
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interplay of secondary and tertiary folding in abiotic foldamers†
Daniela
Mazzier
ab,
Soumen
De
 b,
Barbara
Wicher
c,
Victor
Maurizot
b and
Ivan
Huc
*ab
b,
Barbara
Wicher
c,
Victor
Maurizot
b and
Ivan
Huc
*ab
aDepartment of Pharmacy, Centre for Integrated Protein Science, Ludwig-Maximilians-Universität, Butenandtstraße 5-13, D-81377 Munich, Germany. E-mail: ivan.huc@cup.lmu.de
bCBMN Laboratory, Université de Bordeaux, CNRS, IPB, Institut Européen de Chimie et Biologie, 2 rue Escarpit, 33600 Pessac, France
cDepartment of Chemical Technology of Drugs, Poznan University of Medical Sciences, Grunwaldzka 6, 60-780 Poznan, Poland
First published on 10th June 2019
Abstract
The first true abiotic tertiary folded structures, i.e. at the exclusion of any aliphatic amino acid, have recently been introduced under the form of aromatic oligoamide helix–turn–helix foldamers stabilized by hydrogen bonds in organic solvents. We present an investigation of the interplay of secondary and tertiary folding and of some cooperative effects in these systems. A solid phase synthesis approach to the preparation of these sequences was developed to facilitate systematic variation. Flexible pyridine-based units were introduced in various proportions in replacement of more rigid quinoline-based units. Conformational behaviour was assessed in solution by NMR, in the solid state by X-ray crystallography, and computationally through molecular dynamics simulations. Altogether, our results demonstrate that tertiary folding stabilizes otherwise flexible secondary structures, and that the disruption of tertiary folds upon adding polar solvents follows different mechanisms depending on whether secondary structures are inherently stable or not. These findings constitute a solid basis on which to further increase the size and complexity of abiotic folded structures and to eventually orchestrate folding dynamics and responsiveness.
Introduction
Foldamer research is based on the premise that non-natural backbones will give access to folded structures and functions beyond the reach of biopolymers. Researchers in the field have been successful in eliciting folding of secondary motifs such as single helices or sheets in a great variety of synthetic oligomers.1–4 However, the ab initio design of tertiary structures is found to be far more complex and constitutes a major challenge ahead. Motivation to meet this challenge comes from the fact that biopolymer functions emerge at the tertiary structure level. In comparison to a full length protein, a simple α-helix cannot achieve much. Tertiary folds may thus be the level at which foldamers will reveal their full potential.Current progress in protein design5–10 allows one to be optimistic about its extension to foldamers. Some important milestones have indeed been achieved. For example, α-peptidic tertiary folds and α-helix bundles have been shown to tolerate several β-amino-acids.11–16 Furthermore, helix bundles comprised of β-amino-acids17,18 or β-ureas,19 at the exclusion of any α-amino acid have also been described. In contrast, until recently, progress in the area of abiotic backbones was less advanced and limited to a few reports describing structures produced by connecting several secondary elements without well-defined interactions over extended surfaces between them.20–24 Yet, there are good reasons to invest efforts in abiotic foldamer development. Because their chemical nature is by definition remote from that of biopolymers, the structures that can be obtained and by extension their properties and functions are also expected to be distinct. In this respect, aromatic foldamers,25–27i.e. foldamers with aryl groups in their main chain, represent a promising and rapidly developing family of abiotic backbones. Illustrations of their distinctiveness with respect to biopolymers include their strong folding propensity in all kinds of media other than water and the production of novel shapes such as helices with a reduced diameter at the end of the sequence and a wider diameter in the centre of the sequence that eventually give rise to cavities suitable for molecular recognition.28–31
We recently reported the ab initio computational design, solution phase synthesis and structural characterization of the first true abiotic tertiary structures.32 We used the well-characterized helically folded oligoamides of 8-amino-2-quinolinecarboxylic acid Q (Fig. 1) as secondary modules.33,34 Upon introducing monomers X and Y at specific positions in a sequence, the aryl-OH groups borne by these monomers were shown to promote the formation of well-defined inter-helix arrays of hydrogen-bonds. When two helices were combined with a turn unit T (Fig. 1), we observed that sequences such as 5b fold into a helix–turn–helix structure with high stability in non-polar organic solvents. Remarkably, tertiary folding proved to follow an all-or-nothing mechanism: the progressive addition of a polar solvent that competes for hydrogen bonding caused a sharp transition towards a non-hydrogen-bonded helix–turn–helix structure.
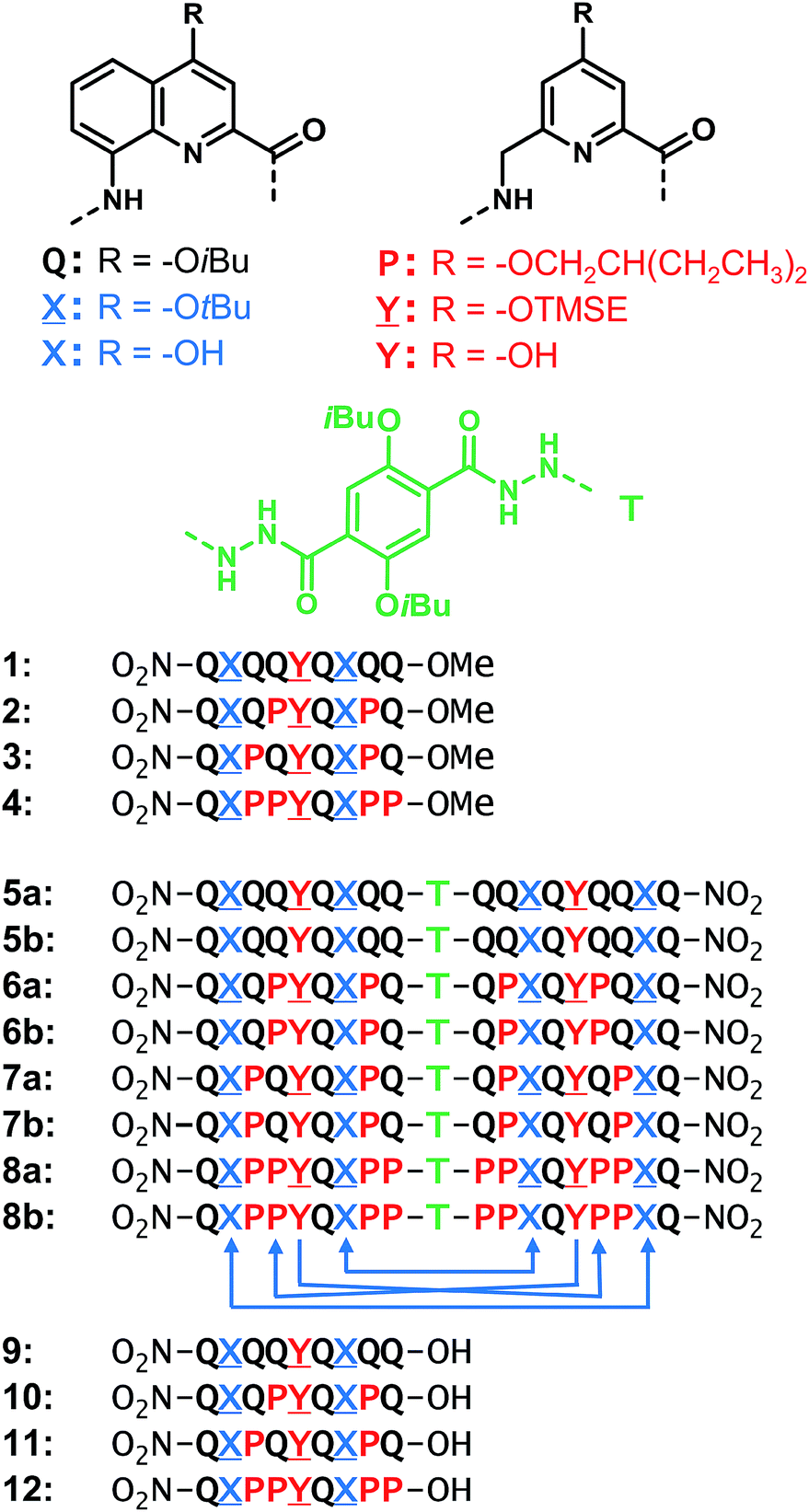 | ||
Fig. 1 Structures of units Q, X, P, Y, T and foldamer sequences. ![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) and and ![[Y with combining low line]](https://www.rsc.org/images/entities/char_0059_0332.gif) are the protected precursors of hydrogen bonding units X and Y. All flexible units P, , or Y are shown in red. Sequences are labelled ‘a’ when protected and ‘b’ when deprotected. Sequences end with 8-nitro groups; this group is noted in the replacement of the NH group at terminal Q units. The T unit constitutes an inversion of C → N sequence polarity; sequences that contain T thus have two N termini. The arrows below 8b indicate the hydrogen bonds pattern between the helices. TMSE = 2-trimethylsilylethyl. are the protected precursors of hydrogen bonding units X and Y. All flexible units P, , or Y are shown in red. Sequences are labelled ‘a’ when protected and ‘b’ when deprotected. Sequences end with 8-nitro groups; this group is noted in the replacement of the NH group at terminal Q units. The T unit constitutes an inversion of C → N sequence polarity; sequences that contain T thus have two N termini. The arrows below 8b indicate the hydrogen bonds pattern between the helices. TMSE = 2-trimethylsilylethyl. | ||
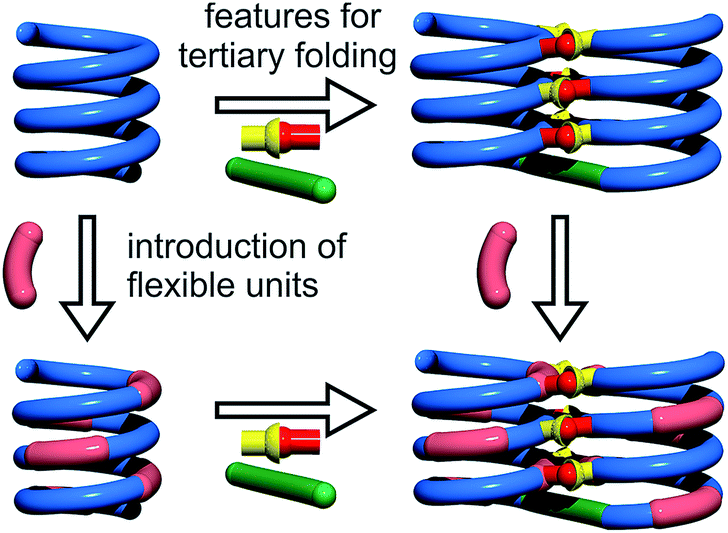
Cooperativity lies at the core of protein functions in that it allows for key dynamics and structural changes to take place. Controlling tertiary structure dynamics through cooperative effects may thus bring critical advantages to implement function. We thus endeavoured to investigate further this aspect in an abiotic context. In the following, we report that abiotic tertiary folds based on aromatic oligoamides tolerate a great deal of destabilization of their secondary helical modules. Specifically, we first developed a new synthetic approach based on solid phase synthesis and post-synthetic fragment assembly that allows for systematic variations of the sequences. Upon introducing an increasing number of flexible units in the helices that reduce inherent conformational stability, we show that the tertiary structures still fold and promote helicity in the secondary units. We also find that the destabilizing effect of polar solvents leads to different unfolding pathways depending on the number of flexible units. In addition, computations show that thermal denaturation of the tertiary structures is expected to occur, but at temperatures higher than what can be reasonably reached in experiments. Altogether, these results validate a rational approach to design tertiary structure dynamics, starting from rigid modules and progressively adding degrees of freedom to the system.
Results and discussion
Foldamer sequence design and synthesis
For the design of the helix–turn–helix conformation of 5b, an aliphatic amine unit, 6-aminomethyl-4-hydroxy-2-pyridinecarboxylic acid Y, was introduced as structural analogue of X.32Y, which lacks the benzenic ring of quinolines, is required to prevent steric clashes between the helices while it imparts the same strand curvature as X. In addition, its aliphatic methylene unit brings an additional rotatable bond that enhances flexibility. We have already shown that the introduction of several of these aliphatic amine monomers into aromatic oligoamides may promote non-canonical conformations,35 and enhance conformational dynamics. Although these aliphatic amine units possess an overall poor folding propensity, they adopt the canonical aromatic helix conformation in the presence of a sufficient number of aromatic units.36–38By analogy with Y which replaces X, we designed aliphatic unit P to replace Q (Fig. 1). We expected that this substitution would enhance flexibility, handedness dynamics, and folding entropy: P units should alter helix stability, yet this effect may be compensated when helices are held together in a tertiary motif. Thus, sequences 1–4 were designed so as to adopt single helical conformations having an increasing number – one, three or five – of aliphatic amine units (Fig. 1). Sequences 2 and 3 have the same number of P units and differ only by their position in the sequence. Sequences 2 and 4 contain adjacent aliphatic amine units, respectively PY or PPY and PP, but the other two sequences only possess non-adjacent P or Y monomers. In order to evaluate the effect of tertiary folding on the stability of the secondary helical modules sequences, deprotected 6b, 7b and 8b were designed (Fig. 1) as potential helix–turn–helix analogues of 5b. In principle, these compounds altogether allow one to build so-called double mutant cycles in order to assess cooperative effects (Fig. 2).39,40 If compared to reference compound 1, a stable isolated helical secondary motif (side chains are all protected), compounds 2–4 are made more flexible through the introduction of P units, and compound 5b has the features that promote tertiary folding (turn unit and hydroxyl groups). In compounds, 6b, 7b and 8b, both flexible units and features to promote tertiary folding are present.
 | ||
| Fig. 2 Double mutant cycle designed to assess cooperative effects in abiotic tertiary structures. The green rod and the red-and-yellow connector depict the turn unit and hydrogen bonding functions, respectively. | ||
For their use in the solid phase synthesis (SPS) of the sequences, each new monomer (, P and ) was produced as an Fmoc-amine protected carboxylic acid. Their preparation is presented in detail in the ESI.† Monomers and also possess a TFA labile protection of their 4-hydroxy group, i.e. tert-butyl ether and trimethylsilyl ethyl ether, respectively. P is functionalized with a 2-ethylbutan-1-ol solubilizing group. Fmoc-Q-OH has been described previously.38 Because the direct acid chloride activation of Fmoc--OH and Fmoc-P-OH, as required for coupling onto aromatic amines, proved to be troublesome, Fmoc-Q-OH and Fmoc-PQ-OH dimers were pre-assembled and incorporated as such during SPS. The preparation of oligomers 9–12 was then carried out using previously reported SPS methods.38,41 The use of a SASRIN resin allowed for the cleavage from the resin while preserving side chain protection using hexafluoro-isopropanol (see ESI†). Acid terminated oligomers 9–12 were then converted into methyl esters 1–4, respectively, under mild conditions using trimethylsilyl-diazomethane. Protected sequences 5a–8a, were synthesized in solution by coupling foldamer acids 9–12 with the terephthalic acid bis-hydrazide turn unit (T). Finally, deprotected compounds 5b–8b were obtained after TFA treatment of 5a–8a, respectively.
Secondary folding
The behaviour of oligomers 1–4 in solution was studied by NMR spectroscopy. The 1H NMR spectra of 1 and 2 in CDCl3 exhibited features typical of the expected single helical folding. The diastereotopic motifs of the CH2 signals of the isobutoxy side chains of the Q units indicate a slow helix handedness inversion on the NMR timescale (Fig. 3a and b). For 1 no coalescence was observed upon heating up to 55 °C (Fig. S1†) but for 2 signals start to coalesce at 45 °C (Fig. S2†). In contrast, the 1H NMR spectrum of 3, that differs from 2 only for the position of P units in the sequence, shows broad signals at 25 °C (Fig. 3c). Upon heating these broad signals sharpen and, upon cooling, all signals coalesce before splitting into sharp diastereotopic motifs (Fig. S3†). In the case of compound 4, the broad 1H NMR spectrum at 25 °C (Fig. 3d) sharpens upon heating, but becomes broader without showing the emergence of any diastereotopic pattern at low temperature (Fig. S4†). These results reflect an enhancement of conformational dynamics when the number of P units increases, and also depending on their position. | ||
| Fig. 3 Extracts of 1H NMR spectra in CDCl3 at 25 °C of 1 (a), 2 (b), 3 (c) and 4 (d) showing side chain and main chain CH2 protons. Part of 2D TOCSY NMR spectra in CDCl3 of 1 (e), 2 (f), 3 (g) and 4 (h) showing NH–CH2 scalar couplings. Red brackets in (a)–(c) indicate pairs of main chain diastereotopic benzylic CH2 protons as observed in (e)–(g), respectively. | ||
Helix folding was also assessed from the chemical shift values of the benzylic methylene protons of each P or monomer. Within an aromatic helix, these protons are diastereomeric and may be both strongly anisochronous and significantly upfield shifted.36 Distinct signals are then observed provided helix handedness inversion is slow, notably in 2D NMR (TOCSY) experiments monitoring CH2–NH scalar couplings (Fig. 3e–h). The spectra of compounds 1–3 indeed showed that the signals of these protons are spread over almost three ppm from 4.3 to 1.8 ppm, and that anisochronicity (Δδ) can be as high as 1.4 ppm. This contrasts with the spectrum of 4 where no anisochronicity (i.e. fast dynamics or no chiral conformation) and less significant upfield shifts (signals clustered between 4 and 5 ppm) are observed (Fig. 3h). Solution data thus suggest different stabilities of the helical conformations 1–3 and no strong evidence of the helical folding of 4. A crystallographic solid state structure provided valuable information, showing 4 with part of its sequence folded in a helix, and part of it unfolded (Fig. 4a). Though this structure probably simply represents one of many conformations available to 4 in solution, it is illustrative of the additional flexibility introduced by the insertion of several aliphatic amine units in the backbone and of the fact that its helical folding may not be complete.
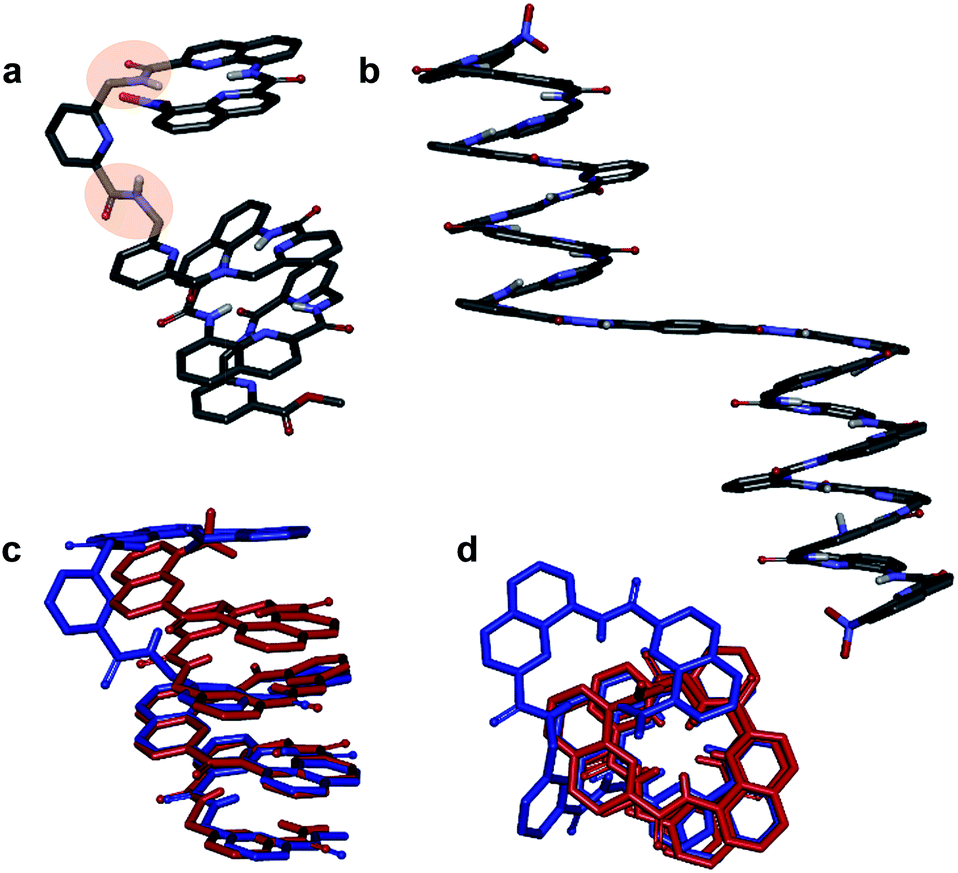 | ||
| Fig. 4 Crystal structures of compounds 4 (a) and 8a (b). Overlay of the structure of 4 (blue) and of the M helix fragment of 8a (red) (c and d). 90° kinks in the backbone of 4 occur at main chain methylene groups and are highlighted in (a). Side chains of Q, P, T, and , included solvent molecules and hydrogen atoms other than NH have been omitted for clarity. | ||
We also studied the solution phase folding of protected sequences 5a–8a. All these compounds comprise two potentially helical segments that can each have P or M handedness, separated by a turn unit T. The 1H NMR spectra of sequences 5a and 6a show two sets of signals consistent with the coexistence of PM and PP/MM conformers in slow exchange on the NMR timescale (Fig. S5a and b†). Based on earlier studies32,42 we assume the favoured and most abundant species to be PM (see also below). Compound 7a shows a broad NMR spectrum suggesting a faster exchange between PM and PP/MM conformations (Fig. S5c†), consistent with the dynamics of oligomer 3. One set of sharper NMR signals are observed in the case of 8b (Fig. S5d†) indicating a fast exchange regime, as confirmed by the absence of diasterotopic motifs in the signals of isobutoxy side chains. An X-ray crystal structure of 8a shows the most stable PM conformer (Fig. 4b). As opposed to 4, 8a crystallized with both its segment folded into a canonical aromatic helix despite the high number of aliphatic amine units. The overlay of the X-ray structures of 4 and 8a makes it clear that conformation variations occur due to rotations at the methylene moieties of P units (Fig. 4c and d).
Tertiary folding
We then explored the folding propensity of the corresponding deprotected sequences 5b–8b. Compounds 5b–7b all show one set of sharp NMR signals (Fig. 5a–c) consistent with well-defined helix–turn–helix motifs in which the handedness of the two helical segments are not independent from each other. A solid state structure of 7b was obtained. The data quality for 7b is low and conclusions drawn from the structure shall be treated with some care but the structure still represent a valuable piece of information regarding main chain shape and its nearest environment. The asymmetric unit of 7b in the crystal contains two symmetry independent molecules (Fig. 6a–d). Both molecules adopted the expected PP or MM helix–turn–helix fold with inter-helix O⋯O distances in the range expected for hydrogen bonds between OH groups of X and Y units as a donors and amide carbonyls as acceptors (Table S1†).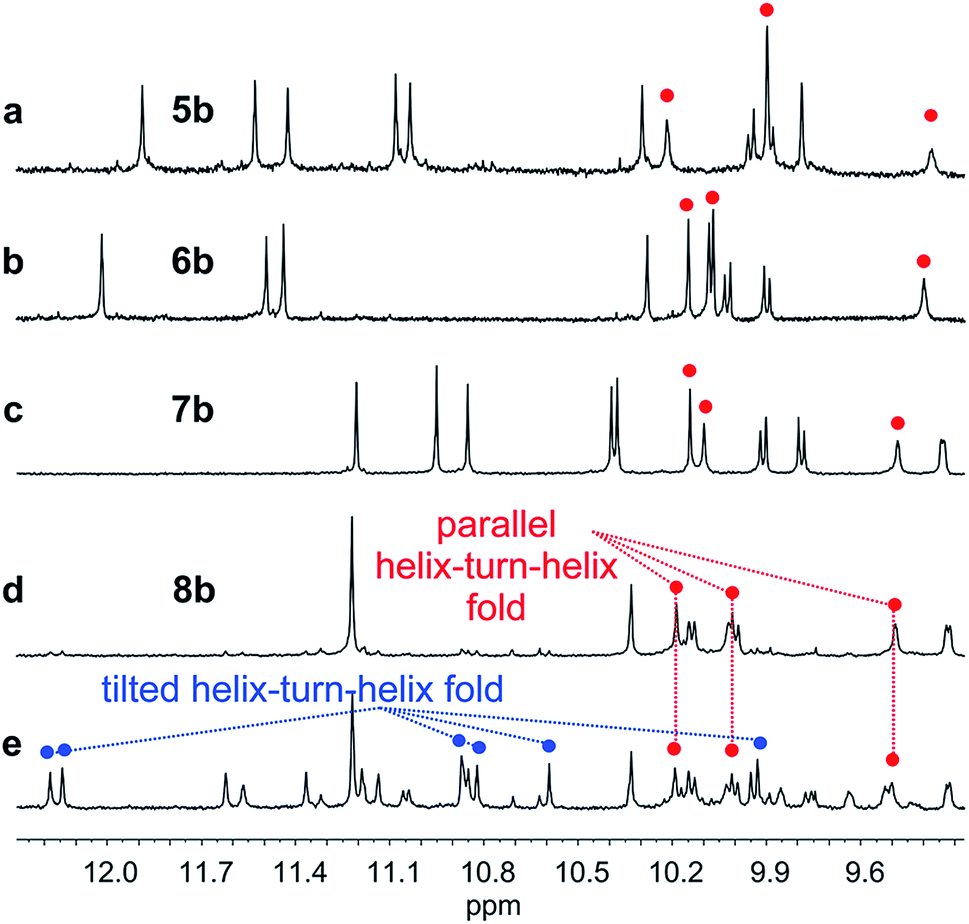 | ||
| Fig. 5 Extract of 1H NMR spectra (500 MHz, CDCl3) showing the NH and OH resonances of 5b (a), 6b (b), 7b (c) and 8b freshly dissolved crystals (d) and at equilibrium (e). The red and blue dots indicate signals corresponding to OH protons. | ||
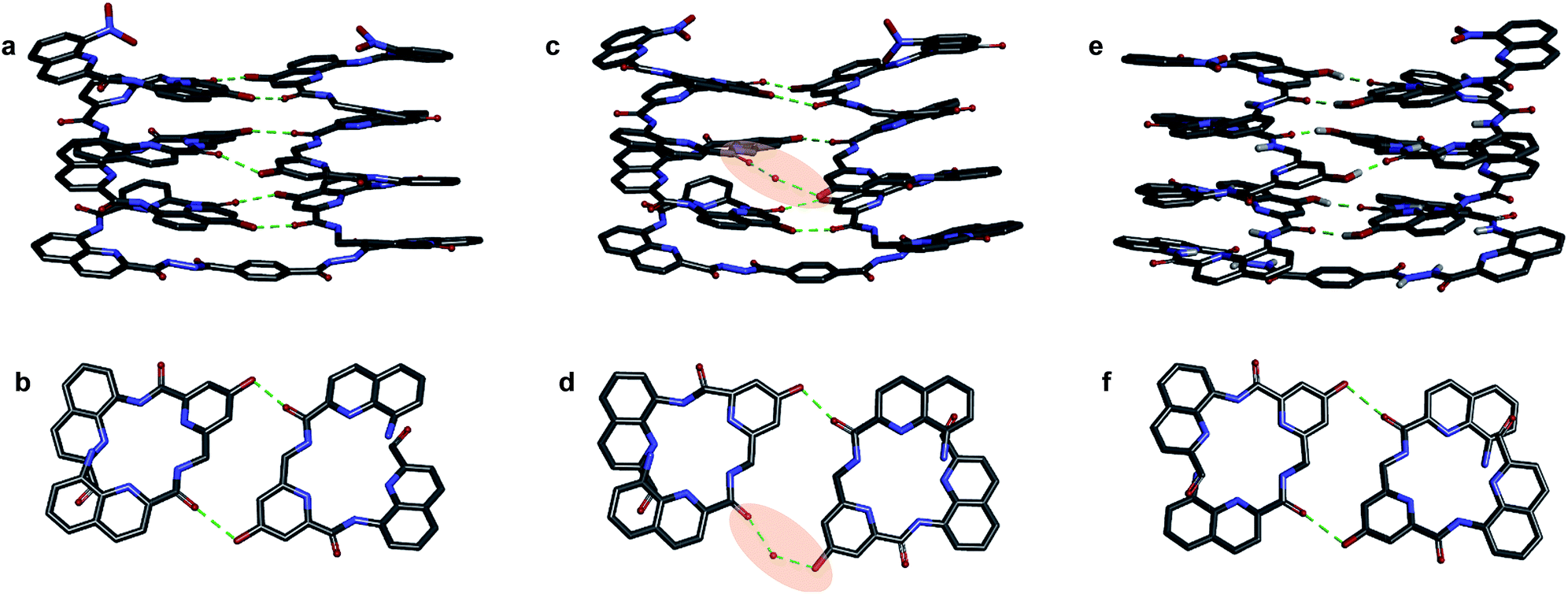 | ||
| Fig. 6 Crystal structures of compounds 7b (a–d) and 5b32 (e and f). Two symmetry independent molecules of 7b are present in the asymmetric unit (a, b and c, d). They differ by the presence of an H-bonded water molecule (highlighted). In all representations, side chains of Q, P and T, included solvent molecules and most hydrogen atoms have been omitted for clarity. | ||
One of the two independent molecule showed no noticeable differences with the crystal structure of 5b32 (Fig. 6e and f), confirming the equivalent contribution to curvature of P and Q units. For the second independent molecule of 7b, the structure is overall identical except for an electron density peak that could be interpreted as a water molecule inserted in the array of hydrogen bonds, bridging the OH of a Y unit and the carbonyl amide of the other helix (Fig. 6c and d). Such water-mediated hydrogen-bonded bridges are common in biopolymer structures. Here, it hints at possible mechanisms through which polar solvents disrupt the hydrogen-bonded helix–turn–helix fold. It may also be related to the non-optimal nature of hydrogen bonds within this fold: helices have been shown to assemble into other structures such as trimers or tilted dimers when they are not connected by the turn unit.32
Oligomer 8b which contains the largest number of P units behaves differently. Its NMR spectrum at equilibrium in CDCl3 is also sharp but it consists of more than one set of signals (Fig. 5e), indicating the coexistence of different conformations in solution. Changing the solvent to toluene led to variations of the proportions between these species (Fig. S24 and 25†). Crystals of 8b could be grown from toluene/hexane but they proved to be unsuitable for X-ray diffraction analysis. Nevertheless, an NMR spectrum of freshly dissolved crystals in CDCl3 showed one set of sharp signals (Fig. 5d) corresponding to the major species observed at equilibrium in CDCl3.43 The signals of other species progressively emerge over the course of hours (Fig. 5d, e and S22†). The 1H NMR spectrum of the major species of 8b resembles those of 5b–7b (Fig. 5a–d). Signals corresponding to OH protons hydrogen bonded at the helix–helix interface were all found in the same 9.5 to 10.2 ppm window (Fig. S10–S13†). Based on this, we tentatively assigned the major species in CDCl3 solutions of 8b to a canonical helix–turn–helix structure similar to those of 5b–7b. For the minor species present in equilibrated solutions of 8b, OH proton signals were found in the 10.5–12.5 ppm window suggesting other folding modes with stronger hydrogen bonds. Such downfield shifted OH signals were previously observed in the trimers and tilted dimers that prevail in the absence of turn unit.32 The spectrum of 8b does not change with concentration (data not shown), ruling out the coexistence of aggregated species. We thus assigned the other species to tilted helix–turn–helix folds. Indeed, energy minimized molecular models (Fig. S31 and S32†) show that the presence of four flexible P units close to the turn allows for considerable conformational freedom. Upon unfolding the central PPTPP segment of 8b into an extended conformation, it can behave as a long turn between helices in a titled helix–turn–helix structure. In this conformation the two helix axes are not anymore parallel, yet all hydroxy groups are involved in hydrogen bonds.
The prevalence of PP or MM helix–turn–helix structures in 5b–8b and the corresponding sharp sets of 1H signals much differ from the dynamic behaviour of their protected precursors. AB diastereotopic patterns for 5b–8b indicate slow exchange of helix handedness on the NMR time scale, in contrast with e.g.7a and 8a. In addition, PM species present in solutions of the protected precursors are absent once the tertiary motifs form. In the case of 8a, other species coexist but the very slow dynamics much contrasts with the fast dynamics of 4 and 8a. Altogether, and referring to the double mutant cycle shown in Fig. 2, these results suggest that helix–turn–helix folds are stable even when the helical segments taken individually are very dynamic or, possibly, not fully helical. Such cooperativity is reminiscent to that observed in protein tertiary structure folding.
Effects of polar solvent and heat
Tertiary folding was further evaluated through polar solvent addition as well as heating. Thus, the effect of the progressive addition of DMSO-d6, a solvent that competes for hydrogen bonding, was assessed by 1H NMR by monitoring chemical shift variations (Fig. S14–S23†). It should be noted that DMSO only has a weak effect on aromatic helix stability: Qn helices are very stable in all non-protic solvents and extremely stable in protic solvents.34 The prime effect of adding DMSO-d6 is to disrupt inter-helix hydrogen bonds. For compound 5b, a sharp transition between two states state was observed suggesting an all-or-nothing phenomenon: the addition of up to 13% DMSO had little effect whereas the helix bundle went from folded to completely disrupted upon increasing DMSO content up to 28% DMSO. This can be understood if the two helical segments behave as rigid bodies that can establish all six hydrogen bonds between them, or none, i.e. that there is a strong chelate effect between the hydrogen bonds. The addition of DMSO then leads to hydrogen bond disruption but not to complete unfolding as the secondary modules remain intact even though their respective handedness become independent (Fig. 7a). In contrast, the transition observed for compounds 6b–8b was more progressive and, for the two most flexible sequences, changes started with less than 5% DMSO. Furthermore, at high DMSO content, signals are broad and do not split into two sets of signals as for 5b. It thus appears that enhancing helix flexibility increases sensitivity to polar solvent, and allows for partial unfolding to occur. This suggests a progressive unfolding of the tertiary motif and simultaneous destabilization of secondary structure (Fig. S18–S23†). As shown in Fig. 7, we thus relate the different behaviour of the oligomers to the rigidity of each helix backbone, that is, to the ratio and sequence of P and Q monomers. Although tertiary fold disruption undergoes a sharper transition for rigid helices because of a chelate effect, cooperativity between secondary and tertiary folds, i.e. the stabilization of secondary folds in the tertiary structure, becomes apparent only for the more flexible sequences.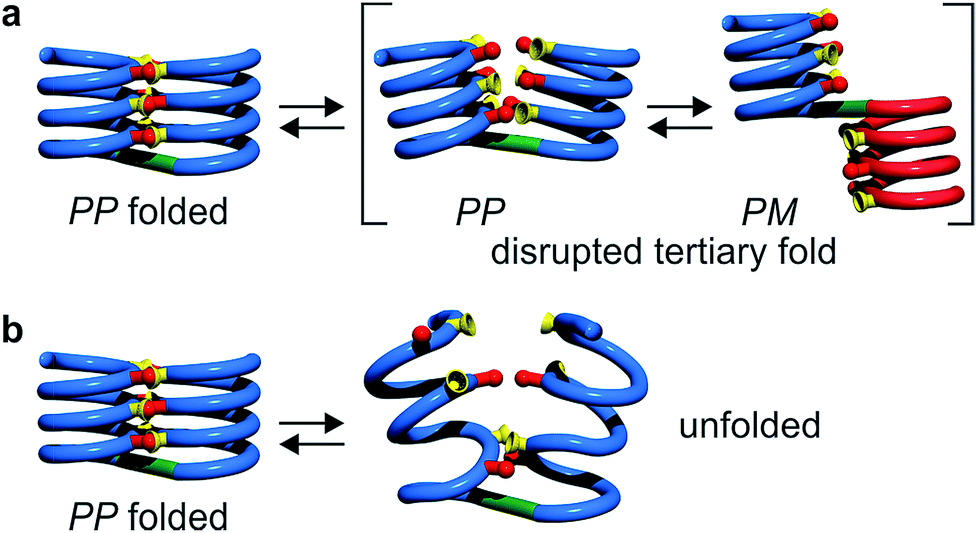 | ||
| Fig. 7 Tertiary structure disruption (a) or unfolding (b) occur upon adding a polar solvent, depending on whether the secondary helical modules are inherently stable (a) of have gained stability in the tertiary fold (b). | ||
Another possible consequence of flexible units was an enhancement in the folding entropy that would result in the possibility to denature, or “melt” the structures upon heating. We measured NMR spectra in CDCl3 up to 55 °C (Fig. S6–S9†) and, in the case of 7b, in C2D2Cl4 up to 80 °C, unfortunately this temperature was not high enough to observe any denaturation. However, some variations of chemical shift values were detected in particular for OH protons: upon heating, signals shifted upfield suggesting an increase of averaged hydrogen bond distances.
We then performed some molecular dynamic (MD) studies to evaluate the effect of temperatures higher than can be reached experimentally. MD simulations were performed under stochastic dynamic conditions at different temperatures using the program MacroModel. Simulations were carried out for 1 ns using an energy minimized initial structure. Such a short time is insufficient for a correct equilibration of the system and MD simulation data collected at very high temperatures should be considered with caution. Yet some relevant information could be deduced from the results.
Single helical compounds 1 and 4 showed a marked difference of stability upon heating (Fig S30†): increasing the number of P units is highly destabilizing at high temperature, in agreement with solution data. We then modelled the behaviour of helix–turn–helix structures 5b and 8b (Fig. 8, see also ESI†). During the simulations, O⋯O hydrogen bond distances between the two helices were monitored and structures sampled every 10 ps. At a temperature of 300 K, the folded helix–turn–helix motif is highly conserved over time for both compounds (Fig. 8a and f). H bonds fluctuate but were present for most of the simulations, yet the central H bonds between Y units were found to be less stable (Fig. S28f and S29f†). This may be related to the inserted water molecule observed in the crystal structure of 7b (Fig. 6c and d). Another feature of hydrogen bond fluctuation concerns the concerted behaviour of individual hydrogen bond pairs that occur at different levels of the helices. At moderate temperatures, it is frequently seen that one hydrogen bond of a given pair would break, or the other, but not both simultaneously (Fig. S28f, g, l and S29f, h†).
 | ||
| Fig. 8 Stochastic dynamic simulations of 5b in CHCl3 over 1 ns at 300 K (a), 400 K (b), 500 K (c), 600 K (d) and 700 K (e). Stochastic dynamic simulations of 8b in CHCl3 over 1 ns at 300 K (f), 400 K (g), 500 K (h), 600 K (i) and 700 K (j). In each case, ten structures sampled every 100 ps are overlaid for each temperature. Side chains of Q, P and T have been omitted for clarity. | ||
Upon increasing temperature by increments of 100 K, motions are evident and H bonds between the two helices break frequently (Fig S28 and S29†). Different behaviours were observed for the two compounds. For 5b, the helical conformation of the backbone is mostly conserved, even at high temperature, when the tertiary fold is disrupted (hydrogen bonding distances > 5 Å), a behaviour comparable to that caused by the addition of DMSO. In addition, the conformation of each helical segment does not much differ from that of isolated helix 1 at the same temperature. For compound 8b, partial helix unfolding start to be observed at 500 K (Fig. 8g), together with tertiary structure disruption. Furthermore, this disruption occurs by the free termini of the tertiary fold, and the helical structure near the turn unit is mostly conserved. This contrasts with the melting of single helical 4 that is more extensive at the same temperatures (Fig. S30f–j†). Altogether, the simulations confirm the conclusions drawn from the effect of adding DMSO (Fig. 7). When secondary structures are robust as for 5b, helix–helix interactions can be disrupted without losing the secondary structures. For flexible sequences such as 8b, the melting of tertiary and secondary folds occurs concomitantly. There is thus evidence of cooperative folding: the most flexible secondary structures are stabilized within the tertiary fold, but more subject to melting upon increasing temperature. Yet this melting occurs at high in silico temperatures; the temperatures that can be implemented experimentally were too low to cause melting.
Conclusion
The rational design of abiotic tertiary folded structures as large, complex, and functionally efficient as proteins represents an exciting yet formidable challenge for chemistry. The possibility to elicit protein-like structures and properties in media completely different from water is intriguing and will certainly be enabling. First steps towards this goal are being made using aromatic oligoamide helix–turn–helix motifs stabilized by hydrogen bonds. From the start, it is important not to limit these designs to mere structures but to include dynamic aspects as well, for example how the folding of tertiary and secondary structures relate to one another and exchange with fully or partially unfolded states. We have shown here that completely abiotic structures show different types of unfolding behaviour depending on whether individual secondary modules are inherently stable or not. Both heating and polar solvents may disrupt helix–helix interactions. However, concomitant unfolding of the helices occurs only for more flexible helices that gain stability within the tertiary fold. Heating is predicted to be conducive of complete tertiary structure melting but only at temperatures above what can be reached experimentally. It might be inferred from this that the helix–turn–helix structures studied here may be tolerant towards even more flexible units than those introduced until now. For example, the P units may be replaced by 2-amino-ethoxy-acetic acid groups that have the same length and an ether oxygen atom instead of the pyridine endocyclic nitrogen atom. The findings reported here constitute a solid basis on which to further increase the size and complexity of these abiotic structures. Efforts towards more complex objects are in progress in our laboratory and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council under the European Union's Seventh Framework Programme (Grant Agreement No. ERC-2012-AdG-320892). It benefited from the facilities and expertise of the Biophysical and Structural Chemistry platform at IECB, CNRS UMS3033, INSERM US001, Université de Bordeaux. We thank Dr Brice Kauffmann for assistance with X-ray data collection, and Dr Post Sai Reddy for helping optimize monomer preparation.Notes and references
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS PubMed.
- R. V. Nair, K. N. Vijayadas, A. Roy and G. J. Sanjayan, Eur. J. Org. Chem., 2014, 2014, 7763–7780 CrossRef CAS.
- J. Dou, A. A. Vorobieva, W. Sheffler, L. A. Doyle, H. Park, M. J. Bick, B. Mao, G. W. Foight, M. Y. Lee, L. A. Gagnon, L. Carter, B. Sankaran, S. Ovchinnikov, E. Marcos, P.-S. Huang, J. C. Vaughan, B. L. Stoddard and D. Baker, Nature, 2018, 561, 485–491 CrossRef CAS.
- P.-S. Huang, S. E. Boyken and D. Baker, Nature, 2016, 537, 320–327 CrossRef CAS.
- S. E. Boyken, Z. Chen, B. Groves, R. A. Langan, G. Oberdorfer, A. Ford, J. M. Gilmore, C. Xu, F. DiMaio, J. H. Pereira, B. Sankaran, G. Seelig, P. H. Zwart and D. Baker, Science, 2016, 352, 680–687 CrossRef CAS PubMed.
- B. Dang, H. Wu, V. K. Mulligan, M. Mravic, Y. Wu, T. Lemmin, A. Ford, D.-A. Silva, D. Baker and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10852–10857 CrossRef CAS.
- N. H. Joh, T. Wang, M. P. Bhate, R. Acharya, Y. Wu, M. Grabe, M. Hong, G. Grigoryan and W. F. DeGrado, Science, 2014, 346, 1520–1524 CrossRef CAS.
- D. Hilvert, Annu. Rev. Biochem., 2013, 82, 447–470 CrossRef CAS PubMed.
- K. L. George and W. S. Horne, Acc. Chem. Res., 2018, 51, 1220–1228 CrossRef CAS.
- W. S. Horne, J. L. Price, J. L. Keck and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 4178–4180 CrossRef CAS.
- C. Mayer, M. M. Müller, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 6978–6981 CrossRef CAS PubMed.
- Z. E. Reinert, G. A. Lengyel and W. S. Horne, J. Am. Chem. Soc., 2013, 135, 12528–12531 CrossRef CAS PubMed.
- N. A. Tavenor, Z. E. Reinert, G. A. Lengyel, B. D. Griffith and W. S. Horne, Chem. Commun., 2016, 52, 3789–3792 RSC.
- M. Drewniak, E. Węglarz-Tomczak, K. Ożga, E. Rudzińska-Szostak, K. Macegoniuk, J. M. Tomczak, M. Bejger, W. Rypniewski and Ł. Berlicki, Bioorg. Chem., 2018, 81, 356–361 CrossRef CAS.
- D. S. Daniels, E. J. Petersson, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 1532–1533 CrossRef CAS.
- E. J. Petersson, C. J. Craig, D. S. Daniels, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 5344–5345 CrossRef CAS PubMed.
- G. W. Collie, K. Pulka-Ziach, C. M. Lombardo, J. Fremaux, F. Rosu, M. Decossas, L. Mauran, O. Lambert, V. Gabelica, C. D. Mackereth and G. Guichard, Nat. Chem., 2015, 7, 871–878 CrossRef CAS PubMed.
- N. Delsuc, M. Hutin, V. E. Campbell, B. Kauffmann, J. R. Nitschke and I. Huc, Chem.–Eur. J., 2008, 14, 7140–7143 CrossRef CAS PubMed.
- N. Delsuc, S. Massip, J.-M. Léger, B. Kauffmann and I. Huc, J. Am. Chem. Soc., 2011, 133, 3165–3172 CrossRef CAS PubMed.
- W. Ichinose, J. Ito and M. Yamaguchi, Angew. Chem., Int. Ed., 2013, 52, 5290–5294 CrossRef CAS PubMed.
- A. Lamouroux, L. Sebaoun, B. Wicher, B. Kauffmann, Y. Ferrand, V. Maurizot and I. Huc, J. Am. Chem. Soc., 2017, 139, 14668–14675 CrossRef CAS.
- H.-Y. Hu, J.-F. Xiang, Y. Yang and C.-F. Chen, Org. Lett., 2008, 10, 69–72 CrossRef CAS PubMed.
- I. Huc, Eur. J. Org. Chem., 2004, 2004, 17–29 CrossRef.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- I. Saraogi and A. D. Hamilton, Chem. Soc. Rev., 2009, 38, 1726–1743 RSC.
- Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef CAS.
- Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed.
- W. Wang, C. Zhang, S. Qi, X. Deng, B. Yang, J. Liu and Z. Dong, J. Org. Chem., 2018, 83, 1898–1902 CrossRef CAS PubMed.
- S. Saha, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2018, 57, 13542–13546 CrossRef CAS.
- S. De, B. Chi, T. Granier, T. Qi, V. Maurizot and I. Huc, Nat. Chem., 2018, 10, 51–57 CrossRef CAS PubMed.
- H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448–3449 CrossRef CAS.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337–6339 RSC.
- N. Delsuc, F. Godde, B. Kauffmann, J. M. Leger and I. Huc, J. Am. Chem. Soc., 2007, 129, 11348–11349 CrossRef CAS PubMed.
- D. Sanchez-Garcia, B. Kauffmann, T. Kawanami, H. Ihara, M. Takafuji, M. H. Delville and I. Huc, J. Am. Chem. Soc., 2009, 131, 8642–8648 CrossRef CAS PubMed.
- M. Vallade, P. Sai Reddy, L. Fischer and I. Huc, Eur. J. Org. Chem., 2018, 2018, 5489–5498 CrossRef CAS.
- B. Baptiste, C. Douat-Casassus, K. Laxmi-Reddy, F. Godde and I. Huc, J. Org. Chem., 2010, 75, 7175–7185 CrossRef CAS PubMed.
- S. L. Cockroft and C. A. Hunter, Chem. Soc. Rev., 2007, 36, 172–188 RSC.
- L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC.
- S. J. Dawson, X. Hu, S. Claerhout and I. Huc, Methods Enzymol., 2016, 580, 279–301 CAS.
- V. Maurizot, C. Dolain, Y. Leydet, J.-M. Léger, P. Guionneau and I. Huc, J. Am. Chem. Soc., 2004, 126, 10049–10052 CrossRef CAS PubMed.
- This species also prevails after freeze-drying a sample following incubation in TFA/CH2Cl2 (Fig. S24†). Note crystals of this species grow from toluene even though it does not predominate in this solvent (Fig. S25 and S26†).
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods, characterization of new compounds, detailed of the crystallographic and NMR investigations, and of molecular dynamics simulations. CCDC 1901969–1901971. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01322a |
| This journal is © The Royal Society of Chemistry 2019 |