
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe regulation of nanomaterials and nanomedicines for clinical application: current and future perspectives
Rachel
Foulkes
a,
Ernest
Man
b,
Jasmine
Thind
a,
Suet
Yeung
a,
Abigail
Joy
a and
Clare
Hoskins
 *ab
*ab
aSchool of Pharmacy and Bioengineering, Keele University, Keele, ST5 5BG, UK. E-mail: clare.hoskins@strath.ac.uk; Tel: +44 (0)0141 5482796
bDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1RD, UK
First published on 9th July 2020
Abstract
The use of nanomaterials in biomedicine has increased over the past 10 years, with many different nanoparticle systems being utilised within the clinical setting. With limited emerging success in clinical trials, polymeric, metallic, and lipid based nanoparticles have all found a place in medicine, with these generally providing enhanced drug efficacy or therapeutic effect compared to the standard drug treatments. Although there is great anticipation surrounding the field of nanomedicine and its influence on the pharmaceutical industry, there is currently very little regulatory guidance in this area, despite repeated calls from the research community, something that is critical to provide legal certainty to manufacturers, policymakers, healthcare providers and the general public. This is reflected in the lack of an international definition of what these materials are, with several bodies, including the National Institute of Health, USA, the European Science Foundation and the European Technology Platform, having differing definitions, and the FDA having no clear definition at all. The uncertainty created by the lack of consistency across the board may ultimately impact funding, research and development of such products negatively thus destroying public acceptance and perception of nano-products. This review aims to discuss the use of nanomaterials within the clinical setting, why regulation of these materials is so important, and the challenges faced in regulating these materials generally, as well as the current regulation used in different nations.
 Clare Hoskins | Dr Clare Hoskins (FRSC, CSci) is a Reader in the School of Pure and Applied Chemistry, University of Strathclyde. She leads a vibrant interdisciplinary research group focussing on the development of a range of multifunctional nanoparticles and their translation into medical therapies and agricultural products. She has published >40 peer reviewed articles, 1 patent and her research has been supported with over £2 M funding. Clare is the Elected Secretary of the RSC Chemical Nanosciences and Nanotechnology Network, and a Committee Member of the UK and Ireland Controlled Release Society. In 2019 Clare was awarded the Academy of Pharmaceutical Sciences ‘Emerging Scientist’ sponsored by Pfizer and also the North Staffordshire Medical Institute Researcher Award. |
Introduction
The broad definition of nanotechnology is based upon the use and application of materials within the nanometre range. This incredibly minute range provides many benefits across a wide range of applications1 including for electronics,2 sunscreens,3 cosmetics,4 energy storage5 and drug delivery.6 The size of these particles often confers unique and desirable properties when in their nanoscale form, including chemical, physical and biological properties, that may be beneficial over their larger equivalents.7 Nanoscale medicines can be highly beneficial considering that many biological significant molecules such as water, antibodies, proteins, glucose, enzymes, haemoglobin and receptors all fit within this range (Fig. 1).8 The application of nanotechnology within the field of medicine was expected to have a revolutionary impact on healthcare. Despite this, the expectation has not matched the initial hype, though most working in the field contribute this to the fact that nanomedicine is still in its infancy and lack of clarity over regulation for clinical use is greatly hindering their translation.9,10 Although we have very little knowledge or data regarding the pharmacokinetics, pharmacodynamics and toxicity of many nanomaterials in humans, there are several conceivable benefits of such technologies. There remains great anticipation surrounding the field of nanomedicine and its influence on the pharmaceutical industry, however regulatory guidance in this area is urgently required, which is critical to provide legal certainty to manufacturers, policymakers, healthcare providers and the general public.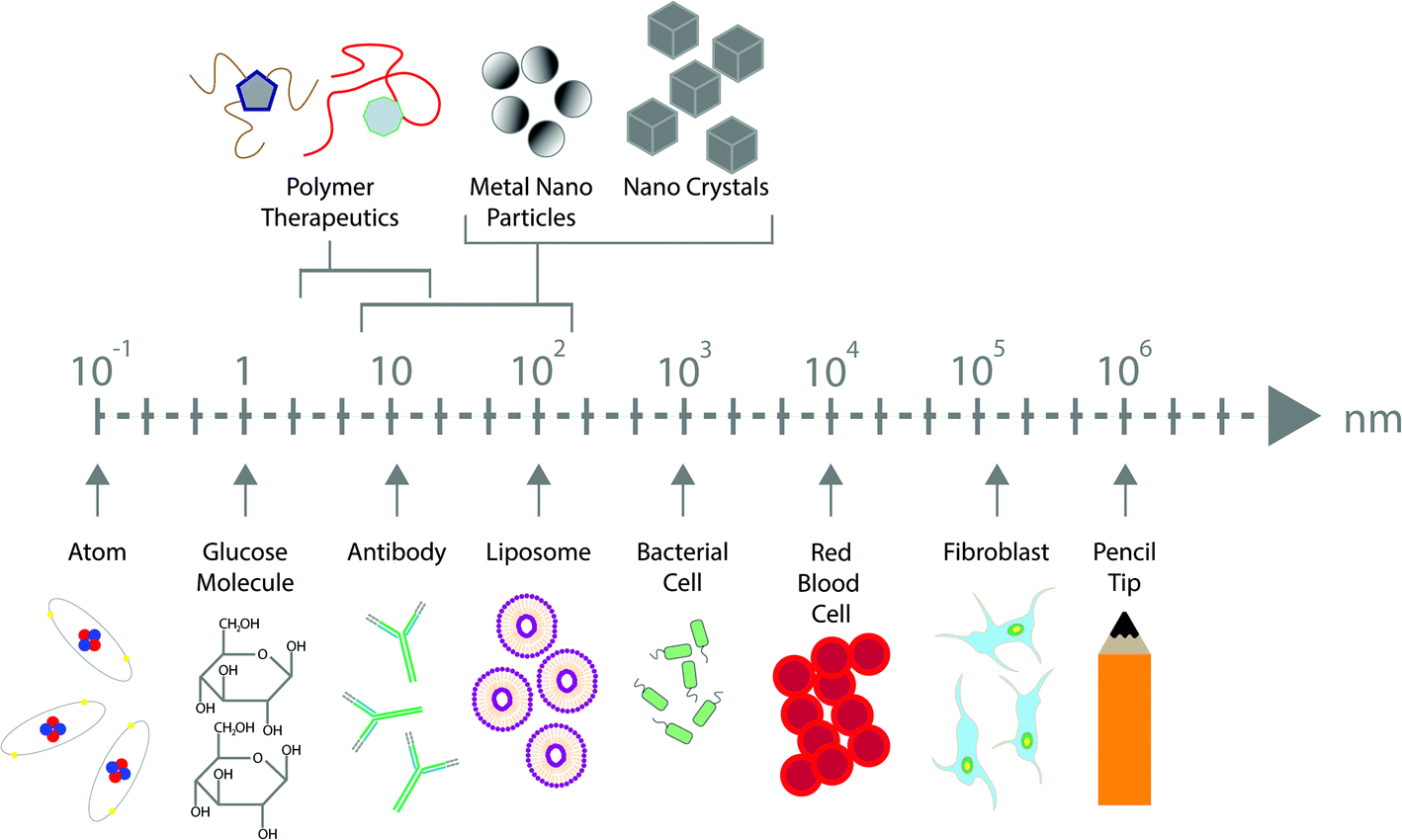 | ||
| Fig. 1 Schematic representation of nanomaterial size in comparison to other biological molecules. | ||
Properties of nanomaterials
Nanomaterials have several properties that make them suitable for a variety of clinical applications. One of the major benefits of nanoparticles is their small size of 10–200 nm allowing them to circulate the body without disrupting blood flow, as well as being able to avoid clearance by both the renal and complement systems.11 The size of clinically used nanomaterials is also relevant in treatments for cancer, as it was first thought that the enhanced permeability and retention effect was one of the ways that nanoparticles could successfully penetrate tumour tissues.12 However, this concept has been greatly contested within the scientific community, with very strong evidence emerging which suggests that active transport mechanisms dominating nanoparticle trafficking resulting in tumour accumulation.13 Opinion on this is still divided amongst many in the community, however, as greater attention is paid to tumour microenvironment, it is becoming more clear that localised pressure within the tumour site would not be conducive to passive targeting through EPR, and is perhaps the major limiting barrier to this phenomenon. Another key property of nanoparticles is their electronic and optical properties, particularly possessed by the metal nanoparticles. These properties are based on the principle of surface plasmon resonance; where free electrons in the metal nanoparticles oscillate.14 Some metallic metal oxide nanoparticles are also magnetic, allowing them to be used for several applications such as imaging, cell separation, targeting and drug delivery. Nanomedicines are generally simple and cheap to manufacture on the small scale, however, difficulty with scale up and stability on large scale manufacture has been widely experienced.15 Once manufactured, nanomaterials are relatively simple to sterilise before clinical use, with the majority being syringe filtered below the molecular cut off for biological contaminants such as bacteria.The need for nanomedicine regulation
Although there is a great deal of excitement surrounding the emerging field of nanomedicine, there is currently a lack of guidance in this field. Many nanomedicines work by direct interaction with genetic materials, or by interaction with biomolecules that are required for normal genome function and cell division,16 all of which can cause genotoxicity and mutagenicity.17 Such toxicity to nanomedicines is mediated by the inflammatory response of neutrophils and macrophages by the production of reactive oxygen and nitrogen species which cause oxidative and nitrosative stress.18 The accumulation of such free radicals can cause extensive damage to the body.19 There are several ways in which this damage can occur, including inducing oxidative DNA damage leading to strand breakage, protein denaturation and lipid peroxidation causing cancer, causing damage to mitochondrial membranes leading to cell death and necrosis, and transcription of genes responsible for carcinogenesis and fibrosis.20 When administered intravenously, there is a wealth of data which shows accumulation of these particles within the liver, and translocation to areas such as the central nervous, cardiovascular and renal systems (Fig. 2).21 For particles that have no ability for tracing after administration, there are simply too many unknowns which may pose potential threats against safety. Currently, the precise interactions of many nanomedicines with biological systems is not yet fully understood, therefore making understanding, identifying or drawing conclusions about the physicochemical and toxicological properties of nanomedicines difficult. However, without standardised regulatory guidance in this area, very little is set to change. It has to also be acknowledged that ‘one-size’ certainly does not fit all in this process as the unique properties observed at the nano-scale are highly dependant upon nanoparticle type, surface properties, administration route and importantly nanoparticle morphology which can be diverse (Fig. 3) – something which is certainly holding up the regulatory process.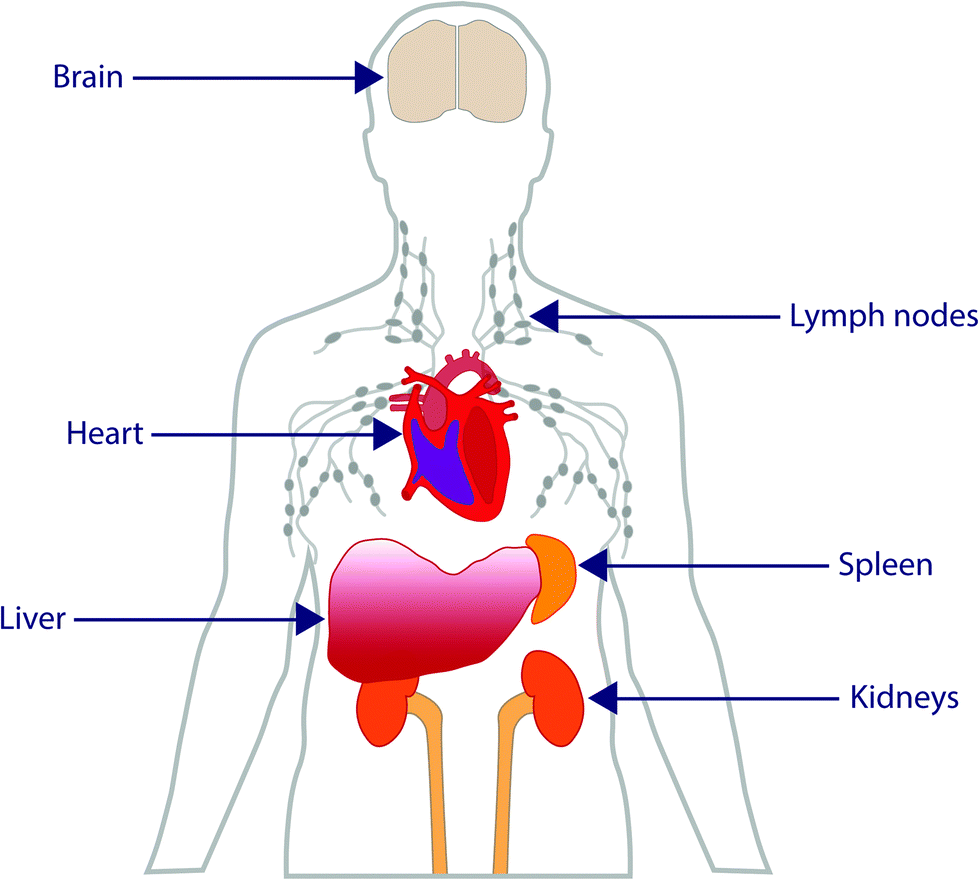 | ||
| Fig. 2 Schematic representation of the main areas of nanoparticle translocation and accumulation after administration. | ||
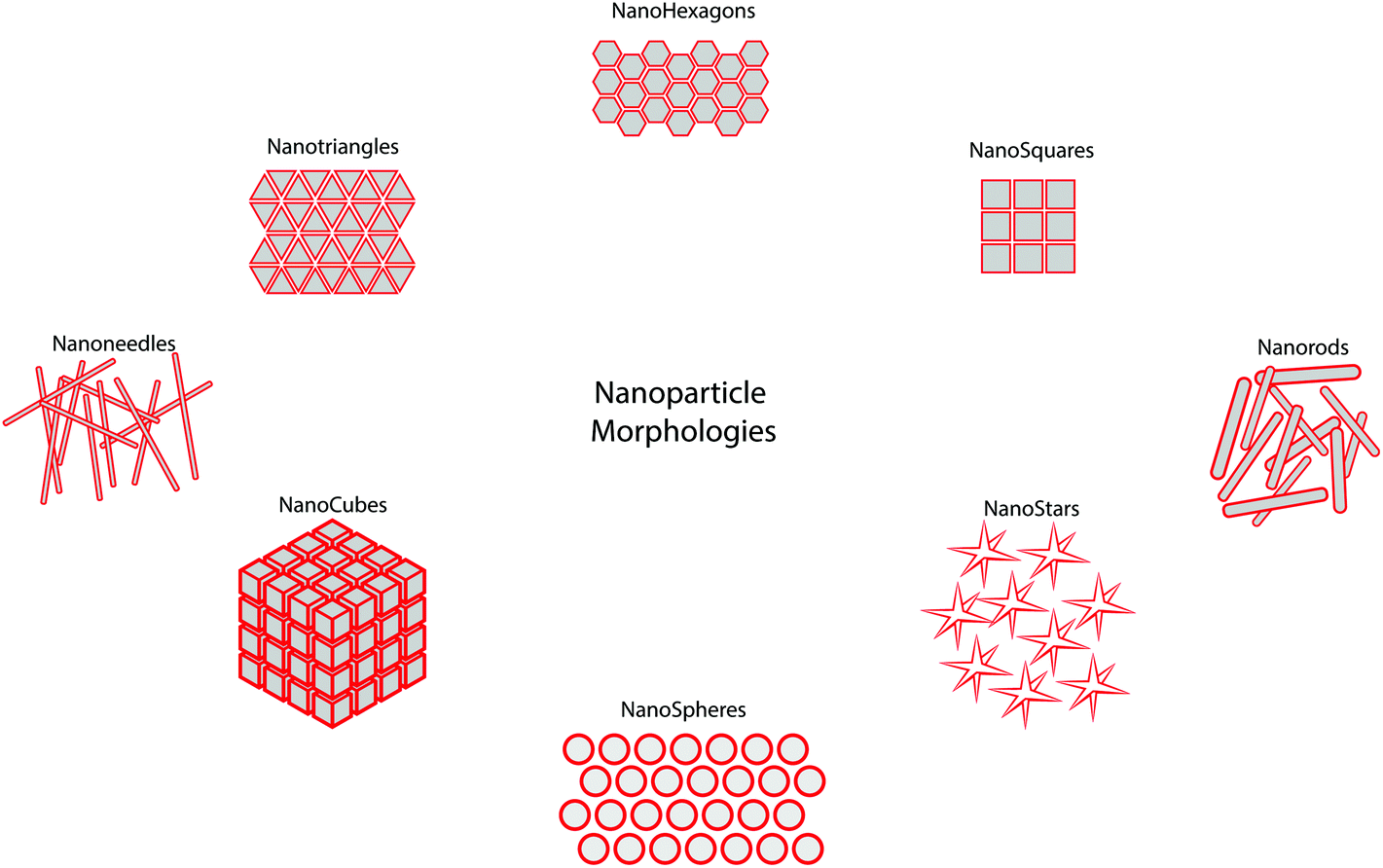 | ||
| Fig. 3 Schematic representation of the diverse morphology of nanomaterials reported for clinical application. | ||
The regulatory agencies are right to be cautious, in the past, market approval has been gained for nanoparticles used in medical imaging, which subsequently have been withdrawn after the emergence of unanticipated patient events after administration.22 Sinerem®, an ultra-small super paramagnetic iron oxide (USPIO) contrast agent for magnetic resonance imaging, was declined a recommendation for marketing authorisation and withdrawn from the market in 2008 by the European Medicines Agency (EMA) due to concerns raised in clinical trials. These concerns involved severe adverse reactions involving muscle pains, particularly in the lower back, and, more worryingly, allergic reactions which resulted in one death. It was therefore concluded that the risks associated with this particular nanomolecule far outweighed any potential benefits and so it was denied marketing authorisation.23
However, this over cautious approach appears to be manifesting as great inertia within the field, often the benchmark checks required for approval are still opaque and align with the regulation for small drug molecules (Fig. 4) which do not accurately reflect the nanomaterials potential. Guidance is critical as without it, manufacturers, healthcare providers, the public and policymakers are without clarity and legal certainty. The US Food and Drug Administration (FDA), Environmental Protection Agency (EPA), and Health and Consumer Protection Directorate of the European Commission have taken initiatives in order to deal with potential risks posed by nanoparticles.24 Initiatives within local (or semi local) communities have been put together and funded such as the REFINE project, which seeks to define the criteria for regulatory needs for nanomedicines and nanomaterials for clinical use.25
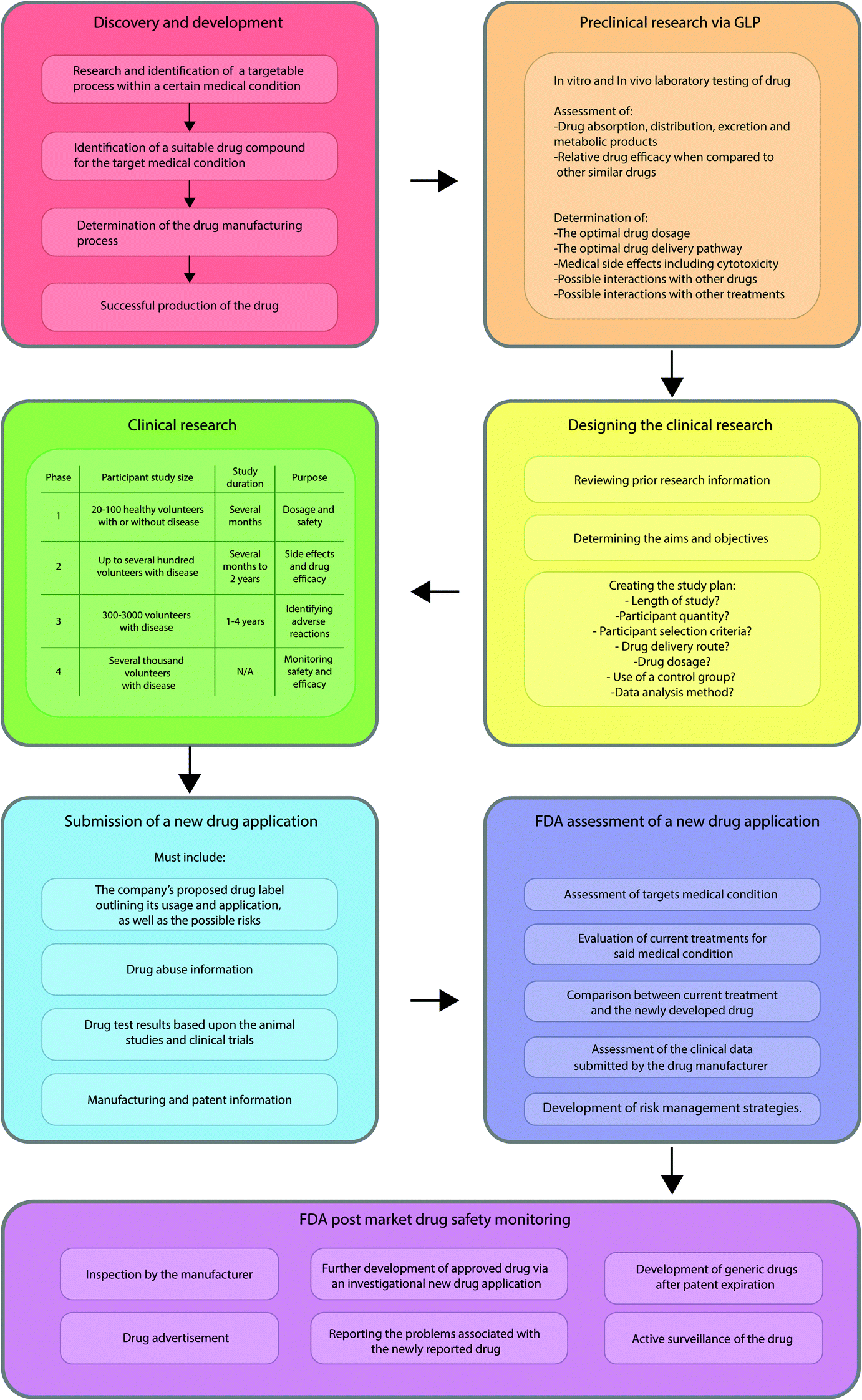 | ||
| Fig. 4 Flow diagram showing well defined approval process for small drug molecules. | ||
However, many feel that no firm and consistent lines have been drawn in order for the uniformity of regulation worldwide, or indeed guarantee that regulatory agencies will act upon such guidance. In their white paper published in 2019, the REFINE project outlines their objectives, including ‘Development and validation of new analytical or experimental methods’.26 A sentiment of need which is echoed across the community, as those nano-based interventions reach clinical and subsequently fail due to lack of consistent or appropriate pre-clinical testing models.27
Another factor to consider when contemplating the impact of nanomedicines, is in their possible environmental impact, after use, upon disposal, and during production.28 It is widely accepted that conventional pharmaceuticals are eventually recovered in the environment and so it is expected that nanomedicines will behave no differently, therefore there is a chance that they could negatively affect the environment.29 The FDA cite the lack of data to determine the safety to humans and the environment, thus they are struggling to formulate a criterion to ensure safe and efficacious development of nano-products, whether they are a drug, device or biologic. The FDA released a first draft guidance in June 2011 as a response to criticism for their lack of nanoparticle regulation, however a final guidance document has not yet been generated for nanoparticles in medicine.30 Despite the great need for a formal regulatory document, the FDA continues to ignore already collated data on toxicity profiles, rather they are taking a precautionary approach to the regulation of Nanomedicine, perhaps in hope to prevent future negative public opinion, treating them as an equal counterpart to their bulk equivalent. This is only negatively impacting the development of nanomedicine and inhibiting future use of these medicines as this uncertainty impacts future funding, research and development whilst destroying public acceptance. This may lead to a delay in the commercialisation of nano-products.31 In the assessment of medical products in the USA and the EU, there are inclusion and exclusion criteria based on estimated environmental effects. In the EU, all marketing authorization applications are required to undergo an environmental risk assessment and a pre-screening stage involving a rough estimation of the predicted environmental concentration for surface water with the acceptable limit being 0.01 ppb. Therefore, if the estimated environmental concentration is below this and no other environmental concerns are raised no further actions are taken for the product in terms of environmental risk assessment. In the USA, the FDA use an environmental assessment for new drug applications unless they are exempt from this, however, if the expected concentration in the environment exceeds 1 ppb, an exemption cannot be made.
Regulatory challenges for nanomedicines
The main challenges faced in the regulation of nanomedicines is outlined in Fig. 5. Arguably, the biggest issue for the regulation of nanomedicines is the fact that regulatory bodies such as the FDA use safety data based on the bulk materials, which do not display the same pharmacodynamic and pharmacokinetic activity as nanomedicines.32 This means data collected on safety and efficacy will not be representative of what could actually occur when the nanomedicine is used in clinical situations once they have achieved marketing authorisation. This leads to issues in creating regulations on safety and efficacy parameters of nanomedicines as a non-nano version may pass regulatory standards when a nanomedicine might not.31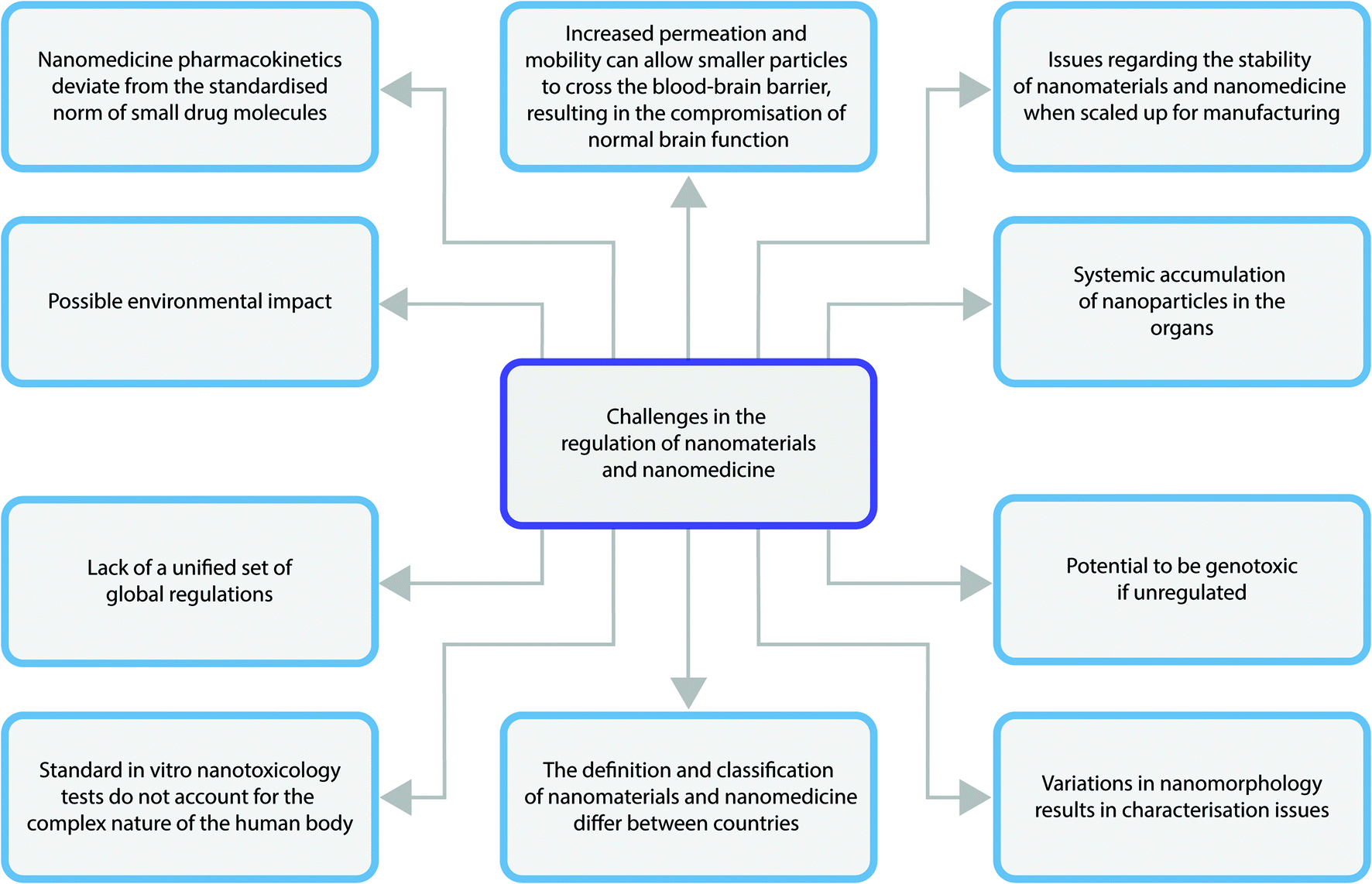 | ||
| Fig. 5 Diagram highlighting the major challenges faced in the regulation of nanomaterials. | ||
Another huge challenge experienced is in the nanomedicine classification. They could be classified as medicines or as medical devices and it is not always consistent across the global regulators. This means a nanomedicine could be classed as a medicine in one country and a medical device in another, hence the regulations that must be adhered to will change depending on its classification. As such, the specific safety and efficacy standards it must pass to be on the market will differ and so some countries will be able to use a nanomedicine that may not have passed regulatory standards in another country.33
In 2009 Rannard and Owen gave the warning that one size does not fit all in terms of nanomedicine based upon their clinical need, application route and physiology,34 but this has largely been ignored by the current regulatory frameworks. The complexity in the structure, form, size and clinical application of nanomedicines challenges regulatory bodies to characterise and categorise nanomedicines. For instance, dynamic light scattering can be used as an estimation of hydrodyanamic size, however this technique equates the particulates scattering light to spherical forms, so for rod shaped materials this is not an accurate metrology technique.35 Additionally, other techniques commonly used for size measurement may render the nanomaterial in a different form than would be experienced in the human body. An example of this is the use of transmission electron microscopy. Here, samples are dried and this may effect their shape or size compared to their solution phase. Protein corona are widely reported to form upon injection of nanomaterials into the bloodstream,36 therefore all size reports may greatly underestimate ultimate size when in the physiological environment. Even in the literature there is no clarity over the best nanometrology or characterisation standards.37 Until there is rigorous clinical regulatory guidance or intervention, preclinical nanomedicine development characterisation will remain unchanged.
Often scale up and manufacturing process is hit or miss for nanomaterial and nanomedicine stability.38 Hence, stringent protocols and assurances are required for approval. There is a need to identify and control manufacturing processes at critical points, it is fundamental to create Critical Quality Attributes (CQA) to enhance the understanding of the nanomedicine manufacturing concept.39 Due to their high complex structures and properties, it is difficult to establish a strong and consistent manufacturing process, which defines the nanomedicines’ quality, efficacy, stability as well as safety.40 A detailed clarification of CQA would mean the identification and analysis of nanomedicine properties in small-scale manufacturing process, thus facilitate the understanding of large-scale manufacturing process.41 In relation to such concerns consortiums and government agencies around the world were set up to provide researchers with semi-regulatory testing facilities, these include the US and EU Nanotechnology Characterisation Laboratories. Thought often such resources are not accessed by researchers until much further down their development pipeline at the end of preclinical testing.
Nanotoxicology and cellular response is another challenge faced by regulators.42 There have been many proposals on plausible ways to measure nanotoxicity. Traditional toxicity measures, with a key example of this being large scale animal testing as seen in the past for small drug molecules, have been reduced and deemed unethical, too costly and impractical when applied to the measurement of nanotoxicity.43In vitro toxicity methods are used as a first approach for the assessment of nanoparticles. It is an efficient way in terms of cost and time and provides more control on experiment conditions when compared to animal testing. However, many of the common 2-dimensional assays used, neglects the complexity of the human body,44 which uses compensation mechanisms and pathological responses to combat toxins as well as complicated metabolic activities. Moreover, there is increasing evidence that the traditional assays used for in vitro testing of small compounds are not fit for purpose for nanomaterials.45 Many nanoparticles interact with the reagents of in vitro assays, or interfere with the detection mechanism and false positives or invalid data is generated. Nanomaterial properties, such as high adsorption capacity, optical properties, catalytic activity, acidity or alkalinity, magnetic properties and dissolution, are all likely to promote interaction with in vitro testing reagents or measurements.46,47 As a result, new assays to measure the toxicity of nanomaterials as well as nanomedicines are required before proper regulatory guidance is written, a factor which is severely hindering progress in this area.44
When developing nanomedicines for clinical use, the mechanism and action of drug delivery requires considerable preclinical safety data before approval, including that of adverse effects.48 The toxic effects of a high drug dose in the nano form may be that of toxicity of a particular cell or organ (something that may be lethal in patients with chronic kidney disease or diabetes) or the emergence of antibiotic resistance. Additionally, the size of the particles may pose a threat to patients given that they are more mobile than their larger counterparts. This allows them to cross the blood–brain barrier, potentially compromising brain function long term, or at least causing oedema.49 This factor is perhaps one of the most important, and without sufficient data to overwhelmingly convince that the new form of medicine is safe, can lead to approvals which may later be revoked.
Defining pharmacokinetics of nanomedicines is posing a major challenge in their regulation.50,51 This is due to the fact that they deviate from the normal and expected course experienced by small drug molecules. A result of this is that they are bioavailable for a sustained period of time thus, if nanomedicine products were ever to be used over the counter, there may be a high health hazard to the public. Regulatory bodies must assess whether or not any given nanomedicine should enter the market under strict supervision or be available as an over the counter products. It is, however, very difficult to come to a conclusive answer regarding this matter due to the lack of toxicity information and data currently available.
Nanomedicines and their follow-ons, nanosimilars, have been introduced into the market over the last decade.40 The challenges of pharmaceutical development and manufacturing process not only applies to nanomedicines but also their follow-on products. On the other hand, their growing awareness have challenged regulatory bodies to evaluate their existing regulations. In the EU, follow-on products could be approved by EMA as an abridged application under the category of generic or hybrid. However, the unknown critical quality attributes of nanomedicine deemed the generic approach invalid. The term ‘nanosimilar’ was also deemed more appropriate due to their complexity. The regulation of generics application is determined by the equation PE + BE = TE. PE is the pharmaceutical equivalence, BE is bioequivalence and TE is therapeutic equivalence. All are challenged heavily by nanomedicines as well as nanosimilars. PE indicates the identical active ingredients found in the same composition. However, it is difficult to isolate or identify the ‘active ingredient’ from many nanomedicines because it exists not as a homo-molecular structure but as complex nanoparticulate structures.47 In addition, without an established CQA, it is challenging to identify which parameters are identical or novel and this still remains a grey area.
An additional issue surrounding the regulation of nanomedicines is the question of who should hold the responsibility of formulating the guidelines for nanomedicines. This decision involves a consultative process that involves many stakeholders made up of academics and clinicians. In relation to this, there is a further immediate need to establish regulatory, high calibre laboratories to a federal level along with risk assessment of personnel, guidelines and technical standards needs to be developed. Often key bodies lack of scientific expertise around the topic due to how new the technology is and how diverse nanomedicines are in mode of action. It is difficult to create adequate regulations when there is limited knowledge of nanomedicines and so any regulations made may not be suitable to maintain patient safety and regulate the use of nanomedicines in a clinical setting.51 In many ways, this infrastructure is already in place with strong national consortiums and national characterisation laboratories, however, the translation of information and guidance suggestions from these bodies is not yet integrated into regulatory frameworks.
All challenges noted are highly limiting on the future of nanomedicine, hindering the process of production of safe, high quality nanomedicine products, and may be leading to ineffective control of nanoparticles due to the lack of nanomedicine-specific safety protocols. Without clear and consistence guidance coming from the governing agencies, it is highly unlikely the breakthrough success of nano will be realised to its full potential.
Nanomedicines already approved for clinical use
A range of nanomaterials and nanotechnologies have already gained regulatory approval and been deployed in the clinical setting for various drug types, including antifungals, anticancer and in pain management, a few examples are outlined in Table 152,53 with a more extensive list outlined recently by Anselmo and Mitragotri.54| Clinical use | Name | Approved for | Class of nanomedicine | Use |
|---|---|---|---|---|
| Cancer | Doxil | Ovarian cancer/HIV associated Kaposi's sarcoma | Liposome | Drug delivery |
| NBTXR3/Hensify | Locally advanced sarcoma | Metallic | Radiation enhancer | |
| Vyxeos | Myeloid leukemia | Bilamellar liposomes | Combination therapy | |
| Abraxane | Pancreatic cancer, breast cancer, non-small cell lung cancer | Albumin bound | Drug delivery | |
| Onivyde | Pancreatic cancer | Liposome | Drug delivery | |
| DaunoXome | HIV associated Kaposi's sarcoma | Liposome | Drug delivery | |
| Myocet | Breast cancer | Liposome | Drug delivery | |
| Antifungal | AmBisome | Crytococcal meningitis, aspergillus, candida infections and visceral leishmenaisis | Liposome | Drug delivery |
| Other | Patisiran/ONPATTRO | Transthyretin amyloidosis | Lipid | siRNA delivery |
| Diafer | Iron deficient anemia | Metallic | Iron replacement | |
| Diprivan | Anaesthesia | Liposome | Anaesthetic |
Amphotericin B is an antifungal agent that cannot be used alone due to poor water solubility, low tolerance and side effects exhibited by patients.55 Amphotericin B was first formulated in deoxycholate, forming a mixed micellar dispersion (Fungizone).56 Studies into the toxicity of Fungizone on human cells determined that this particular nanoparticle system may not be suitable for use. Forster, Washington and Davis determined that Fungizone showed toxicity towards erythrocytes and was determined to be due to the fast diffusion rate of Amphotericin B out of the micelles that it forms.57 Dolberg and Bissell determined that Fungizone at the recommended dose was able to decrease the synthesis of DNA, reduce the number of cells and change the number of transport molecules in chick embryo fibroblasts at 10 days old.58 Hence, other nanoparticle systems have been tested, with Fungizone often used as a standard for toxicity. Since then multiple studies into nanotechnology driven formulations of amphotericin B have entered clinical trial with success. These include Abelcet®, Amphotec® and AmBisome®. AmBisome® was the first nanomedicine approved in Europe is AmBisome®, where amphotericin B is encapsulated into a liposome, which has gone on to great success.
Anti-cancer drugs often possess poor physicochemical properties such as poor aqueous solubility and due to their potent nature after administration result in high systemic toxicity. Hence major efforts have gone into formulation of such compounds, which has dominated much of the nanomedicine research over the past 30 years. The first cancer nanomedicine to gain FDA approval in 1995 was a liposome based doxorubicin hydrochloride formulation (Doxil®) for treatment of Kaposi's sarcoma in patients with human immunodeficiency virus (HIV).59 Since then other lipid based formulations have been approved such as Daunorubicin®60 and Myocet®.61 Other success stories for cancer nanomedicine include protein drug conjugates such as Abraxane® which was approved in the USA in 2005.62 Abraxane® is an albumin-paclitaxel nanoparticle approved for a number of cancers including pancreatic and metastatic breast cancers. Virosomes are also licensed for use in clinical settings in some countries, for example in the Philippines the use of Rexin-G® for solid tumours has been used since 2007 due to its ability to specifically target exposed collagen which is commonly found in metastatic tumours.63 More recently, Rexin-G was fast tracked by the FDA to become a second line treatment for pancreatic cancer.64 One new focus within chemotherapy driven nanomedicines, is on the development of combination therapies within on nanoplatform. Combination treatment has proven to result in increased efficacy against multiple cancers. In particular in cancers which are hard to treat the development of combination therapies has given real hope. In particular Vyxeos® has proven very successful in the treatment of adult acute myeloid leukemia.65 Vyxeos® is a liposomal formulation of daunorubicin and cytarabine. In the phase 3 trials, Vyxeos® demonstrated superior overall survival and reduced risk of death in patients compared to those who were administered the two drugs in a combination regime with no nanotechnology.66 It is forecast that more focus on combination therapy will result in a greater number of such products reaching trial and requiring regulation. Nanotechnology offers real promise in this arena as those patients who are already sick can barely tolerate chemotherapy regimes on only one drug. The protection from systemic toxicity of these potent compounds offered by nanotechnology and site specificity are key to the success which is being experienced in this domain.
DepoDur®, approved in 2004 is another type of nanomedicine which has gained approval for chronic pain management.67 Formed of morphine sulphate encapsulated within multivesicular liposome, which results in a more sustained drug release.64 The intent was to reduce those patients who required opiod treatments to single dose formulations, in order to prevent misuse, addiction and overdose. Other nanotechnology formulations include polyethylene glycoylated (PEGylated) proteins, polypeptides and aptamers such as Cimzia® and Micera®. Cimzia is a PEGylated antibody indicated for Crohn's disease approved in 2008, whilst Mircera® is indicated for anaemia associated with chronic renal failure in adults.64 Nanocrystals have also licenced for clinical use as nanomedicines, Emend® is currently used as an antiemetic due to its increases dissolution rate and subsequent increased bioavailability compared to standard antiemetic formulations of aprepitant.64 Metal-based nanoformulations such as Feraheme® have also been licenced due to their prolonged steady release of the drug, allowing less frequent dosing for patients with anaemia in chronic kidney disease.68
As more knowledge was gained in the field, diversification of treatment condition and indeed cargo type were explored. In particular, nanomedicine has had great success in the delivery of small interfering RNA (siRNA). ONPATTRO® is one example of such success, with its approval for the treatment of the autosomal dominant disease hATTR amyloidosis.69 ONPATTRO® are lipid based nanoparticles which where approved by the FDA in 2018 and were the first RNA based therapeutic approved for clinical use.70 Given that siRNA are particularly difficult to administer alone, the use of nanotechnology within these formulations is the enabling factor. This approval has opened the field wide up to many applications where biologics may be used and delivered efficiently.
Nanomedicine approval and marketing has not come without criticism. There is still a wealth of unknowns when it comes to toxicity profiling, accumulation and clearance of many of the nanotechnologies. There are two potential risks based on this. The first, as commented on already, as was the case with Sinerem®, market approval and clinical use is not always plain sailing and new unknown adverse events can manifest within the patient population after widespread use which ultimately lead to withdrawal. This perhaps again due to the approval testing requirements following the route for small molecule drugs, where a more bespoke testing for nanomedicines are required. Secondly, there is a huge cost implication. Nanotechnologies for medicine have been widely criticised globally for their cost. For example, Abraxane® which was first approved in the USA and subsequently the UK, was not licenced by the UK National Health Service due to its high cost at point of need – despite its major clinical advantages in pancreatic cancer patients, who otherwise had a dismal prognosis. Gradually over time, this has been approved, however, lessons need to be learned from these experiences. As nanotechnologies pass through the clinical trial process and indeed enter the market. Late stage failure, results in huge costs which need to be recuperated elsewhere. If regulation was bespoke and appropriate, this would enable better refinement at preclinical study level, reducing failure rate either later in the clinical trials or indeed after marketing and clinical use.
Global strategies to nanomedicine regulation
The EMA applies General Medicinal Product legislation on regulating nanomedicines. At the same time, it creates a specialized multidisciplinary expertise to evaluate nanomedicines using current risk/benefit-analysis principles. It has also established a definition of nanomedicine and published a list of specific guidance for nanomedicine which could be browsed on their guidance webpage. In 2009, the European Nanomedicines Expert Group was formed by the EMA to meet the increasing need for evaluation of nanomedicines from stakeholders. Established academics and regulatory science specialists from the Expert Group met with regulatory specialists from other regulatory agencies such as the FDA.71USA
Until now, the FDA are regulating nanotechnology products, including nanomedicines, using the current statutory and regulatory authorities as well as product-specific standards under its jurisdiction. Throughout the years, the FDA has issued guidance for nanomaterials on food, cosmetics and animal food. However, there is no published specific guidance for nanomedicine. In 2017, FDA produced a draft guidance on drug products, including biological products, that contain nanomaterials. In addition, the FDA does not attempt to categorize nanotechnology as safe or harmful but evaluate each nanotechnology on a case-by-case basis.72 It should be noted that FDA identified several attributes concerning their regulatory approach. Nanomedicine products would be assessed in a product-specific way. Manufacturers are advised to consult with FDA when developing their nanotechnology products to establish a mutual understanding on regulatory issues. Consultation with the FDA is encouraged so that help on reviewing safety information and post-marking safety designs could be given to manufacturers. Even after approval, post-market monitoring would be continued by FDA to protect consumers. Premarket review is required, and for nanotechnology that are not subject to premarket review, FDA would offer guidance and advice to corresponding manufacturers.72 Ultimately, the responsibility to assure the safety of nanomedicines as well as their adherence to all applicable legal requirements lies on the manufacturers. Other institutes have also contributed to the regulation of nanomedicines, such as the Nanotechnology Characterization Laboratory of the National Cancer Institute (NCL-NCI) who have been contributing for more than 10 years.The FDA formed the Nanotechnology Task Force and Nanotechnology Interest Group comprised of representatives from many regulatory centres in order to tackle the issue of regulating nanotechnology worldwide. Despite this, the FDA is yet to produce a clear set of guidelines, rather the Task Force has concluded that pre-existing regulations are comprehensive enough to ensure the safe production of nanomedicines as these products undergo pre-market testing and approval under the New Drug Application process. This conclusion is based upon the assumption that regulatory requirements already in place would detect toxicities in nano-products.31 Despite this fact, the FDA has not changed their regulatory requirements and nanomedicines continue to be regulated according to existing guidelines for their larger counterparts. This lack of action in the changing landscape has resulted in great criticism of the FDA. As a result, nano-formulations comprising of existing approved building blocks appear to fast track through the system not undergoing the new drug approval or full premarket approval scrutiny. This strategy is extremely risky and only time will tell whether appropriate.
UK
Medicines within the UK are regulated by the Medicines and Healthcare products Regulatory Authority (MHRA). No clear guidance has been published in relation to nanomedicine approval and in common with the FDA, these appear to being treated on a case-by-case basis. Researchers developing nanomedicines are encouraged to liaise with the MHRA Innovation Office for guidance and steering through the process. In common with the US, other organizations such as the European Nanomedicine Characterization Laboratory (EU-NCL) which are based across the UK and EU provide and constantly refine knowledge on preclinical characterization assays of nanomedicine.73EU
Within the EU, progress has been made with task forces and consortiums being put together to define the formal meaning of the word nanomaterial, with various reports and recommendations coming out from these which touch on food, environment and health. Other initiatives which have already been mentioned such as the EU-NCL and REFINE project have been funded through government awards to contribute to the advances within this field.74–76 Unlike the UK, the regulatory body in the EU, the European Medicines Union (EMU) have published a range of specific preliminary guidelines for a range of nanomedicine preparation standards.71,73,77 However, these are only at the public consultation stage and no formal regulatory guidance is currently in place. The EU-NCL work closely with the regulatory bodies as they do in the UK to inform and influence decision making on the regulation and potential danger of such products.Canada
Health Canada has established a Working Definition of Nanomaterials, where it “considers any manufactured product, material, substance, ingredient, device, system or structure to be nanomaterial if it is at or within the nanoscale (1–100 nm) in at least one spatial dimension, or is smaller or larger than the nanoscale in all spatial dimensions and exhibits one or more nanoscale phenomena”. Regarding the approval of nanotechnology products, Canada relies on existing regulatory frameworks. Health Canada advises manufacturers to consult with the responsible regulatory authority during the early development process to identify and assess the product's risks and properties.78 Health Portfolio Nanotechnology Working Group is established in Canada for the gathering and discussion of issues related to nanotechnology, which consists of representatives from regulatory bodies like Health Canada and the Canadian Institutes of Health Research (CIHR). A general guidance on nanotechnology-based health products and food have also been issued by the Health Canada.79Japan
Medicines in Japan are regulated by the Ministry of Health, Labour and Welfare (MHLW)/the Pharmaceuticals and Medical Devices Agency (PMDA). The Japanese regulatory bodies have yet to come up with a definition as well as nanomedicine-specific regulations for nanomedicines.80 In 2016, a guideline for the development of liposome drug products were issued. Nanomedicines are regulated under the Pharmaceutical Affairs Law framework, which is a general medicinal product legislation, on a case-by-case basis. It should be noted that regulators and reviewers are assembling and analysing nanomedicine data. The MHLW/PMDA have also collaborated with the EMA on issuing reflection papers, notably on the development of block-copolymer micelle medicinal products and nucleic acids (siRNA)-loaded nanotechnology-based drug products.Others
Although there is little regulation regarding this field in Asia, countries such as India, Japan, China and Thailand are currently in the process of determining governance and regulatory policies to address the growing issues in the field of nanotechnology. In India, the Department of Science and Technology, and the Government of India have created a group to regulate nanotechnology and draft a set of guidelines creating a three-tiered governance framework which has been implemented to assist policy makers in developing a pathway for regulation of nanomedicine. This ensures further growth of this technology whilst also addressing risks associated with nanomedicine.Conclusions and future outlook
Despite the lack of specific regulation guidance over 50 nanomedicines have reached the market and this number grows more steadily. These predominantly lie in cancer therapy, owing to the stubborn toxic compounds required and very challenging tumour landscape which hinders effective drug treatment. The most notable of these include the liposomal preparations Doxil®, AmBisome® with more recent success with albumin-drug nanoparticles such as Abraxane®, polymeric micelles such as Eligard® to name a few.Lack of formal regulation of nanomedicines and nanomaterial production for health related applications is a global issue. The inconsistency across different government agencies determines some nanomedicines as medical devices and others as medicines. What is deemed fit for purpose in one jurisdiction does not translate to others, and whilst small molecules often are not licenced globally for this reason, the nanomedicine community require urgent coherence across the governance sector to enable development to continue in line with expectation. The formation of clusters and working groups has not amounted to action to date, nanomaterials are not new and the need and urgency with which treatments for some diseases or conditions cannot be met under the current regulatory structure.
Whilst there have been some efforts across academic communities and government agencies to form National Characterisation Laboratories, more explicit and stringent guidance is needed from the main governing bodies such as the FDA and MHRA. Many diseases do not discriminate due to race or location, hence a global consortium for the regulation of nanomaterials should form to push forward these agendas and issue formal guidance to the research communities. Billions of dollars of investment have been funnelled into nanomedicine development over the past two decades, and unless there is clear leadership and guidance from the regulatory bodies, these efforts will not result in products coming to the market and future investment will be placed elsewhere.
Conflicts of interest
The authors have no conflict of interest.References
- http://www.britishsocietynanomedicine.org/what-is-nanomedicine/ [accessed 14/06/19].
- N.-C. Yeh, Technovation, 2013, 33, 108 CrossRef.
- D. Hanigan, L. Truong, J. Schoepf, T. Nosaka, A. Mulchandani, R. L. Tanguay and P. Westerhoff, Water Res., 2018, 139, 281–290 CrossRef CAS PubMed.
- A. Mihranyan, N. Ferraz and M. Strømme, Prog. Mater. Sci., 2012, 57, 875–910 CrossRef CAS.
- E. Pomerantseva, F. Bonaccorso, X. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, 6468 CrossRef PubMed.
- Y. Min, J. M. Caster, M. J. Eblan and A. Z. Wang, Chem. Rev., 2015, 115, 11147–11190 CrossRef CAS PubMed.
- C. Ventola, P. T., 2012, 37, 512–517 Search PubMed.
- R. Seigneuric, L. Markey, D. Nuyten, C. Dubernet, C. Evelo, E. Finot and C. Garrido, Curr. Mol. Med., 2010, 10, 640–652 CrossRef CAS PubMed.
- J. P. Martins, J. das Neves, M. de la Fuente, C. Celia, H. Florindo, N. Günday-Türeli, A. Popat, J. L. Sanots, F. Sousa, R. Schmid, J. Wolfram, B. Sarmento and H. A. Santos, Drug Delivery Transl. Res., 2020, 10, 726–729 CrossRef PubMed.
- H. He, L. Liu, E. E. Morin, M. Liu and A. Schwendeman, Acc. Chem. Res., 2019, 52, 2445–2461 CrossRef CAS PubMed.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef CAS PubMed.
- S. K. Golombek, J. N. May, B. Theek, L. Appold, N. Drude, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2018, 130, 17–38 CrossRef CAS PubMed.
- S. Sindhwani, A. M. Syed, J. Ngai, J. B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, A. B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed.
- Y. Xia and J. Halas, MRS Bull., 2005, 30, 338–348 CrossRef CAS.
- R. Paliwal, R. J. Babu and S. Palakurthi, AAPS PharmSciTech, 2014, 15, 1527–1534 CrossRef CAS PubMed.
- X.-Q. Zhang, X. Xu, N. Bertrand, E. Pridgen, A. Swami and O. C. Farokhzad, Adv. Drug Delivery Rev., 2012, 64, 1363–1384 CrossRef CAS PubMed.
- N. Singh, B. Manshian, G. J. Jenkins, S. M. Griffiths, P. M. Williams, T. G. Maffeis, C. J. Wright and S. H. Doak, Biomaterials, 2009, 30, 3891–3914 CrossRef CAS PubMed.
- B. Smolkova, M. Dusinska and A. Gabelova, Food Chem. Toxicol., 2017, 109, 780–796 CrossRef CAS PubMed.
- V. Lobo, A. Patil, A. Phatak and N. Chandra, Pharmacogn. Rev., 2010, 4, 118–126 CrossRef CAS PubMed.
- A. Manke, L. Wang and Y. Rojanasakul, BioMed Res. Int., 2013, 942916 Search PubMed.
- A. Kermanizadeh, D. Balharry, H. Wallin, S. Loft and P. Møller, Crit. Rev. Toxicol., 2015, 45, 837–872 CrossRef CAS PubMed.
- M. Modoa, J. Kolosnjaj-Tabib, F. Nicholls, W. Linga, C. Wilhelm, O. Debargee, F. Gazeauc and O. Clement, Contrast Media Mol. Imaging, 2013, 8, 439–455 CrossRef PubMed.
- https://www.ema.europa.eu/documents/withdrawal-report/withdrawal-assessment-report-sinerem_en.pdf [Accessed 3 Feb. 2019].
- R. Faulkner and N. Jasper, Global Environ. Pollut., 2012, 12, 30–35 CrossRef.
- http://refine-nanomed.eu/ [Accessed 10/06/20].
- B. Halamoda-Kenzaoui, H. Box, M. van Elk, S. Gaitan, R. E. Geertsma, E. Gainza Lafuente, A. Owen, A. Del Pozo, M. Roesslein and S. Bremer-Hoffmann, The REFINE White Paper, Publications office of the European Union, Luxemburg, 2019 Search PubMed.
- D. Landesman-Milo and D. Peer, Bioconjugate Chem., 2016, 27, 855–862 CrossRef CAS PubMed.
- I. Mahapatra, J. R. A. Clark, P. J. Dobson, R. Owen, I. Lynch and J. R. Lead, Environ. Sci.: Nano, 2018, 5, 1873–1889 RSC.
- A. Baun and S. F. Hansen, Nanomedicine, 2008, 3, 605–608 CrossRef PubMed.
- Food and Drug Administration, Drug Products, Including logical Products, that Contain Nanomaterials Guidance for Industry, 2017. Available from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/drug-products-including-biological-products-contain-nanomaterials-guidance-industry [Accessed 11/19/19].
- R. Bawa, Curr. Drug Delivery, 2011, 8, 227–234 CrossRef CAS.
- R. Bawa, G. F. Audette and B. Reece, Handbook of Clinical Nanomedicine: Law, Business, Regulation, Safety, and Risk, Pan Stamford Series on Nanomedicine, CRC Press, Taylor and Francis, Boca Raton, Florida, USA, 2016, vol. 2 Search PubMed.
- B. Kelly, 2010, Nanomedicines: regulatory challenges and risks ahead, [online] Cov.com. Available at: https://www.cov.com/-/media/files/corporate/publications/2010/10/nano medicines regulatory.pdf [Accessed 5 Feb. 2019].
- S. Rannard and A. Owen, Nano Today, 2009, 4, 382–384 CrossRef CAS.
- A. N. Cleland, J.-L. Fraikin, P. Meinhold and F. Monzon, Drug Dev. Delivery, 2016, 16(3), 20–26 Search PubMed.
- J. Y. Oh, H. S. Kim, L. Palanikumar, E. M. Go, B. Jana, S. A. Park, H. Y. Kim, K. Kim, J. K. Seo, S. K. Kwak, C. Kim, S. Kang and J.-H. Ruy, Nat. Commun., 2018, 9, 4548 CrossRef PubMed.
- H. S. Leong, K. S. Butler, J. C. Brinker, M. Azzawi, S. Conlan, C. Dufes, A. Owen, S. Rannard, C. Scott, C. Chen, M. A. Dobrovolskaia, S. V. Kozlov, A. Prina-Mello, R. Schmid, P. Wick, F. Caputo, P. Boisseau, R. M. Crist, S. E. McNeil, B. Fadeel, L. Tran, S. Foss Hansen, N. B. Hartmann, L. P. W. Clausen, L. M. Skjolding, A. Baun, M. Ågerstrand, Z. Gu, D. A. Lamprou, C. Hoskins, L. Huang, W. Song, H. Cao, X. Liu, K. D. Jandt, W. Wen, B. Y. S. Kim, K. E. Wheeler, A. J. Chetwynd, I. Lynch, M. Moghimi Sayed, A. Nel, T. Xia, P. S. Weiss, B. Sarmento, J. das Neves, H. A. Santos, L. Santos, S. Mitragotri, S. Little, D. Peer, M. M. Amiji, M. J. Alonso, A. Petri-Fink, S. Balog, A. Lee, B. Drasler, B. Rothen-Rutishauser, S. Wilhelm, H. Acar, R. G. Harrison, C. Mao, P. Mukherjee, R. Ramesh, L. R. McNally, S. Busatto, J. Wolfram, P. Bergese, M. Ferrari, R. H. Fang, L. Zhang, J. Zheng, C. Peng, B. Du, M. Yu, D. M. Charron, G. Zheng and C. Pastore, Nat. Nanotechnol., 2019, 14, 629–635 CrossRef CAS PubMed.
- M. S. Muthu and B. Wilson, Nanomedicine, 2012, 7, 307–309 CrossRef CAS PubMed.
- S. Soares, J. Sousa, A. Pais and C. Vitorino, Front. Chem., 2018, 6, 360 CrossRef.
- S. Muhlebach, Adv. Drug Delivery Rev., 2018, 131, 122–131 CrossRef CAS PubMed.
- V. Sainz, J. Conniot, A. Matos, C. Peres, E. Zupanoio, L. Moura, L. Silva, H. Florindo and R. Gaspar, Biochem. Biophys. Res. Commun., 2015, 468, 504–510 CrossRef CAS PubMed.
- J. Paradise, AMA J. Ethics, 2019, 21, E347–E355 CrossRef PubMed.
- A. Kroll, M. Pillukat, D. Hahn and J. Schnekenburger, Eur. J. Pharm. Sci., 2009, 72, 370–377 CAS.
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS PubMed.
- A. M. Dickinson, J. M. Godden, K. Lanovyk and S. S. Ahmed, Appl. In Vitro Toxicol., 2019, 5, 114–122 CrossRef.
- C. Hoskins, L. Wang, W. P. Cheng and A. Cuschieiri, Nanoscale Res. Lett., 2012, 7, 77 CrossRef PubMed.
- B. Fluhmann, I. Ntai, G. Borchard, S. Simoens and S. Muhlebach, Eur. J. Pharm. Sci., 2019, 128, 73–80 CrossRef CAS PubMed.
- S. Siegrist, E. Cörek, P. Detampel, J. Sandström, P. Wick and J. Huwyler, Nanotoxicology, 2019, 13, 73–99 CrossRef CAS PubMed.
- H. S. Sharma, S. Hussain, J. Schlager, S. F. Ali and A. Sharma, Acta Neurochir. Suppl., 2010, 106, 359–364 Search PubMed.
- V. Agrahari and P. Hiremath, Nanomedicine, 2017, 12, 819–823 CrossRef CAS PubMed.
- V. Limaye, G. Fortwengel and D. Limaye, Int. J. Drug Regul. Aff., 2014, 2, 33–41 CrossRef.
- https://www.accessdata.fda.gov/scripts/cder/daf/ [Accessed 10/06/20].
- https://products.mhra.gov.uk/ [Accessed 10/06/20].
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 Search PubMed.
- F. Meunier, H. G. Prentice and O. Ringdén, J. Antimicrob. Chemother., 1991, 28, 83–91 CrossRef PubMed.
- H. A. Gallis, R. H. Drew and W. W. Pickard, Rev. Infect. Dis., 1990, 12, 308–329 CrossRef CAS PubMed.
- D. Forster, C. Washington and S. S. Davis, J. Pharm. Pharmacol., 1998, 40, 325–328 CrossRef PubMed.
- D. Dolberg and M. J. Bissell, In Vitro, 1974, 10, 26–29 CrossRef CAS PubMed.
- T. Cooley, D. Henry, M. Tonda, S. Sun, M. O'Connell and W. Rackoff, Oncologist, 2007, 12, 114–123 CrossRef CAS PubMed.
- https://drugs.ncats.io/drug/ZS7284E0ZP [Accessed 10/06/20].
- https://www.ema.europa.eu/en/medicines/human/EPAR/myocet-liposomal-previously-myocet [Accessed 10/06/20].
- https://www.drugs.com/history/abraxane.html [Accessed 10/06/20].
- V. Weissig, T. K. Pettinger and N. Murdock, Int. J. Nanomed., 2014, 9, 4357–4373 CrossRef CAS PubMed.
- S. P. Chawla, V. S. Chua, L. Fernandez, D. Quon, W. C. Blackwelder, E. M. Gordon and F. L. Hall, Mol. Ther., 2010, 18, 435–441 CrossRef CAS PubMed.
- K. T. J. Chen, R. Gilabert-Oriol, M. B. Bally and A. W. Y. Leung, Pharm. Res., 2019, 36, 125 CrossRef PubMed.
- https://vyxeospro.com/clinical-data/efficacy/ [Accessed 07/07/20].
- P. C. Nagle and J. C. Gerancher, Tech. Reg. Anesth. Pain Manag., 2007, 11, 9–18 CrossRef.
- C. L. Ventola, P. T., 2017, 42, 742–755 Search PubMed.
- https://www.onpattro.com/ [Accessed 07/07/20].
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Baros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. A. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. van der Meel and P. R. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- R. Pita, F. Ehmann and M. Papaluca, AAPS J., 2016, 18(6), 1576–1582 CrossRef CAS PubMed.
- FDA, Nanotechnology Fact Sheet, 2018. Available: https://www.fda.gov/ScienceResearch/SpecialTopics/Nanotechnology/ucm402230.htm [Accessed 12/12/19].
- S. Bremer-Hoffmann, B. Halamoda-Kenzaoui and S. Borogs, Join, 2018, 3, 1 Search PubMed.
- H. Rauscher, G. Roebben, V. Amenta, A. B. Sanfeliu, L. Calzolai, H. Emons, C. Gaillard, N. Gibson, T. Linsinger, A. Mech, L. Q. Pesudo, K. Rasmussen, J. R. Sintes, B. Sokull-Klüttgen and H. Stamm, Towards a review of the EC Recommendation for a definition of the term “nanomaterial” Part 1: Compilation of information concerning the experience with the definition, JRC Scientific Quality Report, Publications office of the European Union, Luxemburg, 2014 Search PubMed.
- G. Roebben, H. Rauscher, V. Amenta, K. Aschberger, A. B. Sanfeliu, L. Calzolai, H. Emons, C. Gaillard, N. Gibson, U. Holzwarth, R. Koeber, T. Linsinger, K. Rasmussen, B. Sokull-Klüttgen and H. Stamm, Towards a review of the EC Recommendation for a definition of the term “nanomaterial” Part 2: Assessment of collected information concerning the experience with the definition, JRC Scientific Quality Report, Publications office of the European Union, Luxemburg, 2014 Search PubMed.
- H. Rauscher, G. Roebben, A. B. Sanfeliu, H. Emons, N. Gibson, R. Koeber, T. Linsinger, K. Rasmussen, J. R. Sintes, B. Sokull-Klüttgen and H. Stamm, Towards a review of the EC Recommendation for a definition of the term “nanomaterial” Part 3: Scientific-technical evaluation of options to clarify the definition and to facilitate its implementation, JRC Scientific Quality Report, Publications office of the European Union, Luxemburg, 2015 Search PubMed.
- https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/multidisciplinary/multidisciplinary-nanomedicines [Accessed 23/02/20].
- Health Canada, Nanotechnology-based Health Products and Food, 2011, Available: https://www.canada.ca/en/health-canada/services/drugs-health-products/nanotechnology-based-health-products-food.html [Accessed 27/02/20].
- Health Canada, Current Issues with Nanomedicines in Canada, 2010, Available: https://www.ema.europa.eu/en/documents/presentation/presentation-nanomedicines-current-initiatives-canada-duc-vu-health-canada_en.pdf [Accessed 23/01/20].
- MHLW/PMDA, Current Initiative in Japan for nanomedicines, 2010, Available: https://www.ema.europa.eu/en/documents/presentation/presentation-nanomedicines-current-initiatives-japan-kumiko-sakai-kato-toru-kawanishi-national/mhlw_en.pdf. [Accessed 23/01/20].
| This journal is © The Royal Society of Chemistry 2020 |