
Second-order programming the synthesis of metal–organic frameworks†
Mitchell G.
Fishburn
 a,
Dayne R.
Skelton
a,
Shane G.
Telfer
b,
Pawel
Wagner
c and
Christopher
Richardson
*a
a,
Dayne R.
Skelton
a,
Shane G.
Telfer
b,
Pawel
Wagner
c and
Christopher
Richardson
*a
aSchool of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: chris_richardson@uow.edu.au
bMacDiarmid Institute for Advanced Materials and Nanotechnology, Institute of Fundamental Sciences, Massey University, Palmerston North 4442, New Zealand
cIntelligent Polymer Research Institute, University of Wollongong, Wollongong, NSW 2522, Australia
First published on 10th September 2020
Abstract
Herein we report a new second-order coordinate-covalent programming strategy for metal–organic framework synthesis. We show controlled heterofunctional copolymerisation turns on ‘in lattice’ linking to deliver highly porous frameworks in a single step process.
The synthesis of metal–organic frameworks (MOFs) has seen significant advances over the past two decades. These include an understanding of reticular chemistry,1 coordination modulation2–4 and a gamut of available post-synthetic methods.5,6 However, MOF formation still hinges on a single-step synthesis to organise molecular components into a periodic lattice. This is programmed from information encoded within the components, together with the experimental conditions. Thus, advancing the synthesis of MOFs requires planned control over covalent, coordinate and metal–organic reactivity.
Matzger and co-workers sought to exercise simultaneous control over covalent and coordinate chemistry to direct MOF phase selection.7 They showed that even transient reversible reactions of the linkers with additives in situ can direct crystallisation towards new MOF phases. In situ reactions offer shorter synthetic sequences and are generally advantageous saving time, lowering cost and raising yield. Furthermore, a small number of examples exist showing different MOF structures result from pre-formed versus in situ generated ligands.8–10 We expect the next evolution of this concept is synergistic, irreversible bond formation to develop MOFs with improved structures and properties.
Although many in situ ligand transformations are unplanned and lead to new MOFs and coordination polymers through the wonder of serendipity11,12 there are examples of planned covalent-coordinate programming.13,14 This is where the organic component(s) first undergo covalent chemistry generating a new linker before incorporation into the lattice, as shown schematically in Fig. 1a.
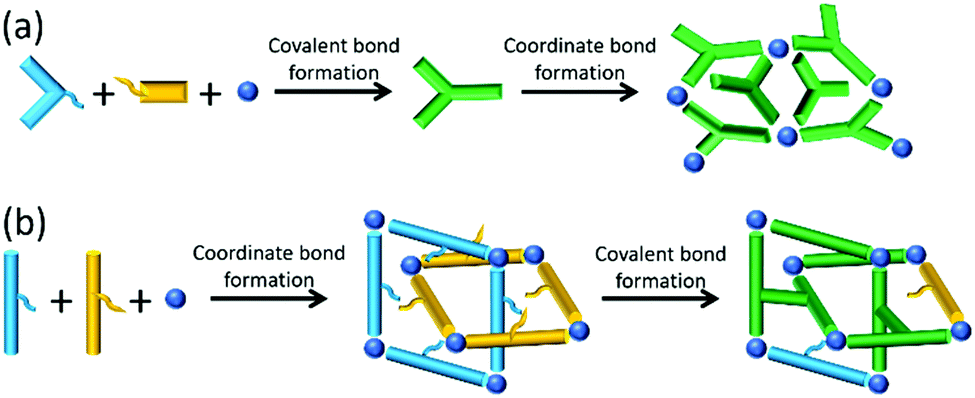 | ||
| Fig. 1 Two paths for in situ transformation in the synthesis of coordination-based materials. (a) The covalent-coordinate approach where covalent bond formation precedes framework formation; (b) the coordinate-covalent approach where framework formation precedes covalent bond formation. | ||
We hypothesised that the microscopic reverse of this process presents potentially powerful yet untapped programming for MOF synthesis. Such coordinate-covalent programming is a far more difficult challenge as it requires the organic components to be unreactive under solvothermal conditions but to become reactive once assembled within the framework (Fig. 1b). We considered these could be ‘proximity-driven’ reactions and could also be considered, in a MOF context, as ‘intra-framework’. Herein we report the realisation of this strategy through judicious selection of compatible molecular components.
The Huisgen [3+2] cycloaddition of azides and alkynes to give 1,2,3-triazoles under copper(I) catalysis has become the archetypal ‘click’ reaction.15 The reaction is well-used in MOF chemistry in ligand synthesis and in post-synthetic modification (PSM).16 However, this reaction has been known for nearly sixty years17 and proceeds under uncatalysed thermally-promoted conditions to give a mixture of 1,4- and 1,5-triazole regioisomers.
Our strategy to demonstrate coordinate-covalent programming was linking adjacent bridging ligands within interpenetrated frameworks using azide–alkyne cycloaddition chemistry. In our experience, zinc frameworks with many functionalised linear biphenyldicarboxylate ligands crystallise as a pair of interpenetrated lattices (Table S1, ESI†). The secondary building units (SBUs) of these frameworks are hexacarboxylate tetrazinc clusters “Zn4O(CO2)6” and the linear biphenyldicarboxylate ligands reticulate the SBUs into an interpenetrated cubic-like arrangement (Fig. 2a, Movie S1, ESI†). Recently, we classified the relative arrangements these lattices assume.18
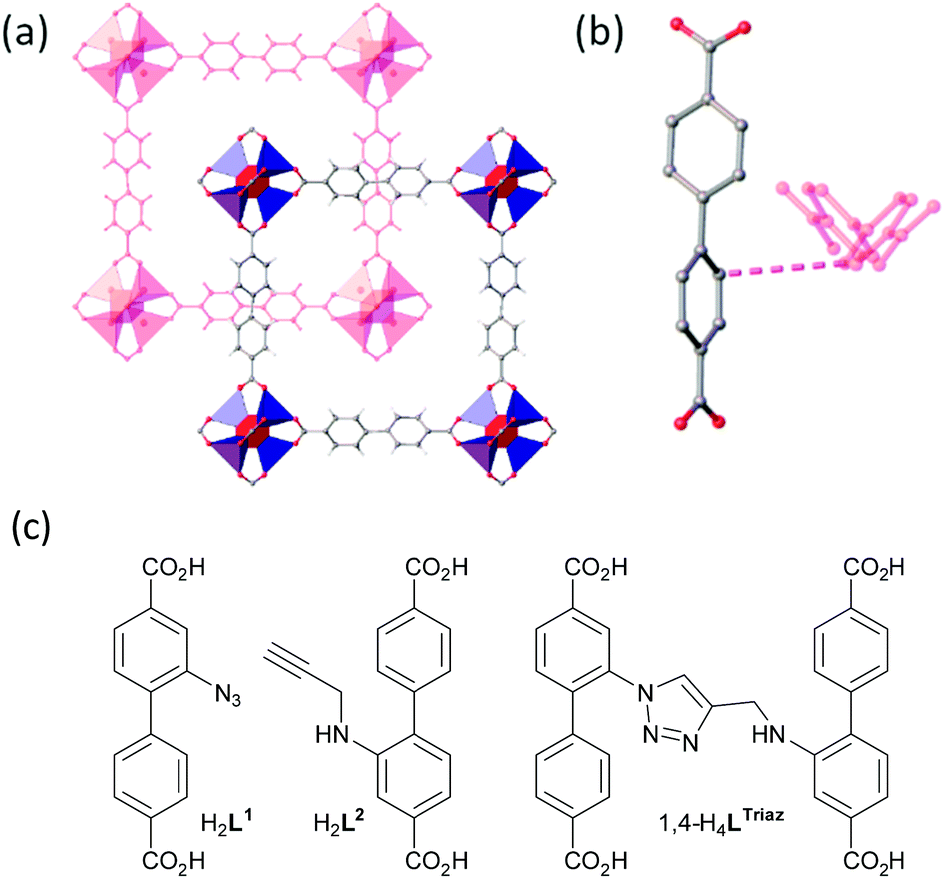 | ||
| Fig. 2 (a) A view of the interpenetrated lattices with zinc and oxido atoms in SBUs shown as polyhedra; (b) a close-up view showing the ∼4 Å distance, as indicated by the dashed line, between biphenyl linkers in interpenetrated lattices; (c) the structures of H2L1, H2L2 and 1,4-H4LTriaz. Hydrogen atoms not shown in (b) for clarity. | ||
Considering the typical separation of ca. 4 Å between the closest points of ligands in the interpenetrating lattices (Fig. 2b), we decided to attach an azide substituent directly to one biphenyldicarboxylic acid backbone and to give the partner alkyne-containing biphenyldicarboxylic acid a larger more flexible tether. Thus, 2-azido-[1,1′-biphenyl]-4,4′-dicarboxylic acid and 2-(prop-2-yn-1-ylamino)-[1,1′-biphenyl]-4,4′-dicarboxylic acid (hereafter H2L1 and H2L2, respectively) (Fig. 2c) were synthesised for this study (ESI†). Fig. 2c also shows the structure of the 1,4-regioisomer for the cycloaddition product of H2L1 and H2L2, 1,4-H4LTriaz.
To maximise the conversion of ‘in-lattice’ linking, we reasoned that equimolar numbers of L1 and L2 should be present in the lattice. Initial experiments determined that greater amounts of H2L2 were required in the feed ratio in order to achieve this. Under optimised conditions, copolymerisation of H2L1 (1.00 eq.) and H2L2 (1.13 eq.) with five equivalents of Zn(NO3)2 in dry DMF gave light brown crystals. PXRD data were consistent with the pattern expected for a functionalised IRMOF-9 compound (Fig. S15, ESI†). Digestion in DMSO-d6 and DCl and analysis by 1H NMR spectroscopy revealed a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 1,4- to 1,5-triazole regioisomers in the MOF and a L1:L2:LTraiz ratio (where LTriaz refers to the combination of regioisomers) of 1.04:1.00:1.52, respectively (Fig. S8, ESI†). This gives the multivariate MOF a ligand composition of [Zn4O(L1)0.63(L2)0.59(LTriaz)0.89] (WUF-50) and represents a linking density of 60%. A random statistical distribution of linkers would result in a maximum linking density of 50%, with the remaining 50% being unproductive azide–azide and alkyne–alkyne co-location. A result higher than 50% crosslinking suggests a greater proportion of azide–alkyne co-location and we posit there is size matching of functionalities as the lattice forms.
:1 ratio of 1,4- to 1,5-triazole regioisomers in the MOF and a L1:L2:LTraiz ratio (where LTriaz refers to the combination of regioisomers) of 1.04:1.00:1.52, respectively (Fig. S8, ESI†). This gives the multivariate MOF a ligand composition of [Zn4O(L1)0.63(L2)0.59(LTriaz)0.89] (WUF-50) and represents a linking density of 60%. A random statistical distribution of linkers would result in a maximum linking density of 50%, with the remaining 50% being unproductive azide–azide and alkyne–alkyne co-location. A result higher than 50% crosslinking suggests a greater proportion of azide–alkyne co-location and we posit there is size matching of functionalities as the lattice forms.
The identification of triazole products by 1H NMR spectroscopy was aided by synthesising regioisomerically-pure 1,4-H4LTriaz using copper-catalysed conditions. Regioisomer confirmation was achieved by single crystal X-ray diffraction (SCXRD) analysis of the methyl ester derivative, 1,4-Me4LTriaz (Fig. S19, ESI†). The uncatalysed thermal reaction gave a 2:1 mixture of 1,4:1,5-Me4LTriaz regioisomers (Fig. S7, ESI†).
Evidence that this reaction follows the coordinate-covalent path rather than the established covalent-coordinate route came from analysis of the supernatant solution from the MOF synthesis, which showed approximately 1% H4LTriaz. Similarly, a control reaction under MOF-forming conditions using Me2L1 and Me2L2 showed less than 2% conversion. These results clearly establish the zinc(II) ions are ineffective catalysts and triazoles are not formed in significant amounts under the conditions used for MOF synthesis and point squarely to following the coordinate-covalent route of Fig. 1b.
We considered that lowering the reaction temperature would regulate the reaction outcome by inhibiting thermally-promoted secondary covalent bond formation, thereby giving less conversion to triazoles. This proved true as incubation of H2L1 and H2L2 with three equivalents of Zn(NO3)2 at 75 °C in dry DMF gave crystals with a much-reduced LTriaz content, [Zn4O(L1)1.28(L2)1.12(LTriaz)0.30], representing a 20% linking density, as determined from 1H NMR analysis after MOF digestion. Linking density rises to 42% upon continued heating for an additional 24 hours at 75 °C in the reaction solution and to 60% by heating in fresh dry DMF solution at 120 °C for 2 hours. Notably, the latter result is the same as that achieved in the optimised direct synthesis.
We also sought to determine if a crystalline phase would be formed directly from 1,4-H4LTriaz under the same reaction conditions. Therefore, 1,4-H4LTriaz was heated with five equivalents of Zn(NO3)2 in dry DMF at 100 °C overnight. This resulted in the formation of a precipitate and heating was continued out to 36 hours, which led to the formation of fawn-coloured wedge-shaped crystals (WUF-53). These were separated from the powdery material and subjected to a cell determination using SCXRD. This was consistent with an interpenetrated IRMOF-9-type framework, and PXRD supported bulk phase purity (Fig. S18, ESI†). Analysis by 1H NMR spectroscopy after digestion in DMSO-d6 and DCl showed the crystals contained a 1:2 mixture of bpdc-NH2 and 1,4-LTriaz linkers (Fig. S11, ESI†), respectively, allowing the framework to be formulated as [Zn4O(1,4-LTriaz)1.20(bpdc-NH2)0.60]. We have observed C–N bond cleavage in amine-functionalised bpdc ligands under the same reaction conditions.18 Thus, the bpdc-NH2 linkers derive from decomposition of 1,4-H4LTriaz during the MOF-forming reaction (Fig. 3). Further support comes from 1H NMR analysis of a control reaction of 1,4-Me4LTriaz under identical conditions, which established that a 34% conversion to Me2bpdc-NH2 occurred. Our rationalisation is that H2bpdc-NH2, formed from the breakdown of some 1,4-H4LTriaz (Fig. 3), templates the formation of the interpenetrated lattices, and remaining 1,4-H4LTriaz linkers join and grow the lattice. This series of experiments suggests that linked pcu-like lattices cannot be made from only 1,4-LTriaz linkers.
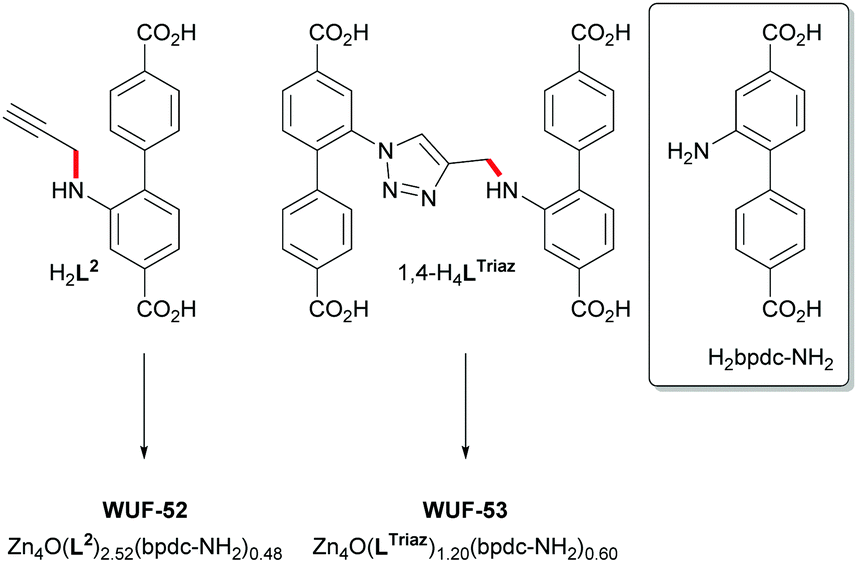 | ||
| Fig. 3 Ligand structures and framework formulations for WUF-52 and WUF-53. The red bond in the structures of H2L2 and 1,4-H4LTriaz indicates the disconnection point to liberate H2bpdc-NH2 (structure shown at right) during the MOF syntheses. | ||
For comparison, the MOFs from H2L1 and H2L2 (WUF-51 and WUF-52, respectively) were prepared under identical conditions to WUF-50. Analysis by PXRD and 1H NMR digestion analysis showed the azide-tagged ligands were incorporated into the interpenetrated lattices undamaged. However, the alkyne-tagged L2 ligands underwent ∼16% C–N bond cleavage to bpdc-NH2 (Fig. S10, ESI†), giving a multivariate MOF of formulation [Zn4O(L2)2.52(bpdc-NH2)0.48]. The SCXRD structures of WUF-51 and WUF-52 were also determined and confirmed the expected interpenetrated lattices (ESI†).
The set of MOFs were studied using simultaneous thermogravimetric-differential scanning calorimetry (TG-DSC) (Fig. S22–S28, ESI†). The TG trace of WUF-51 (Fig. 4a and Fig. S25, ESI†) shows a 6.9% mass loss centred at 176 °C corresponding to exothermic elimination of nitrogen from the azide groups (calc. 7.5%). The DSC trace indicates two near-simultaneous processes during the mass loss event and digestion and 1H NMR analysis showed a near quantitative PSM to 9H-carbazole-2,7-dicarboxylate linkers (Fig. 4b and Fig. S12, ESI†), forming a new MOF, PS-WUF-54 (PS = post-synthetic). This reactivity is known for H2L1 but has not been reported as a MOF PSM.19 The TG-DSC trace for WUF-50 (Fig. 4a and Fig. S23, ESI†) shows thermal events that occur with only very small mass loss between 170 °C and 220 °C. Subsequent 1H NMR analysis showed these were the simultaneous Huisgen cycloaddition to form more LTriaz linkers and carbazole dicarboxylate formation arising from azide decomposition. This solventless heating method increased the triazole linking density from 20% to 52% and PXRD analysis confirmed crystallinity was retained.
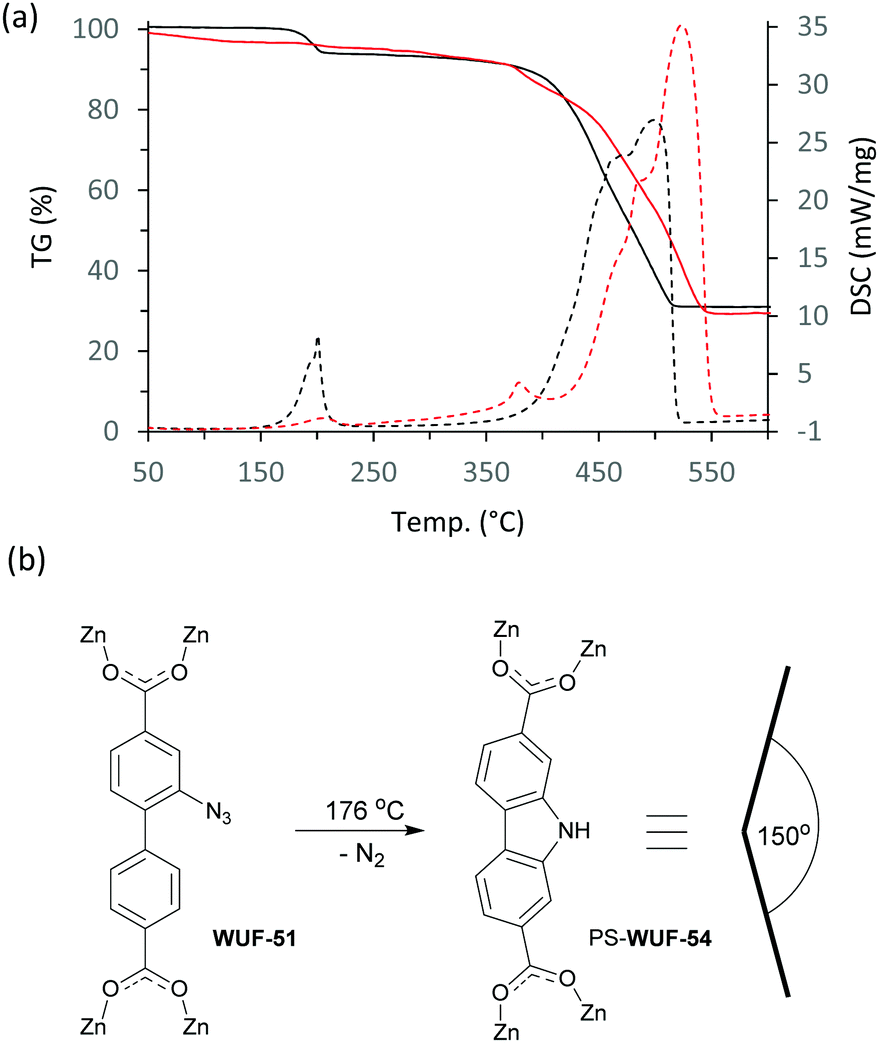 | ||
| Fig. 4 (a) TG-DSC traces of WUF-50 (red) and WUF-51 (black) with solid lines showing the TG data and the dashed lines the DSC data; (b) the molecular transformation of WUF-51 to PS-WUF-54 together with a representation of the approximate angular preference for the carbazole dicarboxylate linker. | ||
Nitrogen adsorption measurements at 77 K showed all the MOFs were porous with type I isotherms, consistent with their expected microporous structures (Fig. 5 and Fig. S29–S33, ESI†). The accessible surface area calculated by the Brunauer–Emmet–Teller (BET) method for WUF-50 (2206 m2 g−1) shows it falls between its parent MOFs, WUF-51 (2350 m2 g−1) and WUF-52 (2076 m2 g−1). Interestingly, the BET surface area for WUF-53 (1700 m2 g−1) is considerably lower as a result of the greater incorporation of larger 1,4-LTriaz linkers. Although PS-WUF-54 retains a type I isotherm, the surface area reduces to 740 m2 g−1. We suspect the reason for this is damage from the release of significant lattice strain that must result changing from linear L1 to angled carbazole linkers (∼150°) (Fig. 3b).
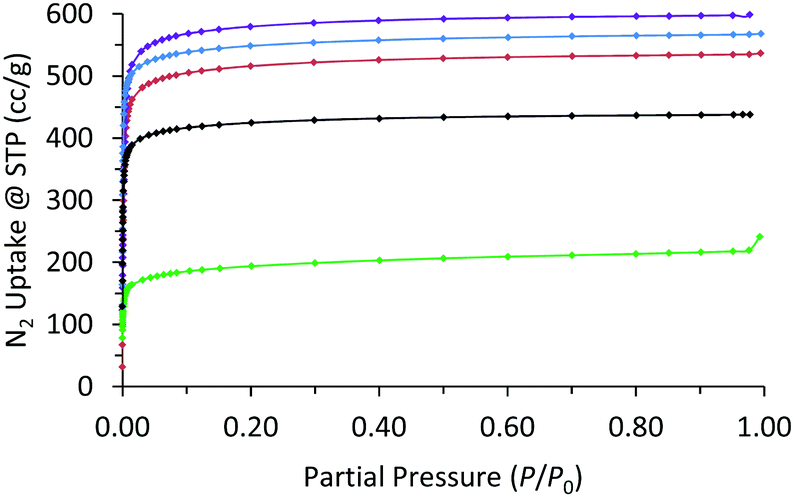 | ||
| Fig. 5 N2 adsorption isotherms at 77 K of WUF-50 (blue), WUF-51 (purple), WUF-52 (red), WUF-53 (black), PS-WUF-54 (green). | ||
In conclusion, we have provided the first example of a deliberate second-order coordinate-covalent synthetic programme for MOFs. Our findings show temperature control is crucial to modulating and maximising conversion in this programmed chemistry. The multivariate approach pursued here begets quantitative linking due to unproductive functional group matching in the lattice. We hope our findings stimulate the pursuit of synthesising advanced MOFs by similar coordinate-covalent programming, particularly where control over precise placement of functional groups in the lattice can be achieved.
Conflicts of interest
There are no conflicts to declare.Notes and references
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry: Metal-Organic Frameworks and Covalent Organic Frameworks, Wiley, 2019 Search PubMed.
- R. J. Marshall, C. L. Hobday, C. F. Murphie, S. L. Griffin, C. A. Morrison, S. A. Moggach and R. S. Forgan, J. Mater. Chem. A, 2016, 4, 6955–6963 RSC.
- D. Bara, C. Wilson, M. Mörtel, M. M. Khusniyarov, S. Ling, B. Slater, S. Sproules and R. S. Forgan, J. Am. Chem. Soc., 2019, 141, 8346–8357 CAS.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643–6651 CrossRef CAS.
- S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 CrossRef CAS.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- A. Dutta, K. Koh, A. G. Wong-Foy and A. J. Matzger, Angew. Chem., Int. Ed., 2015, 54, 3983–3987 CrossRef CAS.
- C. E. Rowland, N. Belai, K. E. Knope and C. L. Cahill, Cryst. Growth Des., 2010, 10, 1390–1398 CrossRef CAS.
- K. E. Knope and C. L. Cahill, CrystEngComm, 2011, 13, 153–157 RSC.
- C.-C. Chang, Y.-C. Huang, S.-M. Huang, J.-Y. Wu, Y.-H. Liu and K.-L. Lu, Cryst. Growth Des., 2012, 12, 3825–3828 CrossRef CAS.
- X.-M. Zhang, Coord. Chem. Rev., 2005, 249, 1201–1219 CrossRef CAS.
- X.-M. Chen and M.-L. Tong, Acc. Chem. Res., 2007, 40, 162–170 CrossRef CAS.
- X. J. Kong, T. He, Y. Z. Zhang, X. Q. Wu, S. N. Wang, M. M. Xu, G. R. Si and J. R. Li, Chem. Sci., 2019, 10, 3949–3955 RSC.
- U. S. F. Arrozi, V. Bon, S. Krause, T. Lübken, M. S. Weiss, I. Senkovska and S. Kaskel, Inorg. Chem., 2020, 59, 350–359 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- P.-Z. Li, X.-J. Wang and Y. Zhao, Coord. Chem. Rev., 2019, 380, 484–518 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633–645 CrossRef.
- A. Khansari, M. R. Bryant, D. R. Jenkinson, G. B. Jameson, O. T. Qazvini, L. Liu, A. D. Burrows, S. G. Telfer and C. Richardson, CrystEngComm, 2019, 21, 7498–7506 RSC.
- X.-C. Yi, F.-G. Xi, Y. Qi and E.-Q. Gao, RSC Adv., 2015, 5, 893–900 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and 1H and 13C spectra of for ligands and their precursors; crystal structure tables and descriptions (CCDC 2022084 and 2022085). Procedures for control reactions, PXRD patterns, DSC-TGA traces, gas adsorption isotherms and BET calculations. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc05451h |
| This journal is © The Royal Society of Chemistry 2020 |