
Unravelling a long-lived ligand-to-metal cluster charge transfer state in Ce–TCPP metal organic frameworks†
Sizhuo
Yang
a,
Wenhui
Hu
a,
James
Nyakuchena
a,
Christian
Fiankor
b,
Cunming
Liu
c,
Eli Diego
Kinigstein
c,
Jian
Zhang
b,
Xiaoyi
Zhang
c and
Jier
Huang
 *a
*a
aDepartment of Chemistry, Marquette University, Milwaukee, Wisconsin 53201, USA. E-mail: jier.huang@marquette.edu
bDepartment of Chemistry, University of Nebraska-Lincoln, Lincoln, Nebraska 68588, USA
cX-ray Science Division, Argonne National Laboratory, Argonne, Illinois 60349, USA
First published on 13th October 2020
Abstract
Metal organic frameworks (MOFs) have emerged as promising photocatalytic materials for solar energy conversion. However, a fundamental understanding of light harvesting and charge separation (CS) dynamics in MOFs remains underexplored, yet they are key factors that determine the efficiency of photocatalysis. Herein, we report the design and CS dynamics of the Ce–TCPP MOF using ultrafast spectroscopic methods.
The direct conversion of water or CO2 by sunlight into fuel is a promising approach to address global energy and environmental issues.1 However, it remains a great challenge to drive such a reaction in an efficient way and an appropriate catalyst is highly desired to promote the reaction at a reasonable rate. As an emerging class of porous materials, metal organic frameworks (MOFs) offer a new opportunity by taking advantage of both homogenous and heterogeneous catalysis for photocatalysis.2 MOFs are created by assembling metal-containing secondary building units (SBUs) with organic linkers.3 With flexible metal SBUs and organic linkers, MOFs are able to integrate light-harvesting materials and catalysts into a single matrix.4 In addition, the high crystallinity and porous nature of MOFs may facilitate charge transport and diffusion of reactants during the photocatalytic reaction.5 Due to these reasons, a large number of MOFs with built-in photosensitizers (PSs) and molecular catalysts have been used for water splitting and CO2 reduction.6 However, the majority of these works are centered on their catalytic performance and stability rather than fundamental mechanism. The commonly accepted catalytic pathway initiates with light absorption by the PS, which is followed by charge separation (CS) events either through oxidative quenching of the PS by electron transfer (ET) to the catalyst or reductive quenching through ET from the electron donor. As a result, it is essential to gain thorough knowledge of the fundamental aspects of the light harvesting and CS processes before we explore MOFs for photocatalytic applications.6c,7 Indeed, the presence of a long-lived CS state in a few MOFs following photoexcitation has been reported previously.7a,8 We have also reported the formation of a long-lived CS state in a zeolitic imidazolate framework based on a Co node and 2-methyl imidazolate ligand (ZIF-67).9 While these fundamental studies demonstrate the great potential of MOFs as light harvesting and CS materials, these systems largely rely on a ligand that has limited absorption in the visible region7a,8a,e or a transition metal d–d transition with a low extinction coefficient9 as the PS, preventing their further applications in solar energy conversion. In response to these challenges, in this work, we report the excited state and CS dynamics of a porphyrin-based Ce–TCPP MOF by using optical transient absorption (OTA) and X-ray transient absorption (XTA) spectroscopy, where the TCPP ligand (tetrakis(4-carboxyphenyl)porphyrin) has broad absorption and a relatively high extinction coefficient in the UV-visible spectrum. We show that the excitation of Ce–TCPP MOFs leads to the formation of a long-lived CS state with ligand-to-metal cluster charge transfer (LCCT) character, where the mixed-phase structure in Ce–TCPP MOFs was found to play an important role in the formation of this LCCT state.
As shown in Fig. 1a, the 3D Ce–TCPP MOF was synthesized by a solvothermal reaction. The obtained product was characterized by powder XRD (Fig. 1b) and SEM (inset of Fig. 1b) and shows a needle-like shape. After comparing the XRD patterns of the Ce–TCPP MOF with the patterns simulated from the single crystal structure of many TCPP MOFs, it seems that the structure of the Ce–TCPP MOF did not agree with any of these single-phase MOFs. Instead, we found that the XRD patterns agree well with a combination of the XRD patterns of the crystal structures of VETTUK10 and CAU-19 (Fig. 1b),11 where the patterns of the former are more prominent. These results suggest that the crystalline structure of Ce–TCPP MOFs is dominated by a structure similar to VETTUK with a slight contribution from CAU 19. The porosity of Ce–TCPP MOFs was confirmed by N2 sorption measurement (Fig. S1, ESI†) and thermogravimetric analysis (TGA) (Fig. S2, ESI†). The average BET surface area is 342.67 m2 g−1 based on two measurements. This value is relatively small but within the range of literature results of similar TCPP-based MOFs (330–600 m2 g−1).11 The TGA results show two characteristic steps, including a weight loss below 100 °C and one at 360–500 °C, which can be attributed to the evaporation of water molecules and the decomposition of the MOF with the dissociation of the Ce–O bond, respectively.11 The formation of Ce–TCPP MOFs was further supported by the Fourier-transform infrared (FT-IR) spectra (Fig. 1c), where we observed the disappearance of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1700 cm−1, which is present in TCPP resulting from free –COOH, the shift of CC valence vibration of phenyl rings from 1559 cm−1 to 1526 cm−1, and two new peaks at 1587 cm−1 and 1400 cm−1, which can be assigned to asymmetric and symmetric vibrational stretching of COO−, respectively.12 These results together suggest the successful coordination of the carboxyl group in TCPP with the Ce metal ion in Ce–TCPP MOFs.
O stretching at 1700 cm−1, which is present in TCPP resulting from free –COOH, the shift of CC valence vibration of phenyl rings from 1559 cm−1 to 1526 cm−1, and two new peaks at 1587 cm−1 and 1400 cm−1, which can be assigned to asymmetric and symmetric vibrational stretching of COO−, respectively.12 These results together suggest the successful coordination of the carboxyl group in TCPP with the Ce metal ion in Ce–TCPP MOFs.
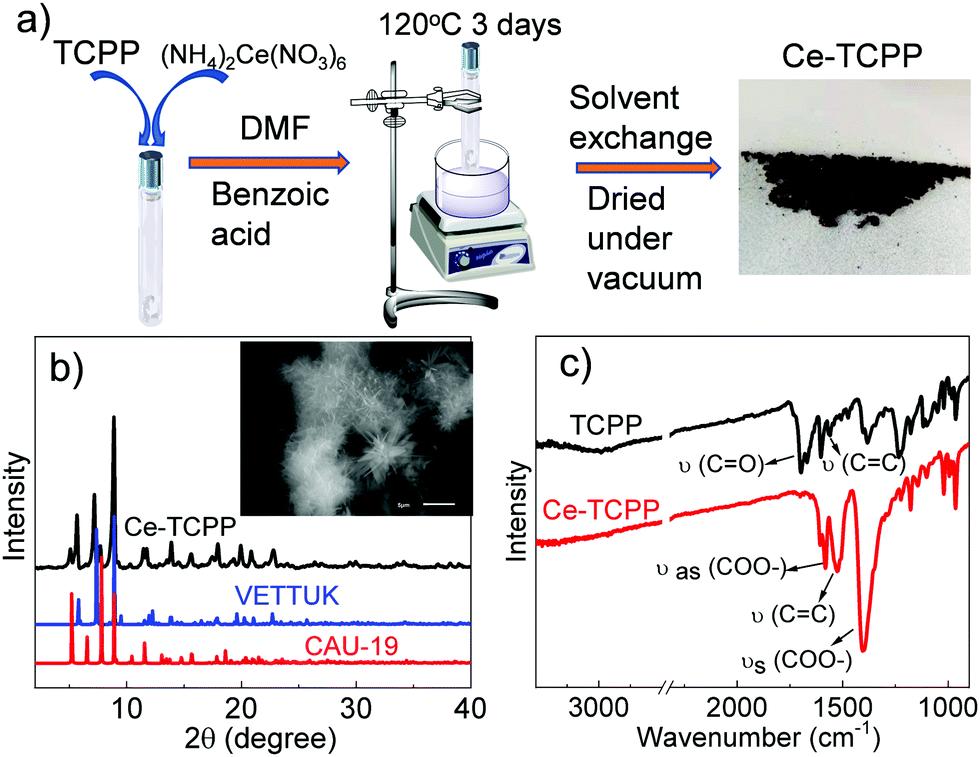 | ||
| Fig. 1 (a) Synthetic scheme of Ce–TCPP; (b) XRD patterns of Ce–TCPP and patterns simulated from the single crystal structure of similar TCPP MOFs; (c) FTIR spectra of TCPP and Ce–TCPP. Inset of (b) is the SEM image of Ce–TCPP MOFs. | ||
In addition to the bulk structure, the local coordination environment at the Ce center in Ce–TCPP MOFs was confirmed by X-ray absorption spectroscopy (XAS). The X-ray absorption near edge structure (XANES) spectra of Ce–TCPP MOFs and two reference samples, i.e., hexagonal CeCl3·7H2O (Ce3+) and cubic CeO2 (Ce4+), as well as the starting material (NH4)2Ce(NO3)6, are shown in Fig. 2a. The main feature at ∼5725.5 eV in the spectrum of CeCl3 is the absorption white line corresponding to the dipole allowed transition from Ce 2p to 5d mixed with the 4f1 final state,13 suggesting that Ce in CeCl3 is in a trivalent state (Ce3+). In contrast, the XANES spectrum of CeO2 and (NH4)2Ce(NO3)6 exhibits two distinct features, where the features for CeO2 occur at ∼5731 eV and 5737.5 eV and those for (NH4)2Ce(NO3)6 occur at 5729 eV and 5738 eV, which can be attributed to the mixed-valence behavior of tetravalent Cerium (Ce4+) in its final state (4f15dt2gL and 4f05d; L denotes the oxygen 2p hole).14 Unlike the reference samples, the XANES spectrum of Ce–TCPP MOF shows the main absorption edge at 5725.5 eV and a weak feature at 5737.5 eV, which can be assigned to the 4f1 and 4f0 absorption peaks, respectively, suggesting the co-existence of a Ce3+/Ce4+ valence state in the Ce–TCPP MOF. This is further supported by the energy difference (∼12 eV) between these two features, which agrees well with the Coulomb interaction of Ce 2p and Ce 4f orbitals.15 As suggested by the previous literature report on similar Ce–MOFs,11 the Ce center in the Ce–TCPP MOF is dominated by Ce3+ that can be attributed to the reduction of Ce4+ by the redox-active linkers (i.e., TCPP).
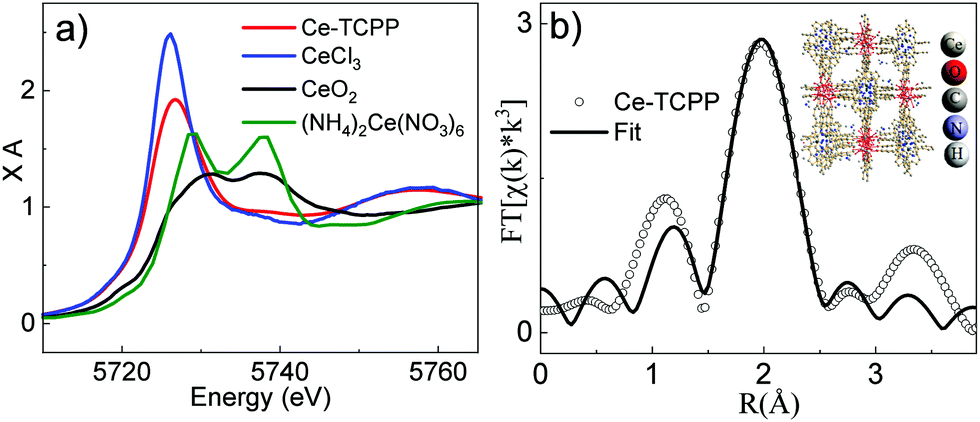 | ||
| Fig. 2 (a) XANES spectra of Ce–TCPP MOFs (red), CeCl3 (blue), CeO2 (black) and (NH4)2Ce(NO3)6 (olive). (b) The Fourier-transformed XAS spectrum of Ce–TCPP in R-space. The inset shows the fitting model. The data are shown as open dots and FEFF fits are shown as solid lines. | ||
To gain a deeper insight of the local coordination environment, we quantitatively fitted the extended X-ray absorption fine structure (EXAFS) spectrum of Ce–TCPP (Fig. S3a, ESI†) using the Demeter XAS analysis package. As the XRD results suggest that the structure of Ce–TCPP MOF is featured by a combination of VETTUK and CAU-19, the fitting models based on the crystal structure of both VETTUK10 and CAU-1911 were used to fit the data. It is interesting to note that the EXAFS spectrum can be adequately fit based on the crystal structure of VETTUK (fitting parameters listed in Table S1, ESI†) but significant deviation was observed based on CAU-19. This can be explained by the fact that the crystalline structure of the Ce–TCPP MOF is dominated by a structure similar to VETTUK, which is consistent with the XRD results. The EXAFS data and the resulting best fit in R-space and k-space are presented in Fig. 2b and Fig. S3b (ESI†), respectively. From the best fitting, the bond distance of Ce to O in Ce–TCPP was found to be between 2.22 Å and 2.61 Å. The Ce–O distances are within the range of distances reported in the literature for Ce(IV) and Ce(III) clusters (2.206–2.234 Å for core Ce–O, 2.429 Å to 2.563 Å and 2.364 to 2.765 Å for the remaining Ce–O),16 suggesting the validity of our fitting model.
Fig. 3a shows the UV-visible absorption spectrum of TCPP in methanol (black plot), and the diffuse reflectance (DR) spectra of TCPP in the solid state (blue plot) and Ce–TCPP MOFs (red plot). It was found that TCPP in methanol solution exhibits an intense Soret band centered at 415 nm (S0–S2 transition) and relatively weaker Q bands (S0–S1 transition) that spread over a wide range in the visible region with four distinct peaks at 512 nm (Qy), 547 nm (Qy), 588 nm (Qx) and 645 nm (Qx).17 In contrast, Ce–TCPP MOFs show broad absorption extended to ∼700 nm. This feature is likely due to scattering because it was also observed in the DR spectrum of TCPP in the solid state. The Soret and Q bands of Ce–TCPP MOFs show a prominent red-shift compared to TCPP, which can be attributed to the planarity change caused by the deformation of TCPP during incorporation: the confirmed non-planarity of TCPP in the Ce–TCPP MOF destabilizes the porphyrin HOMOs while the LUMOs were not significantly affected.18 Moreover, the strong coupling of the well-arranged TCPP monomers (J-aggregation)17b may also contribute to the red-shift, which has been observed in other porphyrin-based MOFs reported previously.19
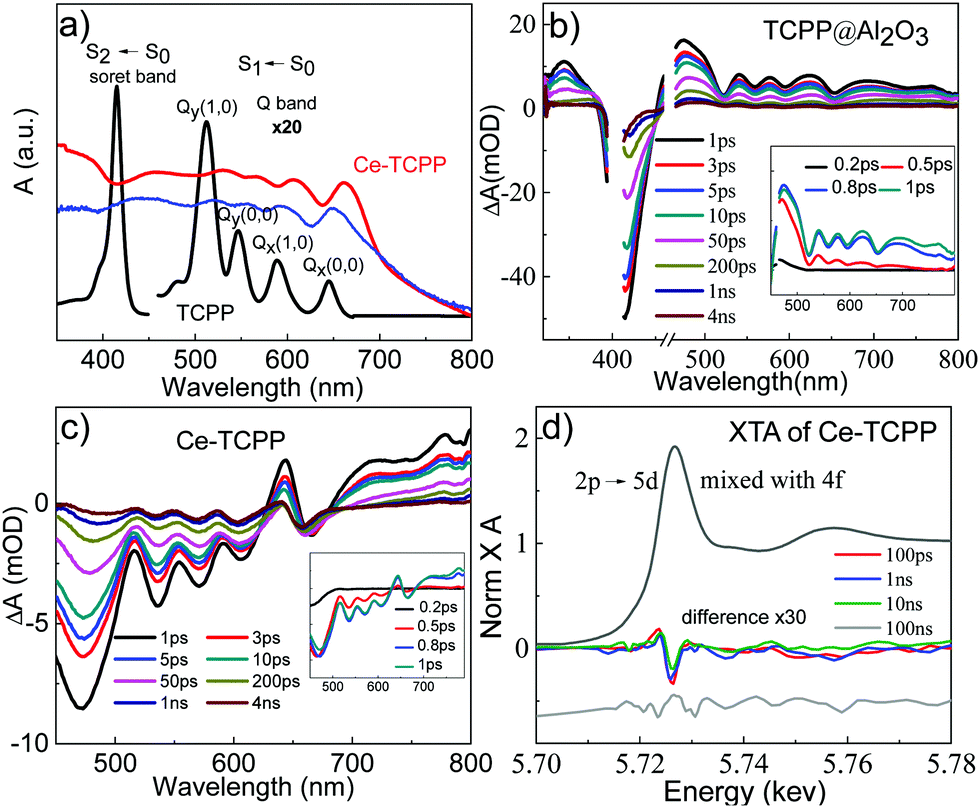 | ||
| Fig. 3 (a) UV-visible absorption spectrum of TCPP in methanol and the diffuse reflectance spectrum of TCPP in the solid state (blue) and Ce–TCPP (red). OTA spectra of TCPP/Al2O3 (b) and Ce–TCPP (c) following 400 nm excitation. The insets show the early time OTA spectra. (d) XANES spectrum of Ce–TCPP MOFs at the Ce L3-edge (black plot) and the difference XANES spectra at 100 ps (red), 1 ns (blue), 10 ns (green), and 100 ns (gray) obtained by subtracting the laser-off spectrum from the laser-on spectrum. The difference spectrum at 100 ns delay is shifted manually to provide an offset for a clearer view. | ||
Femtosecond OTA spectroscopy was performed with a selective excitation of the TCPP Soret band to examine the excited state (ES) dynamics of the Ce–TCPP MOFs. To better understand the effect of the porous nature on the ES dynamics in the MOFs, we first measured the OTA spectra of TCPP on an Al2O3 thin film, which represents a control sample for intrinsic ES dynamics of TCPP in a heterogeneous environment. As shown in Fig. 3b and Fig. S4a (ESI†), the OTA spectra of TCPP/Al2O3 thin film consist of a negative band centered at 415 nm and a broad positive absorption feature from 450 nm overlapping with several distinct bleach signals at 520 nm, 563 nm, 596 nm and 653 nm. These features have been well studied and can be attributed to the TCPP Soret band ground state bleach (GSB), ES absorption (ESA), and Q band GSB, respectively. The recovery of the Soret GSB and the decay of ES follow the same kinetics (Fig. S4b, ESI†), together with the presence of the isosbestic point at 451 nm, suggesting that the decay of ES molecules to their GS is the only relaxation process in TCPP/Al2O3 after excitation.
Compared to TCPP, the OTA spectra of Ce–TCPP MOFs are dramatically different. As shown in Fig. 3c, the OTA spectra of Ce–TCPP exhibit the Soret and Q band GSB with a red-shift in the range of 450 nm to 670 nm, which is consistent with its UV-visible ground state DR reflectance spectrum. However, the positive features pertaining to ESA are missing in the region < 600 nm even at early times (<1 ps) (inset of Fig. 3c). We attribute this difference to the ultrafast (<200 fs) formation of a new CS state evolved from singlet ES (i.e., S2 and S1) (Fig. S4c, ESI†), as superfast CS was also observed upon Q band excitation (Fig. S4d, ESI†). The CS was further confirmed by observation of a transient feature in the region >700 nm as the fingerprint absorption of one electron oxidized TCPP*+.20 These results, similar to previous reports accounting for electron transfer (ET) processes in MOFs,19,21 imply that fast ET occurs from TCPP ligand to Ce metal cluster.
To gain more insight on the nature of the CS state, we measured the OTA spectra of three reference samples including CAU-19, a Ce-based MOF that has the same crystal structure as VETTUK (denoted as Ce-VETTUK), and a physical mixture of these two MOFs. The details of the synthetic procedure and structural characterization (XRD, FTIR, DR spectrum, and XAS) of these two MOFs are presented in the ESI and Fig. S5–S8 (ESI†). The OTA spectra of CAU-19 (Fig. S9a, ESI†), Ce-VETTUK (Fig. S9b, ESI†), and the physical mixture of both MOFs (Fig. S9c, ESI†) largely look similar to that of TCPP/Al2O3, consisting of a Soret GSB at <480 nm, broad ESA, and multiple Q band GSB signals. However, it is notable that the Q band GSB bands, which remain positive in the spectra of TCPP/Al2O3 at all time delays, become negative in the spectra of CAU-19 at ∼100 ps, Ce-VETTUK at ∼50 ps, and the physical mixture of CAU-19 and Ce-VETTUK as early as 1 ps (Fig. S9 and S10, ESI†). These results together suggest that the CS state observed in the mixed phase Ce–TCPP MOF also occurs in these reference samples, where the formation of CS is fastest in the mixed-phase Ce–TCPP, which then decreases following the order of the physical mixture of CAU-19 and Ce-VETTUK > Ce-VETTUK > CAU-19 (Fig. S11, ESI†). These results suggest that the mixed-phase crystal structure in Ce–TCPP plays an important role in facilitating CS. While the specific roles of each phase remain unclear, similar phase facilitated CS has been observed in other mixed-phase materials.22 Nevertheless, we can conclude that the presence of a long-lived CS state after ultrafast ET in the mixed-phase Ce–TCPP is expected to be beneficial for applications in photocatalysis.
To gain further insight on the nature of the long-lived CS in the mixed-phase Ce–TCPP, we directly probed the electron density changes at the Ce center following selective excitation of the TCPP ligand using XTA spectroscopy. Fig. 3d shows the XANES spectrum of Ce–TCPP at the Ce L3 edge and the difference spectra obtained by subtracting the GS (laser-off) spectrum from the spectrum collected at different delay times (100 ps, 1 ns, 10 ns and 100 ns) following 400 nm laser excitation. The positive feature observed at 5723.7 eV indicates that the edge energy of Ce shifts to a lower energy, suggesting the formation of a reduced Ce center in Ce–TCPP. This is further supported by the negative feature observed at 5725.5 eV: the decreased number of empty 4f orbitals prohibits the excitation of 2p core electrons, resulting in the decreased absorption intensity. The intensity of this negative feature decreases gradually from 100 ps to 10 ns until 100 ns where no transient signal was observed, suggesting that this is a long-lived transient species (>10 ns). These results together confirmed the formation of a long-lived CS state due to ligand-to-metal cluster charge transfer (LCCT) after photoexcitation of Ce–TCPP, consistent with the OTA results above. As the Ce centers in Ce–TCPP are dominated by Ce3+, which is a stable oxidation state, the nature of the long-lived CS state is likely the reduction of a small fraction of Ce4+ by the TCPP ligands, which is similar to the mechanism of forming Ce3+ centers in Ce–TCPP MOF from Ce4+ in the starting material, (NH4)2Ce(NO3)6.23
In summary, we have synthesized mixed-phase Ce–TCPP constructed from the free-base TCPP ligand and Cerium ammonium nitrate. Using OTA spectroscopy, we show that ultrafast ET occurs from the TCPP ligand to Ce center in Ce–TCPP MOFs following excitation of the TCPP ligand, forming a long-lived CS state. Systematic OTA studies on three control samples revealed that the presence of a mixed-phase structure in the Ce–TCPP MOF is responsible for the ultrafast formation of the CS state. The nature of this CS state is LCCT, as confirmed by XTA, where the reduction of Ce centers was observed due to excitation of the TCPP ligand. The observed ultrafast charge transfer process that results in the formation of a long-lived CS state (>10 ns) is expected to be beneficial for photocatalysis and thus implies the potential application of Ce–TCPP MOFs in solar energy conversion.
This work was supported by the National Science Foundation (CBET-1706971). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award No. DE-AC02-06CH11357. Sizhuo Yang acknowledges the John J. Eisch fellowship during the 2018–2019 academic year.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS; (b) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- (a) P. Moroz, L. Royo Romero and M. Zamkov, Chem. Commun., 2019, 55, 3033–3048 RSC; (b) S. Ye, C. Ding, M. Liu, A. Wang, Q. Huang and C. Li, Adv. Mater., 2019, 31, 1902069 CrossRef CAS; (c) S. Ye, C. Ding, R. Chen, F. Fan, P. Fu, H. Yin, X. Wang, Z. Wang, P. Du and C. Li, J. Am. Chem. Soc., 2018, 140, 3250–3256 CrossRef CAS; (d) P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef CAS; (e) P. Hu, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2014, 4, 2649–2652 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- (a) S. Yang, B. Pattengale, S. Lee and J. Huang, ACS Energy Lett., 2018, 3, 532–539 CrossRef CAS; (b) C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS; (c) M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. D. Vos and N. Stock, Chem. Commun., 2015, 51, 12578–12581 RSC.
- (a) A. Dhakshinamoorthy, Z. H. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC; (b) J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS.
- (a) N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS; (b) R. Li, W. Zhang and K. Zhou, Adv. Mater., 2018, 30, 31 Search PubMed; (c) A. M. Rice, G. A. Leith, O. A. Ejegbavwo, E. A. Dolgopolova and N. B. Shustova, ACS Energy Lett., 2019, 4, 1938–1946 CrossRef CAS.
- (a) L. Hanna, P. Kucheryavy, C. Liu, X. Zhang and J. V. Lockard, J. Phys. Chem. C, 2017, 121, 13570–13576 CrossRef CAS; (b) S. M. Shaikh, A. Chakraborty, J. Alatis, M. Cai, E. Danilov and A. J. Morris, Faraday Discuss., 2019, 216, 174–190 RSC.
- (a) L. Hanna, C. L. Long, X. Zhang and J. V. Lockard, Chem. Commun., 2020, 56, 11597–11600 RSC; (b) E. Caballero-Mancebo, B. Cohen, S. Smolders, D. E. De Vos and A. Douhal, Adv. Sci., 2019, 6, 1901020 CrossRef CAS; (c) J. Yu, J. Park, A. Van Wyk, G. Rumbles and P. Deria, J. Am. Chem. Soc., 2018, 140, 10488–10496 CrossRef CAS; (d) M. Alvaro, E. Carbonell, B. Ferrer, F. X. LlabrésiXamena and H. Garcia, Eur. J. Chem., 2007, 13, 5106–5112 CrossRef CAS; (e) M. de Miguel, F. Ragon, T. Devic, C. Serre, P. Horcajada and H. García, ChemPhysChem, 2012, 13, 3651–3654 CrossRef CAS.
- B. Pattengale, S. Yang, J. Ludwig, Z. Huang, X. Zhang and J. Huang, J. Am. Chem. Soc., 2016, 138, 8072–8075 CrossRef CAS.
- S. George, S. Lipstman and I. Goldberg, Cryst. Growth Des., 2006, 6, 2651–2654 CrossRef CAS.
- T. Rhauderwiek, N. Heidenreich, H. Reinsch, S. Oien-Odegaard, K. A. Lomachenko, U. Rutt, A. V. Soldatov, K. P. Lillerud and N. Stock, Cryst. Growth Des., 2017, 17, 3462–3474 CrossRef CAS.
- A. Lin, A. A. Ibrahim, P. Arab, H. M. El-Kaderi and M. S. El-Shall, ACS Appl. Mater. Interfaces, 2017, 9, 17961–17968 CrossRef CAS.
- T. Sugimoto, B. Joseph, E. Paris, A. Iadecola, T. Mizokawa, S. Demura, Y. Mizuguchi, Y. Takano and N. L. Saini, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89 Search PubMed.
- G. Jacobs, P. M. Patterson, L. Williams, E. Chenu, D. Sparks, G. Thomas and B. H. Davis, Appl. Catal., A, 2004, 262, 177–187 CrossRef CAS.
- (a) B. T. Thole, G. Vanderlaan, J. C. Fuggle, G. A. Sawatzky, R. C. Karnatak and J. M. Esteva, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 5107–5118 CrossRef CAS; (b) A. Kotani, K. O. Kvashnina, P. Glatzel, J. C. Parlebas and G. Schmerber, Phys. Rev. Lett., 2012, 108, 5 CrossRef.
- (a) L. Mathey, M. Paul, C. Coperet, H. Tsurugi and K. Mashima, Chem. – Eur. J., 2015, 21, 13454–13461 CrossRef CAS; (b) R. Das, R. Sarma and J. B. Baruah, Inorg. Chem. Commun., 2010, 13, 793–795 CrossRef CAS.
- (a) Y. Venkatesh, M. Venkatesan, B. Ramakrishna and P. R. Bangal, J. Phys. Chem. B, 2016, 120, 9410–9421 CrossRef CAS; (b) B. D. McCarthy, E. R. Hontz, S. R. Yost, T. Van Voorhis and M. Dincă, J. Phys. Chem. Lett., 2013, 4, 453–458 CrossRef CAS.
- (a) A. B. J. Parusel, T. Wondimagegn and A. Ghosh, J. Am. Chem. Soc., 2000, 122, 6371–6374 CrossRef CAS; (b) R. E. Haddad, S. Gazeau, J. Pecaut, J. C. Marchon, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2003, 125, 1253–1268 CrossRef CAS.
- X. X. Li, C. H. Gong, G. G. Gurzadyan, M. F. Gelin, J. X. Liu and L. C. Sun, J. Phys. Chem. C, 2018, 122, 50–61 CrossRef CAS.
- (a) A. Harriman, J. Chem. Soc., Faraday Trans., 1981, 77, 1281–1291 RSC; (b) P. M. Keane and J. M. Kelly, Photochem. Photobiol. Sci., 2011, 10, 1578–1586 RSC.
- M. Gutierrez, B. Cohen, F. Sanchez and A. Douhal, Phys. Chem. Chem. Phys., 2016, 18, 27761–27774 RSC.
- (a) A. Li, Z. Wang, H. Yin, S. Wang, P. Yan, B. Huang, X. Wang, R. Li, X. Zong, H. Han and C. Li, Chem. Sci., 2016, 7, 6076–6082 RSC; (b) N. A. Rodríguez, R. Parra and M. A. Grela, RSC Adv., 2015, 5, 73112–73118 RSC; (c) A. Henning, V. K. Sangwan, H. Bergeron, I. Balla, Z. Sun, M. C. Hersam and L. J. Lauhon, ACS Appl. Mater. Interfaces, 2018, 10, 16760–16767 CrossRef CAS; (d) T. Ferron, M. Waldrip, M. Pope and B. A. Collins, J. Mater. Chem. A, 2019, 7, 4536–4548 RSC.
- (a) Cerium and Cerium Compounds. In Kirk-Othmer Encyclopedia of Chemical Technology, pp. 1–23; (b) E. J. Schelter, Nat. Chem., 2013, 5, 348 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1–S8, Tables S1–S3. See DOI: 10.1039/d0cc04116e |
| This journal is © The Royal Society of Chemistry 2020 |