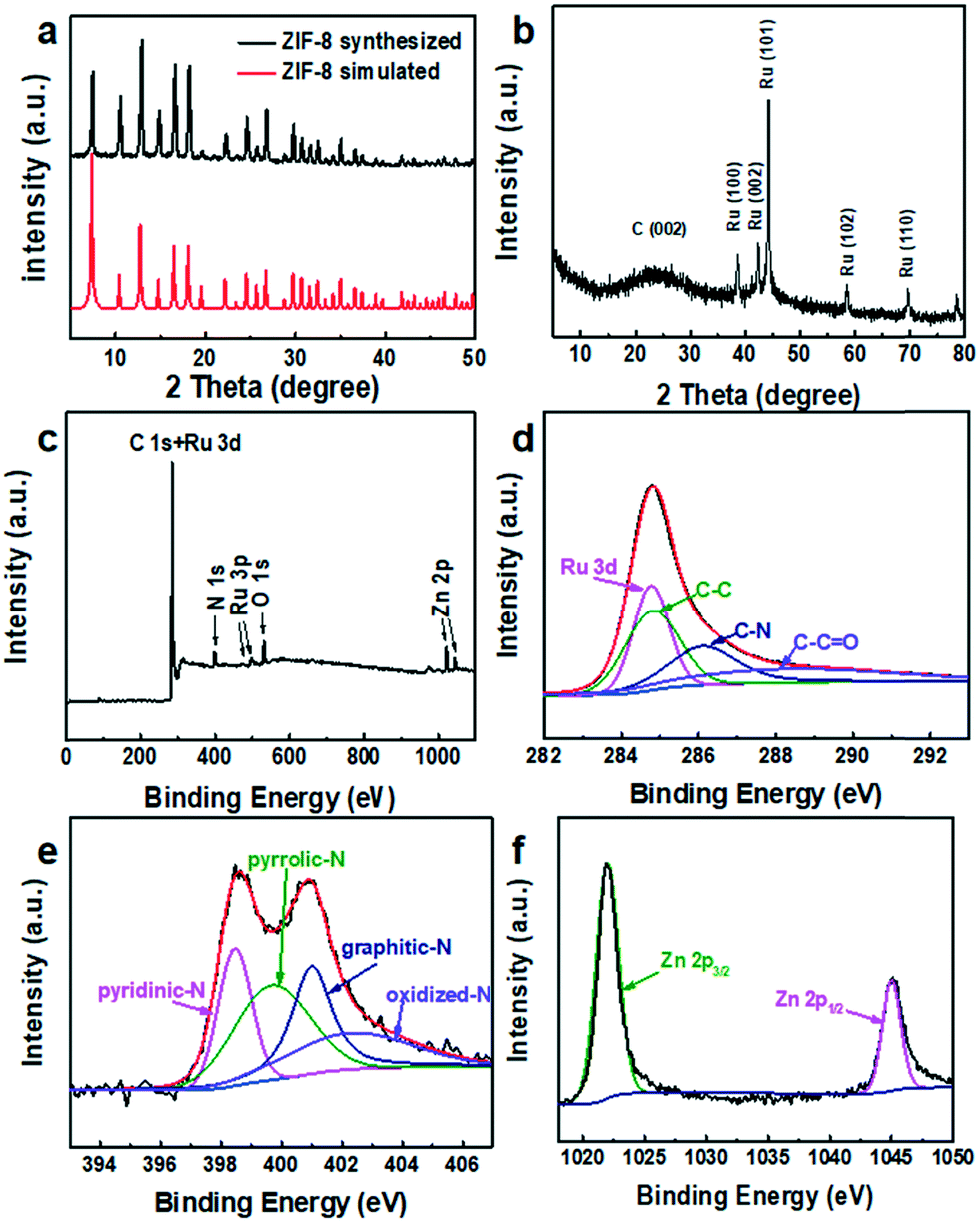 | ||
| Fig. 4 (a) XRD patterns of ZIF-8, (b) XRD patterns, (c) XPS survey spectrum and corresponding high-resolution spectra of Ru 3d + C 1s (d), N 1s (e), and Zn 2p (f) of NC@Ru-3. | ||
To obtain information on the compound surface including chemical composition and element bonding configurations, X-ray photoelectron spectroscopy was carried out for NC@Ru. As shown in Fig. 4c, C, N, O, Ru and Zn were observed. In the C 1s and Ru 3d spectra of NC@Ru (Fig. 4d), the fitting peaks were observed at 284.8, 286.1 and 288.4 eV, which can be ascribed to the overlap of C–C and Ru 3d, C–N, and C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively.39Fig. 4e shows the high-resolution spectra of N 1s, which present 4 types of N coordination environments at 398.3 eV for pyridinic-N, 399.7 eV for pyrrolic-N, 401.0 eV for graphitic-N, and 403.7 eV for oxidized-N. Meanwhile, the N content was determined to be 6.41 at%. N doping in the porous carbon polyhedrons is beneficial for stabilizing metallic Ru, as well as regulating the electronic structure and surface permeability of the NC@Ru catalyst.43 The presence of O and Zn signals at 529.9 and 1022.0 eV in the XPS survey (Fig. 4c) is unavoidable due to the surface oxidation and incomplete gasification, respectively. In addition, the positions of the Zn 2p3/2 and 2p1/2 peaks of NC@Ru (1022.1 and 1045.1 eV) (Fig. 4f) shifted positively in comparison to the standard binding energy of Zn (1021.8 and 1044.8 eV), respectively, indicating that the Ru atoms filled some vacancies, which used to be the Zn atoms.
O, respectively.39Fig. 4e shows the high-resolution spectra of N 1s, which present 4 types of N coordination environments at 398.3 eV for pyridinic-N, 399.7 eV for pyrrolic-N, 401.0 eV for graphitic-N, and 403.7 eV for oxidized-N. Meanwhile, the N content was determined to be 6.41 at%. N doping in the porous carbon polyhedrons is beneficial for stabilizing metallic Ru, as well as regulating the electronic structure and surface permeability of the NC@Ru catalyst.43 The presence of O and Zn signals at 529.9 and 1022.0 eV in the XPS survey (Fig. 4c) is unavoidable due to the surface oxidation and incomplete gasification, respectively. In addition, the positions of the Zn 2p3/2 and 2p1/2 peaks of NC@Ru (1022.1 and 1045.1 eV) (Fig. 4f) shifted positively in comparison to the standard binding energy of Zn (1021.8 and 1044.8 eV), respectively, indicating that the Ru atoms filled some vacancies, which used to be the Zn atoms.
The standard curve method was used to measure the products from electrolysis (Fig. 5a and b). A series of standard ammonia and hydrazine colorimetric solutions in different concentrations were measured using an ultra-violet-visible spectrophotometer (Fig. 5c and d) to quantitatively analyze the yields of ammonia and hydrazine. The electrolyte before and after electrolysis was mixed with Nessler's reagent to detect the production of ammonia, while hydrazine was determined by the Watt and Chrisp method.
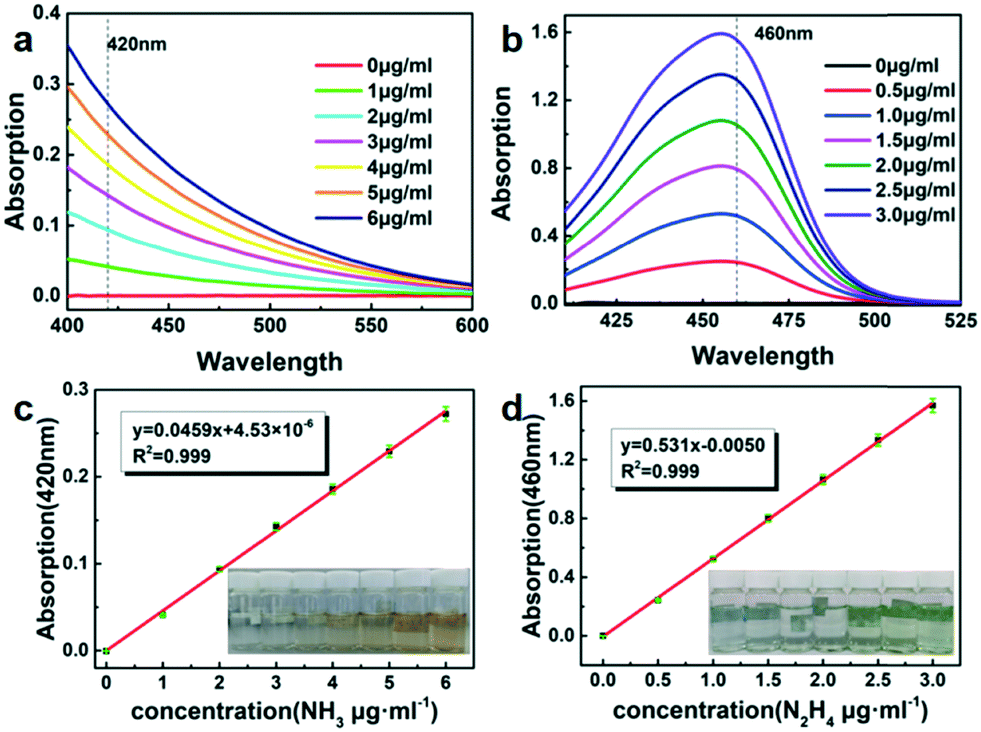 | ||
| Fig. 5 (a) UV-vis curves of Nessler's reagent tests with NH4+ ions. (b) UV-vis curves of Watt and Chrisp method tests with hydrazine. (c) Calibration curve used for the estimation of NH3. (d) Calibration curve used for the estimation of hydrazine. | ||
During electrocatalysis, N2 gas was continuously fed to the working cathode and Ar gas was used as the blank control. Meanwhile, since the gas flow velocity would not affect the yield of ammonia,21 it was set as 50 mL min−1. The linear sweep voltammetry (LSV) test was performed after gas charging for 30 min and 60 cycles of voltammetry scanning to stabilize the working electrode. The LSV curves (Fig. 6a) show different current densities in Ar and N2 from −0.1 V (vs. RHE), which may be due to the nitrogen reduction reaction. In the meantime, the different current densities during electrolysis corresponding to the applied potential are shown in Fig. 6b to reveal more information about the actual electrolysis process. As shown in Fig. 6c and d, the current density of the working electrode in an N2 atmosphere was more negative than that in Ar, and ammonia was detected in the N2-saturated electrolyte, while not in the Ar electrolyte, which further suggests that the NRR occurred.
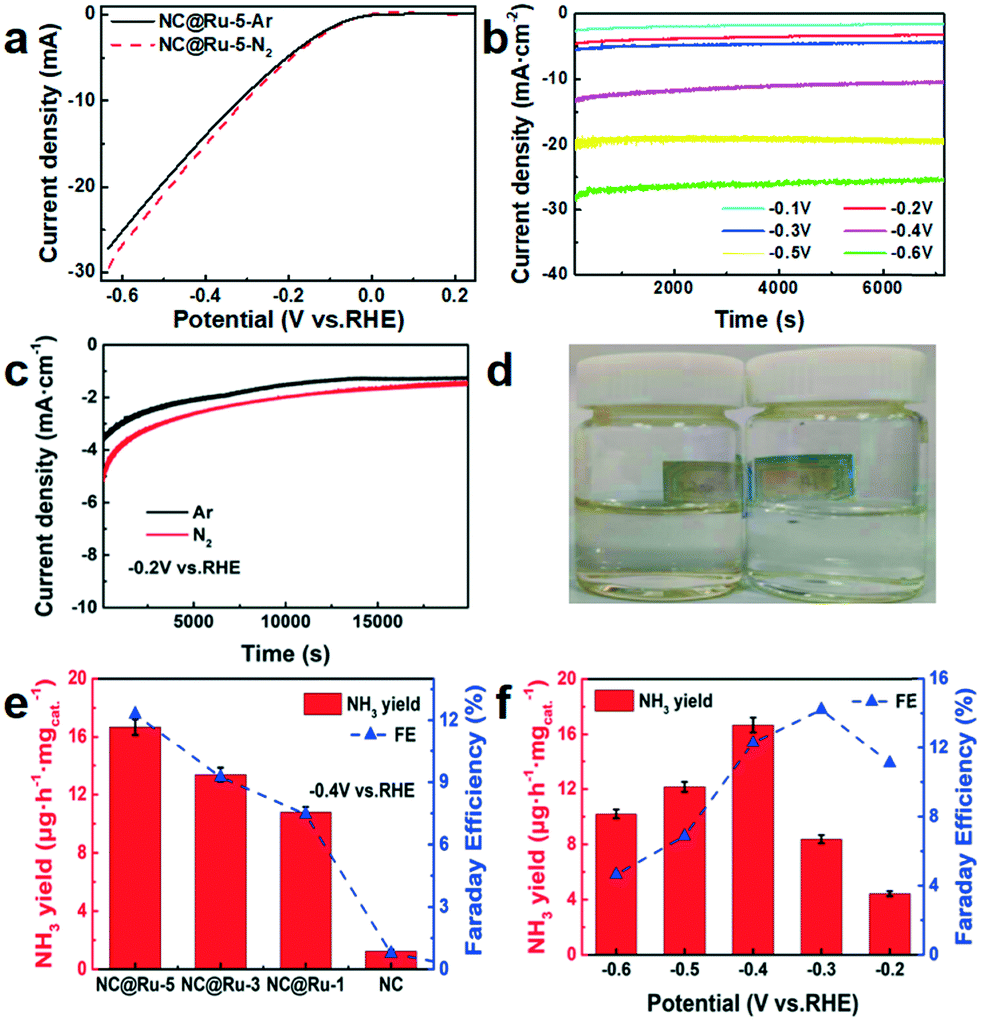 | ||
Fig. 6 (a) Linear sweep voltammetry curves in N2-saturated and Ar-saturated electrolyte using the NC@Ru-5 catalysts. (b) Current density of the NC@Ru-5 electrode during electrolysis at different voltages in N2-saturated electrolyte for 2 h. (c) Current density of the NC@Ru-5 electrode during electrolysis in N2-saturated and Ar-saturated electrolyte at −0.2 V vs. RHE for 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s. (d) Photograph of N2-saturated (left) and Ar-saturated (right) electrolyte mixed with potassium sodium tartrate and Nessler's reagent after electrolysis. (e) Yield rate of NH3 with different working electrodes at −0.4 V vs. RHE. (f) Yield rate of NH3 with different potentials using NC@Ru as the working electrode. 000 s. (d) Photograph of N2-saturated (left) and Ar-saturated (right) electrolyte mixed with potassium sodium tartrate and Nessler's reagent after electrolysis. (e) Yield rate of NH3 with different working electrodes at −0.4 V vs. RHE. (f) Yield rate of NH3 with different potentials using NC@Ru as the working electrode. | ||
The yields of hydrazine were <0.1 μg mL−1 in each test, which was below the detection limit of the instrument. The yield of ammonia was 0 in an Ar atmosphere (Fig. 6c and d), which shows the nitrogen source of NH3 originated from the N2 input instead of the carbon paper or NC@Ru. On the other hand, the yield of ammonia for NC@Ru was more than 8 times that of NC (Fig. 6e). Simultaneously, a higher content of Ru resulted in a higher ammonia production and Faraday efficiency, which implies the Ru dispersed in NC can significantly enhance its catalytic performance in the NRR.
To study the relation between the ammonia yield and potential, a series of electrolytes in different potentials (ranging from −0.1 to −0.6 V) were measured to determine the concentration of ammonia. As shown in Fig. 6f, from −0.1 to −0.4 V (vs. RHE), the ammonia yield rate increased as the potential became more negative and reaches the highest yield rate of 16.68 μg h−1 mgcat.−1 at −0.4 V. However, as the potential became more negative, the yield rate decreased mainly on account of the competition from the HER. Also, the Faraday efficiency increased from −0.1 to −0.3 V (vs. RHE) and achieved 14.23% at −0.3 V (vs. RHE). Then it decreased from −0.4 to −0.6 V (vs. RHE) rationally due to the competition from the HER. The current density–time curve during electrolysis is also presented in Fig. 6c to show the favourable stability of NC@Ru.
Based on the above studies, NC@Ru exhibits an effective catalytic effect on the electrochemical NRR possibly due to the following reasons: 1) its N-doped carbon framework provides ideal electroconductibility, dispersion and protection for the Ru atoms, which are probably the active sites of the NRR. 2) ZIF-8 still retains some of its original structure after carbonization, which may be hydrophobic, and thus it can inhibit the HER and improve the selectivity of ammonia formation. 3) Sufficient pyridinic N in the N-doped carbon framework can adsorb nitrogen strongly. Also, the neighboring Ru atom can accelerate the dissociation of the NN bond.
Overall, the process of NRR catalysed by NC@Ru can be described as follows: firstly, N2 is adsorbed by pyridinic N in the N-doped carbon framework. Then a Ru atom nearby assists the dissociation of the NN bond. Next, H2O in the surface of NC@Ru is electrolysed and releases an activated H. Afterwards, the activated H reacts with the activated NN to form an N–H bond and break the NN bond. Finally, the activated H from water electrolysis reacts with an activated nitrogen molecule and adds H to N2 one by one to generate ammonia.
Conclusions
In summary, we synthesized a new Ru-dispersed N-doped nanocarbon framework to electrocatalyse the nitrogen reduction reaction. The rate of NH3 production was as fast as 16.68 μgNH3 h−1 mgcat.−1 at −0.4 V (vs. RHE) with a faradaic efficiency of 14.23% at −0.3 V (vs. RHE) at room temperature and ambient pressure in 0.1 M KOH electrolyte. More importantly, we found a simplified and feasible way to synthesize the dispersed noble metal N-doped carbon framework, showing high stability and effectiveness, which provides a promising way to decrease the cost of noble metal catalysts during the NRR kinetics. This is a innovate approach to reduce the price of NRR catalysts, enhance their catalytic efficiency and increase their durability, which is also beneficial to the environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key R&D Program of China (No. 2016YFC1102802).References
- U.S. Geological Survey, Mineral Commodity Summaries 2016, U.S. Geological Survey, U.S. Government Publishing Office, Washington, DC, 2016, p. 118 Search PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- M. Appl, Ammonia principles and industrial practice, Wiley-VCH, New York, 2007 Search PubMed.
- S. A. Topham, Catalysis science and technology, ed. J. R. Anderson and M. Boudart, Springer-Verlag, Berlin-Heidelberg, 1985, pp. 1–50 Search PubMed.
- J. M. Modak, Resonance, 2011, 16, 1159 CrossRef.
- J. A. Pool, E. Lobkovsky and P. Chirik, Nature, 2004, 427, 527 CrossRef CAS PubMed.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Science, 2005, 307, 555 CrossRef CAS PubMed.
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836–841 CrossRef CAS PubMed.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2016, 55, 3942–3946 CrossRef CAS PubMed.
- S.-J. Yuan, J.-J. Chen, Z.-Q. Lin, W.-W. Li, G.-P. Sheng and H.-Q. Yu, Nat. Commun., 2013, 4, 2249 CrossRef PubMed.
- R. D. Milton, S. Abdellaoui, N. Khadka, D. R. Dean, D. Leech, L. C. Seefeldt and S. D. Minteer, Energy Environ. Sci., 2016, 9, 2550–2554 RSC.
- K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed.
- K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed.
- J. S. Anderson, G. E. Cutsail, J. Rittle, B. A. Connor, W. A. Gunderson, L. Zhang, B. M. Hoffman and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 7803–7809 CrossRef CAS PubMed.
- R. Lan, K. A. Alkhazmi, I. A. Amar and S. Tao, Appl. Catal., B, 2014, 152–153, 212–217 CrossRef CAS.
- D. Bao, Q. Zhang, F.-L. Meng, H.-X. Zhong, M.-M. Shi, Y. Zhang, J.-M. Yan, Q. Jiang and X.-B. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- Y. Abghoui, A. L. Garden, J. G. Howalt, T. Vegge and E. Skúlason, ACS Catal., 2016, 6, 635–646 CrossRef CAS.
- Y. Liu, Y. Su, X. Quan, X. Fan, S. Chen, H. Yu, H. Zhao, Y. Zhang and J. Zhao, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS.
- L. Cong, Z. Yu, F. Liu and W. Huang, Catal. Sci. Technol., 2019, 9, 1208–1214 RSC.
- R. Zhao, H. Xie, L. Chang, X. Zhang, X. Zhu, X. Tong, T. Wang, Y. Luo, P. Wei, Z. Wang and X. Sun, EnergyChem, 2019, 1, 100011 CrossRef.
- G.-F. Chen, X. Cao, S. Wu, X. Zeng, L.-X. Ding, M. Zhu and H. Wang, J. Am. Chem. Soc., 2017, 139, 9771–9774 CrossRef CAS PubMed.
- Y.-C. Hao, Y. Guo, L.-W. Chen, M. Shu, X.-Y. Wang, T.-A. Bu, W.-Y. Gao, N. Zhang, X. Su, X. Feng, J.-W. Zhou, B. Wang, C.-W. Hu, A.-X. Yin, R. Si, Y.-W. Zhang and C.-H. Yan, Nat. Catal., 2019, 2, 448–456 CrossRef CAS.
- H. Huang, L. Xia, X. Shi, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 11427–11430 RSC.
- R. Zhang, X. Ren, X. Shi, F. Xie, B. Zheng, X. Guo and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 28251–28255 CrossRef CAS PubMed.
- Y. Zhang, W. Qiu, Y. Ma, Y. Luo, Z. Tian, G. Cui, F. Xie, L. Chen, T. Li and X. Sun, ACS Catal., 2018, 8, 8540–8544 CrossRef CAS.
- J. Han, X. Ji, X. Ren, G. Cui, L. Li, F. Xie, H. Wang, B. Li and X. Sun, J. Mater. Chem. A, 2018, 6, 12974–12977 RSC.
- X. Wu, L. Xia, Y. Wang, W. Lu, Q. Liu, X. Shi and X. Sun, Small, 2018, 14, 1803111 CrossRef PubMed.
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S.-Z. Qiao, Adv. Mater., 2019, 31, 1902709 CrossRef PubMed.
- C. Chen, D. Yan, Y. Wang, Y. Zhou, Y. Zou, Y. Li and S. Wang, Small, 2019, 15, 1805029 CrossRef PubMed.
- Z. Wang, X. Hu, Z. Liu, G. Zou, G. Wang and K. Zhang, ACS Catal., 2019, 9, 10260–10278 CrossRef CAS.
- Y. Gao, Z. Han, S. Hong, T. Wu, X. Li, J. Qiu and Z. Sun, ACS Appl. Energy Mater., 2019, 2, 6071–6077 CrossRef CAS.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- Y. Yang, Z. Lin, S. Gao, J. Su, Z. Lun, G. Xia, J. Chen, R. Zhang and Q. Chen, ACS Catal., 2017, 7, 469–479 CrossRef CAS.
- V. Kordali, G. Kyriacou and C. Lambrou, Chem. Commun., 2000, 1673–1674 RSC.
- Y. Lu, J. Li, T. Tada, Y. Toda, S. Ueda, T. Yokoyama, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2016, 138, 3970–3973 CrossRef CAS PubMed.
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- H. Gao, J. Zang, X. Liu, Y. Wang, P. Tian, S. Zhou, S. Song, P. Chen and W. Li, Appl. Surf. Sci., 2019, 494, 101–110 CrossRef CAS.
- Y. Wang, X. Cui, J. Zhao, G. Jia, L. Gu, Q. Zhang, L. Meng, Z. Shi, L. Zheng, C. Wang, Z. Zhang and W. Zheng, ACS Catal., 2019, 9, 336–344 CrossRef CAS.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS.
- M. Dabiri, R. Nikbakht and S. K. Movahed, J. Photochem. Photobiol., A, 2019, 379, 159–170 CrossRef CAS.
- J. Huang, C. Du, J. Nie, H. Zhou, X. Zhang and J. Chen, Electrochim. Acta, 2019, 326, 134982 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy02500f |
| This journal is © The Royal Society of Chemistry 2020 |