
Enhancing the activity of photocatalytic hydrogen evolution from CdSe quantum dots with a polyoxovanadate cluster†
Emily H.
Edwards‡
a,
Alex A.
Fertig‡
a,
Kevin P.
McClelland
a,
Mahilet T.
Meidenbauer
a,
Saikat
Chakraborty
a,
Todd D.
Krauss
 *ab,
Kara L.
Bren
*a and
Ellen M.
Matson
*a
*ab,
Kara L.
Bren
*a and
Ellen M.
Matson
*a
aDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA. E-mail: krauss@chem.rochester.edu; bren@chem.rochester.edu; matson@chem.rochester.edu
bThe Institute of Optics, University of Rochester, Rochester, New York 14627, USA
First published on 29th June 2020
Abstract
We report the improvement of photocatalytic proton reduction using molecular polyoxovanadate-alkoxide clusters as hole scavengers for CdSe quantum dots. The increased hydrogen production is explained by favorable charge interactions between reduced forms of the cluster and the charge on the quantum dots arising from the capping ligands.
Semiconducting nanocrystals, or quantum dots (QDs), have emerged as leading photocatalysts for visible light-driven charge-transfer reactions.1–4 Inspired by the creation of a solar fuel in natural photosynthesis, one of the most widely studied photocatalytic reactions using QDs is the reduction of protons to form hydrogen (H2).5–7 When paired with sacrificial electron donors, cadmium selenide (CdSe) QDs are capable of photocatalytic proton reduction from the nanoparticle surface (i.e. without a co-catalyst).8 The yield of H2 production can be significantly increased in the presence of a transition metal co-catalyst which facilitates charge separation9 and decreases the barrier for proton reduction.2,10–12 For example, in combination with a homogeneous nickel catalyst (“Ni-DHLA”; DHLA = dihydrolipoic acid), CdSe QDs produce H2 with turnover numbers (TONs) up to 600
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 over two weeks in aqueous solution.13
000 over two weeks in aqueous solution.13
Traditionally, research into the optimization of photocatalytic proton reduction with QDs has focused on improvements to the reduction co-catalyst. By comparison, less work has been dedicated to studying the subsequent charge balancing reactions that must occur through reduction of the QD. For cadmium chalcogenides, the large effective mass of the hole relative to that of the electron makes hole transfer a likely rate-limiting step,3,14,15 limiting catalytic efficiency (Fig. 1). Some methods to improve hole-transfer rates have used modified ligands to delocalize the hole wave function.2,3,12,16 However, as a result of the chemical instability of these ligands,17 few studies have reported on their application in enhancing photocatalysis.18–20 Shelling the QD to form a type-II heterostructure is another viable method of separating the electron and hole, but this approach decreases catalytic efficiency by localizing one carrier to the core of the nanocrystal.21 Thus, a robust and efficient method of hole extraction from QDs remains necessary for improving photocatalytic systems.
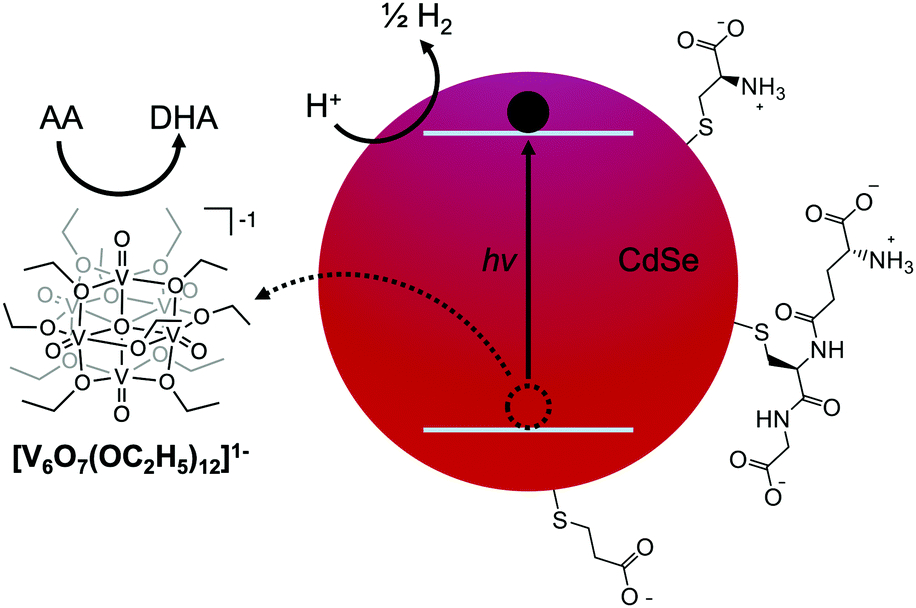 | ||
| Fig. 1 Illustration of the general photocatalytic scheme. Upon excitation by visible light, the electron reduces protons at the surface of the QD, while the hole is extracted by a POV-alkoxide cluster. Ascorbic acid (AA) is used to regenerate the reduced form of the cluster in solution. Capping ligands were chosen to promote solubility in aqueous solutions, and include cysteine (top), glutathione (middle), and 3-mercaptopropionic acid (bottom). DHA indicates dehydroascorbic acid. | ||
A possible solution to enable efficient hole extraction is the coupling of QDs to either bulk or molecular metal oxides. Watson and coworkers have demonstrated that the mid-bandgap states in doped V2O5 nanowires facilitate fast (<500 fs) hole transfer from CdSe QDs.22 The authors propose that the resulting charge-separated state could be used to delay charge recombination and improve photocatalytic proton reduction.22,23 In order to study homogeneous photocatalytic systems, we hypothesized that polyoxovanadate (POV) clusters could be used as an alternative to V2O5 nanowires for efficient hole extraction. While most POV clusters are isolated in their most-oxidized state (i.e. electron-deficient), the polyoxovanadate-alkoxide (POV-alkoxide) clusters explored in this study (Fig. 1) are reduced analogues of these metal oxide assemblies, rendering them well-suited to serve as hole-transfer reagents in photocatalytic schematics (Fig. S1, ESI†).24 Indeed, POV-alkoxide clusters have been shown to reductively quench molecular chromophores for reactions such as water oxidation.25 However, they have not been studied in combination with QD photosensitizers.
To test our hypothesis that charge transfer between photoexcited CdSe QDs and the POV-alkoxide cluster, [V6O7(OC2H5)12]1−, is possible, steady-state photoluminescence quenching experiments were conducted. Given previous activity of mid-sized CdSe QDs as photocatalysts, QDs with a first excitonic absorbance peak at 525 nm (±5 nm) (correlating with a diameter of approximately 2.6 nm26) capped with trioctylphosphine (TOP) ligands were synthesized (Fig. S2, ESI†).27 Their luminescence was monitored in the presence of increasing equivalents of [V6O7(OC2H5)12]1− (Fig. 2 inset). The photoluminescence of the QDs was efficiently quenched in the presence of the POV-alkoxide clusters, with a 96% decrease in emission intensity at 10 equivalents. The nonlinear behavior of the Stern–Volmer analysis suggests that the clusters quench the QD fluorescence through both static and dynamic mechanisms (Fig. S3, ESI†).28 It is worth noting that there is a small overlap between the absorbance spectrum of [V6O7(OC2H5)12]1− and the emission of CdSe QDs, so we cannot definitively rule out the possibility of an energy transfer quenching mechanism (Fig. 2). However, given the low absorbance of the clusters at these concentrations, contributions from such a pathway are predicted to be negligible in comparison to those of charge transfer (Fig. S4; see ESI† for details).
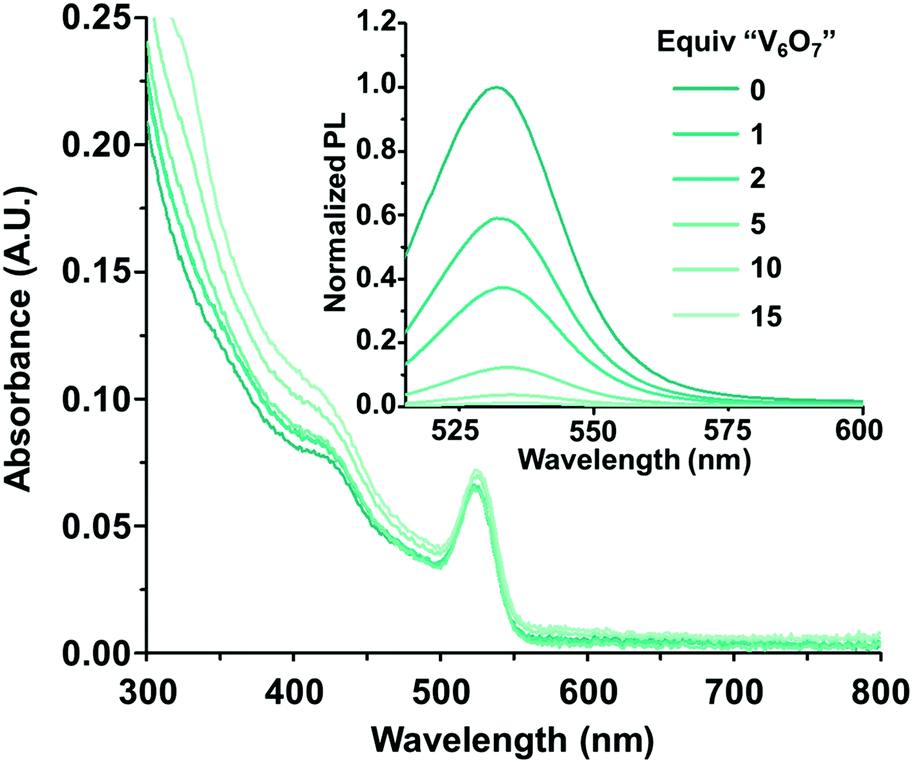 | ||
| Fig. 2 The absorbance and photoluminescence (inset) spectra of CdSe–TOP with varying equivalents of [V6O7(OC2H5)12]1− (“V6O7”). All samples contained 1 μM CdSe with 0–15 equivalents of [V6O7(OC2H5)12]1− in dichloromethane and the photoluminescence was normalized to the absorbance at the excitation wavelength (500 nm). | ||
Given the promising results from photoluminescence quenching (vide supra), we sought to probe the proton reduction characteristics upon the addition of [V6O7(OC2H5)12]1− to a solution of glutathione-capped (CdSe–GSH) QDs and sacrificial electron donor ascorbic acid (Fig. 3). For this study, we opted to not include an external co-catalyst in an effort to simplify the already-complicated system, ensuring that any changes in catalysis were due to cluster/QD interactions. To establish a baseline for photoactivity of QDs under our conditions, a mixture of ascorbic acid and CdSe–GSH QDs, dissolved in ethanol and water (1:1), was irradiated with green (530 nm ± 10 nm) light (Fig. 3 and Fig. S5, ESI†). Over 48 hours, the multicomponent system produces 110 μmol (±42 μmol) of H2. In the absence of ascorbic acid, negligible hydrogen evolution is observed (Fig. S6, ESI†). The rate of H2 evolution remains constant over this time frame, indicating minimal degradation of the QDs, with an average rate of H2 evolution of 2.22 μmol h−1 (±0.78 μmol h−1). This correlates to a quantum yield (QY) of 8.0% (±2.4%) (see ESI† for details).
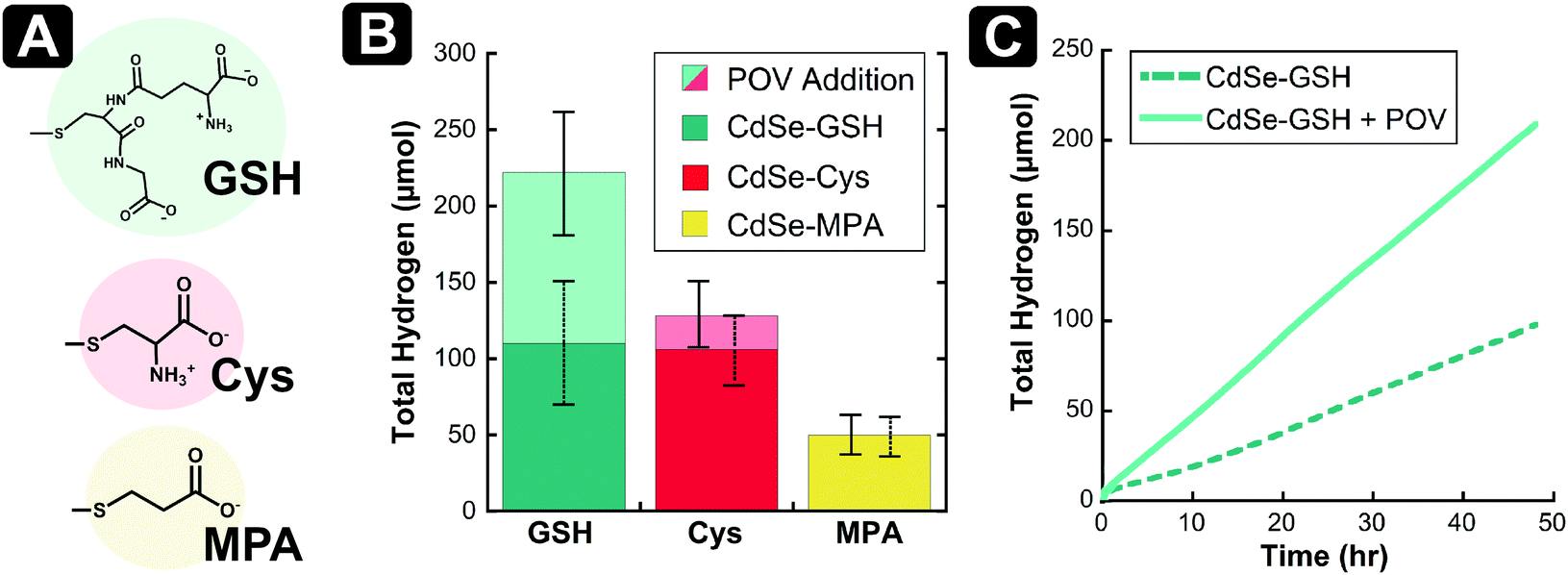 | ||
| Fig. 3 (A) Structures of the capping ligands used on the surface of CdSe QDs; (B) total H2 evolution after 48 hours from CdSe–GSH, CdSe–Cys, and CdSe–MPA in the presence of 0.5 M ascorbic acid, in a 1:1 EtOH:H2O mixture, being irradiated by 530 nm light at 15 °C. Upon addition of 100 μM [V6O7(OC2H5)12]1−, the positive change in total H2 production for CdSe–GSH and CdSe–Cys is shown (n = 3); (C) representative trials of H2 evolution over time for the CdSe–GSH H2 evolution system described in (B), showing improvement in the presence of [V6O7(OC2H5)12]1−. | ||
Upon addition of [V6O7(OC2H5)12]1− (100 μM) to the photocatalytic system described above, both the average H2 evolved in 48 hours and the rate of catalysis are approximately doubled (Fig. 3 and Fig. S5, ESI†). The total H2 produced improves to 222 μmol (±37 μmol) with an improvement in rate to 4.61 μmol h−1 (±0.85 μmol h−1), and an increase of 125% in average QY to 18% (±4.5%). To evaluate whether [V6O7(OC2H5)12]1− might act as a H2-production catalyst, which would complicate its intended use as an electron mediator in this system, independent electrochemical analysis of the POV-alkoxide cluster was performed. Upon titration of 40 mM aqueous phosphate buffer (pH = 6) to a solution of 1 mM [V6O7(OC2H5)12]1− and 0.1 M [nBu4N]PF6 in acetonitrile, no change in the electrochemical response of the cluster was observed, indicating that this reduced POV-alkoxide cluster is not active as a proton reduction catalyst (Fig. S7, ESI†). Taken together, these results support our hypothesis that POV-alkoxide clusters efficiently extract the hole from CdSe QDs, acting as a hole shuttle and resulting in improved production of H2.
Activity enhancement from the introduction of the POV-alkoxide cluster to the GSH-capped CdSe QDs prompted further exploration of the photocatalytic system. Given previously reported ligand-dependence on the photocatalytic activity of QDs,12,29–31 additional thiolate ligands were selected to determine if the mechanism of enhancement by [V6O7(OC2H5)12]1− could be generalized to other QD-ligand systems. Cysteine (Cys) and 3-mercaptopropionic acid (MPA) were chosen as additional water-soluble ligands to cap QDs (Fig. 3A and Fig. S8, ESI†). The impact of the capping ligands on the first excitonic absorbance is small (<10 nm).
It has been previously reported that the degree of passivation of surface Cd2+ ions dramatically influences the yield of proton reduction for QDs in the absence of a co-catalyst.8,32,33 Given the differences in the molecular structure of the three thiolate ligands investigated in this work, we anticipated that surface coverage would vary between GSH-, Cys- and MPA-capped QDs. Thus, to establish new baselines for H2 production for each system, we evaluated the photocatalysis for the CdSe QDs in the absence of POV-alkoxide clusters. The CdSe–Cys and CdSe–MPA systems have slow H2 evolution in the first ten hours of catalysis (Fig. S9 and S10, ESI†). After this induction period, H2 generation is linear over the remainder of the experiment. This is in contrast to the GSH-capped QDs, which produce H2 at a constant rate throughout the 48 hours of irradiation (Fig. 3C). In total, the Cys-capped CdSe QDs produce a similar amount of H2 to those capped with glutathione ligands (106 μmol ± 22 μmol), while the CdSe–MPA analogues produce significantly less H2 in the same time period (50 μmol ± 12 μmol) (Fig. S11, ESI†). Since proton reduction has been reported to occur at solvated Cd surface-sites,8 these differences in H2 production without POV-alkoxide clusters suggest that MPA-capped QDs have a higher packing density than Cys- or GSH-capped QDs. This may be due to differences in the available binding modes of the ligands, or the ligand exchange procedures used.
Upon introduction of [V6O7(OC2H5)12]1− to the photocatalytic experiments with Cys- and MPA-capped QDs, interesting trends were observed. As with GSH, Cys-capped CdSe QDs showed a boost in activity with the addition of POV-alkoxide clusters (Fig. 3 and Fig. S9, ESI†). In the presence of clusters, the average H2 evolved increases to 128 μmol (±20 μmol) from 106 μmol (±22 μmol). In contrast, negligible change in the total production of H2 is observed upon addition of POV-alkoxide cluster to the MPA-capped CdSe QDs (Fig. 3 and Fig. S10, ESI†; 50 μmol ± 12 μmol vs. 48 μmol ± 10 μmol with [V6O7(OC2H5)12]1−).
We justify the observed differences in H2 production as resulting from differences in the interactions between the QDs and the POV-alkoxide clusters. The ligands chosen to probe the photocatalytic reactivity of these QDs have a different charge at pH = 4.5, thus providing a different surface chemistry for the photosensitizer. Glutathione is a zwitterionic tripeptide containing cysteine, glutamate, and glycine (pKas: 2.1 (COOH), 3.5 (COOH), 8.8 (NH3+)). It is anticipated that this ligand binds to the surface of the QD through the thiol moiety, allowing for the remaining charged regions of the ligand to interact with the environment.34 At pH = 4.5, the amine group located on the GSH ligand will remain protonated (i.e. positively-charged) and may interact with the anionic charge-states of the POV-alkoxide cluster.35 As charge transfer from the POV-alkoxide to the QD occurs, the anionic cluster cycles through higher oxidation states (e.g. [V6O7(OC2H5)12]n; n = 0, 1+, 2+), at which point the charge attraction with the amine group becomes less favorable. The diminished coulombic interaction between the anionic POV-alkoxide cluster and the positively charged residues at the QD surface drives more oxidized forms of the cluster away from the QD surface, preventing charge recombination via back electron transfer. The dissociated, oxidized form of the POV-alkoxide cluster subsequently reacts with ascorbic acid, resulting in re-reduction of the vanadium oxide assembly.
Supporting this mechanistic hypothesis, photocatalytic experiments with CdSe–Cys dots also showed an increase in the amount of H2 produced in the presence of [V6O7(OC2H5)12]1−. Cysteine, an amino acid (pKas: 1.8 (COOH), 10.7 (NH3+)), has also been used as a zwitterionic capping ligand for QDs.12,36 The cysteine ligands offer a similar positively charged site for electrostatic interactions to occur, attracting the reduced cluster to the surface of the QD. We believe, however, that the smaller size of cysteine relative to glutathione leads to a more densely packed surface which inhibits the adsorption of [V6O7(OC2H5)12]1−, reducing the impact the clusters have on catalysis.
In contrast, CdSe–MPA showed no enhancement of H2 production in the presence of [V6O7(OC2H5)12]1−. The absence of a positively charged residue in the MPA capping ligands would disfavor interaction between the QD and the reduced, anionic forms of the POV-alkoxide cluster. Instead, the more oxidized (cationic) states of the POV-alkoxide cluster (e.g. [V6O7(OC2H5)12]n; n = 1+, 2+) would be attracted to the QD surface. In these oxidation states, the cluster would energetically prefer to accept reducing equivalents from the QD, no longer functioning as a hole scavenging reagent. Also, as noted above, MPA-capped CdSe QDs appear to have fewer uncoordinated sites, which could further inhibit interaction with the POV-alkoxide clusters and hole transfer from the QD.
Here, we have used POV-alkoxide clusters to efficiently extract photogenerated holes from CdSe QDs in order to significantly increase the volume of H2 produced without the addition of a proton reduction co-catalyst. CdSe QDs capped with GSH show the largest increase in both the rate of reaction and the total amount of H2 produced with the addition of POV-alkoxide clusters. Cys-capped QDs show a moderate increase in the amount of H2 produced when clusters are present, while QDs capped with MPA show no significant increase in the amount of H2 produced following addition of the vanadium oxide cluster. These results provide insight into the electrostatic interaction required for charge transfer between the photosensitizer and the cluster, which will aid in designing more efficient systems for the photocatalytic production of H2.
This work is supported by the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy, Grant No. DE-FG02-09ER16121. E. H. E. Acknowledges support from NIH training grant T32-GM118283.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Huang, X.-B. Li, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2018, 24, 11530–11534 CrossRef CAS PubMed.
- M. S. Kodaimati, K. P. McClelland, C. He, S. Lian, Y. Jiang, Z. Zhang and E. A. Weiss, Inorg. Chem., 2018, 57, 3659–3670 CrossRef CAS PubMed.
- R. D. Harris, S. Bettis Homan, M. Kodaimati, C. He, A. B. Nepomnyashchii, N. K. Swenson, S. Lian, R. Calzada and E. A. Weiss, Chem. Rev., 2016, 116, 12865–12919 CrossRef CAS PubMed.
- D. J. Norris and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 16338–16346 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Nat. Rev. Chem., 2018, 2, 160–173 CrossRef CAS.
- J. Zhao, M. A. Holmes and F. E. Osterloh, ACS Nano, 2013, 7, 4316–4325 CrossRef CAS PubMed.
- V. V. Matylitsky, L. Dworak, V. V. Breus, T. Basché and J. Wachtveitl, J. Am. Chem. Soc., 2009, 131, 2424–2425 CrossRef CAS PubMed.
- F. F. Schweinberger, M. J. Berr, M. Döblinger, C. Wolff, K. E. Sanwald, A. S. Crampton, C. J. Ridge, F. Jäckel, J. Feldmann, M. Tschurl and U. Heiz, J. Am. Chem. Soc., 2013, 135, 13262–13265 CrossRef CAS PubMed.
- M. J. Berr, F. F. Schweinberger, M. Döblinger, K. E. Sanwald, C. Wolff, J. Breimeier, A. S. Crampton, C. J. Ridge, M. Tschurl, U. Heiz, F. Jäckel and J. Feldmann, Nano Lett., 2012, 12, 5903–5906 CrossRef CAS PubMed.
- E. A. Weiss, ACS Energy Lett., 2017, 2, 1005–1013 CrossRef CAS.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS PubMed.
- K. E. Knowles, M. D. Peterson, M. R. McPhail and E. A. Weiss, J. Phys. Chem. C, 2013, 117, 10229–10243 CrossRef CAS.
- P. V. Kamat, J. A. Christians and J. G. Radich, Langmuir, 2014, 30, 5716–5725 CrossRef CAS PubMed.
- S. Lian, D. J. Weinberg, R. D. Harris, M. S. Kodaimati and E. A. Weiss, ACS Nano, 2016, 10, 6372–6382 CrossRef CAS PubMed.
- R. D. Harris, V. A. Amin, B. Lau and E. A. Weiss, ACS Nano, 2016, 10, 1395–1403 CrossRef CAS PubMed.
- J. R. Lee, W. Li, A. J. Cowan and F. Jäckel, J. Phys. Chem. C, 2017, 121, 15160–15168 CrossRef CAS.
- O. M. Pearce, J. S. Duncan, N. H. Damrauer and G. Dukovic, J. Phys. Chem. C, 2018, 122, 17559–17565 CrossRef CAS.
- C. M. Wolff, P. D. Frischmann, M. Schulze, B. J. Bohn, R. Wein, P. Livadas, M. T. Carlson, F. Jäckel, J. Feldmann, F. Würthner and J. K. Stolarczyk, Nat. Energy, 2018, 3, 862–869 CrossRef CAS.
- T. X. Ding, J. H. Olshansky, S. R. Leone and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 2021–2029 CrossRef CAS PubMed.
- J. Cho, A. Sheng, N. Suwandaratne, L. Wangoh, J. L. Andrews, P. Zhang, L. F. J. Piper, D. F. Watson and S. Banerjee, Acc. Chem. Res., 2019, 52, 645–655 CrossRef CAS PubMed.
- J. L. Andrews, J. Cho, L. Wangoh, N. Suwandaratne, A. Sheng, S. Chauhan, K. Nieto, A. Mohr, K. J. Kadassery, M. R. Popeil, P. K. Thakur, M. Sfeir, D. C. Lacy, T.-L. Lee, P. Zhang, D. F. Watson, L. F. J. Piper and S. Banerjee, J. Am. Chem. Soc., 2018, 140, 17163–17174 CrossRef CAS PubMed.
- L. E. VanGelder, A. M. Kosswattaarachchi, P. L. Forrestel, T. R. Cook and E. M. Matson, Chem. Sci., 2018, 9, 1692–1699 RSC.
- M.-P. Santoni, G. La Ganga, V. Mollica Nardo, M. Natali, F. Puntoriero, F. Scandola and S. Campagna, J. Am. Chem. Soc., 2014, 136, 8189–8192 CrossRef CAS PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- F. Qiu, Z. Han, J. J. Peterson, M. Y. Odoi, K. L. Sowers and T. D. Krauss, Nano Lett., 2016, 16, 5347–5352 CrossRef CAS PubMed.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer, US, Boston, MA, 2006, pp. 277–330 DOI:10.1007/978-0-387-46312-4_8.
- A. Das, Z. Han, M. G. Haghighi and R. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16716–16723 CrossRef CAS PubMed.
- C. M. Chang, K. L. Orchard, B. C. M. Martindale and E. Reisner, J. Mater. Chem. A, 2016, 4, 2856–2862 RSC.
- M. Green, J. Mater. Chem., 2010, 20, 5797–5809 RSC.
- M. J. Greaney, E. Couderc, J. Zhao, B. A. Nail, M. Mecklenburg, W. Thornbury, F. E. Osterloh, S. E. Bradforth and R. L. Brutchey, Chem. Mater., 2015, 27, 744–756 CrossRef CAS.
- W. D. Kim, J.-H. Kim, S. Lee, S. Lee, J. Y. Woo, K. Lee, W.-S. Chae, S. Jeong, W. K. Bae, J. A. McGuire, J. H. Moon, M. S. Jeong and D. C. Lee, Chem. Mater., 2016, 28, 962–968 CrossRef CAS.
- P. K. Sudeep, S. T. S. Joseph and K. G. Thomas, J. Am. Chem. Soc., 2005, 127, 6516–6517 CrossRef CAS PubMed.
- Z. Aliakbar Tehrani, Z. Jamshidi, M. Jebeli Javan and A. Fattahi, J. Phys. Chem. A, 2012, 116, 4338–4347 CrossRef CAS PubMed.
- F. Shi, S. Liu and X. Su, New J. Chem., 2017, 41, 4138–4144 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, electronic absorption spectra, luminescence quenching analyses, cyclic voltammetry, hydrogen evolution curves, quantum yield calculations and tabulated results collected herein. See DOI: 10.1039/d0cc03163a |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |