
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew avenues for C–B bond formation via radical intermediates
Florian W.
Friese
and
Armido
Studer
 *
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität, Corrensstrasse 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 3rd September 2019
Abstract
This perspective gives an overview on recent findings in the emerging area of C-radical borylation using diborons as radical trapping reagents. Aryl, vinyl and alkyl boronic esters can be accessed via such an approach under mild conditions. These processes are complementary to established transition metal catalysed cross coupling reactions. Radical borylations can be conducted in the absence of a transition metal but some processes require transition metals as catalysts. It will be shown that various readily available C-radical precursors can be used to run these borylations. For a better understanding of the chemistry, mechanistic discussions are also presented and an outlook on this topic will be provided at the end of the article.
1. Introduction
Aryl and alkyl boronates are highly valuable building blocks in chemical synthesis, especially considering their applications in transition-metal catalysed C–C coupling reactions like the Suzuki–Miyaura1 or Chan–Lam reaction.2 Furthermore, the C–B bond in such esters can easily be transformed into various useful functional groups.3,4 Along with their intense applications as reagents in organic synthesis, boronates have found their way as biological active functionalities into medicinal chemistry and are nowadays relevant also in drug discovery.5Historically, the synthesis of alkyl/aryl boronates is accomplished via nucleophilic attack of a metal–organic reagent (Li, Mg) to a boron-based electrophile which is generally not ideal.6 Using the boron reagents as nucleophiles in two electron transition metal catalysis, remarkable advances have been achieved in the past.4,7 Over the last few years, C-radical borylation has been developed as a complementary approach, allowing to access boronates from various readily available feedstock chemicals under mild conditions, beginning with anilines in 2010 (Scheme 1), followed by aryl halides, alkyl and aryl carboxylic acids, alkyl halides, amines and alcohols. Diboron reagents based on tetrahydroxydiboron, which can be used as the free acid or as its esters like B2pin2 (bis(pinacolato)diboron), B2cat2 (bis(catecholato)diboron) and B2neop2 (bis(neopentyl glycolato)diboron) are the most widely applied radical borylation reagents. These compounds feature two empty p(B)-orbitals that interact with both Lewis bases and radical intermediates which is the basis for a successful radical C–B bond forming reaction.
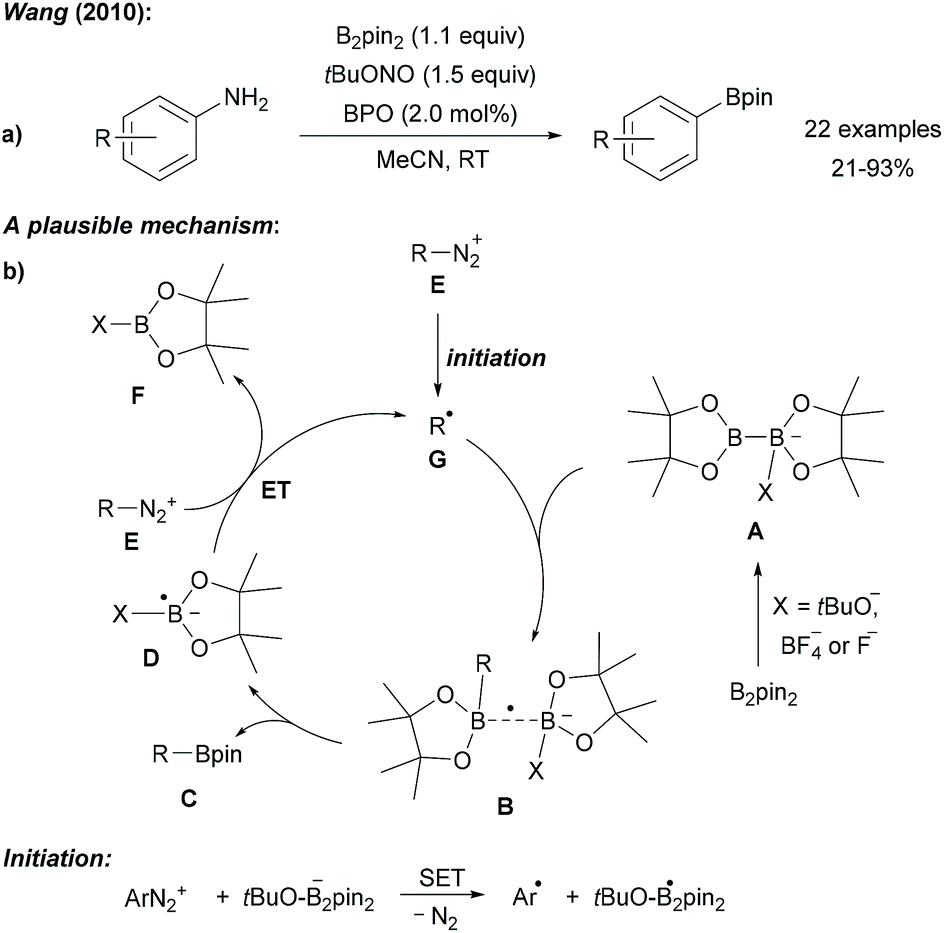 | ||
| Scheme 1 Metal free borylation of anilines via in situ generation of diazonium salts (a). Mechanism (b). | ||
Notably, published review articles cover parts of the current research activities in this highly active field but not in its entirety, especially considering the borylation of sp3-centered radicals.8 It is therefore the aim of this perspective to address the whole field of boronate synthesis using radical pathways. The review is structured based on the nature of the radical precursor and its hybridisation state (sp2 & sp3). Reactions that require metals to mediate the C–B bond forming step are categorised according to the reactive boron species involved, either a diboron reagent or a metal–B complex (see Chapter 4).
2. Radical C(sp2)–B bond formation
2.1. Deaminative borylation (sp2)
Radical deaminative borylation of anilines can be achieved via their quaternary aryl ammonium salts (see Chapter 2.2)9 or via their diazonium salts.Following Sandmeyer-type chemistry, Wang first reported the efficient direct conversion of anilines into aryl pinacol boronic esters by reaction of the aniline with tert-butyl nitrite, B2pin2 and benzoyl peroxide (BPO) as an initiator in MeCN (Scheme 1).10 It was later found that in the absence of BPO at higher reaction temperature substrate scope could be broadened.11 Although in these initial reports a clear mechanistic picture was missing, recent reports in this field allow us to suggest a reasonable mechanism. Anionic Lewis bases such as tert-butanolate generated during the diazotisation of the aniline, coordinate B2pin2 to provide the active ate-complex of type A (Scheme 1).12Via radical anion B, featuring a weak single electron σ bond,13,14 trapping of the aryl radical gives the pinacol boronic ester C and radical anion D. The latter is capable of reducing the diazonium salt E to the aryl radical G under loss of N2 to close the chain. Initiation is either achieved by thermal decomposition of BPO or via ET (electron transfer) from anion A to E (Scheme 1). Alternatively, a nucleophilic ipso-substitution in the diazonium salt with ate-complex A was proposed for such transformations.15 For instance, Yamane suggested an ionic mechanism for the BF3·Et2O mediated borylation of aryl triazenes with B2pin2 as he could not find any evidence for the presence of radicals.16
Yan was the first to employ photo-excited eosin Y to mediate/catalyse the borylation of tetrafluoroborate diazonium salts with B2pin2 (Scheme 2a).17 Ranu further improved that protocol and found that the eosin Y process also works on in situ generated diazonium salts.18 Recently, the eosin Y promoted borylation of BF4 diazonium salts was also conducted as a solvent-free mechanochemical process in a modified ball mill.19 Moreover, Maiti used water soluble CdSe-quantum dots as redox catalysts (or smart initiators) for the radical borylation of diazonium salts with diboron esters in biphasic CH2Cl2/H2O under blue LED irradiation.20
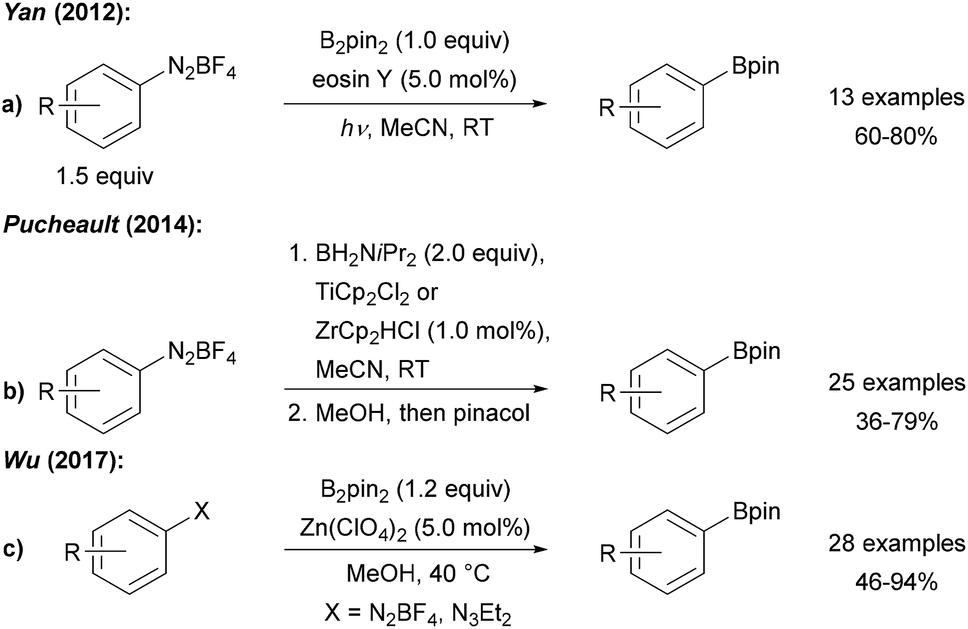 | ||
| Scheme 2 Radical boryl mediated by eosin Y (a), group IV metallocenes (b) or Zn salts (c). | ||
Diazonium salts were found to readily react with various metallocenes (Ti, Zr, Co, Ni, Ru) employing H2B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NiPr2 as boron source to provide the corresponding aryl amino boranes, which were further converted to the pinacol boronic esters prior to isolation, as reported by Pucheault (Scheme 2b).21
NiPr2 as boron source to provide the corresponding aryl amino boranes, which were further converted to the pinacol boronic esters prior to isolation, as reported by Pucheault (Scheme 2b).21
The authors suggested a radical chain reaction initiated by an ET from the metallocene Cp2M to the diazonium salt. The fact that the different metallocenes studied provide similar yields support that mechanistic proposal. Furthermore, Wu disclosed a Zn(ClO4)2 mediated process with B2pin2 as the borylation reagent using preformed BF4 or in situ generated diazonium salts as radical precursors (Scheme 2c).22 The authors suggested that the Zn salt assists initial aryl radical formation.
Efforts devoted to the use of more affordable diboron sources in environmentally friendly solvents have been made. Blanchet and Xiao both utilised diboronic acid in the borylation of aryl diazonium salts derived from anilines by diazotisation with NaNO2/HCl (Scheme 3a–c).23 Wu later showed that aryl diazonium salts can readily be converted to boronic esters using B2pin2 in a water acetone mixture24 and it was found that metal free borylation under blue LED irradiation can also be achieved with methyl arylazo sulfones as C-radical precursors (Scheme 3d).25
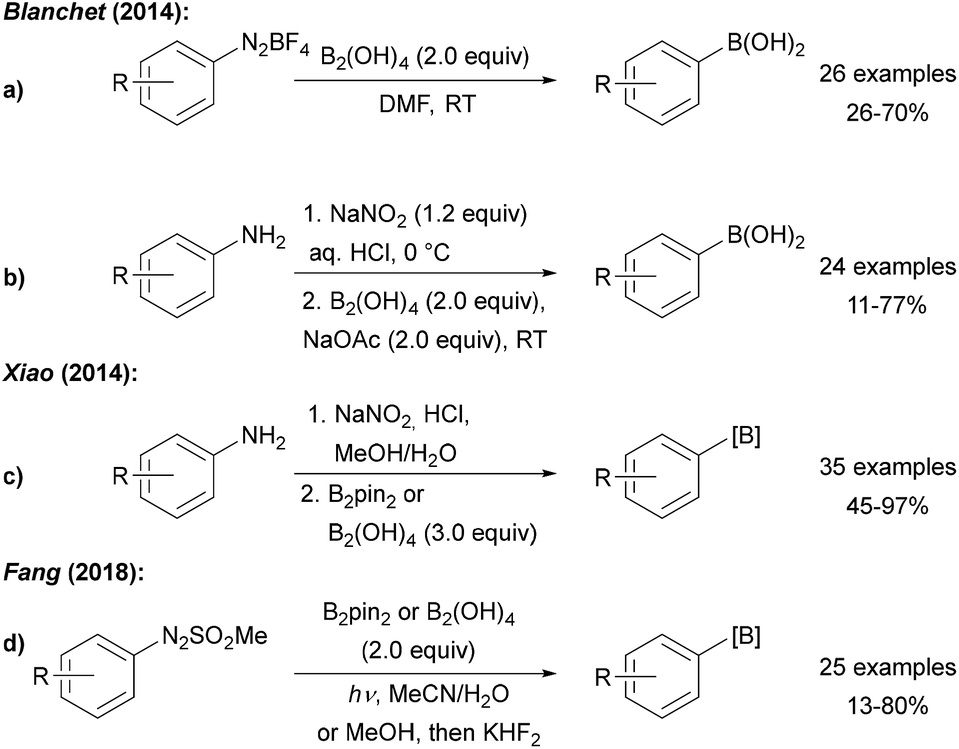 | ||
| Scheme 3 Borylation of anilines and derivatives using diboronic acid reported by Blanchet (a and b), Xiao (c) and Fang (d). | ||
Glorius showed that benzotriazoles, electronically related to aryl diazonium salts, are efficiently converted to aryl boronates under loss of N2 using an Ir photoredox catalyst with B2pin2 as the radical trapping reagent (Scheme 4).26 Determination of the quantum yield (Φ = 64.7) clearly revealed that the Ir complex upon photoexcitation acts as a smart initiator for this efficient radical chain reaction.27
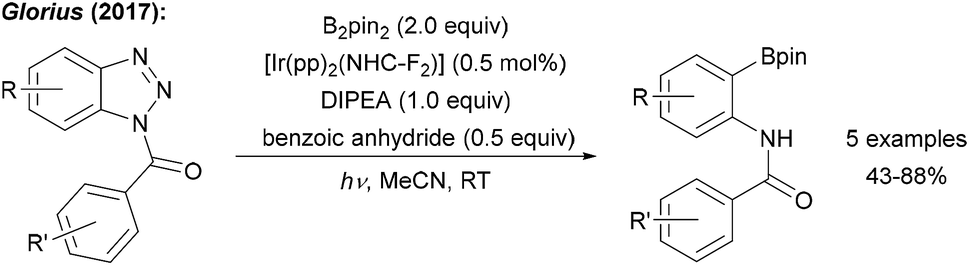 | ||
| Scheme 4 Deaminative borylation of benzotriazoles using a photoredox catalysis with B2pin2 and diisopropylethylamine (DIPEA) under irradiation with blue LEDs. | ||
2.2. Dehalogenative borylation (sp2)
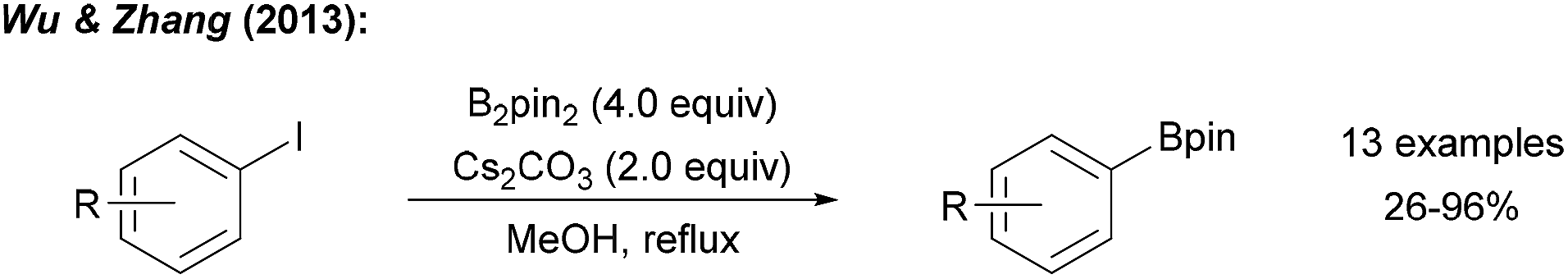 | ||
| Scheme 5 Thermally initiated radical borylation of aryl halides using B2pin2 and a Lewis-basic activator. | ||
A series of experiments provided insights into the possible mechanism of this interesting transformation. Addition of CuI and nBu3P, a typical catalyst system for C–B bond formation,29 did not improve the reaction efficiency, indicating that metal catalysis by Cu-traces is likely not occurring. The presence of TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl), a well-established radical trapping reagent, inhibited the reaction. However, borylation could be restarted upon renewed addition of the diboron reagent. Therefore, authors argued that the process is likely not radical in nature. However, it must be considered that TEMPO might promote the decomposition of B2pin2.30 Starting with para-iodotoluene, formation of toluene and a biphenyl derivative was later observed under these reaction conditions strongly supporting the presence of aryl radicals.12 Since the aryl iodides applied by Wu and Zhang are not particularly activated for a nucleophilic substitution at the ipso-carbon atom, we suggest a radical chain reaction for this transformation (Scheme 6a). We consider this mechanistic scheme of general importance for this perspective and will therefore present it in a generalised form.
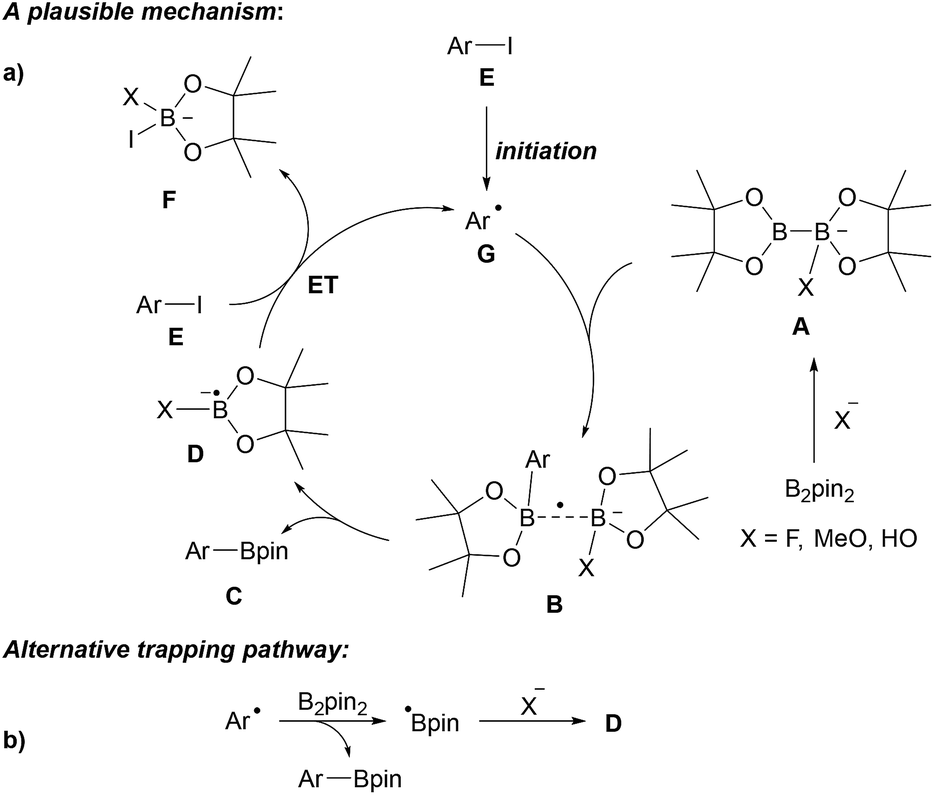 | ||
| Scheme 6 Base mediated borylation of aryl halides and our suggested general mechanism (Ar = aryl) (a). Alternative mechanism (b). | ||
It is well established that anionic Lewis bases coordinate B2pin2 to provide adducts of type A.12 These adducts are activated towards trapping of aryl radicals G to give the corresponding intermediates B. The weak single electron σ bond13,14 in B readily cleaves to give the boronic ester C along with the radical anion D, which can transfer an electron to the starting aryl halide E. C–I bond mesolytic cleavage then leads to the transient aryl radical G and anion F, sustaining the chain reaction. The fate of the second Bpin moiety in the diborane (see F) was confirmed by studies of Lin, Kleeberg and Marder.12 This mechanistic picture is likely valid for most aryl radical borylations with B2pin2 that are conducted in the presence of an ionic activator such as an alcoholate, a carbonate, fluoride or hydroxide in polar solvents. Notably, precoordination is not necessarily required as Wang observed formation of the phenyl boronic ester through thermal decomposition of dibenzoylperoxide (BPO) in the presence of B2pin2 in MeCN in 37% yield,10 albeit benzoate formed in situ as a byproduct might take that role in this particular case. In any case, as an alternative mechanism, direct trapping of the aryl radical with B2pin2 must also be considered (see Scheme 6b).
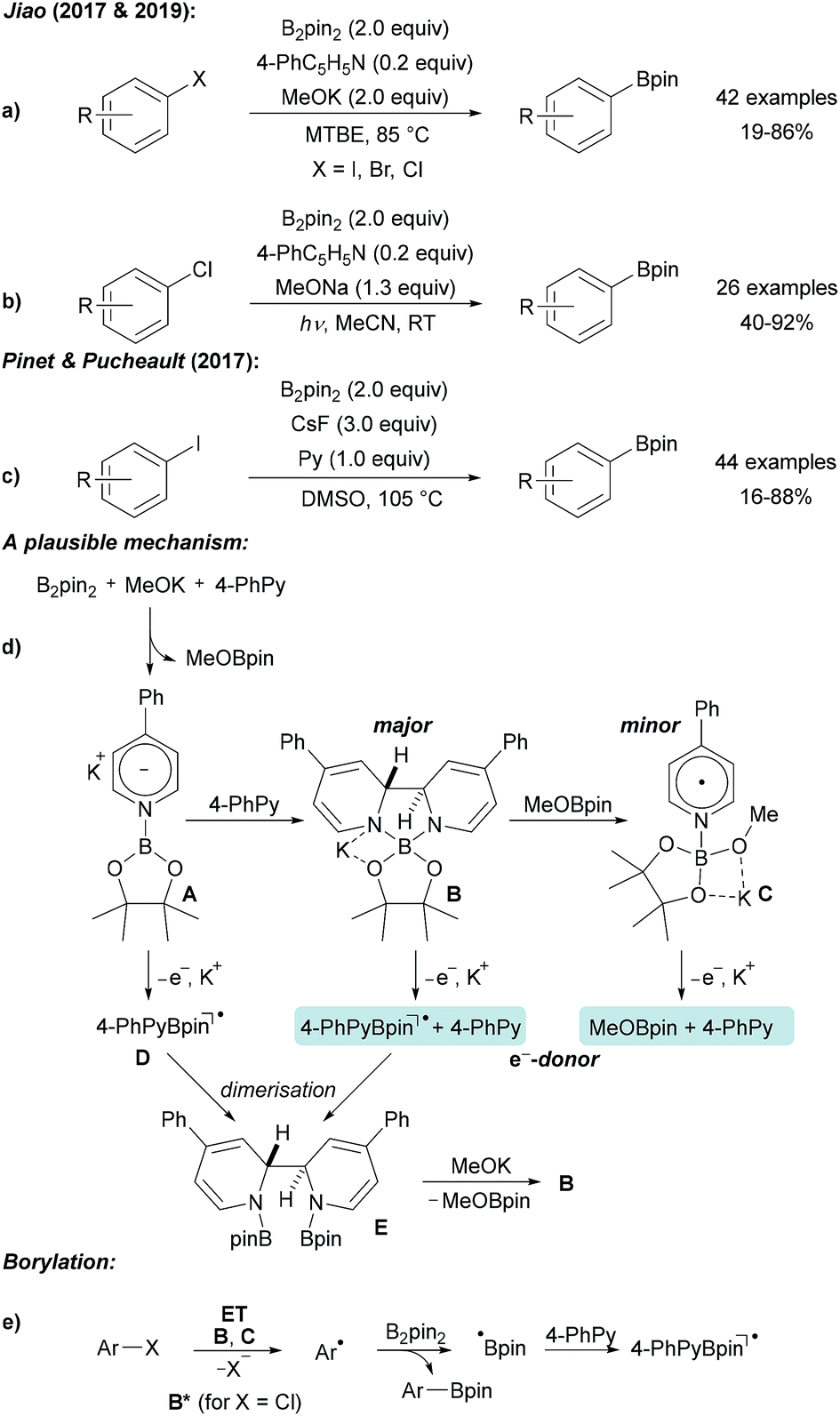 | ||
| Scheme 7 Pyridine mediated borylation of aryl halides enabled by the formation of electron donors derived from diboron and 4-phenyl pyridine (a and b). Related protocol (c) and mechanism (d and e). | ||
The authors first proposed a radical–radical coupling between an aryl radical and a pyridine stabilised boryl radical that was considered as a persistent radical, which is especially true for its 4-cyanopyridine derivative.33–38 Nevertheless, recent findings led to a different mechanistic picture.32,39
B2pin2, 4-phenyl pyridine (4-PhPy) and KOMe react via pyridyl anion A with a second equivalent of 4-PhPy to adduct B (Scheme 7d), which was fully characterised by X-ray diffraction analysis (as its DME complex) and by NMR spectroscopy. In addition, the KOMe adduct of pyridine boryl radical (see C) was detected by EPR spectroscopy. Radical C is formed by the reaction of B with MeOBpin. Both B and C are excellent electron donors and efficiently reduce aryl bromides and iodides to give the corresponding aryl radicals. Through photo excitation, the redox potential of B is significantly decreased thus allowing even the reduction of chloro arenes.32,39 Addition of the thus formed free aryl radicals to B2pin2 gives rise to the borylated product. The boryl radical, generated in the homolytic substitution as a byproduct, likely adds to the 4-PhPy to give D contributing to the regeneration of B. ET from B gives a stabilised boryl radical D that dimerises to E. By the addition of KOMe, B can be regenerated.
A similar process was introduced by Pinet and Pucheault who found that CsF in combination with pyridine upon heating in DMSO also allows preparing boronic esters from aryl iodides and B2pin2 (Scheme 7c).40 Notably, simple pyridine also activates B2pin2 for borylation of aryl iodides, as disclosed by Jiao.31
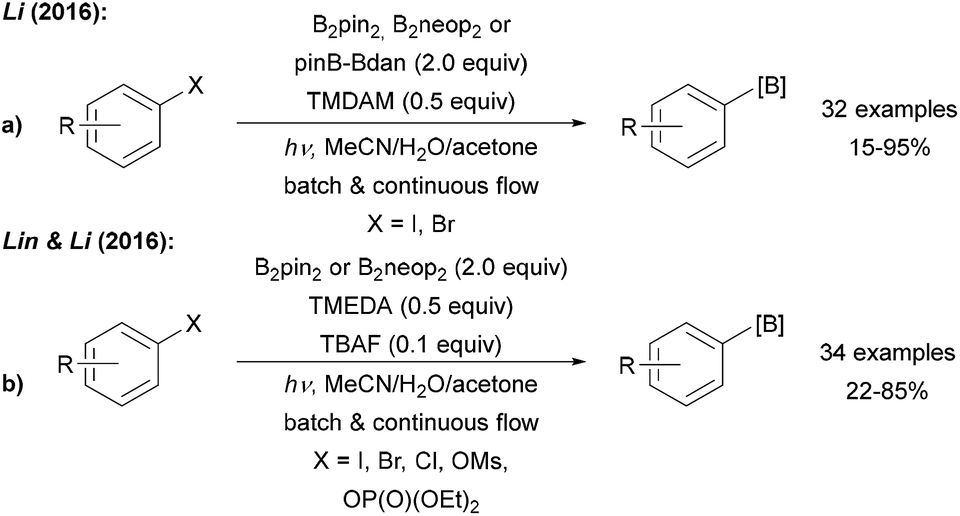 | ||
| Scheme 8 Light mediated borylation of aryl halides, mesylates and phosphates using various diboron reagents under basic conditions with mixed (a) and symmetric (b) diborons. | ||
Larionov was first to use the free diboronic acid for aryl radical borylation upon UV (254 nm) irradiation of aryl halides in MeOH (Scheme 9a).9,43 Along with aryl iodides, bromides, and chlorides also quaternary ammonium salts can be used as formal aryl radical precursors. In contrast to the examples discussed above, no additional base is required. The authors suggested different mechanistic pathways for these transformations as a function of the leaving group at the arene moiety (Scheme 9).
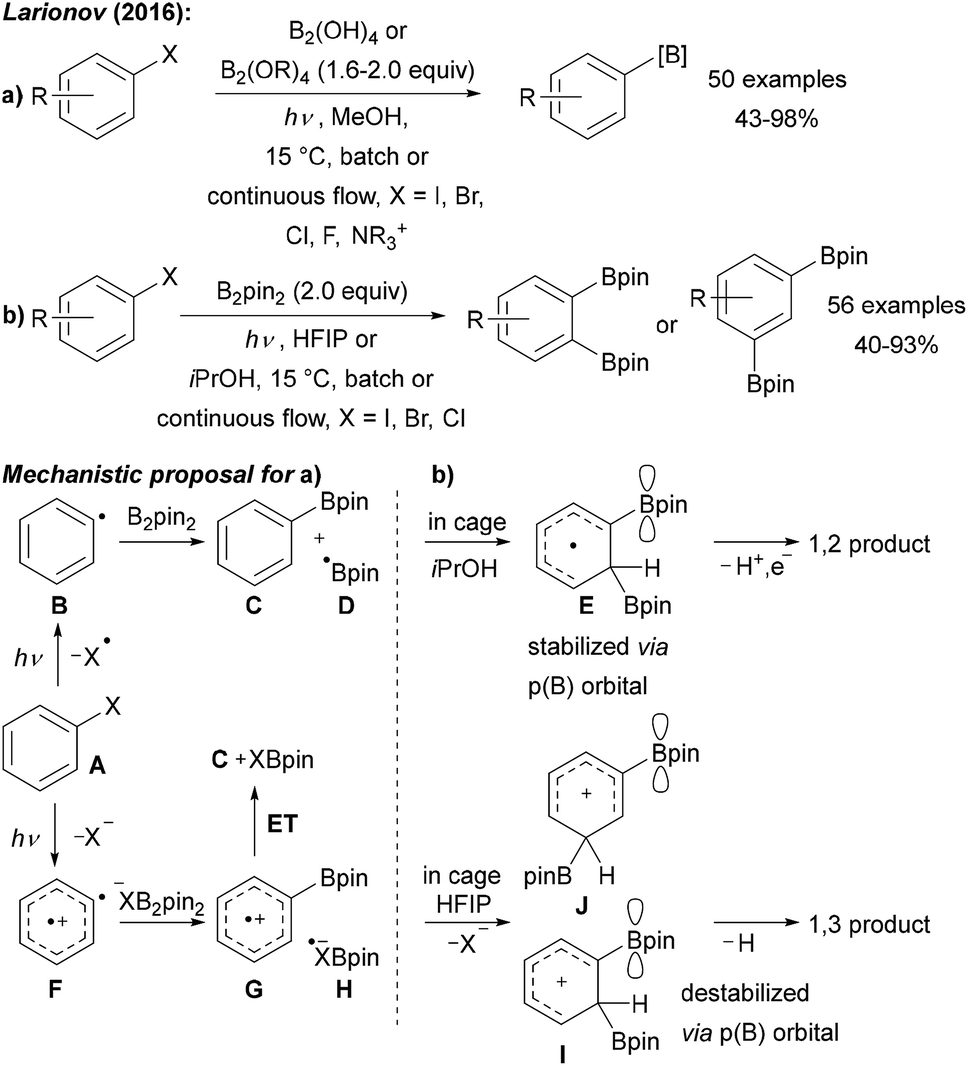 | ||
| Scheme 9 (a) Borylation of aryl halides using various diboron reagents and initiation by UV (254 nm) irradiation. (b) Solvent dependent diborylation of aryl halides. | ||
Upon excitation, bromo- and iodoarenes A preferably homolyse to give the aryl radicals B. However, for fluoroarenes as substrates, heterolysis to a triplet aryl radical cation F was suggested to be the preferred pathway (Scheme 9). The aryl radical intermediates are then trapped with diborane or with a Lewis base complexed diborane to give the boronic ester C and boryl radical D. Radical cation G is formed from triplet F upon reaction with a XB2pin2-adduct anion and G is then reduced to C by H. D might be trapped with the Lewis basic solvent to give a radical adduct that either reduces the aryl halide in the ground or excited state to regenerate the aryl radical.9
Interestingly, changing the solvent to HFIP (hexafluoro-2-propanol) or iPrOH yielded selectively either 1,3- or 1,2-diborylated arenes (Scheme 9b).44 Two different reaction pathways were suggested to explain this interesting reactivity (Scheme 9a).44 In isopropanol, the first C–B bond formation occurs via the aryl radical B to give the boronic ester C and the boryl radical D. Likely enabled by a cage effect, B-radical D then adds to the mono borylated intermediate C to give the p(B)-orbital stabilised cyclohexadienyl radical E which can undergo deprotonation and oxidation to finally afford the isolated 1,2-addition product. The authors suggested that the very polar HFIP supports a heterolytic pathway to generate a triplet aryl radical cation F.45 Trapping with precoordinated B2pin2 gives the intermediate radical cation G that can add to boryl radical anion H to generate the intermediate I upon release of X−. The p(B)-orbital destabilises this cationic intermediate I in case of a 1,2-addition explaining the change in regioselectivity upon switching from iPrOH to HFIP. Finally, deprotonation delivers the 1,3-addition product with good regioselectivity.
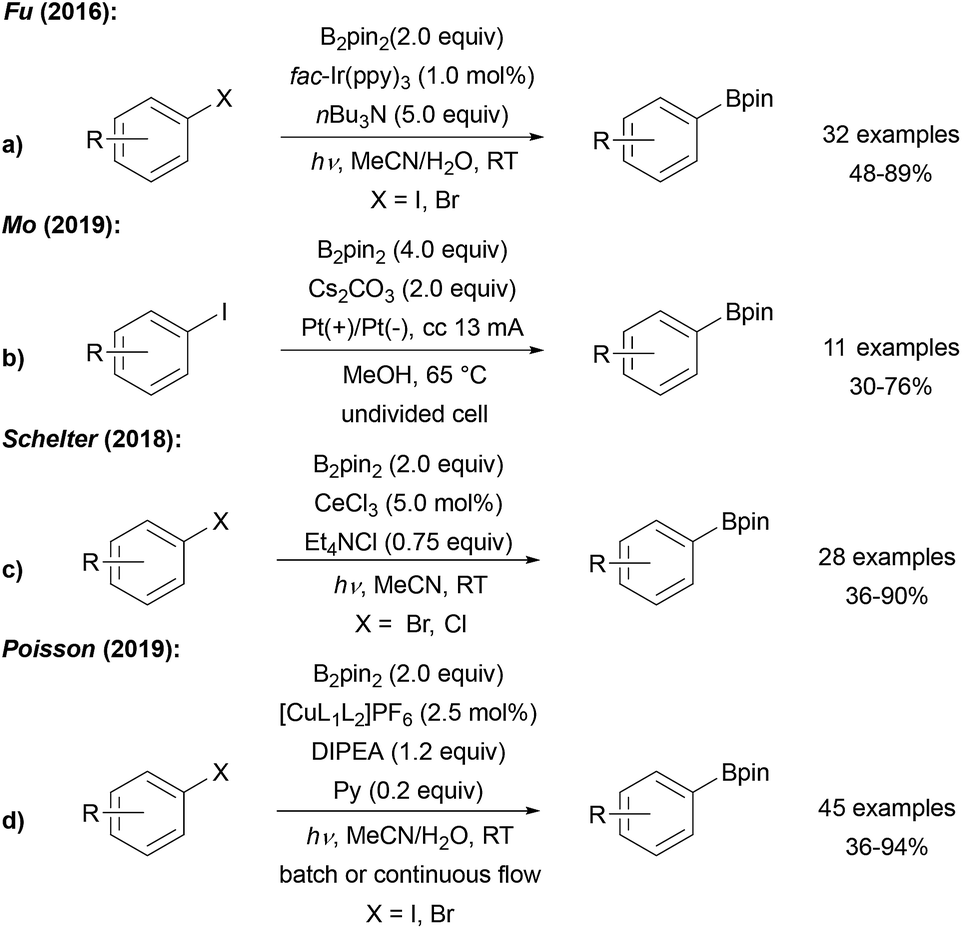 | ||
| Scheme 10 Photoredox catalyst (a), electro chemistry (b), [CeIIICl6]3− (c) or Cu photoredox catalyst ((d), L1 = DPEphos, L2 = DMEGqu) mediated borylation of halo arenes. | ||
The IrIII species gets regenerated from the intermediately formed IrIV complex by SET-reduction with the trialkyl amine. Alternatively, the IrIV complex might also be reduced by radical anion D in a chain reaction (see Scheme 6). In such a scenario, the Ir-complex would then serve as a smart initiator to start a radical chain reaction.27 Along these lines, initiation of the chain can also be achieved electrochemically under basic conditions in MeOH, as reported by Mo (Scheme 10b).47 Borylation of aryl iodides was also realised by Schelter using CeCl3 to initiate/mediate the transformation (Scheme 10c).48 The authors stated that the photo-excited [CeIIICl6]3− is formed in solution under UV light irradiation, which then reduces the aryl halide.
Poisson recently reported a borylation of aryl halides, several vinyl halides and also alkyl halides using a Cu photoredox catalyst with B2pin2 in the presence of DIPEA (EtNiPr2) and pyridine.49 Based on mechanistic studies, ET from a Cu0 species to the halide to generate the corresponding C-radical was suggested. The Cu0 catalyst is then regenerated via reductive quenching of the thus formed photoexcited CuI complex with DIPEA. As in all these redox catalyst mediated borylations of aryl halides (in particular for iodides), a chain reaction has to be considered where the Cu-species acts as a smart initiator and the reducing radical anion D (see Scheme 6) sustains a chain reaction.
Recently, we showed that radical borylation of aryl iodides can readily be achieved under neutral conditions upon blue LED irradiation of the halides in the presence of B2cat2 in DMF (Scheme 11).50 Since the catechol boronic esters obtained as primary products are difficult to handle, transesterification with Et3N and pinacol was conducted. Via radical clock experiments, the rate constant for the trapping of an aryl radical with B2cat2 was measured to be 7.4 × 107 M−1 s−1. For a mechanistic discussion of this interesting transformation we refer to Scheme 14 below.
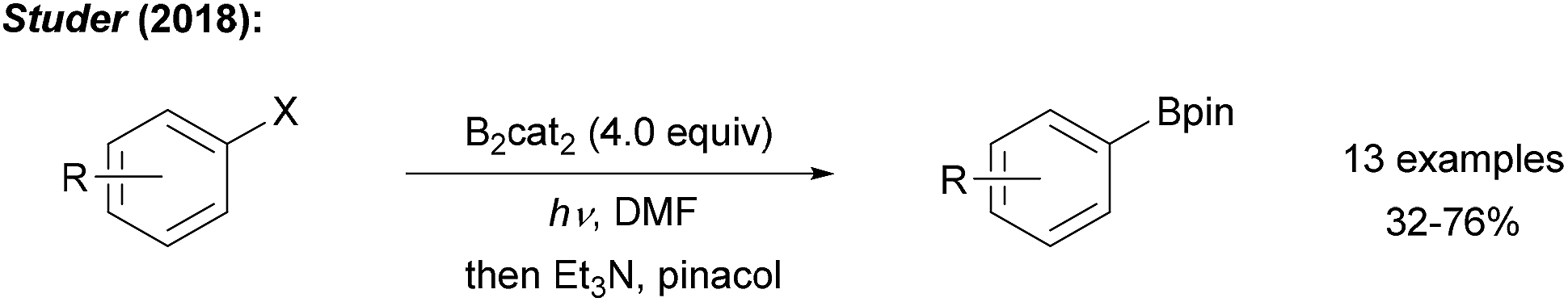 | ||
| Scheme 11 Metal free radical borylation of aryl iodides using B2cat2 in DMF under blue LED irradiation. | ||
2.3. Decarboxylative borylation (sp2)
Guided by the well-known Barton-PTOC-esters,51 Glorius reported that N-hydroxyphthalimide (NHPI) esters can serve as suitable precursors for aryl radicals in borylation reactions (Scheme 12a).52 Easily accessible and stable NHPI esters were reacted with B2pin2 under basic conditions under blue LED irradiation to give various boronic esters in moderate to very good yields. Recently, the same group described the use of aryl 4,4′-difluorobenzophenone oxime esters for photosensitised decarboxylative aryl radical generation.53 Shang and Fu found that a similar transformation is feasible using tert-butyl isonicotinate as the activator for B2pin2 at 110 °C also enabling the borylation of alkenyl NHPI esters (Scheme 12b).54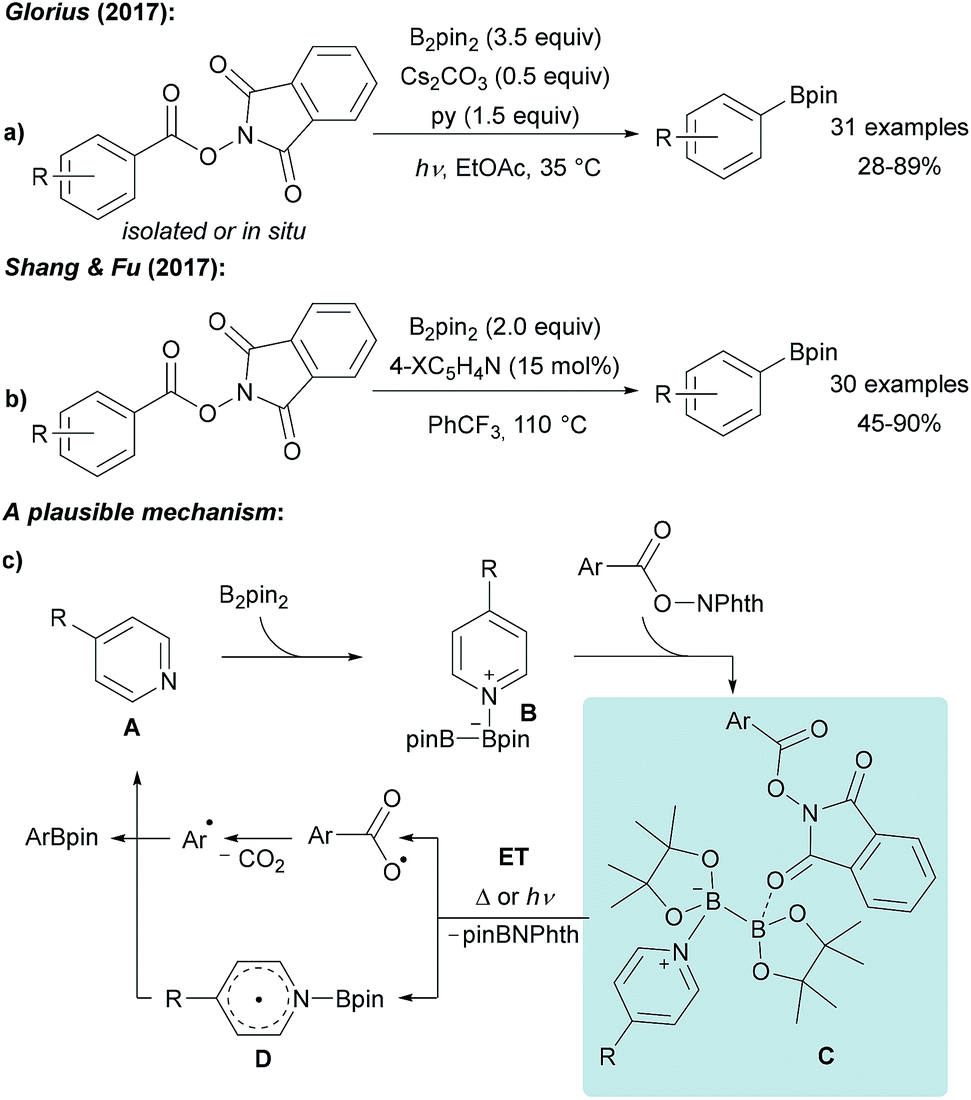 | ||
| Scheme 12 Decarboxylative borylation of NHPI esters using B2pin2 under basic conditions and irradiation with blue LEDs (a) or mediated by tert-butyl nicotinate (X = tBuO2C, (b)). Mechanism (c). | ||
The suggested mechanism involves coordination of pyridine A to B2pin2 generating intermediate B. For the light mediated process, Stern–Volmer fluorescence quenching studies indicated that initiation proceeds via an ET from B to the photo-excited NHPI (Scheme 12c).52 Considering the thermal process, the authors proposed that a dual activation of the diboron is a requirement which is achieved by coordination of the B2pin2/pyridine Lewis acid/base complex with the NHPI ester resulting in intermediate C.54 Intramolecular ET then leads to decarboxylative aryl radical formation and generation of the long-lived radical D.33–37 Subsequent recombination of these radical species yields the corresponding product boronic ester thereby regenerating the pyridine A.
2.4. Deoxygenative borylation (sp2)
The first deoxygenative borylation via sp2-hybridised carbon radicals was disclosed recently by Li (Scheme 13). Aryl triflates were successfully borylated in moderate to good yields upon treatment with B2pin2, TMDAM (N,N,N′,N′-tetramethylmethanediamine) and NaI under UV (254 nm) irradiation.55 The authors suggested a mechanism involving light assisted aryl radical generation by ET from the iodide to the triflate followed by C–O bond mesolytic cleavage and subsequent reaction with B2pin2. In analogy, a vinyl triflate was converted to its boronic ester.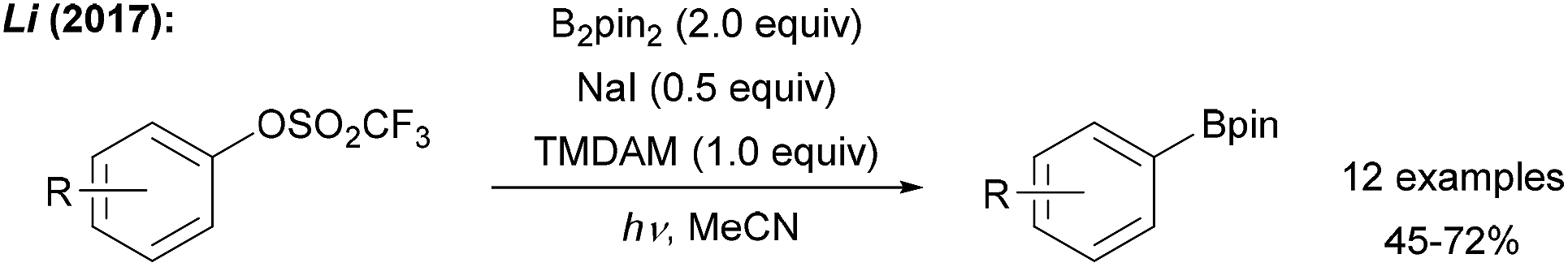 | ||
| Scheme 13 Deoxygenative borylation of aryl triflates under UV irradiation. | ||
3. Radical C(sp3)–B bond formation
As compared to the reaction with aryl radicals, trapping of alkyl radicals with diboron compounds is less efficient and often requires a large excess of the diboron reagent. The trapping of primary alkyl radicals with B2pin2 in DMF was reported;56 however, for the borylation of secondary alkyl radicals under these conditions only trace product formation was achieved. In contrast, diboronic acid reacts rather efficiently with secondary alkyl radicals.56 Presumably the most powerful reagent for the metal free borylation of alkyl radicals is B2cat2 in the presence of a Lewis base (most commonly an amide based solvent), first described by Aggarwal.57 Even selected tertiary alkyl radicals can be borylated under these conditions.57,58 Since the catechol boronic esters are difficult to handle, transesterification with Et3N and pinacol is usually required. The general mechanism for such alkyl radical borylations with B2cat2 is presented in Scheme 14 and specific examples will be discussed in the ensuing chapters. Initiation generates an alkyl radical A from a suitable radical precursor with a leaving group (LG), which adds to B2cat2 to give an adduct radical B. Coordination of the Lewis basic solvent leads to a radical C that features a weak B–B one electron σ bond.13,14 B–B bond cleavage eventually releases product D and the Lewis base stabilised radical E. DFT studies showed that the highest spin density in E is located at the carbon atom of the solvent represented by resonance structure F.50 Atom/group transfer (for iodides, NHPI esters)50,57 or ET (Katritzky salts)59,60 then generates the alkyl radical A from the starting material G sustaining the chain.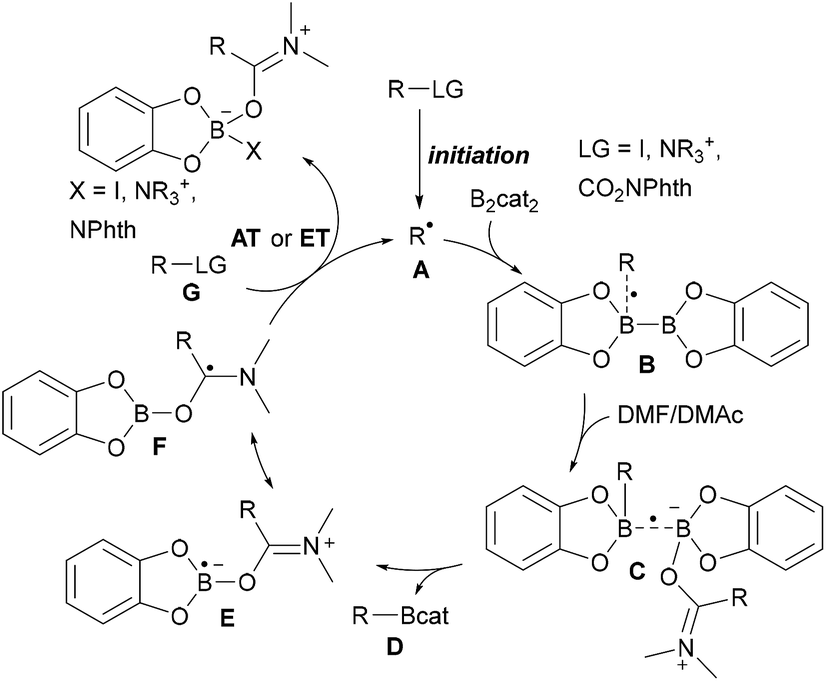 | ||
| Scheme 14 General mechanism for radical borylations involving alkyl radical trapping with B2cat2 in DMF or DMAc. | ||
3.1. Decarboxylative borylation (sp3)
NHPI esters have been shown to be highly valuable alkyl radical precursors.61 Along these lines, Aggarwal reported a metal free borylation of alkyl NHPI esters using B2cat2 in DMAc under blue LED irradiation (Scheme 15a).57 Due to the mild reaction conditions, the protocol tolerates many functional groups and was successfully applied to the borylation of primary, secondary and selected tertiary carboxylic acid derivatives including natural product and drug derived NHPI esters. The borylation of tertiary alkyl radicals seems to be limited to more reactive strained rigid systems. Mechanistically, this valuable process follows the general picture displayed in Scheme 14 (LG = CO2NPhth). Initiation is achieved by dual coordination of DMAc and the NHPI ester to B2cat2. Photo-excitation triggers homolysis of the weak B–B bond of this complex followed by decarboxylation leading to the corresponding alkyl radical (Scheme 15b). Phthalimide transfer from the starting material G to radical F (Scheme 14) and fragmentation of CO2 propagate the chain.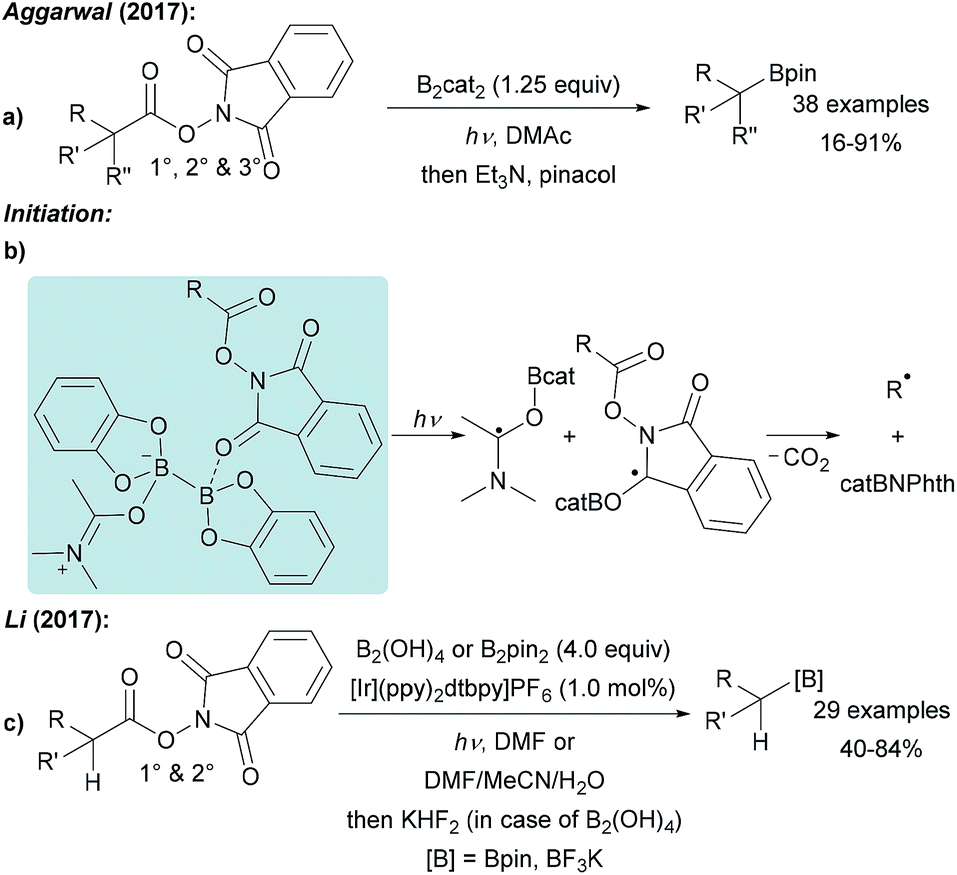 | ||
| Scheme 15 Decarboxylative borylation of alkyl carboxylic ester via the NHPI esters using B2cat2 under blue LED irradiation (a) or with additional photo redox catalyst and B2pin2/B2(OH)4 (c). Initiation (b). | ||
NHPI esters were also borylated by Li using a photoredox catalyst, B2pin2 or B2(OH)4 in DMF or a polar protic solvent mixture under visible light irradiation (Scheme 15c).56 Alkyl radical generation is achieved by ET from the photo-excited Ir complex to the NHPI ester followed by fragmentation and loss of CO2. C–B bond formation occurs as discussed in Scheme 6. Notably, this protocol represents one of the very few examples of a successful borylation of primary and secondary alkyl radicals with a diboron other than B2cat2.
3.2. Dehalogenative borylation (sp3)
Our group first reported a mild metal free borylation of primary and secondary alkyl iodides using B2cat2 in DMF under blue LED irradiation (Scheme 16a).50 These reactions that show excellent functional group tolerance are very easy to conduct and high yielding. Radical clock experiments using the 5-exo-cyclisation of the 5-hexenyl radical62 revealed that the second order rate constant for the borylation of a primary C-radical with B2cat2 in DMF is 3.8 × 106 M−1 s−1 thus representing a very fast transformation (Scheme 16b). According to DFT calculations, the mechanism follows the general proposal presented in Scheme 14 (LG = I). Initiation of these efficient chain reactions is achieved by photolysis of the starting alkyl halides and iodine atom transfer from G to F maintains the chain. This protocol was later extended to the borylation of primary benzylic radicals by Melchiorre (Scheme 16c).63 SN2 reaction of a benzylic chloride or bromide with a dithiocarbonyl anion leads to a dithiocarbamate as in situ generated benzyl radical precursor. Generation of the benzylic radical is achieved by photolysis of the dithiocarbamate in a non-chain process and borylation occurs as discussed in Scheme 14. The authors suggested an ET from F to the thiocarbonyl radical thereby regenerating the dithiocarbonyl anion, which can be used in catalytic amounts (10 mol%).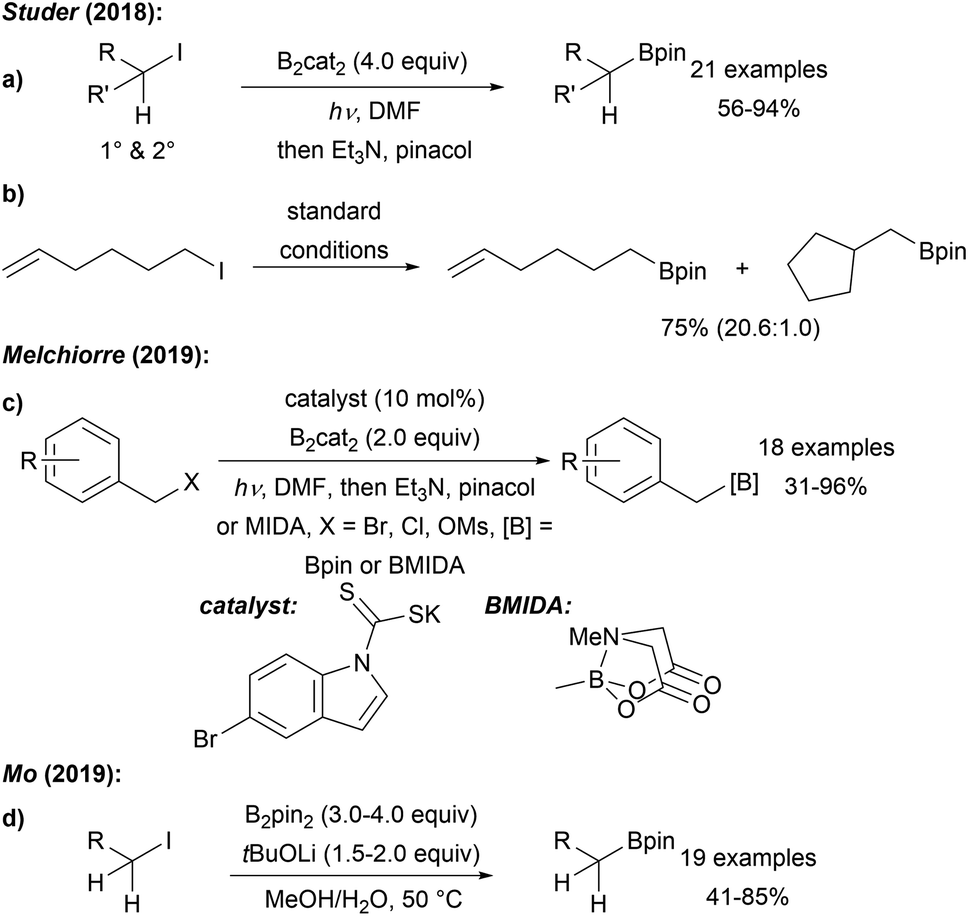 | ||
| Scheme 16 Metal free radical borylation of alkyl iodides (a) and radical probe experiment (b). Dithiocarbonyl anion enabled borylation of benzyl electrophiles (c) using B2cat2 in DMF and blue LED irradiation and base mediated borylation of primary alkyl iodides using B2pin2 (d). | ||
Very recently, Mo disclosed a metal free borylation of primary alkyl iodides using B2pin2 under basic conditions at slightly elevated temperature in MeOH/H2O via a radical pathway (Scheme 16d).64 However, for secondary substrates, the use of B2cat2 was generally necessary.
3.3. Deaminative borylation (sp3)
The use of stable Katritzky salts as alkyl radical precursors was recently disclosed using either Ni complexes or photoredox catalysts for ET-induced C-radical generation.65 The application of such pyridinium salts, that are easily accessible by condensation of alkyl amines with commercial pyrylium salts, in a radical borylation reaction was first reported by Aggarwal (Scheme 17a).59 Treatment of a Katritzky salt with B2cat2 in DMF under blue LED irradiation gives the corresponding alkyl boronic ester in moderate to excellent yield. A similar process was also developed by Glorius and both groups suggested a radical chain mechanism as discussed in Scheme 14.60 Initiation is achieved by light induced intramolecular ET from DMAc coordinated B2cat2 to the Katritzky salt (Scheme 17d), assembled in an electron–donor–acceptor (EDA) complex,66 leading to a dienyl radical and a radical cation. Fragmentation delivers the chain propagating alkyl radical and E (Scheme 14). Chain propagation occurs via ET from E (Scheme 14) to the Katritzky salt. Both groups determined the quantum yield to be significantly larger than 1 (Φ = 7.0 (ref. 59) − 27.8 (ref. 60)) strongly supporting that the transformation proceeds as a radical chain reaction.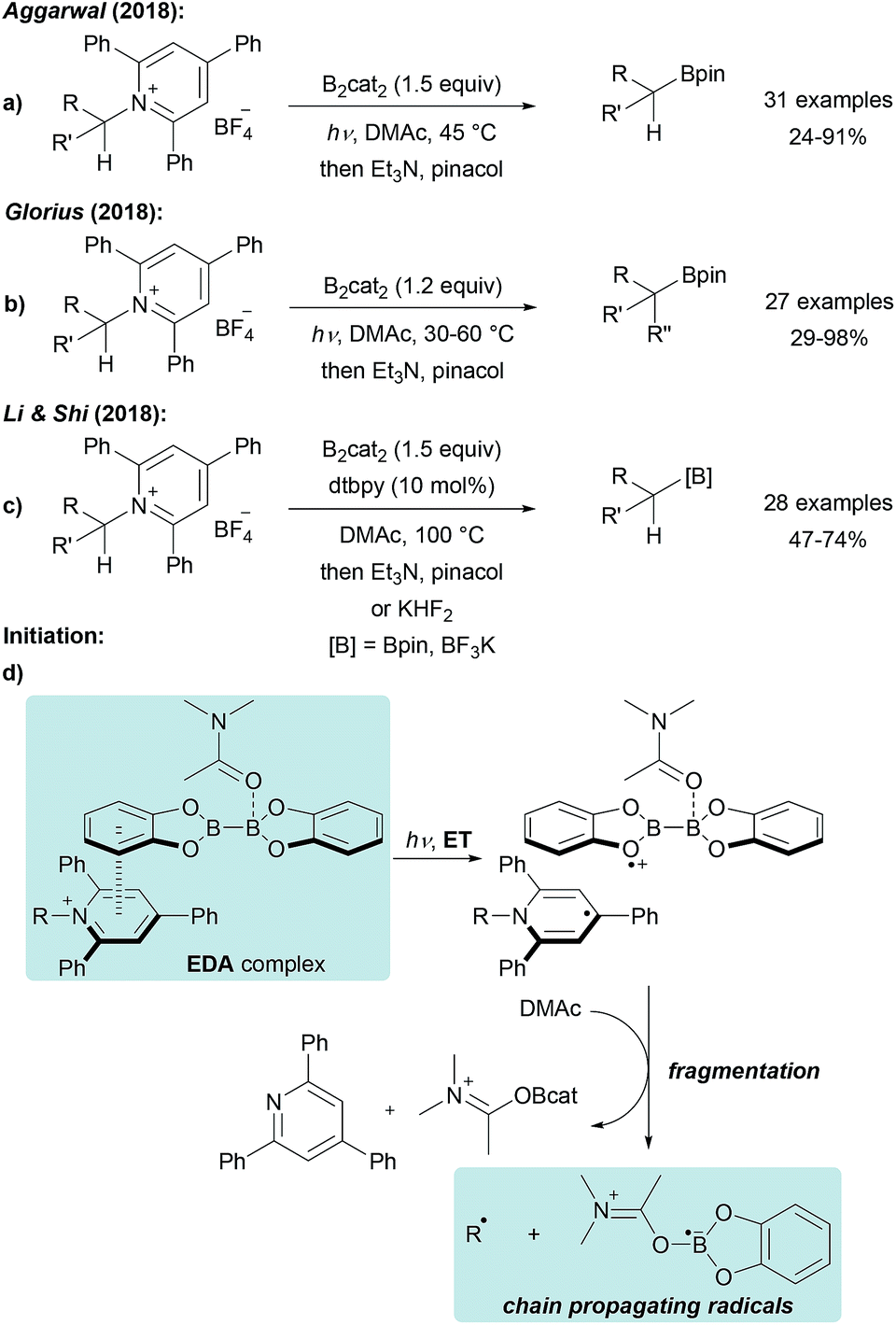 | ||
| Scheme 17 Deaminative borylation of primary alkyl amines via Katritzky salts using B2cat2 under visible light irradiation (a and b) or thermal initiation assisted by dtbpy (c). Initiation (d). | ||
Li and Shi disclosed a related protocol and used the same reagents at elevated temperature in the presence of a catalytic amount of dtbpy (4,4′-di-tert-butyl-2,2′-bipyridine, Scheme 17c).67 The modified protocol was successfully applied to the preparation of various primary and secondary boronic esters. The reaction appeared to be slower in the absence of the bipyridine additive and the authors suggested that dtbpy is involved in the initiation step, assisting the homolysis of B2cat2 in DMAc.
3.4. Deoxygenative borylation (sp3)
Encouraged by previous studies on the borylation of alkyl halides, we decided to search for suitable O-activating groups for the borylation of secondary and tertiary alcohols. Considering secondary alcohols, easily accessible xanthates were chosen and product formation was achieved by using tris(trimethylsilyl)silane (TTMSS) as a chain reducing reagent in combination with B2cat2 and AIBN (azobisisobutyronitrile) in DMF at elevated temperature (Scheme 18a).58 Alternatively, we found that thionocarbamates can be converted into boronic esters with TTMSS and B2cat2 in DMF under blue LED irradiation. For the borylation of tertiary alcohols, readily accessed methyl oxalates were chosen as tert-alkyl radical precursors.58,68 Efficient borylation occurred using an Ir-photoredox catalyst and B2cat2 in DMF under blue LED irradiation (Scheme 18b). Notably, tertiary propargylic alcohols were converted with complete regioselectivity to the corresponding allenyl boronic esters. A silyl radical mediated non-chain radical process was suggested for the xanthate route. The photoredox cycle for the Ir-catalysed process is presented in Scheme 18c. ET from the photoexcited IrIII complex to the oxalate ester gives rise to the alkyl radical A that is trapped by B2cat2 to boronate Dvia adduct radicals B and C. Intermediate radical E is then oxidised by the IrIV complex, thus regenerating the photoredox catalyst.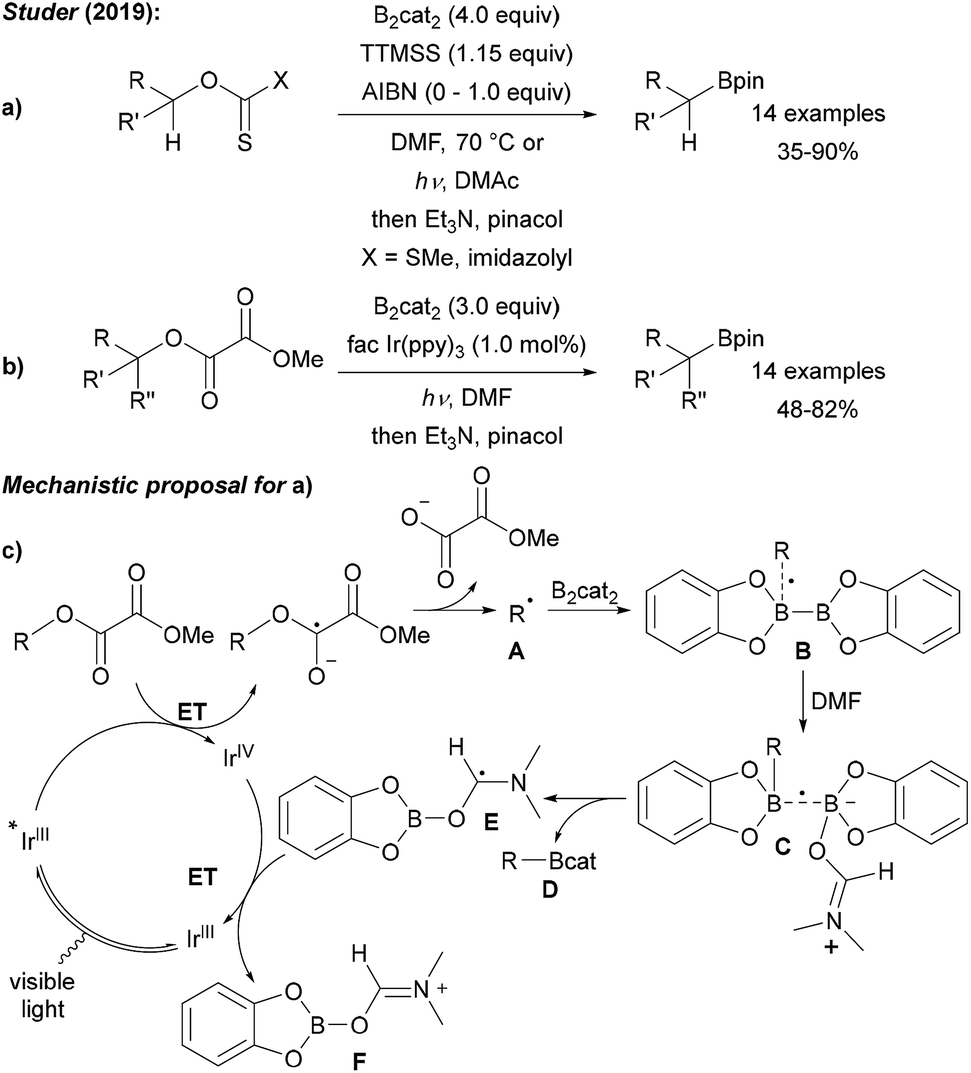 | ||
| Scheme 18 Radical borylation of secondary (a) and tertiary (b) alcohols via its xanthates, thionocarbamates and methyl oxalate esters. Mechanism (c). | ||
3.5. Radical carboboration (sp3)
Our group recently realised the first radical 1,2-carboboration of unactivated alkenes using B2cat2, various iodides (mostly perfluoroalkyl iodides) as radical precursors and irradiation with blue LEDs in DMF (Scheme 19a).14 DFT calculations indicated a radical chain reaction. Upon initiation, the alkyl radical adds regioselectively to the alkene generating a secondary alkyl radical A, which is trapped with B2cat2 as discussed above in Scheme 14. Addition of DMF induces B–B bond cleavage to provide the 1,2-carboboration product B along with intermediate C. Iodine atom transfer (AT) or ET between the iodide and C propagates the chain (Scheme 19b).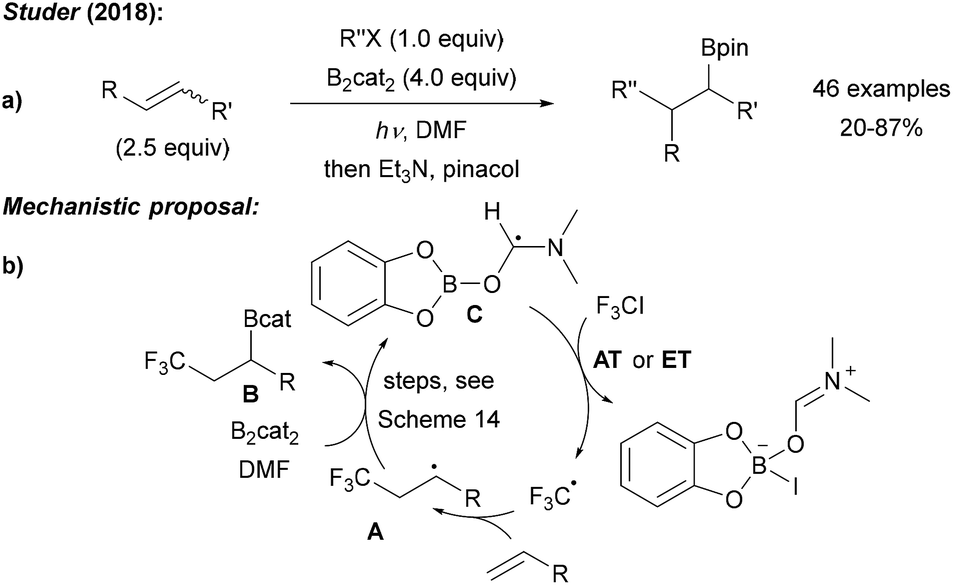 | ||
| Scheme 19 Transition metal free 1,2-carboboration of unactivated alkenes (a) and mechanism (b). | ||
3.6. Radial borylation via C–C bond cleavage (sp3)
The use of cyclobutyl ring opening reactions of imine derivatives for the generation of alkyl radicals was first described by Zard.69 This methodology was recently utilised in the borylative ring opening of cyclic oxime esters using tetrahydroxydiboron in DMAc to yield the borylated nitriles (Scheme 20).70 The authors used B2cat2 as the trapping reagent under visible light irradiation and suggested an initiation sequence according to the proposal of Aggarwal.57 Reductive N–O bond cleavage provides the ring-strained iminyl radical which fragments to the corresponding alkyl radical that is eventually trapped by the diboron to give the targeted distally borylated nitrile.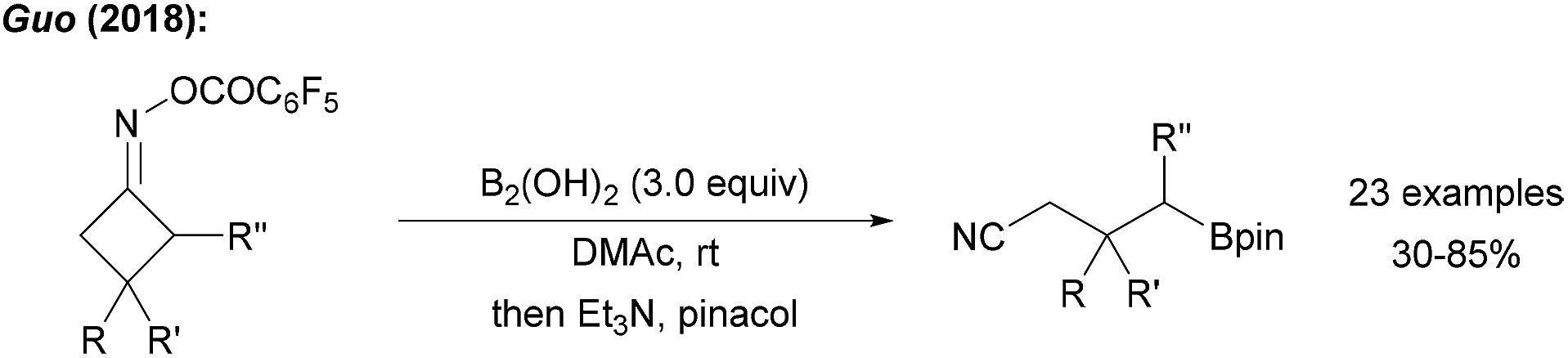 | ||
| Scheme 20 Transition metal free borylation of cycloketone oxime esters. | ||
4. Transition metal assisted borylation (sp2 & sp3)
CuI and NiI catalysed/mediated borylations of aryl halides, alkyl halides and alkyl NHPI esters have been studied extensively in the past. Along these lines, Zn, Fe, Mn, Co-mediated processes have also been reported. Most of these transformations use a diboron reagent in combination with a strong base for its activation and often radical probe experiments were conducted to proof the radical nature of these borylations. The key C–B bond forming step is usually achieved by a metal–boron species as a radical trapping reagent that is generated in situ from the diboron reagent. As for the metal free borylations, a general mechanism can be suggested for the Ni/Cu-catalysed transformations (Scheme 21).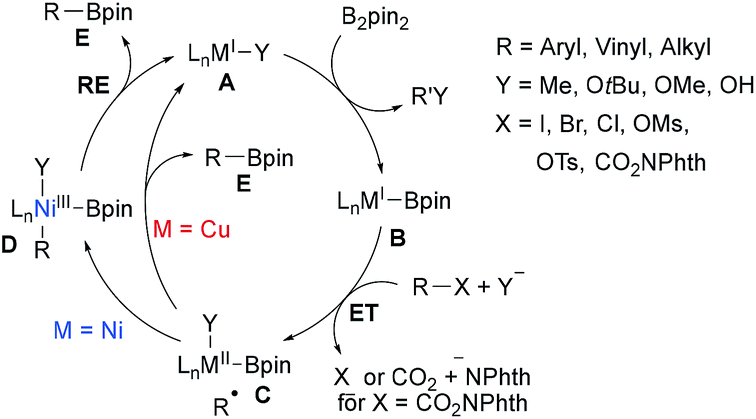 | ||
| Scheme 21 Proposed mechanism for the borylation of alkyl halides, sulfonates and NHPI esters using CuI or NiI salts under basic conditions. | ||
Alkyl/aryl halides and NHPI esters are known to be reduced by low-valent transition metal complexes to generate C-radical intermediates.71 Regarding the borylation, the MI complex A first undergoes ligand exchange with B2pin2, activated by a strong base, resulting in a metal boron complex B. ET from B to the halide or NHPI ester results in an alkyl radical and a persistent metal species C.38,72 For the following radical-metal crossover two pathways have to be considered. For the Ni-catalysed cascade, the C-radical will be trapped by complex C to afford the NiIII complex D, which undergoes reductive elimination (RE) to A releasing the product boronic ester E.73 Considering the Cu-process, C likely undergoes an outer-sphere ligand transfer to directly give product boronate E under regeneration of the CuI complex A, albeit formation of a CuIII species in analogy to the Ni-process cannot be fully ruled out.38,74
4.1. NHPI esters (Ni, Cu)
The Baran group showed that alkyl NHPI esters can be transformed to alkyl boronic esters in a Ni catalysed reaction using B2pin2 (precomplexed with MeLi) in THF/DMF (Scheme 22).75 Even though the authors did not comment on the mechanism in this initial study, alkyl radicals are likely involved since NHPI esters are easily SET-reduced.61 The key C–B bond formation is likely mediated by a Ni species since the trapping of secondary alkyl radicals with B2pin2 is known to be inefficient (see Chapter 6). This approach was recently further extended applying Cu-catalysis. The NHPI esters were borylated using Cu(acac)2, a large excess of LiOH·H2O, MgCl2 and B2pin2 in dioxane/DMF.76 Various NHPI esters derived from primary, secondary and tertiary carboxylic acid including natural products and drugs could be transformed by using this protocol and in most cases the Cu-process outperformed the Ni-process.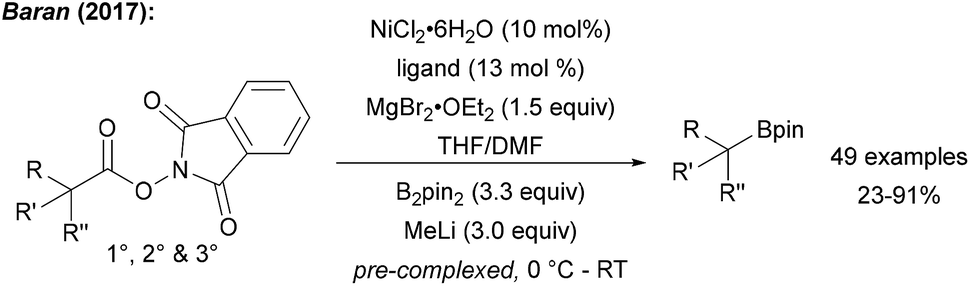 | ||
| Scheme 22 Ni catalysed decarboxylative borylation of various NHPI esters (ligand = 4-4′-dimethoxy-2-2′-bipyridine). | ||
4.2. Alkyl halides (Cu)
Steel, Marder and Liu described a Cu catalysed borylation of primary and secondary halides and tosylates using CuI, a phosphine ligand and a lithium alcoholate as the activator for B2pin2 (Scheme 23a).77 An analogous process was reported by Ito who used CuCl, xantphos and KOtBu for the same transformation (Scheme 23b).78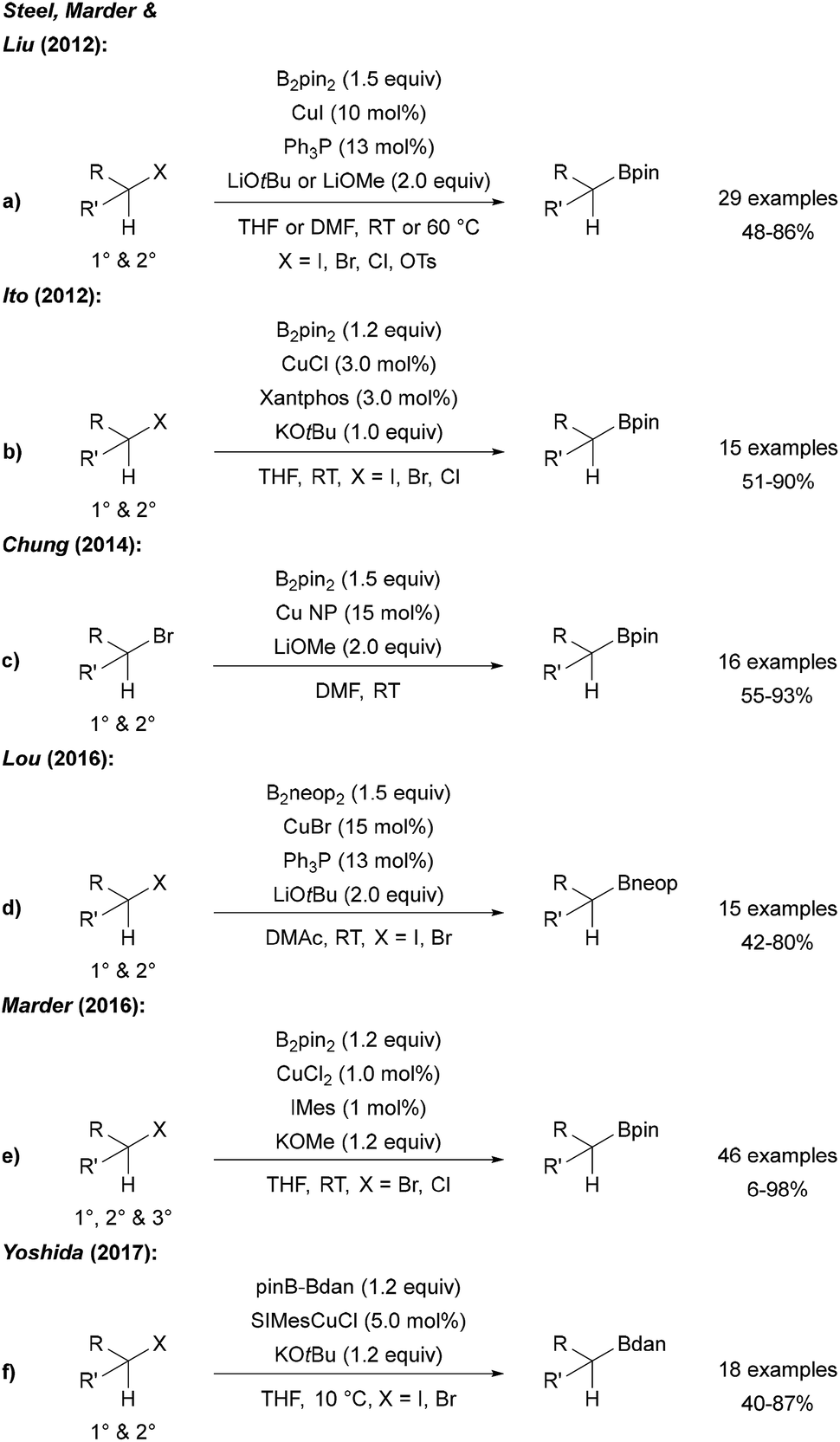 | ||
| Scheme 23 Cu catalysed borylation of alkyl halides using B2pin2 (a, b, c and e), B2neop2 (d) and pinB-Bdan (f). | ||
Cu nanoparticles have been applied to the borylation of alkyl bromides by Chung (Scheme 23c).79 Furthermore, Lou showed that this catalytic system is also applicable to the borylation of halides using B2neop2 as the boron source (Scheme 23d).80 As an alternative catalyst system, CuBr2 with a NHC ligand was introduced for the efficient borylation of alkyl bromides and chlorides (Scheme 23e),81 and Yoshida used an CuI NHC complex in combination with a mixed diboron reagent (pinB-Bdan) for the introduction of the Bdan-moiety (Scheme 23f). In the latter study, the authors also presented the borylation of aryl and vinyl halides using this asymmetric diboron reagent and a CuI phosphine complex.82 Of note, all of these Cu-protocols do not allow borylation of tertiary alkyl halides (except adamantyl iodide leading to the reactive adamantyl radical).
4.3. Alkyl halides (Ni, Fe, Mn, Zn)
Fu reported a very efficient Ni catalysed radical borylation of alkyl halides using NiBr2 and a PyBox ligand.83 Importantly, this catalyst system can also be used for the borylation of tertiary alkyl halides (Scheme 24a). The reaction was suggested to proceed according to the general mechanism discussed in Scheme 21. A similar protocol for the borylation of alkyl halides was developed by Liu (Scheme 24b),84 and Marder reported a ZnII NHC catalysed borylation of alkyl halides (Scheme 24c).85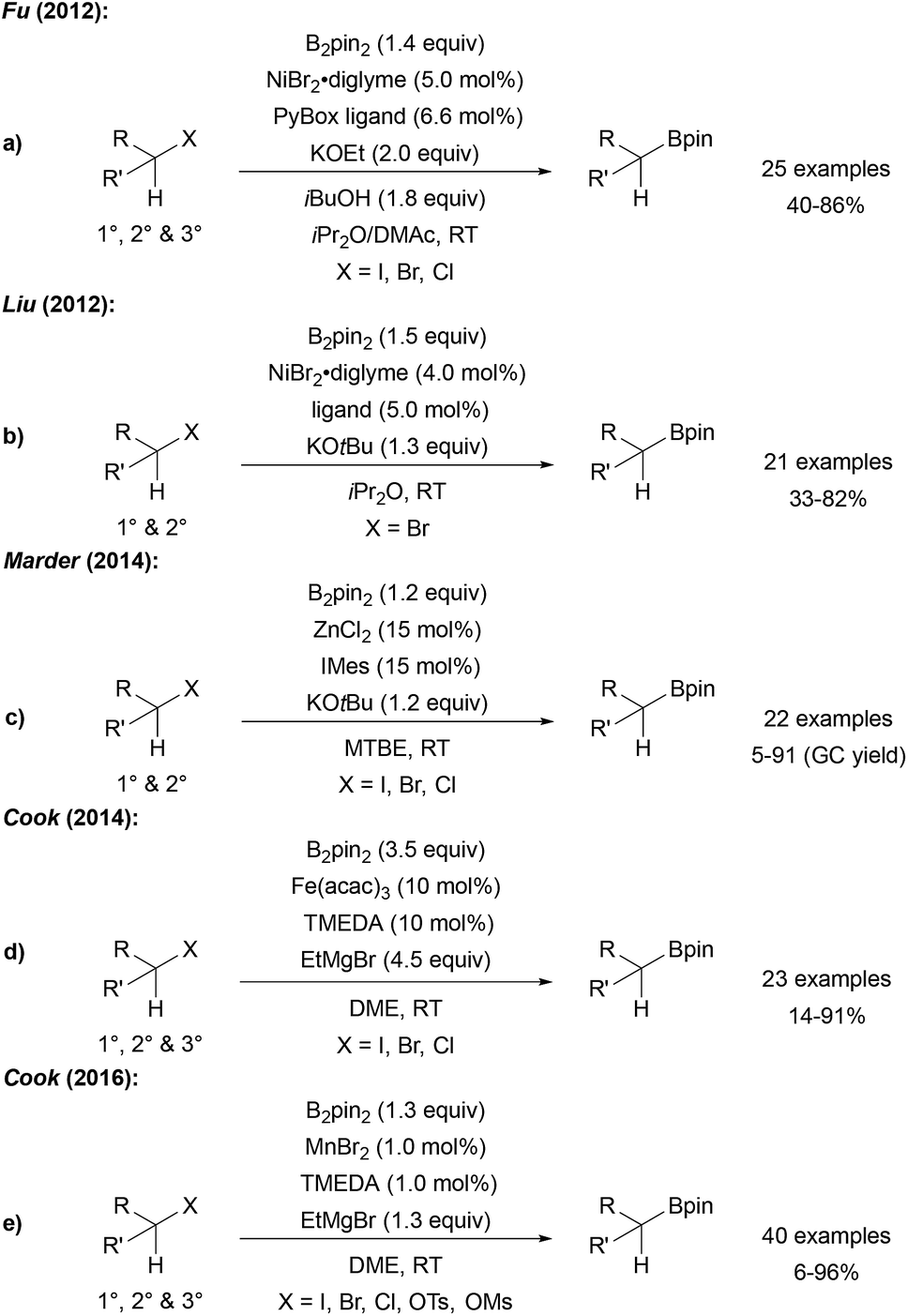 | ||
| Scheme 24 Ni (a and b), Zn (c), Fe (d) and Mn (e) mediated borylation of alkyl halides. | ||
Cyclisation/ring opening experiments revealed a radical based mechanism. Cook disclosed both FeIII and MnII catalysed alkyl halide borylations which are also applicable to the preparation of tertiary alkyl boronates (Scheme 24d and e).86
Comparable results with lower Fe catalyst loadings were described by Bedford.87,88 Considering the Mn-catalysed transformation, cyclisation experiments as radical probes revealed that uncyclised borylated products were formed exclusively at 0 °C. However, at 65 °C mostly cyclised boronic esters were obtained indicating the radical nature of the reaction under the latter conditions.
In contrast to the Ni, Mn and Fe catalysed processes, tertiary alkyl halides except adamantyl iodide did not engage in this reaction. It remains unclear, whether the active boron trapping reagent is a metal–boron species or the free KOtBu-activated diboron reagent.
4.4. Ring opening of cyclic peroxides
Guo reported a borylation of cycloalkylsilyl peroxides using a Cu catalyst system with B2pin2 to yield the corresponding ketones (Scheme 25).89 In the key step, reductively generated tertiary alkoxyl radicals readily undergo ring opening to give a primary alkyl radical that is trapped with a Cu–B complex.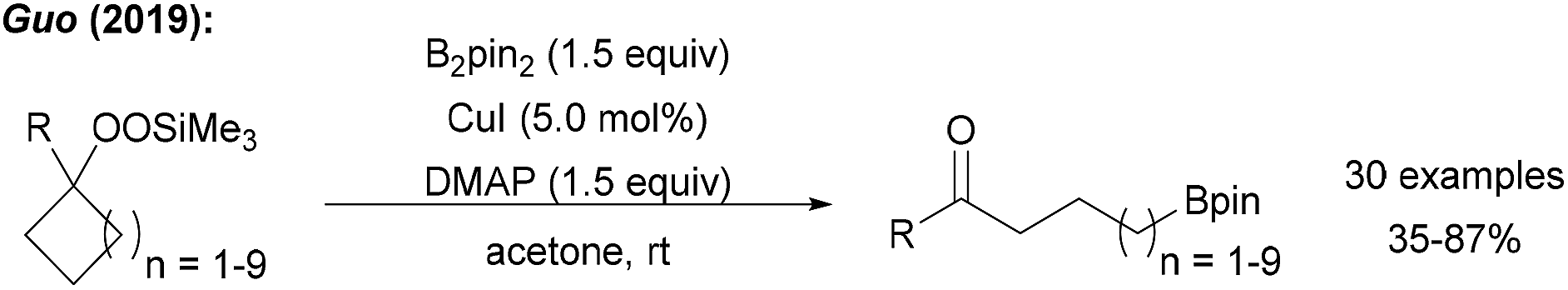 | ||
| Scheme 25 Cu catalysed borylative ring opening of cycloalkylsilyl peroxides. | ||
4.5. Borylation of alkyl halides with concomitant cyclisation
Ito reported the stereo and chemo selective radical cyclisation/borylation using B2pin2, CuCl, a phosphine or phenanthroline ligand and KOtBu (Scheme 26a).74 Shortly thereafter, comparable results were described by Liu (Scheme 26b).90 The cyclisation is a typical signature of a radical transformation. During the investigations on the cyclising borylation towards the synthesis of 3–5 membered rings or the borylation of alkyl bromides bearing terminal alkenes, Ito found that the reaction mechanism changes upon varying the ligand structure and overall reaction conditions.91 Hence, reaction of the alkyl bromide A with the CuCl/PCy3 system provided the borylation product Bvia a primary alkyl radical intermediate (Scheme 26c). However, with CuCl/xantphos as catalyst, A first undergoes a boro cupration to C followed by cyclisation to D in a non-radical pathway.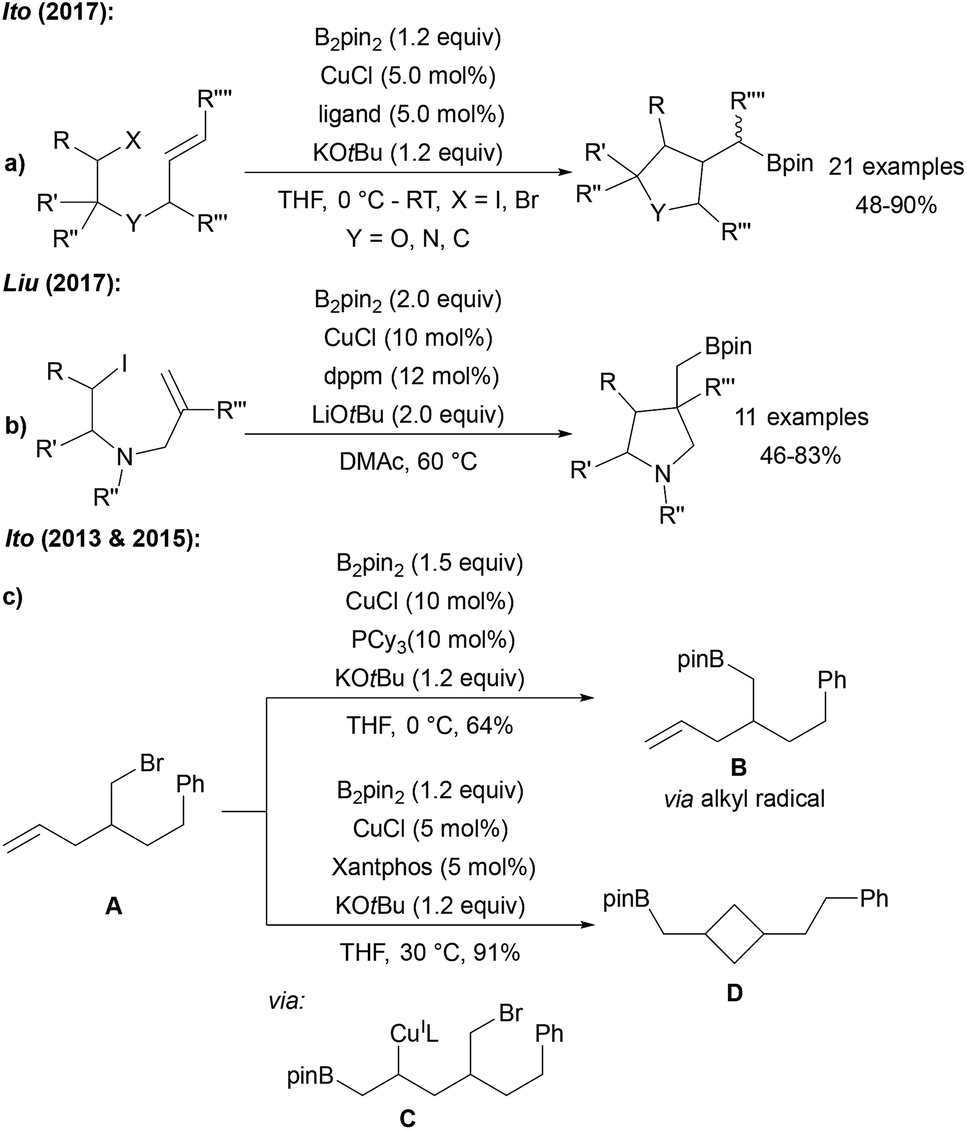 | ||
| Scheme 26 Cu mediated borylative cyclisation of alkyl halides with B2pin2 (ligand = PCy3, BPhen, (2-hydroxyphenyl)diphenylphosphine) via radical pathways (a and b). Non-radical boro-cupration (c). | ||
4.6. Aryl halides
Niwa and Hosoya disclosed a Cu catalysed ipso-borylation of fluoroarenes using a CuI phosphine complex, B2pin2 and CsF in toluene at elevated temperature (Scheme 27a).92 Despite the fact that attempted cyclising borylation failed, the authors proposed a radical type mechanism for this transformation that does not proceed via a free aryl radical. In the initiation step, the fluoroarene is reduced to the radical anion by a CuI–Bpin species. Nucleophilic substitution of the fluoride in the radical anion with CuI–Bpin gives the CuI–F complex and the aryl boronic ester radical anion that undergoes ET to the fluoroarene to provide the product ester thereby propagating the chain. The CuI boryl species is regenerated from the CuI–F complex via ligand exchange with B2pin2.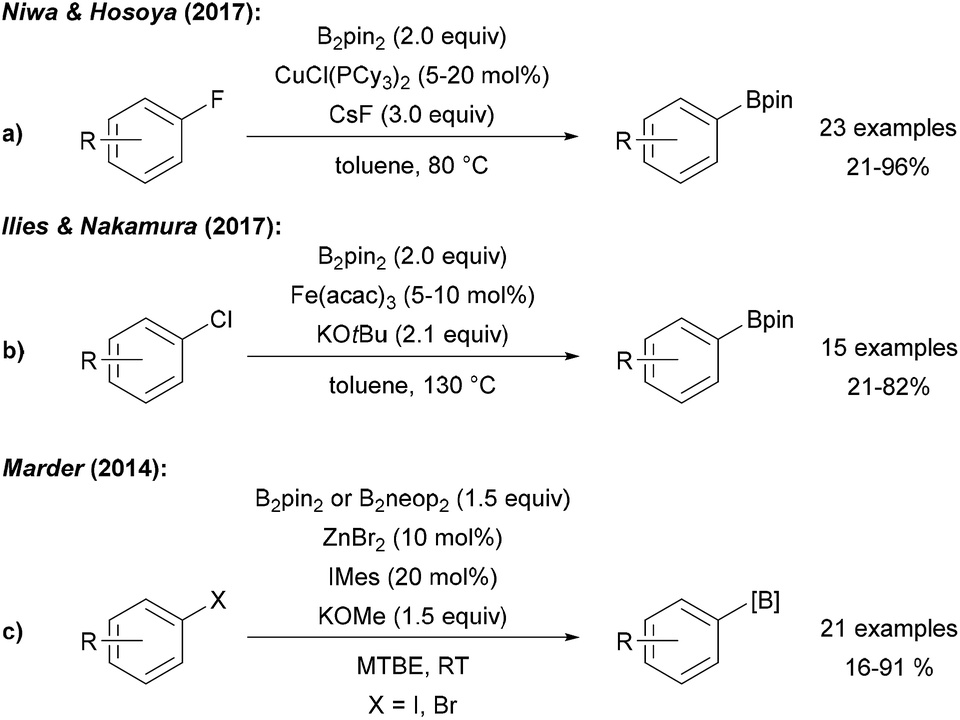 | ||
| Scheme 27 CuI initiated radical substitution of fluoro arenes (a), Fe mediated borylation of aryl chlorides (b) and Zn mediated synthesis of aryl boronates from aryl halides (c). | ||
A protocol for the borylation of aryl chlorides with Fe(acac)3/KOtBu at elevated temperature was reported by Nakamura (Scheme 27b).93 It was suggested that borylation proceeds via oxidative addition of the aryl chloride to an anionic FeII–Bpin species leading to a FeIV aryl complex which is in resonance with a FeIII–Bpin coupled aryl radical. B-group transfer to this aryl radical eventually gives the product boronic ester. Marder disclosed a Zn NHC complex catalysed borylation of aryl iodides and bromides using KOMe and B2pin2/B2neop2 at room temperature (Scheme 27c).94 Based on successful cyclisation experiments, a radical mechanism was considered. It was further described that several aryl halides can be dually C–X and C–H borylated utilising B2pin2, ZnCl2, a bipyridine ligand and KOMe at elevated temperature in MTBE. Reaction via the free aryne was ruled out and a radical mechanism featuring a Zn stabilised ligand radical anion that facilitates the diborylation via a base mediated nucleophilic substitution was suggested.95 Co catalysed borylation of halides, sulfonates and diazo compounds was developed by Hu and Huang using B2pin2, KOMe and a CoII complex. These transformations proceed at elevated temperature in MTBE. Reduction of the CoII complex with MeLi leads to the active Co species that mediates the borylation and aryl boronates were obtained in moderate to good yields. Even though the authors did not provide a clear mechanistic picture, a radical chain process that is initiated by ET from an intermediately generated CoI species is feasible.30
4.7. Ag mediated deoxygenative borylation
Fu recently reported a radical borylation of primary and secondary alkyl tosylates using B2pin2, LiOtBu and catalytic quantities of C2F5CO2Ag in 1,4-dioxane at slightly elevated temperature (Scheme 28).96 The authors suggested that alkyl radical generation is achieved by ET from an in situ generated Ag0 species to the tosylate. C-radical borylation then occurs by the alcoholate/B2pin2 adduct.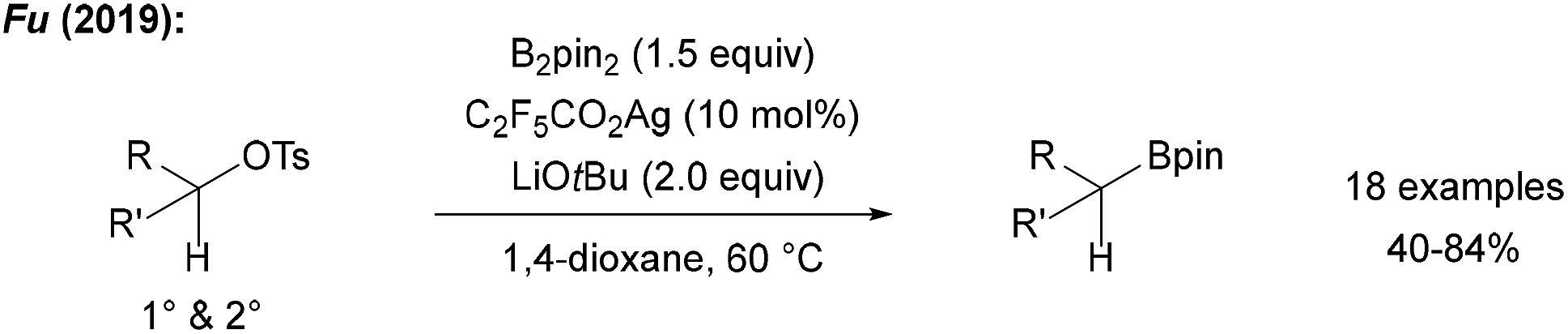 | ||
| Scheme 28 Ag-mediated radical borylation of alkyl tosylates using B2pin2. | ||
5. Addition of boryl radicals to alkenes and alkynes
Recently, it was discovered that NHC boranes engage in hydrogen atom transfer reactions (k = 4 × 104 M−1 s−1 for reduction of secondary alkyl radicals) and can be utilised as reducing reagents in radical chain reactions.97 As compared to the reactions discussed in the previous chapters, the addition of a boryl radical to a CC bond represents a fundamentally different approach to radical C–B bond formation. Initially, NHC boryl radicals were used as initiators in radical polymerisation reactions.98
This C–B bond forming strategy was later also applied to the preparation of low molecular weight compounds (Scheme 29). Curran and Taniguchi first reported the radical 5-endo-dig cyclisation of diynes with NHC boranes providing tricyclic borylated products (Scheme 29a).99 Further studies led to the successful use of NHC boryl radicals for the trans selective hydroboration of alkynes (Scheme 29b).100
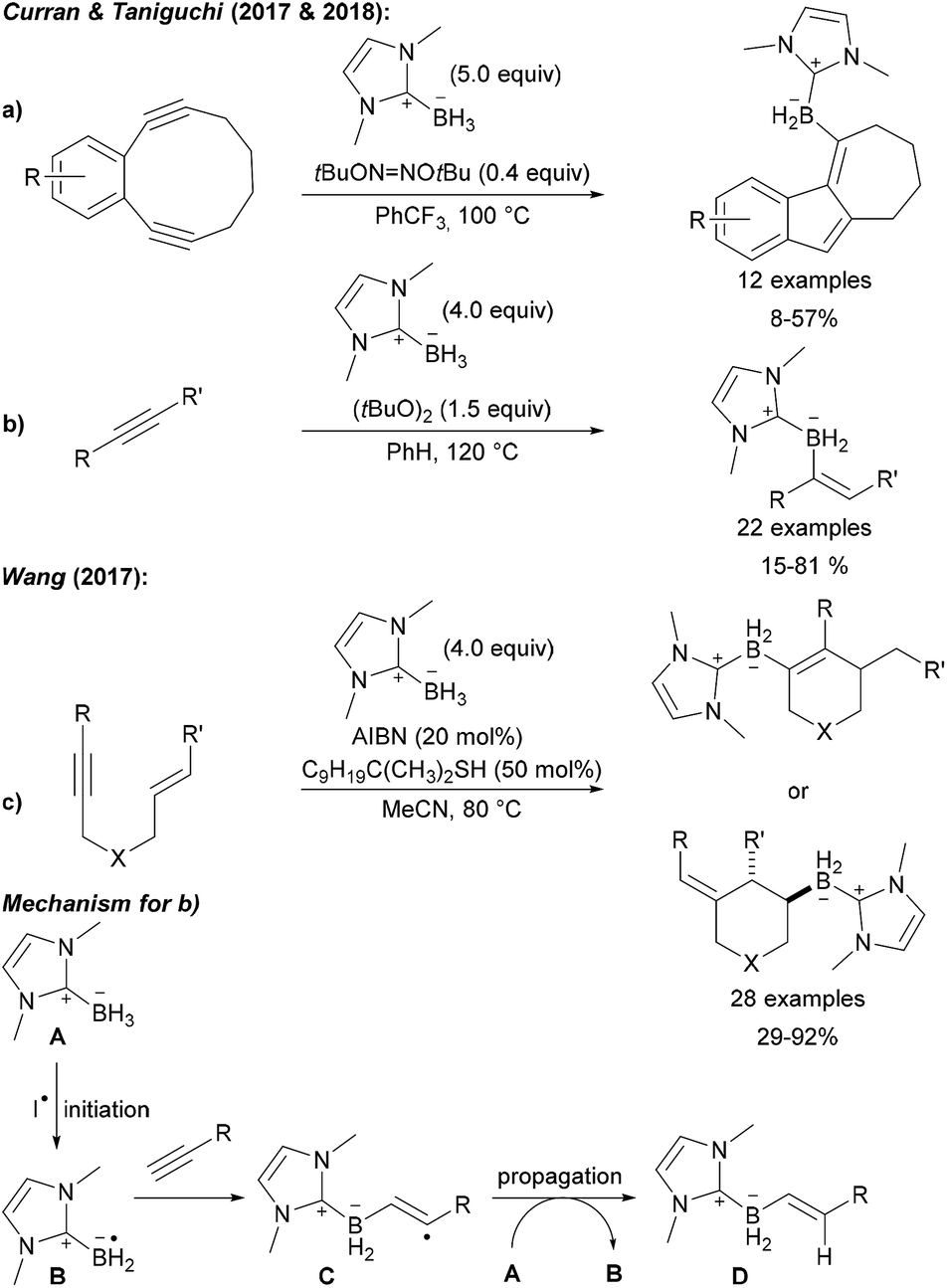 | ||
| Scheme 29 Radical C–B bond formation by intermolecular addition of NHC boryl radicals to alkynes and alkenes. Transannular cyclisation (a), alkyne (b) and enyne (c) hydroboration. Mechanism (d). | ||
Furthermore, Wang disclosed a borylation/cyclisation cascade of 1,6-enynes using NHC boranes as boryl radical precursors (Scheme 29c).101 Depending on the substitution pattern, the boryl radical can either first add to the alkyne or to the alkene leading to two different cyclisation products. The mechanism of these chain reactions involves initiation by hydrogen atom transfer from the NHC borane A to a carbon or O-centred radical derived from the initiator (AIBN, DTBP (di-tert-butyl peroxide), DTHN (di-tert-butyl hyponitrite)) resulting in the boryl radical B (Scheme 29). Addition of B to the alkyne gives the adduct radical C which is reduced by A thus regenerating B to propagate the chain. As shown in Scheme 29a and b, the adduct radical C can first undergo a cyclisation reaction prior to being reduced with the NHC borane A providing the product of a hydroboration with concomitant cyclisation, a reaction that is not possible using classical hydroboration documenting the potential and complementarity of the radical approach.
Ogawa found that radical 1,2-diborylation of alkynes can be achieved upon irradiation of a terminal alkyne, B2pin2 and diphenyl disulfide with UV light (Scheme 30a).102 The authors suggested that disulfide acts as a catalyst for this transformation, but the exact mechanism is not yet understood. However, EPR experiments supported that the diborylation proceeds via a radical process. Trans selectivity could be improved by using Ph3P as a mediator for this transformation (Scheme 30b).103
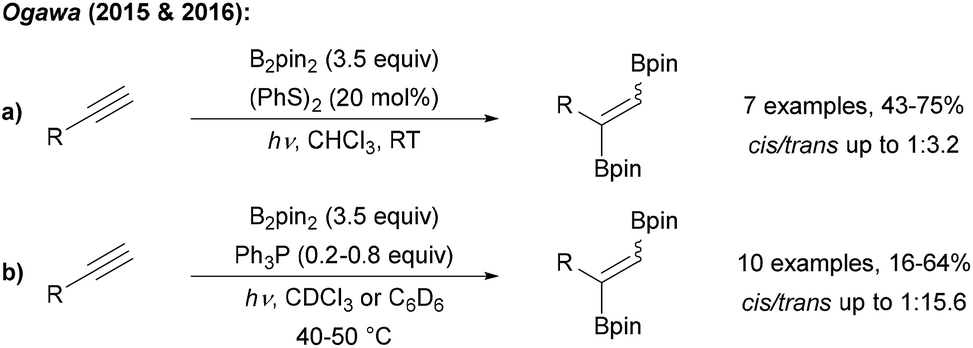 | ||
| Scheme 30 Radical C–B bond formation by addition of boryl radicals to alkynes with disulfides (a) and phosphines (b). | ||
6. Summary & outlook
Over the last decade, radical chemistry has offered novel routes towards synthetically highly valuable aryl and alkyl boronates. The radical strategies are complementary to existing approaches for the preparation of the target compound classes. Reactions are generally conducted under very mild conditions and show high functional group tolerance. Thanks to the radical approach, C–B bond formation can be combined with other typical radical reactions such as cyclisation.As highlighted in this perspective, many challenging transformations have been accomplished and good mechanistic understanding of these processes has been acquired over the years. The diborons used in these radical borylation reactions as C-radical trapping reagents are commercially available. For reactive aryl radicals, borylation does not require the assistance of any transition metal. However, the trapping of less reactive alkyl radicals (in particular tertiary alkyl radicals) with diborons is not efficient and C–B bond formation is usually mediated by a transition metal. In that regard, B2cat2 is an exception and reacts efficiently with various alkyl radicals under transition metal free conditions.
Since C–B bond formation in these transformations generally proceeds via free C-radicals, various radical precursors can be considered in these transformations. Along with the “classical” alkyl/aryl halides also activated carboxylic acids, amines and alcohols have become eligible substrates following the radical approach. Since acids, alcohols and amines are highly abundant and feedstock chemicals, the radical borylation has contributed to sustainable chemistry.
Despite the great recent achievements in this field, there are still various problems to be solved in future. The borylation of non prefunctionalised substrates using a stoichiometric amount of a cheap boron reagent in nontoxic solvents at room temperature is desirable. The synthesis of tertiary boronates via free radicals is still challenging with only a few examples reported so far. One obvious drawback of most of the protocols discussed in this perspective is that the starting materials have to be prepared (halides, NHPI esters, oxalates, Katritzky salts) lowering the atom economy of the overall process. In that regard, methods for the direct borylation of unactivated C(sp3)–H bonds are interesting.104 Considering selective inter- or intramolecular hydrogen atom transfer reactions, which have been widely applied to C(sp3)–H functionalisation,105 radical chemistry should provide an answer to this task.
Furthermore, the metal free borylation of secondary and tertiary alkyl radicals remains rather inefficient, often requiring an excess of the diboron reagent of which only half of the boron atoms are transferred. Therefore, more efficient catalysts and novel boron reagents are needed. A great challenge is the enantioselective radical borylation. While the metal free variants will likely never offer a general solution to this particular problem, metal-mediated enantioselective radical C–B formation should be feasible. Indeed, during the preparation of this perspective, Ito disclosed first examples on the asymmetric radical borylation of various racemic benzyl chlorides by using B2pin2 in combination with a chiral CuI complex (Scheme 31).106
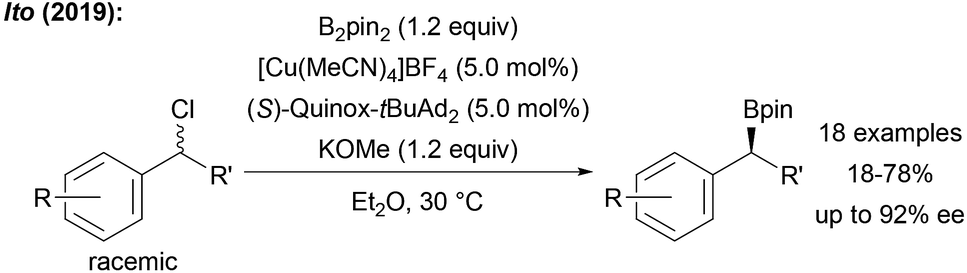 | ||
| Scheme 31 First asymmetric borylation of benzylic chlorides using B2pin2 and a chiral CuI complex. | ||
Considering the many top research teams that are worldwide active in this growing and competitive field it is likely that many of the problems defined above will be solved in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC Advanced Grant agreement no. 692640) for financial support.References
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) V. Polshettiwar, A. Decottignies, C. Len and A. Fihri, ChemSusChem, 2010, 3, 502–522 CrossRef CAS; (c) A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181–5203 RSC; (d) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC; (e) S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Green Chem., 2019, 21, 381–405 RSC.
- J. X. Qiao and P. Y. S. Lam, in Boronic acids. Preparation and applications in organic synthesis, medicine and materials, ed. D. G. Hall, WILEY-VCH, Weinheim, 2nd edn, 2011, vol. 37, pp. 315–361 Search PubMed.
- C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- Boronic acids. Preparation and applications in organic synthesis, medicine and materials, ed. D. G. Hall, WILEY-VCH, Weinheim, 2nd edn, 2011 Search PubMed.
- (a) H. S. Ban and H. Nakamura, Chem. Rec., 2015, 15, 616–635 CrossRef CAS; (b) D. B. Diaz and A. K. Yudin, Nat. Chem., 2017, 9, 731–742 CrossRef CAS.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS.
- J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CAS.
- (a) F. Mo, D. Qiu, Y. Zhang and J. Wang, Acc. Chem. Res., 2018, 51, 496–506 CrossRef CAS; (b) G. Yan, D. Huang and X. Wu, Adv. Synth. Catal., 2018, 360, 1040–1053 CrossRef CAS.
- A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 2985–2988 CrossRef CAS.
- F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1846–1849 CrossRef CAS.
- (a) D. Qiu, L. Jin, Z. Zheng, H. Meng, F. Mo, X. Wang, Y. Zhang and J. Wang, J. Org. Chem., 2013, 78, 1923–1933 CrossRef CAS; (b) D. Qiu, H. Meng, L. Jin, S. Tang, S. Wang, F. Mo, Y. Zhang and J. Wang, Org. Synth., 2014, 91, 106–115 CrossRef CAS; (c) D. Qiu, Y. Zhang and J. Wang, Org. Chem. Front., 2014, 1, 422–425 RSC.
- S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem.–Eur. J., 2015, 21, 7082–7098 CrossRef CAS.
- W. J. Grigsby and P. Power, Chem.–Eur. J., 1997, 3, 368–375 CrossRef CAS.
- Y. Cheng, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221–6225 CrossRef CAS.
- C.-J. Zhao, D. Xue, Z.-H. Jia, C. Wang and J. Xiao, Synlett, 2014, 25, 1577–1584 CrossRef.
- C. Zhu and M. Yamane, Org. Lett., 2012, 14, 4560–4563 CrossRef CAS.
- J. Yu, L. Zhang and G. Yan, Adv. Synth. Catal., 2012, 354, 2625–2628 CrossRef CAS.
- S. Ahammed, S. Nandi, D. Kundu and B. C. Ranu, Tetrahedron Lett., 2016, 57, 1551–1554 CrossRef CAS.
- J. G. Hernández, Beilstein J. Org. Chem., 2017, 13, 1463–1469 CrossRef.
- H. B. Chandrashekar, A. Maji, G. Halder, S. Banerjee, S. Bhattacharyya and D. Maiti, Chem. Commun., 2019, 55, 6201–6204 RSC.
- L. D. Marciasini, M. Vaultier and M. Pucheault, Tetrahedron Lett., 2014, 55, 1702–1705 CrossRef CAS.
- X. Qi, L. -B. Jiang, C. Zhou, J. -B. Peng and X. -F. Wu, ChemistryOpen, 2017, 6, 345–349 CrossRef CAS.
- (a) W. Erb, M. Albini, J. Rouden and J. Blanchet, 2014, 79, 10568–10580; (b) W. Erb, A. Hellal, M. Albini, J. Rouden and J. Blanchet, Chem.–Eur. J., 2014, 20, 6608–6612 CrossRef CAS PubMed.
- X. Qi, H.-P. Li, J.-B. Peng and X.-F. Wu, Tetrahedron Lett., 2017, 58, 3851–3853 CrossRef CAS.
- Y. Xu, X. Yang and H. Fang, J. Org. Chem., 2018, 83, 12831–12837 CrossRef CAS PubMed.
- M. Teders, A. Gómez-Suárez, L. Pitzer, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 902–906 CrossRef CAS.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS.
- J. Zhang, H.-H. Wu and J. Zhang, Eur. J. Org. Chem., 2013, 2013, 6263–6266 CrossRef CAS.
- C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef CAS.
- W. Yao, H. Fang, S. Peng, H. Wen, L. Zhang, A. Hu and Z. Huang, Organometallics, 2016, 35, 1559–1564 CrossRef CAS.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607–610 CrossRef CAS.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124–9128 CrossRef.
- D. Chen, L. Xu, T. Long, S. Zhu, J. Yang and L. Chu, Chem. Sci., 2018, 9, 9012–9017 RSC.
- G. Wang, H. Zhang, J. Zhao, W. Li, J. Cao, C. Zhu and S. Li, Angew. Chem., Int. Ed., 2016, 55, 5985–5989 CrossRef CAS.
- G. Wang, J. Cao, L. Gao, W. Chen, W. Huang, X. Cheng and S. Li, J. Am. Chem. Soc., 2017, 139, 3904–3910 CrossRef CAS.
- J. Cao, G. Wang, L. Gao, X. Cheng and S. Li, Chem. Sci., 2018, 9, 3664–3671 RSC.
- J. Cao, G. Wang, L. Gao, H. Chen, X. Liu, X. Cheng and S. Li, Chem. Sci., 2019, 10, 2767–2772 RSC.
- D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2019, 58 DOI:10.1002/anie.201903726.
- L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711–2722 RSC.
- S. Pinet, V. Liautard, M. Debiais and M. Pucheault, Synthesis, 2017, 49, 4759–4768 CrossRef CAS.
- K. Chen, S. Zhang, P. He and P. Li, Chem. Sci., 2016, 7, 3676–3680 RSC.
- K. Chen, M. S. Cheung, Z. Lin and P. Li, Org. Chem. Front., 2016, 3, 875–879 RSC.
- A. M. Mfuh, B. D. Schneider, W. Cruces and O. V. Larionov, Nat. Protoc., 2017, 12, 604–610 CrossRef CAS.
- A. M. Mfuh, V. T. Nguyen, B. Chhetri, J. E. Burch, J. D. Doyle, V. N. Nesterov, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 8408–8411 CrossRef CAS.
- (a) L. Eberson, M. P. Hartshorn, O. Persson and F. Radner, Chem. Commun., 1996, 2105 RSC; (b) M. Fagnoni and A. Albini, Acc. Chem. Res., 2005, 38, 713–721 CrossRef CAS.
- M. Jiang, H. Yang and H. Fu, Org. Lett., 2016, 18, 5248–5251 CrossRef CAS.
- J. Hong, Q. Liu, F. Li, G. Bai, G. Liu, M. Li, O. S. Nayal, X. Fu and F. Mo, Chin. J. Chem., 2019, 37, 347–351 CrossRef CAS.
- Y. Qiao, Q. Yang and E. J. Schelter, Angew. Chem., Int. Ed., 2018, 57, 10999–11003 CrossRef CAS.
- A. Nitelet, D. Thevenet, B. Schiavi, C. Hardouin, J. Fournier, R. Tamion, X. Pannecoucke, P. Jubault and T. Poisson, Chem.–Eur. J., 2019, 25, 3262–3266 CAS.
- Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS.
- (a) D. H. R. Barton, B. Lacher and S. Z. Zard, Tetrahedron Lett., 1985, 26, 5939–5942 CrossRef CAS; (b) D. H. R. Barton, B. Lacher and S. Z. Zard, Tetrahedron, 1987, 43, 4321–4328 CrossRef CAS.
- L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS.
- T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514–10520 CrossRef CAS.
- W.-M. Cheng, R. Shang, B. Zhao, W.-L. Xing and Y. Fu, Org. Lett., 2017, 19, 4291–4294 CrossRef CAS.
- W. Liu, X. Yang, Y. Gao and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 8621–8627 CrossRef CAS.
- D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770–2773 CrossRef CAS.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS.
- F. W. Friese and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9561–9564 CrossRef CAS.
- J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS.
- F. Sandfort, F. Strieth-Kalthoff, F. J. R. Klauck, M. J. James and F. Glorius, Chem.–Eur. J., 2018, 24, 17210–17214 CrossRef CAS.
- S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS.
- M. Newcomb, Tetrahedron, 1993, 49, 1151–1176 CrossRef CAS.
- D. Mazzarella, G. Magagnano, B. Schweitzer-Chaput and P. Melchiorre, ACS Catal., 2019, 5876–5880 CrossRef CAS.
- Q. Liu, J. Hong, B. Sun, G. Bai, F. Li, G. Liu, Y. Yang and F. Mo, Org. Lett., 2019, 21 DOI:10.1021/acs.orglett.9b01951.
- (a) C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313–5316 CrossRef CAS; (b) W. Guan, J. Liao and M. P. Watson, Synthesis, 2018, 50, 3231–3237 CrossRef CAS; (c) J. Liao, W. Guan, B. P. Boscoe, J. W. Tucker, J. W. Tomlin, M. R. Garnsey and M. P. Watson, Org. Lett., 2018, 20, 3030–3033 CrossRef CAS; (d) F. J. R. Klauck, M. J. James and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12336–12339 CrossRef CAS; (e) Z.-F. Zhu, M.-M. Zhang and F. Liu, Org. Biomol. Chem., 2019, 17, 1531–1534 RSC.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS.
- J. Hu, G. Wang, S. Li and Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 15227–15231 CrossRef CAS.
- (a) D. Rackl, V. Kais, E. Lutsker and A. O. Reiser, Eur. J. Org. Chem., 2017, 2017, 2130–2138 CrossRef CAS; (b) Y. Ye, H. Chen, J. L. Sessler and H. Gong, J. Am. Chem. Soc., 2019, 141, 820–824 CrossRef CAS.
- J. Boivin, E. Fouquet and S. Z. Zard, J. Am. Chem. Soc., 1991, 113, 1055–1057 CrossRef CAS.
- J.-J. Zhang, X.-H. Duan, Y. Wu, J.-C. Yang and L.-N. Guo, Chem. Sci., 2019, 10, 161–166 RSC.
- (a) T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS; (b) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS.
- H. Xu, C. Zhao, Q. Qian, W. Deng and H. Gong, Chem. Sci., 2013, 4, 4022–4029 RSC.
- M. S. Cheung, F. K. Sheong, T. B. Marder and Z. Lin, Chem.–Eur. J., 2015, 21, 7480–7488 CrossRef CAS PubMed.
- H. Iwamoto, S. Akiyama, K. Hayama and H. Ito, Org. Lett., 2017, 19, 2614–2617 CrossRef CAS.
- C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, eaam7355 CrossRef PubMed.
- J. Wang, M. Shang, H. Lundberg, K. S. Feu, S. J. Hecker, T. Qin, D. G. Blackmond and P. S. Baran, ACS Catal., 2018, 8, 9537–9542 CrossRef CAS PubMed.
- C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528–532 CrossRef CAS PubMed.
- H. Ito and K. Kubota, Org. Lett., 2012, 14, 890–893 CrossRef CAS.
- J. H. Kim and Y. K. Chung, RSC Adv., 2014, 4, 39755–39758 RSC.
- X. Lou, Z.-Q. Zhang, J.-H. Liu and X.-Y. Lu, Chem. Lett., 2016, 45, 200–202 CrossRef CAS.
- S. K. Bose, S. Brand, H. O. Omoregie, M. Haehnel, J. Maier, G. Bringmann and T. B. Marder, ACS Catal., 2016, 6, 8332–8335 CrossRef CAS.
- H. Yoshida, Y. Takemoto, S. Kamio, I. Osaka and K. Takaki, Org. Chem. Front., 2017, 4, 1215–1219 RSC.
- A. S. Dudnik and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 10693–10697 CrossRef CAS PubMed.
- J. Yi, J. -H. Liu, J. Liang, J. -J. Dai, C. -T. Yang, Y. Fu and L. Liu, Adv. Synth. Catal., 2012, 354, 1685–1691 CrossRef CAS.
- S. K. Bose, K. Fucke, L. Liu, P. G. Steel and T. B. Marder, Angew. Chem., Int. Ed., 2014, 53, 1799–1803 CrossRef CAS PubMed.
- (a) T. C. Atack, R. M. Lecker and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 9521–9523 CrossRef CAS PubMed; (b) T. C. Atack and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 6139–6142 CrossRef CAS PubMed.
- R. B. Bedford, P. B. Brenner, E. Carter, T. Gallagher, D. M. Murphy and D. R. Pye, Organometallics, 2014, 33, 5940–5943 CrossRef CAS.
- R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485–1493 CrossRef CAS PubMed.
- J.-C. Yang, L. Chen, F. Yang, P. Li and L.-N. Guo, Org. Chem. Front., 2019, 6, 2792–2795 RSC.
- J. Cui, H. Wang, J. Song, X. Chi, L. Meng, Q. Liu, D. Zhang, Y. Dong and H. Liu, Org. Biomol. Chem., 2017, 15, 8508–8512 RSC.
- (a) K. Kubota, E. Yamamoto and H. Ito, J. Am. Chem. Soc., 2013, 135, 2635–2640 CrossRef CAS PubMed; (b) H. Iwamoto, K. Kubota, E. Yamamoto and H. Ito, Chem. Commun., 2015, 51, 9655–9658 RSC.
- T. Niwa, H. Ochiai and T. Hosoya, ACS Catal., 2017, 7, 4535–4541 CrossRef CAS.
- T. Yoshida, L. Ilies and E. Nakamura, ACS Catal., 2017, 7, 3199–3203 CrossRef CAS.
- S. K. Bose and T. B. Marder, Org. Lett., 2014, 16, 4562–4565 CrossRef CAS.
- S. K. Bose, A. Deißenberger, A. Eichhorn, P. G. Steel, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2015, 54, 11843–11847 CrossRef CAS PubMed.
- X. Lu, Z. -Q. Zhang, L. Yu, B. Zhang, B. Wang, T. -J. Gong, C. -L. Tian, B. Xiao and Y. Fu, Chin. J. Chem., 2019, 37, 11–18 CrossRef CAS.
- S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256–11262 CrossRef CAS PubMed.
- (a) S. Telitel, S. Schweizer, F. Morlet-Savary, B. Graff, T. Tschamber, N. Blanchard, J. P. Fouassier, M. Lelli, E. Lacôte and J. Lalevée, Macromolecules, 2013, 46, 43–48 CrossRef CAS; (b) M.-A. Tehfe, J. Monot, M. Malacria, L. Fensterbank, J.-P. Fouassier, D. P. Curran, E. Lacôte and J. Lalevée, ACS Macro Lett., 2012, 1, 92–95 CrossRef CAS; (c) M.-A. Tehfe, J. Monot, M. M. Brahmi, H. Bonin-Dubarle, D. P. Curran, M. Malacria, L. Fensterbank, E. Lacôte, J. Lalevée and J.-P. Fouassier, Polym. Chem., 2011, 2, 625–631 RSC; (d) M.-A. Tehfe, M. Makhlouf Brahmi, J.-P. Fouassier, D. P. Curran, M. Malacria, L. Fensterbank, E. Lacôte and J. Lalevée, Macromolecules, 2010, 43, 2261–2267 CrossRef CAS; (e) J. Lalevée, S. Telitel, M. A. Tehfe, J. P. Fouassier, D. P. Curran and E. Lacôte, Angew. Chem., Int. Ed., 2012, 51, 5958–5961 CrossRef PubMed.
- T. Watanabe, D. Hirose, D. P. Curran and T. Taniguchi, Chem.–Eur. J., 2017, 23, 5404–5409 CrossRef CAS PubMed.
- M. Shimoi, T. Watanabe, K. Maeda, D. P. Curran and T. Taniguchi, Angew. Chem., Int. Ed., 2018, 57, 9485–9490 CrossRef CAS PubMed.
- S.-C. Ren, F.-L. Zhang, J. Qi, Y.-S. Huang, A.-Q. Xu, H.-Y. Yan and Y.-F. Wang, J. Am. Chem. Soc., 2017, 139, 6050–6053 CrossRef CAS PubMed.
- A. Yoshimura, Y. Takamachi, L.-B. Han and A. Ogawa, Chem.–Eur. J., 2015, 21, 13930–13933 CrossRef CAS PubMed.
- A. Yoshimura, Y. Takamachi, K. Mihara, T. Saeki, S.-i. Kawaguchi, L.-B. Han, A. Nomoto and A. Ogawa, Tetrahedron, 2016, 72, 7832–7838 CrossRef CAS.
- R. L. Reyes, T. Iwai, S. Maeda and M. Sawamura, J. Am. Chem. Soc., 2019, 141, 6817–6821 CrossRef CAS PubMed.
- (a) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS; (b) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569–1586 CrossRef CAS.
- H. Iwamoto, K. Endo, Y. Ozawa, Y. Watanabe, K. Kubota, T. Imamoto and H. Ito, Angew. Chem., Int. Ed., 2019, 131, 11229–11234 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |