
A macrocycle directed total synthesis of di-O-methylendiandrin A†
Timothy H.
Barnes
,
Kara F.
Johnson
,
John D.
Gorden
 and
Bradley L.
Merner
*
and
Bradley L.
Merner
*
Department of Chemistry and Biochemistry, Auburn University, Auburn, AL 36830, USA. E-mail: blm0022@auburn.edu
First published on 4th June 2020
Abstract
The total synthesis of the lignan-based cyclobutane di-O-methylendiandrin A has been achieved using diastereoselective, vicinal alkylation and transannular McMurry reactions of a macrocyclic 1,4-diketone as key transformations for establishing relative stereochemistry and furnishing the strained 4-membered ring of the natural product.
Cyclobutane-containing natural products are an important class of compounds, not only for the challenges their strained structures present for chemical synthesis,1 but also due to the promising biological activities they possess, which include anticancer,2 antiviral,3 and antifungal properties.4 In the case of the former, the inherent ring strain (ca. 27 kcal mol−1) of the cyclobutane core requires careful consideration for when it will be assembled during a total synthesis of a complex target. Depending on substitution of the cyclobutane ring, more or less ring strain can be imposed on the 4-membered ring.5 This can lead to ring-opening and fragmentation reactions,6 which, in some cases, are irreversible and result in destruction of the cyclobutane ring system. By far, the most common method for assembling stereochemically rich cyclobutanes is the [2+2] cycloaddition reaction, and this strategy has been employed on numerous occasions.1 While quite powerful and unparalleled in the synthesis of 4-membered rings, the [2+2] cycloaddition reaction is not always well-suited for the construction of architecturally complex natural products, as olefin isomerization can take place during photochemically-driven reactions, resulting in the formation of stereoisomeric products.7 Other strategies that have been recently employed in the synthesis of this class of strained, secondary metabolites include ring-contraction, strain-relief-driven ring-opening (of cyclopropanes), reductive coupling, and transannular reactions.1a Total syntheses that exploit reactive C–H bonds of cyclobutane scaffolds have also been reported.8
Lignan-based, cyclobutane-containing natural products have been known for quite some time, the most recognizable of these being magnosalin (1)9 and the endiandrins (2, 3 and 4, Fig. 1A).10 While the syntheses of these compounds seem well-suited for [2+2] cycloaddition reactions, it is surprising to find that not all of these natural products have been synthesized, given that the corresponding olefin starting materials are known. Our interest in these molecules, and during the development of a research program focused on the utility of macrocyclic 1,4-diketones as key building blocks for complex molecule synthesis, led us to propose a stereocontrolled total synthesis of di-O-methylendiandrin A (3). Drawing inspiration from total syntheses of cyclobutane-containing natural products that have employed either reductive coupling (e.g., echinocidin D (5) and fragranol (7), Fig. 1B),11,12 or ring-contraction/transannular reactions (e.g., aquatolide (6) and hippolachinin A (8), Fig. 1B),13,14 we devised a retrosynthetic analysis that would feature a macrocyclic 1,4-diketone as a key synthetic intermediate to facilitate the stereocontrolled synthesis of di-O-methylendiandrin A (3, Scheme 1).
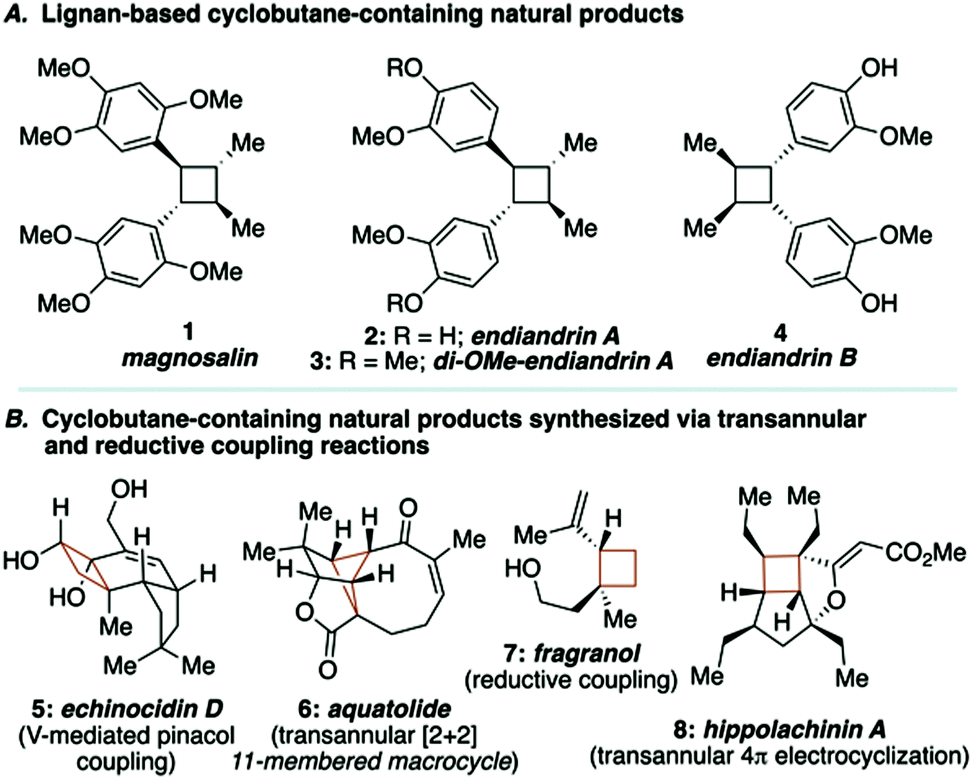 | ||
| Fig. 1 Natural products related to di-O-methylendiandrin A and cyclobutane-containing natural products assembled using transannular or reductive coupling reactions. | ||
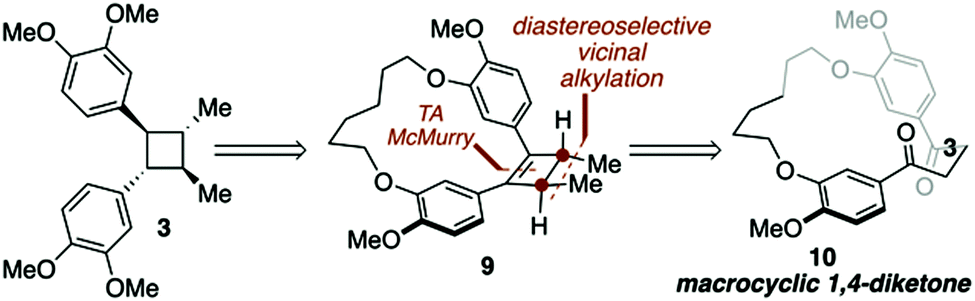 | ||
| Scheme 1 Key disconnections for a macrocycle directed total synthesis of di-O-methylendiandrin A (3). | ||
Recently, we have demonstrated that macrocyclic 1,4-diketones can be converted into highly strained arene-bridged macrocycles using a three-step reaction protocol.15 During these investigations it was discovered that macrocyclic 1,4-diketones, that are also [n.4]metacyclophanes, undergo highly diastereoselective Grignard reactions.16 The selectivity of which is largely controlled by the size of the macrocyclic system employed; however, other factors such as the nature of the organometallic reagent and solvent do contribute to the level of diastereoselectivity observed. Intrigued by these diastereoselective carbonyl addition reactions, we contemplated the possibility of using macrocyclic stereocontrol as a means to introduce vicinal C-methyl stereogenic centers about the 1,4-diketone backbone of macrocycle 10. If this could be achieved, we were optimistic that a transannular McMurry reaction could be employed to furnish a chiral, macrocyclic cyclobutene (9, Scheme 1), which could be converted into lignan-based natural products such as magnosalin (1) and the endiandrins 2 and 3.
The synthetic approach to di-O-methylendiandrin A (3) commenced with the alkylation of isovanillin (11) and 1,6-dibromohexane (12) to afford dialdehyde 13 in 89% yield. A rapid, streamlined protocol for the conversion of dialdehydes such as 13 into macrocyclic 1,4-diketones was reported in 2016.15b Application of this strategy to 13 gave diketone 10 in 17% overall yield (3 steps, Scheme 2), which is significantly lower than what we have reported for an analogous 18-membered macrocycle, derived from 3-hydroxy benzaldehyde.15b Nonetheless, 0.8 gram scale quantities of 10 could be obtained in short order from commercially available materials. Vicinal alkylation of the 1,4-diketone unit present in 10 was achieved using sodium hydride in THF at 0 °C to afford 14 as a single diastereomer in 78% yield. It is noteworthy that employing the same enolate alkylation conditions on an acyclic analogue of 10 resulted in low diastereoselection and low yield (Scheme 2, 15 to 16). Unfortunately, enolate alkylation of 15 leads to the formation of an inseparable mixture of mono-, vicinal, and trimethylated products in ca. 17% yield. The d/l-and meso-isomers have been reported by Jahn and co-workers, and based on comparison of the reported NMR data for these compounds, we believe the isomers are produced in a ca. 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.17 Drewes and co-workers have reported that alkylation of a similar, acyclic 1,4-diketone with LDA and benzyl bromide affords a 4:1 mixture (d/l:meso) of diastereomers; however, we were unable to reproduce this result.18 Oddly, 15 was prone to decomposition under the same conditions that afforded 14 in high yield and diastereoselectivity.
:1 ratio.17 Drewes and co-workers have reported that alkylation of a similar, acyclic 1,4-diketone with LDA and benzyl bromide affords a 4:1 mixture (d/l:meso) of diastereomers; however, we were unable to reproduce this result.18 Oddly, 15 was prone to decomposition under the same conditions that afforded 14 in high yield and diastereoselectivity.
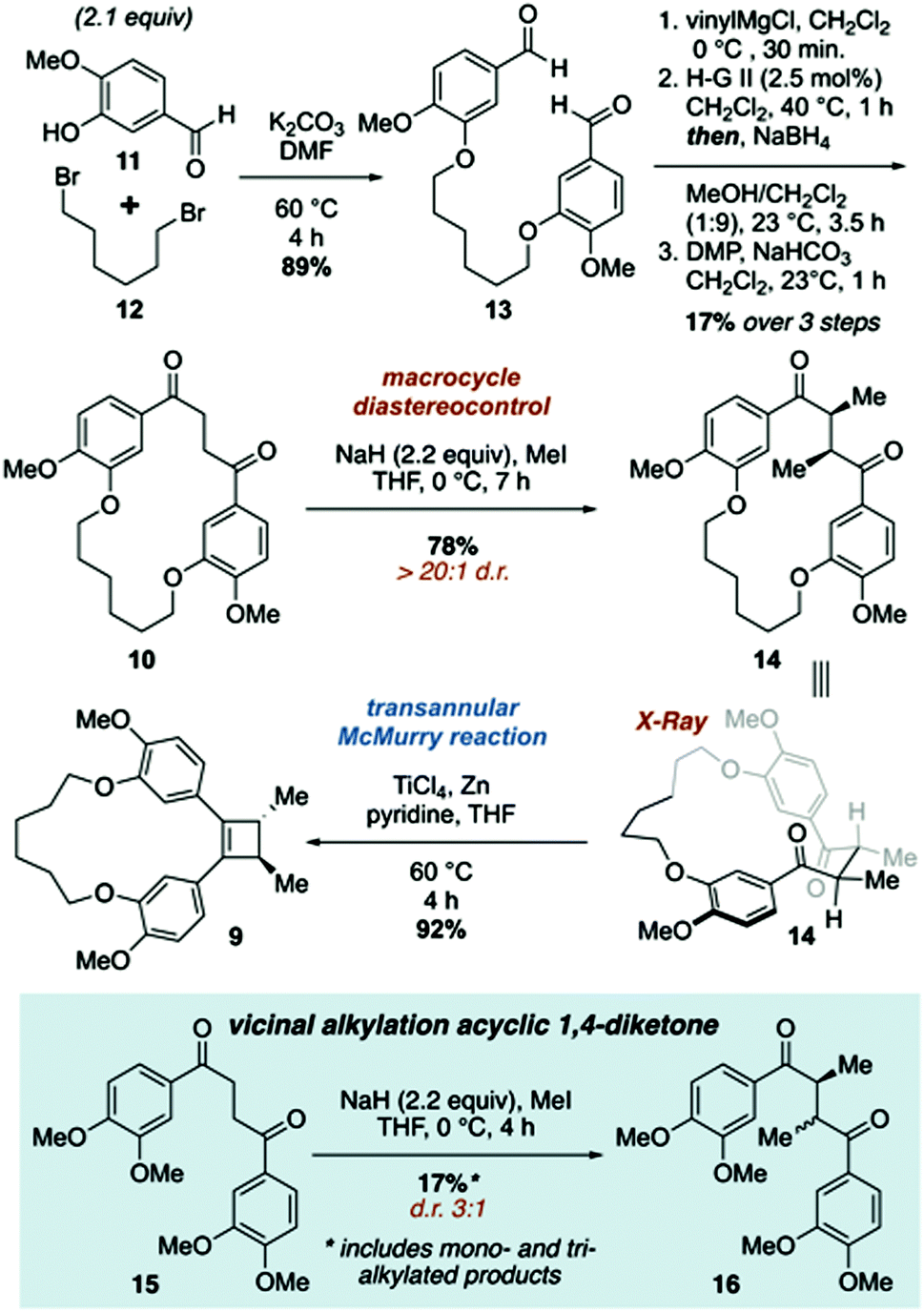 | ||
| Scheme 2 Synthesis of macrocyclic cyclobutene 9 featuring diastereoselective vicinal alkylation and transannular McMurry reactions. | ||
The relative stereochemistry of 14 was difficult to determine from 1H NMR analysis, as the alpha methine proton and C-methyl signals were poorly resolved. Fortunately, crystals suitable for X-ray crystallographic analysis were obtained upon recrystallization of 14 from ethyl acetate and hexanes to unambiguously assign the relative stereochemistry of the C-methyl groups as anti (Fig. 2). The solid-state structure of 14 also reveals the close proximity of the two carbonyls present in the macrocycle. While a direct comparison of the solid state structure to the solution phase structure of 14 is not (entirely) valid, the former does illustrate that there is considerable pre-organization of the ketone units in 14, which would facilitate a transannular McMurry reaction. Indeed, treatment of macrocyclic 1,4-diketone 14 with titanium(IV) chloride, Zn dust and pyridine affords macrocyclic cyclobutene 9 in 92% yield (Scheme 2).
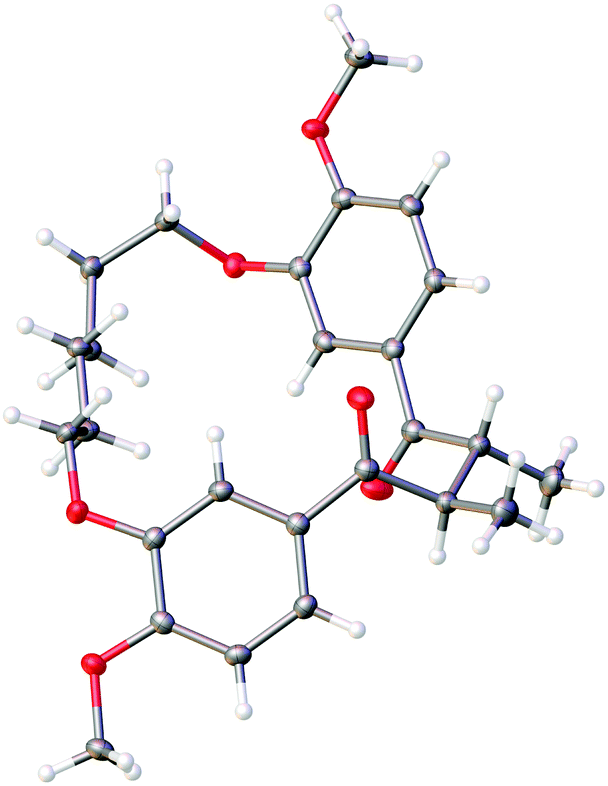 | ||
| Fig. 2 X-ray crystal structure of diketone 14 obtained by recrystallization from ethyl acetate/hexanes. | ||
With cyclobutene 9 in hand, and the relative configuration of the vicinal C-methyl groups established, completing the total synthesis of di-O-methylendiandrin A (3) began with catalytic hydrogenation of the olefin present in 9 to afford a macrocyclic cyclobutane with an anti/anti/syn/syn-relative stereochemical relationship. Having served its purpose for directing the stereochemical outcome of the enolate alkylation reaction and pre-organizing the 1,4-diketone unit for a reductive coupling reaction, the alkyl tether in 9 was cleaved in the presence of BBr3, and the free hydroxyls were subsequently alkylated with methyl iodide to furnish cyclobutane 17 in 60% overall yield (Scheme 3). In order to secure the correct all anti-relative stereochemical relationship of di-O-methylendiandrin A, 17 was subjected to a potassium tert-butoxide-mediated epimerization reaction in DMSO to afford 3 in 95% yield. All spectroscopic data obtained for 3 were identical to those reported by Quinn and co-workers in 2007.10a
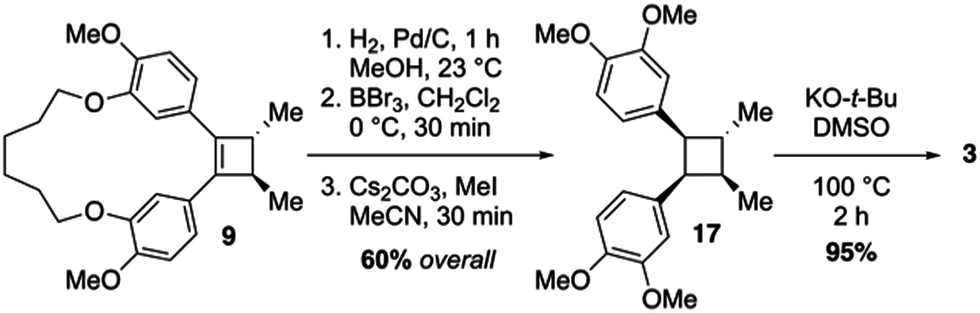 | ||
| Scheme 3 Synthesis of di-O-methylendiandrin A (3) from cyclobutene 9. | ||
In conclusion, a macrocyclic template bearing a 1,4-diketone unit was used to direct a stereocontrolled enolate alkylation reaction, as well as to pre-organize the carbonyl functional groups in a transannular, cyclobutene forming reaction. These strategies have enabled the total synthesis of the lignan-based natural product di-O-methylendiandrin A (3). The utility of these macrocycle-enabled reactions in the synthesis of structurally related natural products are currently being investigated in our laboratories. The results of these studies will be reported in due course.
The authors would like to thank the National Science Foundation (CHE-1654691) and Auburn University for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For recent reviews on the total synthesis of cyclobutane-containing natural products see: (a) J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136 RSC; (b) E. N. Hancock, J. M. Wiest and M. K. Brown, Nat. Prod. Rep., 2019, 36, 1383 RSC; (c) M. Wang and P. Lu, Org. Chem. Front., 2018, 5, 254 RSC.
- A. Cipres, D. P. O’Malley, K. Li, D. Finlay, P. S. Baran and K. Vuori, ACS Chem. Biol., 2010, 5, 195 CrossRef CAS PubMed.
- A. K. Field, A. V. Tuomari, B. McGeever-Rubin, B. J. Terry, K. E. Mazina, M. L. Haffey, M. E. Hagen, J. M. Clark, A. Braitman, W. A. Slusarchyk, M. G. Young and R. Zahler, Antiviral Res., 1990, 13, 41 CrossRef CAS PubMed.
- S.-J. Piao, Y.-L. Song, W.-H. Jiao, F. Yang, X.-F. Liu, W.-S. Chen, B.-N. Han and H.-W. Lin, Org. Lett., 2013, 15, 3526 CrossRef CAS PubMed.
- T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed.
- (a) D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272 CrossRef CAS; (b) E. Leemans, M. D’hooghe and N. De Kimpe, Chem. Rev., 2011, 111, 3268 CrossRef CAS PubMed.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748 CrossRef CAS PubMed.
- For examples see: (a) J. C. Beck, C. R. Lacker, L. M. Chapman and S. E. Reisman, Chem. Sci., 2019, 10, 2315 RSC; (b) W. R. Gutekunst and P. S. Baran, J. Org. Chem., 2014, 79, 2430 CrossRef CAS PubMed.
- Magnosalin isolation and characterization: (a) T. Kikuchi, S. Kadota, K. Yanada, K. Tanaka, K. Watanabe, M. Yoshizaki, T. Yokoi and T. Shingu, Chem. Pharm. Bull., 1983, 31, 1112 CrossRef CAS; (b) S. Malhotra, S. K. Koul, S. C. Taneja, P. Pushpangadan and K. L. Dhar, Phytochemistry, 1990, 29, 2733 CrossRef CAS; (c) R. N. Mahindru, S. C. Taneja, K. L. Dhar and R. T. Brown, Phytochemistry, 1993, 32, 1073 CrossRef CAS; (d) J.-H. Ryu, H. J. Son, S. H. Lee and D. H. Sohn, Bioorg. Med. Chem. Lett., 2002, 12, 649 CrossRef CAS PubMed . For the synthesis of magnosalin and endiandrin A, see: ; (e) M. Riener and D. A. Nicewicz, Chem. Sci., 2013, 4, 2625 RSC . For the synthesis of endiandrin A and di-O-methylendiandrin A, see: ; (f) R. Li, B. Chiyin Ma, W. Huang, L. Wang, D. Wang, H. Lu, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 3097 CrossRef CAS.
- For the isolation and characterization of endiandrin A, see: (a) R. A. Davis, A. R. Carroll, S. Duffy, V. M. Avery, G. P. Guymer, P. I. Forster and R. J. Quinn, J. Nat. Prod., 2007, 70, 1118 CrossRef CAS PubMed . For the isolation and characterization of endiandrin B, see: ; (b) R. A. Davis, E. C. Barnes, J. Longden, V. M. Avery and P. C. Healy, Bioorg. Med. Chem., 2009, 17, 1387 CrossRef CAS PubMed.
- A. Gansäuer, A. Greb, I. Huth, D. Worgull and K. Knebel, Tetrahedron, 2009, 65, 10791 CrossRef.
- M. T. Hovey, D. T. Cohen, D. M. Walden, P. H.-Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2017, 56, 9864 CrossRef CAS PubMed.
- K. Takao, H. Kai, A. Yamada, Y. Fukushima, D. Komatsu, A. Ogura and K. Yoshida, Angew. Chem., Int. Ed., 2019, 58, 9851 CrossRef CAS PubMed.
- N. Winter and D. Trauner, J. Am. Chem. Soc., 2017, 139, 11706 CrossRef CAS PubMed.
- (a) N. K. Mitra, R. Meudom, J. D. Gorden and B. L. Merner, Org. Lett., 2015, 17, 2700 CrossRef CAS PubMed; (b) N. K. Mitra, R. Meudom, H. H. Corzo, J. D. Gorden and B. L. Merner, J. Am. Chem. Soc., 2016, 138, 3235 CrossRef CAS PubMed.
- (a) N. K. Mitra, H. H. Corzo and B. L. Merner, Org. Lett., 2016, 18, 3278 CrossRef CAS PubMed; (b) N. K. Mitra, C. P. Merryman and B. L. Merner, Synlett, 2017, 28, 2205 CrossRef CAS.
- P. R. Jagtap, I. Císařová and U. Jahn, Org. Biomol. Chem., 2018, 16, 750 RSC.
- S. E. Drewes, C. J. Hogan, P. T. Kaye and G. H. P. Roos, J. Chem. Soc., Perkin Trans. 1, 1989, 1585 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1938763. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03302b |
| This journal is © The Royal Society of Chemistry 2020 |