
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtraction of cobalt(II) by methyltrioctylammonium chloride in nickel(II)-containing chloride solution from spent lithium ion batteries
Jiehong Cheng *a,
Tao Lua,
Xiao Wub,
Haojing Zhanga,
Chunyong Zhanga,
Ching-An Pengb and
Shouqiang Huanga
*a,
Tao Lua,
Xiao Wub,
Haojing Zhanga,
Chunyong Zhanga,
Ching-An Pengb and
Shouqiang Huanga
aSchool of Chemical and Environmental Engineering, Jiangsu University of Technology, Changzhou, China 213001. E-mail: Jiehong514@sina.com
bDepartment of Biological Engineering, University of Idaho, Moscow, USA 83844
First published on 23rd July 2019
Abstract
Spent lithium batteries contain valuable metals such as cobalt, copper, nickel, lithium, etc. After pretreatment and recovery of copper, only cobalt, nickel and lithium were left in the acid solution. Since the chemical properties of cobalt and nickel are similar, separation of cobalt from a solution containing nickel is technically challenging. In this study, Co(II) was separated from Ni(II) by chelating Co(II) with chlorine ions, Co(II) was then extracted from the aforementioned chelating complexes by methyltrioctylammonium chloride (MTOAC). The effects of concentrations of chlorine ions in the aqueous phase ([Cl−]aq), MTOAC concentrations in organic phase ([MTOAC]org), ratios of organic phase to aqueous phase (O/A), and the initial aqueous pH on cobalt separation were studied. The results showed that [Cl−]aq had a significant impact on cobalt extraction efficiency with cobalt extraction efficiency increasing rapidly with the increase in [Cl−]aq. The effect of initial pH on cobalt extraction efficiency was not significant when it varied from 1 to 6. Under the condition of [Cl−]aq = 5.5 M, [MTOAC]org = 1.3 M, O/A = 1.5, and pH = 1.0, cobalt extraction efficiency reached the maximum of 98.23%, and nickel loss rate was only 0.86%. The stripping rate of cobalt from Co(II)–MTOAC complexes using diluted hydrochloric acid was 99.95%. By XRD and XRF analysis, the recovered cobalt was in the form of cobalt chloride with the purity of cobalt produced reaching 97.7%. The mode of cobalt extraction was verified to be limited by chemical reaction and the kinetic equation for cobalt extraction was determined to be: R(Co) = 4.7 × 10−3[MTOAC](org)1.85[Co](aq)1.25.
1. Introduction
Lithium ion batteries (LIBs), widely used in portable electronic equipment such as cellular phones, lap-top computers, and other devices using lithium batteries, has resulted in a huge amount of spent LIBs worldwide that need to be dealt with. For instance, the quantity and weight of discarded LIBs in China are expected to surpass 25 billion units and 500 thousand tons by 2020, respectively.1 Considerable metals can be found in spent LIBs (e.g., 5–30% cobalt, 10–30% copper, 5–10% nickel, and 2–12% lithium) varying for different manufacturers and types of batteries.2–5 In some cases, the content of cobalt in the cathodic active material of LIBs could even reach more than 50%.6 Therefore, spent lithium ion batteries containing valuable metallic components can be regarded as a source for the recovery of above-mentioned metals. From the economic and environmental point of view, it is highly desirable to develop ways for sustainable recycling of spent LIBs.5,7,8 Cobalt is considered one of the most valuable raw material in supporting national economic growth and development due to its extremely rare existence on earth and wide uses in military, civilian, and medicinal industries.9,10 A large amount of research efforts have been made on recovering and recycling cobalt and lithium.3,6,9,11–15 Due to high cost and low source supply of cobalt and increased use of cobalt than lithium in making LIBS, recovery of cobalt from LIBs has the merits.16Due to high efficiency on metal recovery, solvent extraction has received a great deal of attention. Solvent extraction has been widely used for the separation and purification of metals from spent LIBs.17,18 However, there are two technical challenges in cobalt extraction. First, the co-existence of small amounts of iron, magnesium, manganese, and nickel in the spent LIBs is prone to have them extracted with cobalt, thereby decreasing separation efficiency. The other issue is that both cobalt and nickel ions exhibit similar physico-chemical properties in aqueous solution which make it difficult to separate and purify cobalt from mixtures containing nickel.18–20 For example, Coll et al.18 used ionic liquid HJMT+–Cy272 (tertiary alkyl (C16–C22) primary ammonium bis 2,4,4-(trimethylpentyl) phosphinate) to extract cobalt with 99% Co(II) recovered from the organic phase; however, 11% Ni(II) was also found in the recovered extracts. It has been reported that organophosphorus extractants (e.g., Cyanex272, D2EHPA or P204, and P507) were employed in the extractive separation of cobalt and nickel from aqueous sulphate media.16,21–24 Cyanex272 has highly selective capacity on cobalt, but it is expensive in China compared to that of MTOAC.25 Moreover, Cyanex272 and P507 all requires saponification the solution prior to extraction for preventing mass production of H+ ions which could inhibit extraction efficiency.18,26 To replace Na+ in the organic phase during stripping process, high concentration of acids must be used.21 Nogueira et al. extracted the interested metals (i.e. Co and Cd) by using nickel salts of the extractants of DEHPA and Cyanex272, but the extractants must be first treated for loading with nickel, which increasing processes step.27 D2EHPA can extract cobalt, nickel, and magnesium from LIBs,22,28 yet it is characteristic of poor selectivity and cannot efficiently separate cobalt from nickel. Combining two or three extraction reagents has also been reported, but these co-extraction methods not only complicated the cobalt extraction process,10,18,28,29 but also reduced phase separation efficiency, thereby increasing stability of cobalt–nickel extract (i.e., reducing stripping rate), which requires the use of acids with high concentration to get cobalt stripped. Therefore, there is a need to look for a cost-efficient reagent that can combine with other compounds for solvent extraction of cobalt from the solution containing nickel Amine extractants (e.g., Aliquat 33, Alamine 336, and Alamine 304) have been examined for solvent extraction of Co metal ions.24,30–32
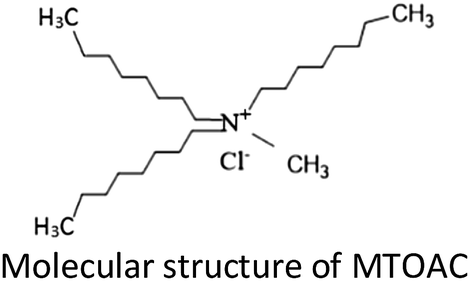
Quaternary ammonium salt (methyltrioctylammonium chloride, MTOAC) is an excellent and inexpensive amine extractant, which is suitable for the recovery of rare metals.33 The MTOAC is strong-base anion exchangers, which require lower concentrations of salting out reagents than other amine extractants. MTOAC is an ion-pair type extractant with a simple inorganic chloride anion. The mechanism with such an anion chloride as exchangers showed that during extraction, a simple inorganic anion was replaced by a complex metallic anion.34 Therefore, it works relatively well in separating cobalt from nickel when it is cobalt not nickel that form stable complex anion with Cl−. In comparison to organophosphorus extractants in cobalt separation, there is small quantities information by using MTOAC for cobalt extraction.28,30,35
In this study, extracting cobalt from the mixed electrode materials is the focus of this research paper, instead of pure cathodes from spent LIBs in previous studies.3,6,13,15,22–25 The spent lithium batteries were dismantled, with mixed electrodes crushed to remove impurities through acid leaching. Iron and manganese were also removed via pretreatment with copper recovery via extraction. The resultant liquid only contained a significant amount of cobalt and a small amount of nickel and lithium, which could improve the cobalt extraction efficiency by MTOAC. As a result, the interference of iron and manganese in extraction can be prevented and the difficulty to separate nickel from cobalt can be diminished. In order to develop a simple, efficient, and economic cobalt extraction process, this paper systematically studied the effect of MTOAC on the extraction and separation of cobalt in solution containing nickel, and discussed the effect of factors of [Cl−]aq, [MTOAC]org, the ratio of organic phase to aqueous phase(O/A), and initial aqueous pH on cobalt recovery efficiency in the extraction process. Furthermore, the constant interface cell method was employed to determine the reaction kinetics in cobalt extraction. The constant interface cell is one of the most applied methods in studying the extraction kinetics, which is easy to use with reliable outcomes, and can provide the net extraction efficiency at any reaction time with equilibrium constants.36 To date, there are no reports on the kinetics of using MTOAC to separate cobalt from nickel containing solutions. As such, knowledge on the cobalt extraction kinetics and activation energy for the MTOAC assisted extraction of cobalt with respect to the mixing speed, extraction temperature, section area, and organic phase concentration is needed to build the theoretical foundation and provide the design parameters for industrialized application of the MTOAC method for cobalt extraction.
2. Experimental methods
2.1 Materials
Aqueous phase is a leachate from spent LIB active electrode powder. The collected and pretreated raw battery materials, after removal of aluminum, iron, magnesium, manganese, and calcium, were leached in the HCl (6 M) + H2SO4 (4 M) + H2O2 (acid mole ratio: HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2SO4:H2O2 = 4:5:1), to obtain leaching solution containing copper, cobalt, nickel, and lithium. The copper was further removed by previously reported procedure.37 The remaining acid leaching solution containing 10.19 g L−1 cobalt, 0.42 g L−1 nickel, and 0.03 g L−1 lithium was used as the raw extraction solution for cobalt. For preparing raw cobalt aqueous solution, a certain amount of NaCl was added to the solution to generate various concentrations of [Cl−]aq.
:H2SO4:H2O2 = 4:5:1), to obtain leaching solution containing copper, cobalt, nickel, and lithium. The copper was further removed by previously reported procedure.37 The remaining acid leaching solution containing 10.19 g L−1 cobalt, 0.42 g L−1 nickel, and 0.03 g L−1 lithium was used as the raw extraction solution for cobalt. For preparing raw cobalt aqueous solution, a certain amount of NaCl was added to the solution to generate various concentrations of [Cl−]aq.
2.2. Experimental procedure
Timing started when the mixer started at a preset mixing speed, and sampling was performed at set intervals from the liquids in the aqueous phase to determine the cobalt level, with the cobalt concentration in the organic phase determined as the difference in cobalt content between the two phases.
All raw experimental data were shown as means of triplicates. All values of the statistical tests were presented as means and standard deviations. Unless otherwise stated, all experiments were carried out under atmospheric pressure in room temperature (25 °C).
2.3 Analytical procedures
The levels of cobalt and nickel in the raw aqueous phase were determined using an inductively-coupled plasma optical emission spectrometer (ICP-AES; model#: PE-2100DV, PerkinElmer). An X-ray fluorescence analyzer (XRF) was used to determine the spent LIB composition (ARL-9800; ARL; Switzerland). X-ray diffraction (XRD) (X'Pert PRO; PANalytical; Holland) was used to detect the component phases of the LIB powder and the recovered cobalt compound powder with Co radiation at a tube voltage of 40 kV and 30 mA of current.2.4 Equations calculating the cobalt extraction rate
where C0 is the cobalt concentration in the solution before extraction (g L−1); C1 is the cobalt concentration in the aqueous phase after extraction (g L−1).
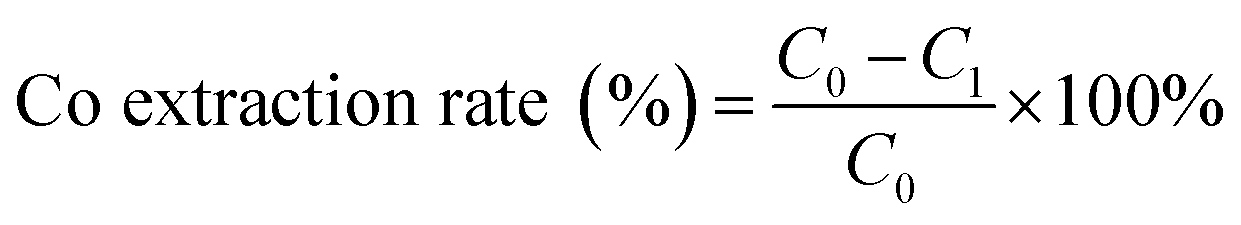
where C2 is the nickel concentration in the solution before extraction (g L−1); C3 is the nickel concentration in the solution after extraction (g L−1).
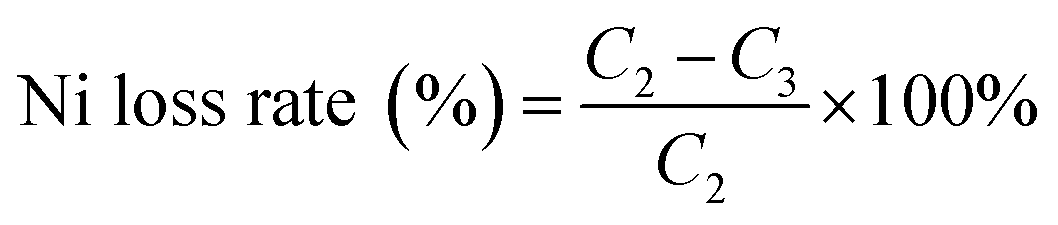
where C4 is the cobalt concentration in the stripping solution (g L−1); C5 is the cobalt concentration in the organic phase solution (g L−1).
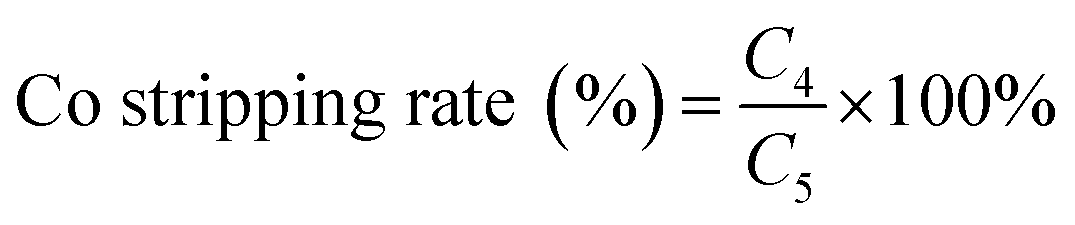
where C6 is the cobalt concentration in the stripping solution (g L−1); C0 is the initial cobalt concentration in the aqueous (g L−1).
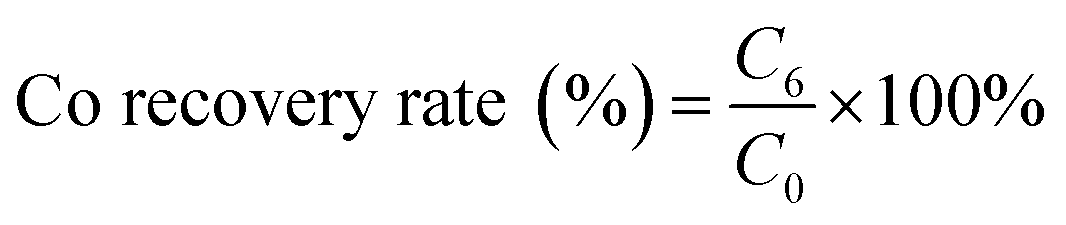
3. Results and discussion
3.1 Cobalt extraction
According to the physicochemical properties of Co and Ni, the stability of anionic complexes formed by Co2+ and Cl− is much higher than that formed by Ni2+ and Cl−. Ni2+ exists mostly in the form of hydration cation [Ni(H2O)62+], making it difficult to form stable complex anion with Cl−.38 Divalent transition metals Co2+ in aqueous chloride solutions readily only form chloro-complexes of CoCl42− by the following reactions:39| Co2+ + iCl− ⇔ CoCli2−i, (i = 1–4) | (1) |
Only the form of CoCl42− can be extracted by MTOAC (R4N+Cl−). After chloro-complexes of CoCl42− were formed in the aqueous solution, Co(II) extraction was operated following the extraction stoichiometry represented as follows:
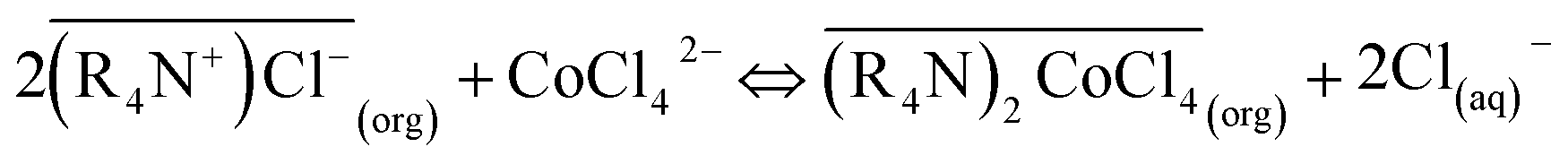 | (2) |
The regenerated organic phase after stripping could be reused in cobalt extraction. After extraction, the stripping agent used was diluted HCl, and the stripping reaction was:
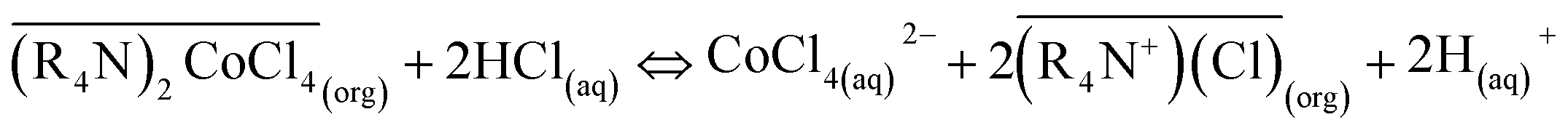 | (3) |
The regenerated organic phase after stripping could be reused in cobalt extraction.
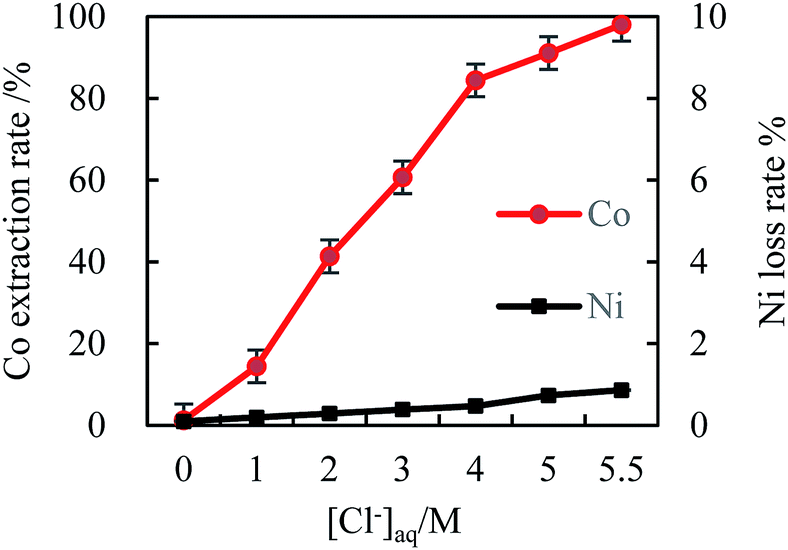 | ||
| Fig. 1 The effect of [Cl−]aq on cobalt extraction efficiency (initial pH 4, [MTOAC]org 0.8 M, O/A 2, oscillating time 90 s). | ||
As shown in Fig. 1, [Cl−]aq played an important role in the extraction process of cobalt from nickel in the solution. The cobalt extraction efficiency increased rapidly with the increase in [Cl−]aq as indicated by the slope of the curve (Fig. 1), while nickel was almost not extracted in the due experimental course with its maximum loss rate below 0.5%. When the Cl− concentration was increased to the maximal value of 5.5 M, the cobalt extraction efficiency reached 87.6%, while the nickel loss rate was only 0.46%. Thus, increasing [Cl−]aq in the solution could effectively separate cobalt from nickel, which was beneficial for recovering high purity cobalt, and recycling nickel at the same time.
From eqn (1), [Cl−]aq could achieve good complexation with almost all the cobalt ions in the same phase, resulting in the formation of cobalt–chloride complexes, which was the key first step in cobalt extraction. Fig. 1 demonstrated that [Cl−]aq needed to be at 5.5 M, which can achieve the highest cobalt extraction efficiency. It indicates that at this concentration, almost all chlorides reacted with Co2+ in the solution to produce cobalt chloride complex anion, CoCl42−.
Also, some past experiments showed that HCl concentration had a positive effect on cobalt extractions.32 Co(II) was more readily to form chlorocomplexes than Ni(II),31 which suggested that HCl was the most suitable agent in the period of acid leaching. Although chloride hydrometallurgy processes have many advantages over the conventional sulphate processes, efficient separation and recovery of valuable metals, especially cobalt and nickel from chloride solutions, have not been reported.40 In this paper, since there was no negative effect of lowering pH on cobalt extraction, using HCl immersion to provide Cl− to the solution reduced the NaCl usage for cobalt extraction, which could ensure sufficient Cl− present to complex cobalt and promote cobalt extraction due to the addition of a certain amount of HCl in the water phase. Therefore, MTOAC was recommended because there were no needs of extractant modification, aqueous pH adjustment, and use of sulfuric acid.
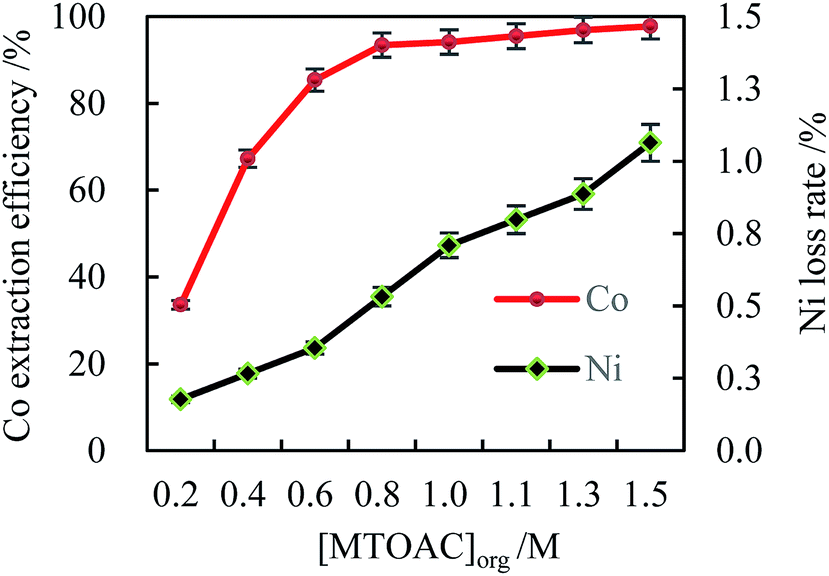 | ||
| Fig. 2 Effect of MTOAC concentration on the cobalt extraction efficiency ([Cl−]aq 5.0 M, initial pH 4, O/A 2, oscillating time 90 s). | ||
As expected, the experimental data illustrated that the MTOAC extractant played a significant role in cobalt extraction in chloride ion aqueous solution (shown in Fig. 1). The Co(II) extraction efficiency was enhanced with the increase of extractant concentration, which agreed with the literature data using amines type extractants in their studies.31,32 When the MTOAC concentration reached a optimal value of 1.3 M, the cobalt extraction efficiency achieved the maximal value of 96.87% while Ni loss rate of 0.89%. When the MTOAC concentration went beyond 1.5 M, the increase in cobalt extraction efficiency was only 0.89%, which was insignificant, while the nickel loss rate was on the rise but at a minimal level, and a remarkable increase in viscosity in the organic phase was observed, which made it difficult to operate the extraction process, and finally stopped the increase in extraction efficiency.
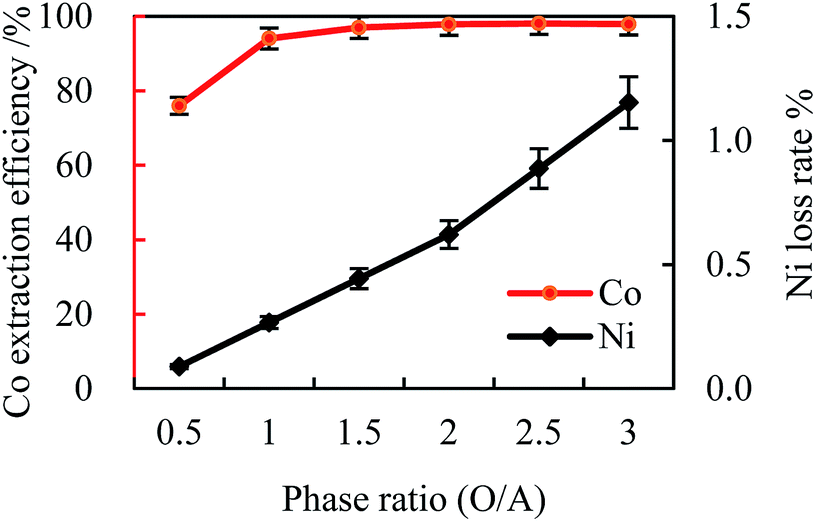 | ||
| Fig. 3 Effect of phase ratio (O/A) on cobalt extraction efficiency ([Cl−]aq 5.0 M, initial pH 4, [MTOAC]org 1.0 M, oscillating time 90 s). | ||
When the O/A was lower than 1.5, increasing phase ratio could increase the cobalt extraction efficiency to around 97.0%, and the nickel loss rate was only 0.44%. However, when the O/A was higher than 1.5, the cobalt extraction efficiency was levelled off with little improvement even when the phase ratio increased from 1.5 to 3.0 (the cobalt extraction efficiency only increased by 0.95%, and the nickel loss rate increased by 0.71%). This observation indicated that keeping increasing the phase ratio had little effect on the cobalt extraction efficiency but increasing the nickel loss rate instead.
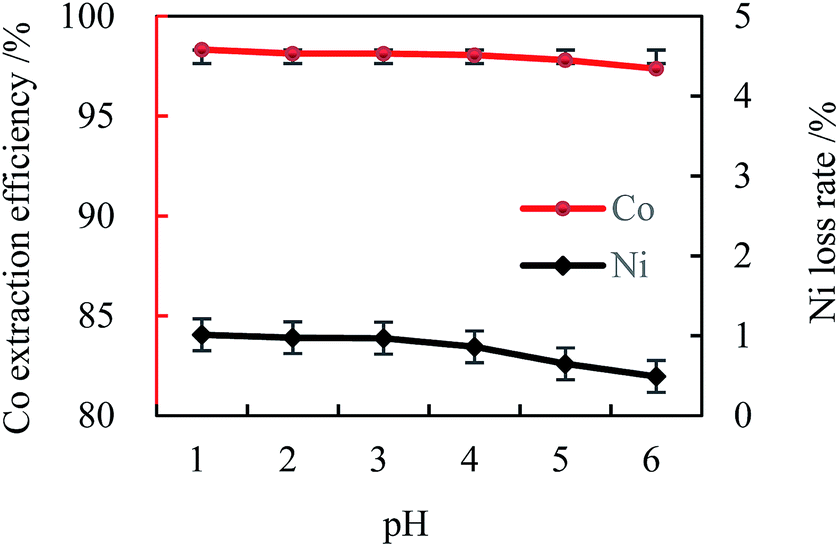 | ||
| Fig. 4 Effect of the initial aqueous pH on cobalt extraction ([Cl−]aq 5.0 M, [MTOAC]org 1.0 M, O/A 1.5,oscillating time 90 s). | ||
The pH-independent extraction could be understood from eqn (2) since H+ was not involved. The above results demonstrated that MTOAC (R4NCl) could provide an acceptable Co extraction and separation, which might be further enhanced by the addition of chloride ions, NaCl, in the aqueous solution in this study.
3.2 Purity analysis of the recovered cobalt products
According to above results, the cobalt loaded in the organic phase was easily stripped with 0.01 M HCl in a single stage and the hydrated CoCl2 powder with high purity was obtained after evaporation of the stripping liquor, as shown in the photos (A) and the XRD results (B) (Fig. 5). The XRF of the cobalt power products was presented in Table 1.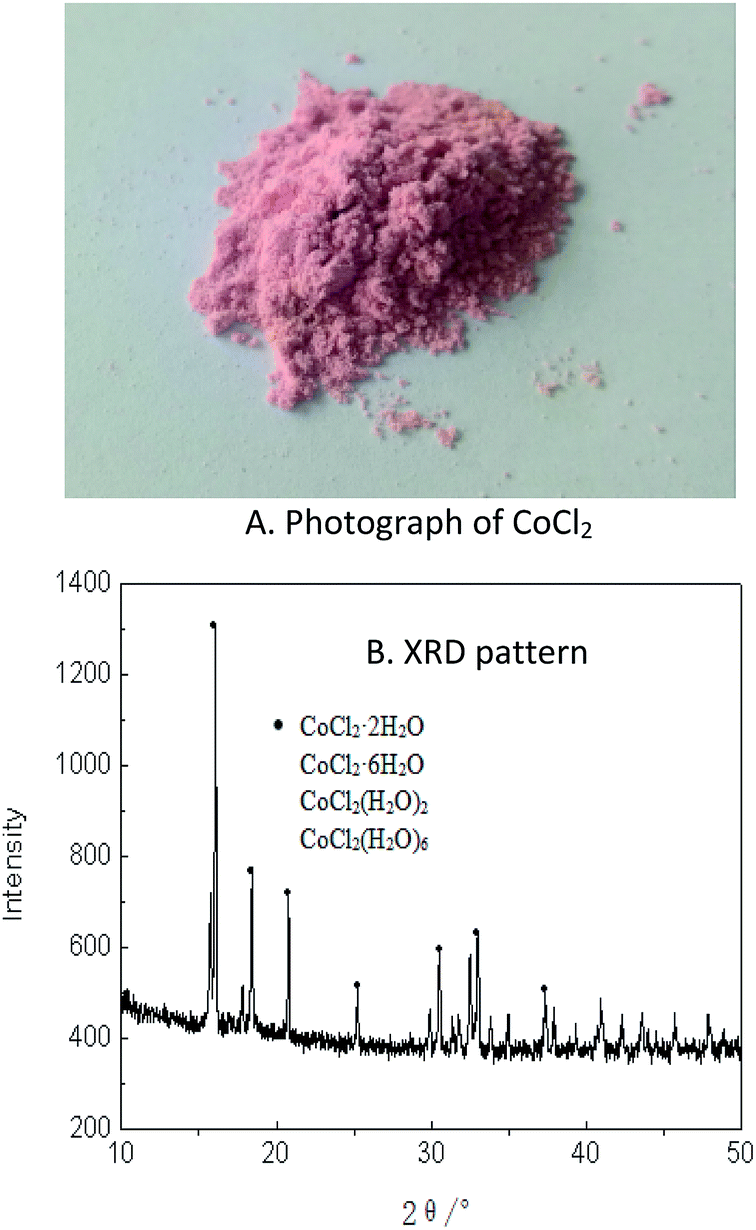 | ||
| Fig. 5 Photograph (A) and XRD pattern (B) of products of CoCl2 and its crystal water obtained by extraction. | ||
| Elements | Co | Cl | Na | S | Si | Fe | Crystal water |
| wt% | 25.60 | 29.75 | 0.54 | 0.029 | 0.021 | 0.001 | 44.06 |
As shown in Fig. 5, the recovered CoCl2 was pink in color, implying that it was hydrated cobalt chloride. The XRD results in Fig. 5 showed that the powder mainly contained CoCl2 with different water content (primarily containing two and six crystal water molecules), and with trace amounts of other elements. The reason of having various water contents is that the crystallization is typically taken under ambient temperature, hence forming water in crystal and solvation form. The mass of all CoCl2 (regardless of water content) amounted to 99.41% in the powder, with other trace elements including Na, S, Si, Fe, etc. Also from Table 1, Co accounted for 97.7% on a mass basis excluding Cl and water, suggesting that it was possible to recover 98.2% of Co with purity of 97.7% from the spent lithium ion batteries with hulls removed. More than 99.8% cobalt can be obtained by further refining by electrolysis.
Because of similar physico-chemical properties,it is extremely difficult to obtain pure cobalt and nickel compounds.18,19 Therefore, separating the nickel–cobalt pair in a simple and cost effective way becomes a challenge. This requires incorporating other processes in cobalt recovery. Some researchers employed other extractants in combination of other processes i.e. chemical, hydro-thermal, hydrometallurgy, etc.,6,18,32 and achieved a compared high extraction efficiency. However, all these processes are technically complicated and time consuming. In comparison, the cobalt extraction method using MTOAC in an HCl leaching system in this study could achieve the first-round extraction efficiency of 98.3%, which was better than all the above processes.
3.3 Extraction equilibrium isotherms of cobalt extraction
The required stages for counter-current extraction are directly related to the operating cost and maintenance requirements. The equilibrium isotherm can show the relative distribution of desired metals between organic and aqueous layers at specific experimental conditions. The McCabe–Thiele diagram for cobalt extraction was therefore employed to predict the number of the cobalt counter-current extraction stages by determining extraction with different A/O ratios through batch-wise extraction experiments. The extraction equilibrium isotherms of cobalt extraction was obtained at 25 °C and shown in Fig. 6.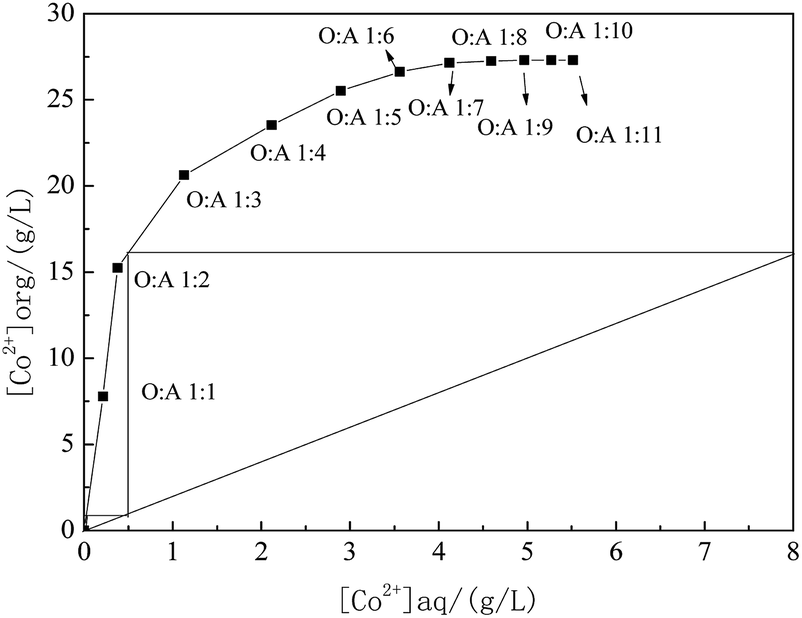 | ||
| Fig. 6 McCabe–Thiele diagram for the extraction of cobalt at different phase ratio. | ||
A McCabe–Thiele graph was constructed based on the counter current theory of extraction. Under the extraction condition with the cobalt content in the residual solution assumed to be around 0.01 g L−1, the McCabe–Thiele operating curve for extracting cobalt was drawn in Fig. 6 for O/A = 1/2 at the following conditions: 62% MTOAC of total concentration; 1.3 M with 5% isopropanol and 37.5% kerosene into organic phase, 8.0 g L−1 cobalt and 5.5 M Cl− into aqueous phase, initial pH 4.0, temperature 25 °C, and contact time 2.5 min.
The O/A ratio was varied within 1/5 to 1/11 while keeping the total volumes of the organic and aqueous phases constant. As revealed in Fig. 6, the McCabe–Thiele diagram indicated that two theoretical counter-current extraction stages would be required to yield a raffinate containing less than 0.01 g L−1 Co and attain a Co extraction efficiency over 99.9% at a phase ratio O/A = 1/6 from an aqueous solution containing 8.0 g L−1 Co2+ at an O/A = 1/2. It was observed that when the aqueous solution volume became higher, the loading of cobalt to the organic phase decreased after a plateau. The curve showed a steeper slope when the cobalt concentration in the oil phase varied from 0 to 15 g L−1, indicating a good cobalt extraction during this period in the current extraction system.
So far, the extraction equilibrium isotherms of using MTOAC to extract cobalt have not been reported. From the extraction equilibrium isotherms of other extractants such as HJMT+–Cyanex272 in extracting cobalt and lithium, a two- or three-stage extraction was required.18,41 Similar stages of extraction were also needed for other extractants such as D2EHPA, which required a three-stage extraction but was still unable to separate cobalt from nickel.28 Swain et al.42 also studied the extraction equilibrium isotherms of Cyanex272 at different volume concentrations, and found that the shape of the extraction isotherms of cobalt was somewhat out of the norm with a noticeable decrease in cobalt loading at higher A/O phase ratios. It was understood that the higher volume ratio of A/O resulted in higher lithium co-extraction, which made the extraction equilibrium for cobalt reverse in direction. In this paper, anionic complexes formed by Co2+ and Cl− in the aqueous phase was more stable than those of Ni2+ and Cl−, which rarely led to Ni co-extraction in the organic phase during the period of cobalt extraction by MTOAC.
3.4 Co extraction kinetics
The cobalt extraction efficiency, RCo, was defined as the number of cobalt ions entering the organic phase through a unit area in a unit time. When the reverse reaction rate could be neglected, the initial cobalt extraction rate could be expressed as below.
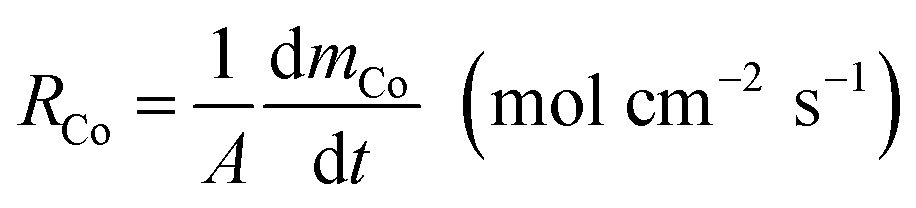 | (4) |
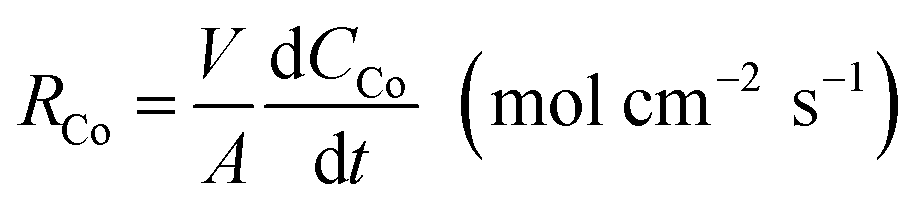 | (5) |
According to eqn (2), cobalt concentration in the organic phase is shown as concentration of (R4N)2CoCl4. So RCo should be expressed as:43
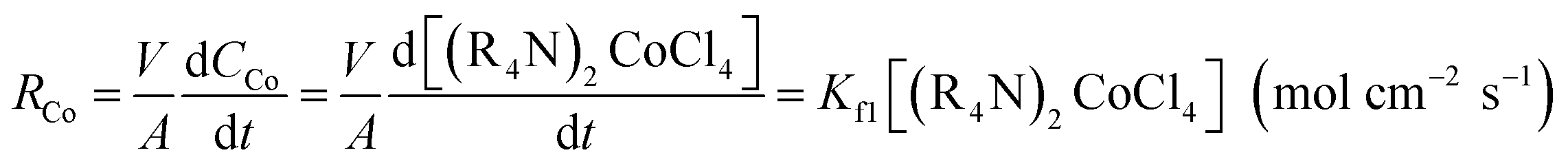 | (6) |
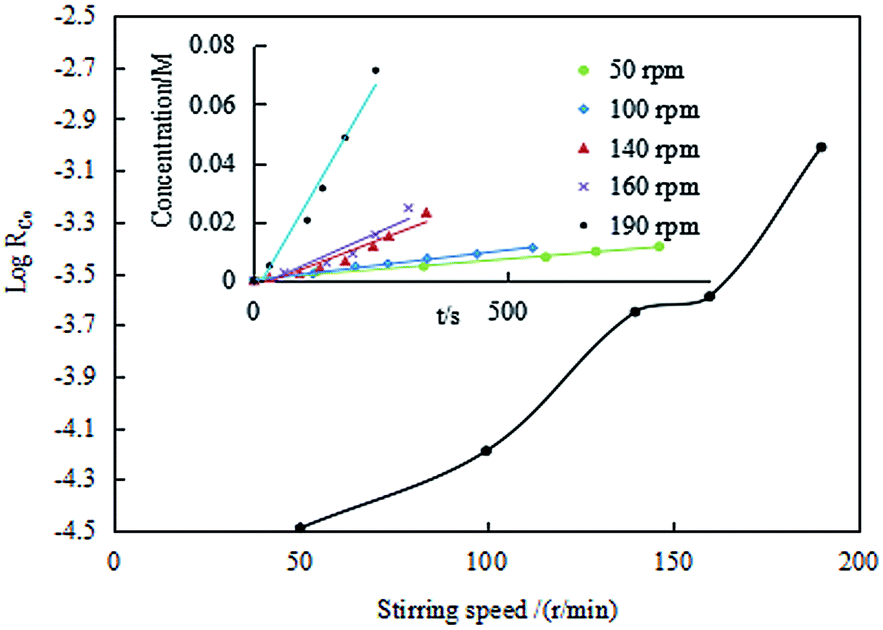
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) RCo on the stirring speed. Under the condition of [Co2+]aq of 0.0106 M, MTOAC concentration of 1.3 M, the O/A of 1, the interface area of 78.50 cm2, and temperature at 30 °C, the extraction regime was identified by the dependence of the Co(II) extraction efficiency on the stirring speed in the extraction kinetic experiments. The effect of stirring speed on the extraction efficiency of Co(II) are showed in Fig. 7.
RCo on the stirring speed. Under the condition of [Co2+]aq of 0.0106 M, MTOAC concentration of 1.3 M, the O/A of 1, the interface area of 78.50 cm2, and temperature at 30 °C, the extraction regime was identified by the dependence of the Co(II) extraction efficiency on the stirring speed in the extraction kinetic experiments. The effect of stirring speed on the extraction efficiency of Co(II) are showed in Fig. 7.
 | ||
| Fig. 7 Effect of stirring speed on extraction efficiency. | ||
Seen from Fig. 7, when the interface contracting area of the two phases was kept constant, the cobalt extraction efficiency was in general in an ascending trend as the stirring speed increased, with the extraction efficiency gradually increasing between 50 rpm and 140 rpm, implying that the extraction efficiency is dependent of the stirring speed. It's also deduced that extraction efficiency is mainly diffusion controlled instead of fully kinetic controlled.
However, when the mixing speed reached between 140 rpm and 160 rpm, the increase in the extraction efficiency was insignificant and almost unchanged, within which it was assumed that the extraction efficiency may be kinetic controlled and the diffusing mass transfer was substantially reduced, causing the cobalt extraction kinetics transitioning into the chemical reaction control type rather than diffusion.44,45 The possible reason might be that chemical reaction rate is slower than diffusion rate. When the mixing speed continued to increase to beyond 160 rpm, a sudden increase in the extraction efficiency was observed, and the mass flow between the two phases was changed from laminar to turbulent flow type, which was no longer in the territory of chemical reaction control. Therefore, for kinetics study, it was appropriate to select the mixing speed between 140 and 160 rpm. In this study, a stirring speed of 140 rpm was therefore chosen for all the following experiments.
Other experiments had to be performed to determine the extraction regime besides the data of extraction efficiency to avoid a wrong conclusion. In this work, relationship of the temperature, interfacial area, [MTOAC]org and [Co2+]aq on extraction efficiency was applied to determine the extraction regime.
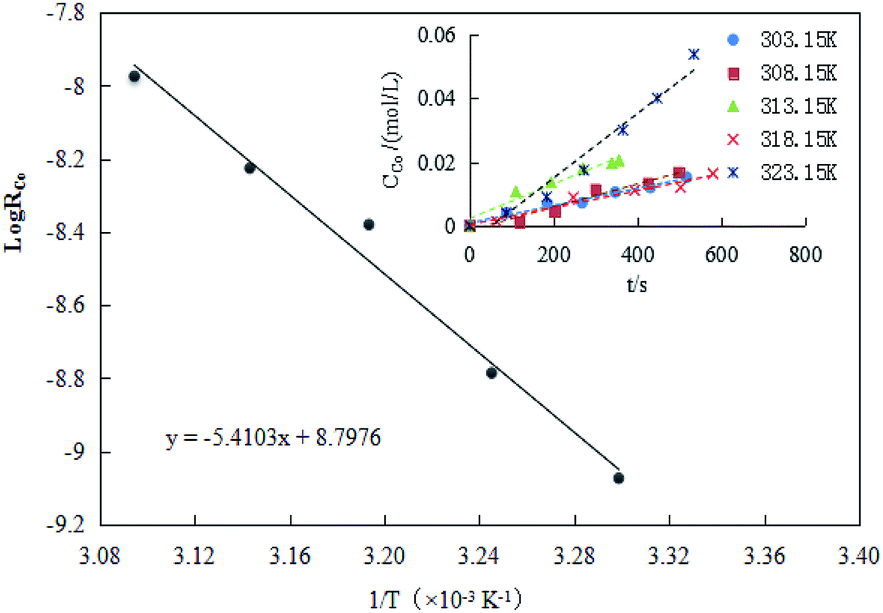
The result of RCo for different temperatures was shown in Fig. 8. As seen in Fig. 8, the extraction efficiency increased as the reaction temperature increased. A linear correlation between lnR0 and 1/T was observed with a slope of −5.4103.
 | ||
| Fig. 8 Arrhenius plot of logRCo vs. 1/T. | ||
The observed activation energy Ea(Co) could be calculated based on the Arrhenius equation, which was 44.98 kJ mol−1. Generally, when the activation energy was higher (e.g., ≥42 kJ mol−1), the cobalt extraction was possibly controlled by the chemical reactions taking place at the interface or near the interface area,while the reverse is true in systems governed by diffusion systems.46 Takacova et al.46 found that the cobalt extraction in sulfuric acid was mainly controlled by the rate of a chemical reaction (Ea(Co) = 43–48 kJ mol−1). Compared to the extraction efficiency obtained under the diffusion control, the effect of temperature on the extraction efficiency was significant. Therefore, considering the temperature effect, the process for cobalt extraction in this study complied with the type that was controlled by chemical reactions.
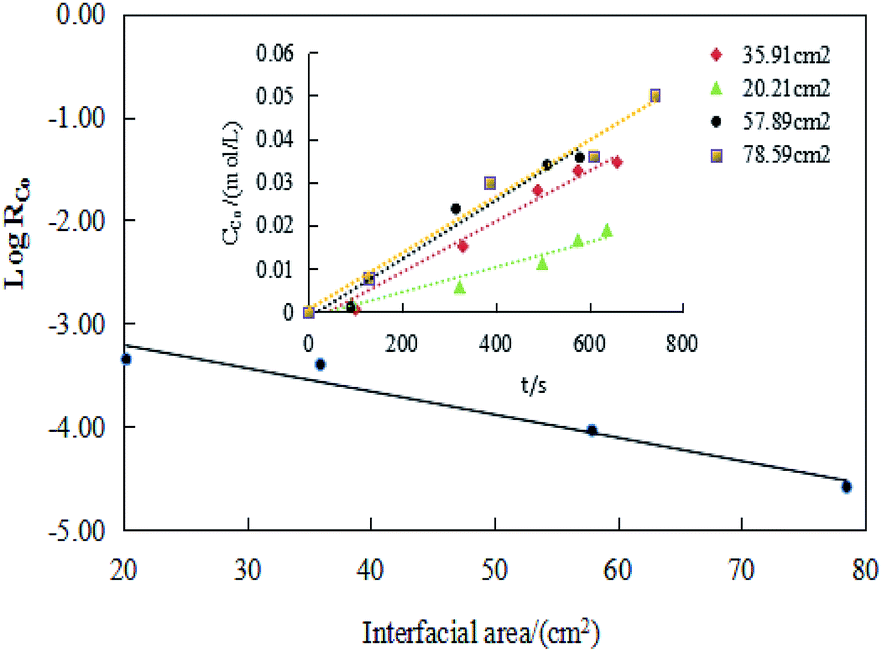
However, it did not determine whether the chemical reaction that controlled the extraction efficiency in a kinetic regime occurred in the bulk phase or at the interface. Thus, the relationship should be clarified between the extraction efficiency and the specific interfacial area (interfacial area/phase volume).
RCo and the interface area. In the former literature, the extraction efficiency is independent of the specific interfacial area, the extraction efficiency decreases with the increasing specific interfacial area.44 Similarly, if the chemical reactions take place at the interfacial zone, the extraction efficiency would be directly proportional to the interfacial area.47 Thus, it can draw a conclusion that the rate controlling reaction takes place in the bulk phase rather than at the interface. Therefore, combining the information obtained about the effect of stirring speed and temperature on cobalt extraction efficiency indicated that the extraction reaction seen in this study should belong to the territory of chemical reaction controlling step.
 | ||
| Fig. 9 Plot of logRCo vs. interfacial area. | ||
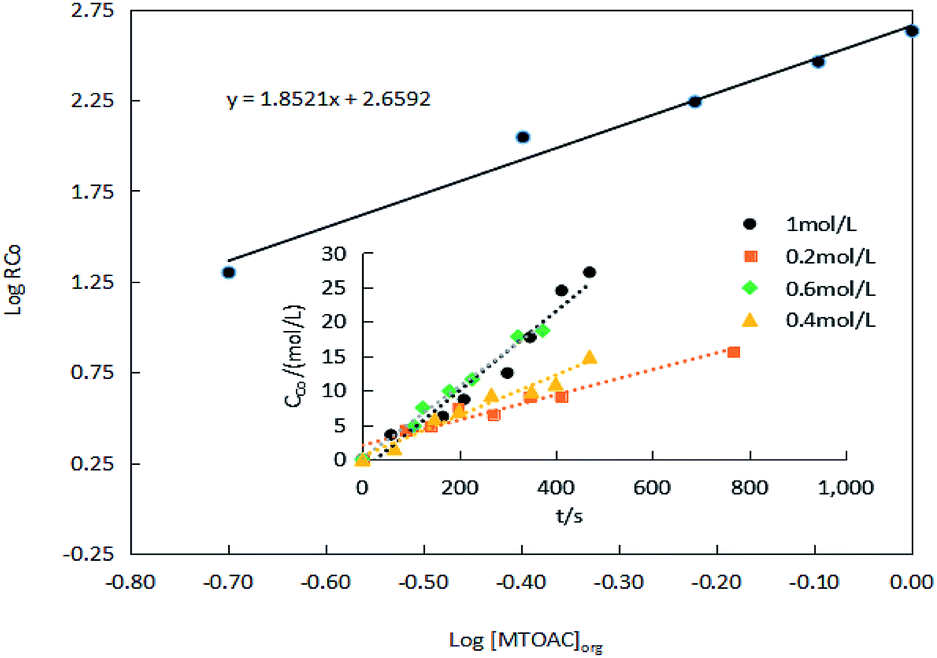
The results of the effect of MTOAC on the extraction efficiency presented in Fig. 10. Fig. 10 revealed that the rate of extraction of Co(II) increases with the increase in MTOAC concentration, and a linear relationship appeared to exist between logRCo and log[MTOAC]org based on linear regression analysis with a slope, α, being 1.85. The slopes of these lines indicate that the extraction efficiency is directly proportional to the extractant MTOAC concentration.
 | ||
| Fig. 10 Plot of logRCo vs. log[MTOAC]org. | ||
(2) Effect of extraction efficiency versus the concentration Co(II) in the aqueous
The results of concentration Co(II)were shown in Fig. 11. Based on Fig. 11, the extraction efficiency increased with the increase of Co(II) concentration in organic phase, and a linear relationship between logRCo and logCCo was observed with a slope, β, of 1.25. The cobalt extraction efficiency constant was calculated as Kf = 4.7 × 10−3.
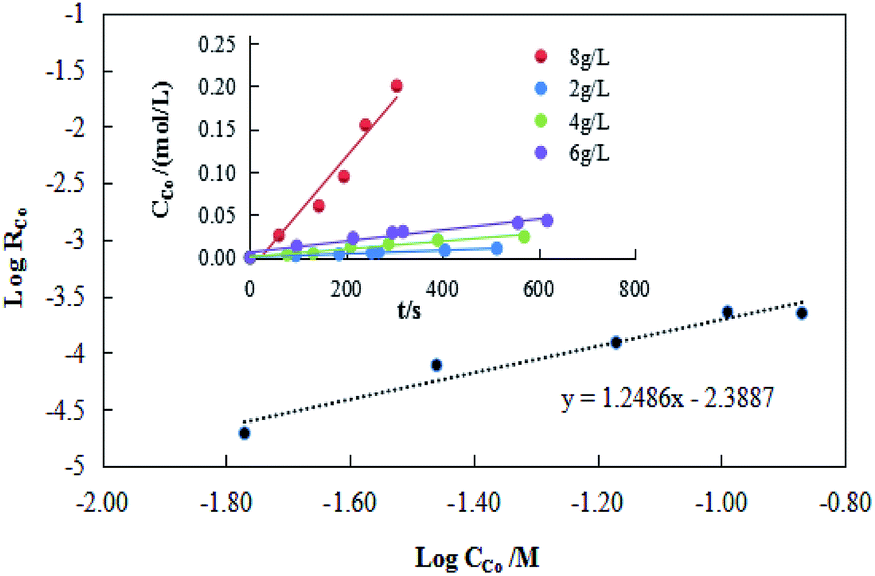 | ||
| Fig. 11 Plot of logRCo vs. logCCo. | ||
According to eqn (1), where Ki is the overall formation constant, is given by
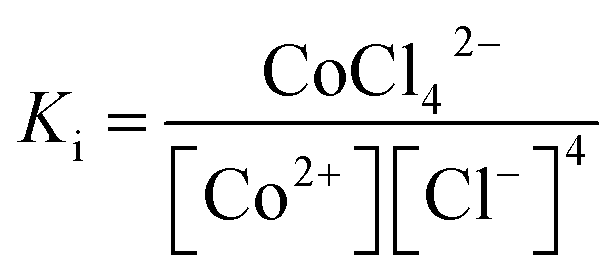 | (7) |
The concentration of CoCl42− in aqueous solution is basically calculated from the following mass balance equations:
| [CoCl42−] = Ki[Co2+][Cl−]4 | (8) |
According to eqn (2), where the overbar refers to the organic phase. The equilibrium constant, K, is given by:
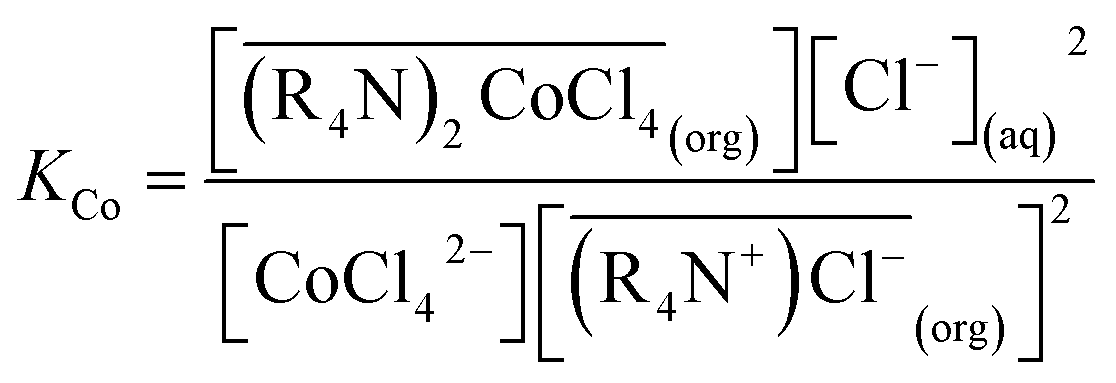 | (9) |
So eqn (6) can be written as:
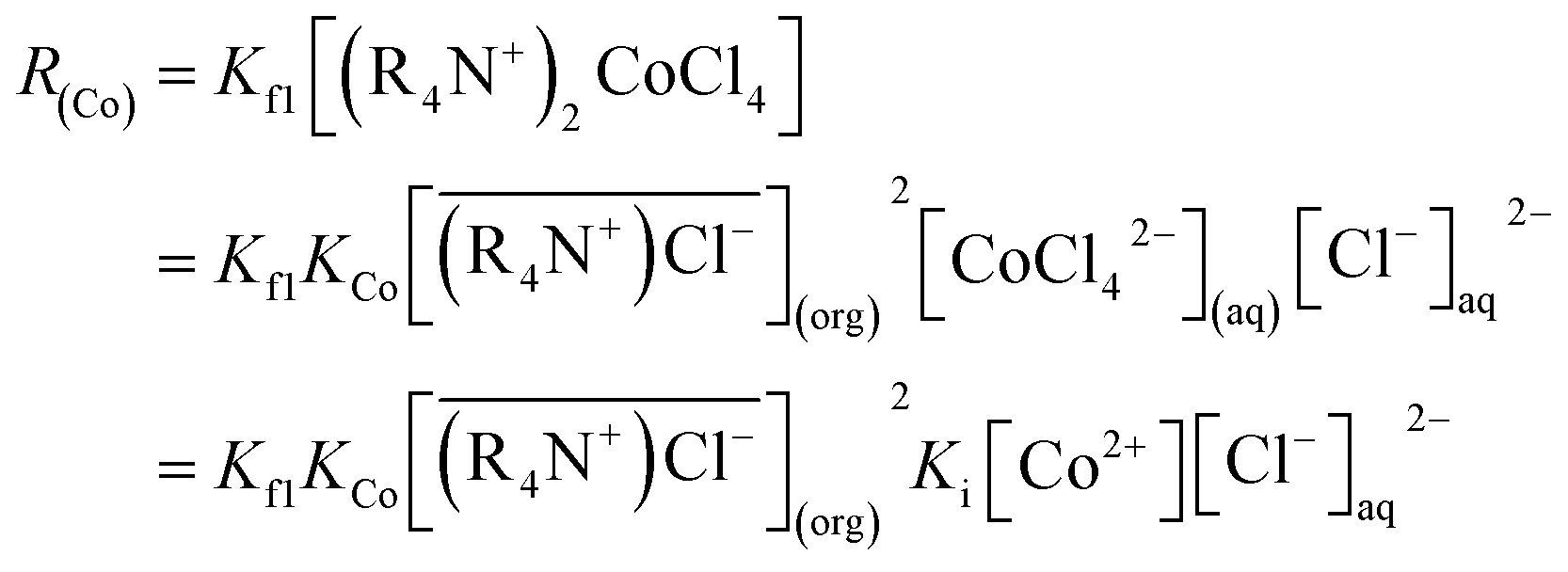 | (10) |
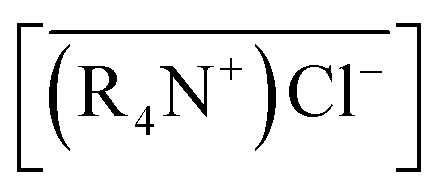 is the initial MTOAC concentration in the organic phase, M; [Co2+]aq is the initial cobalt concentration in the aqueous phase, M; [Cl−]aq is the concentration Cl− ion in the aqueous phase, M.
is the initial MTOAC concentration in the organic phase, M; [Co2+]aq is the initial cobalt concentration in the aqueous phase, M; [Cl−]aq is the concentration Cl− ion in the aqueous phase, M.
Since the concentration of [Cl−]aq is 5.5 M, it can be regarded as a constant. Eqn (10) can be simplified as follows:
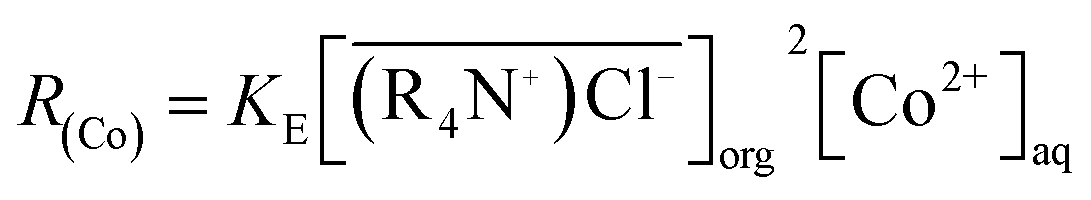 | (11) |
According to the above result, the forward rate equations for the extraction of Co(II) with MTOAC can be written as:
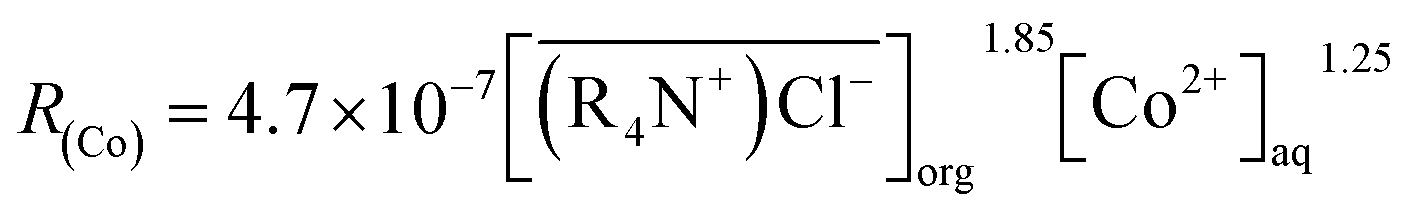 | (12) |
The theoretical cobalt extraction rate R(Co) can be obtained from eqn (2). Since the constant interface experiments were conducted under the condition of Co extraction process during contact time, when the reaction was far from reaching equilibrium, which was verified by Horner et al.33 i.e., the forward reaction rate was much greater than that of reverse reaction, the reverse reaction was considered negligible in our experiments. Thus, the equations for the cobalt extraction rate with MTOAC can be written as:
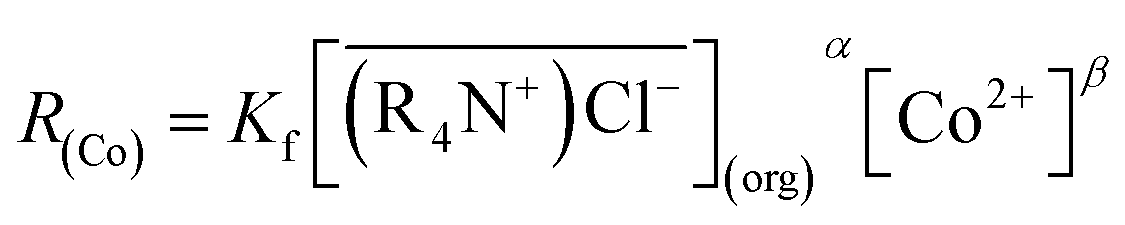 | (13) |
The experimental results showed that the cobalt extraction efficiency increased with the increases in the two-contacting phase area, mixing speed, and the cobalt concentration in the aqueous phase. Therefore, the MTOAC cobalt extraction was likely a process of chemical reaction control, which meant that the steps involved in extracting cobalt using MTOAC were controlled by chemical reactions. The forward extraction efficiency of cobalt increased with the increases in cobalt concentration in the aqueous phase and the MTOAC concentration. Comparing the result of eqn (13), above mechanism is consistent with the rate eqn (12) obtained from experimental results. Therefore, the kinetic equation describing the cobalt extraction efficiency using MTOAC at 30 °C was: 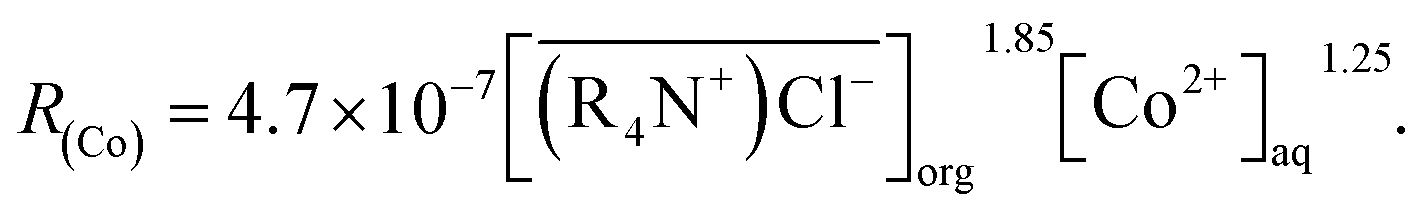
4. Conclusions
4.1 Cobalt extraction
The Cl− concentration played an important role in cobalt extraction, while the initial pH was less influential. Under the conditions of 25 °C, normal pressure, and ≤1 h time for extraction and stripping, the maximum extraction efficiency of 98.32% for cobalt (Co) and the minimum loss rate of 0.86% for nickel were achieved in a single stage with the optimized operating parameters being [Cl−]aq = 5.5 M, [MTOAC]org = 1.3 M, O/A = 1.5, and pH = 1.0. The XRD and XRF results showed that the recovered cobalt was in the form of cobalt chloride with the cobalt purity reaching 97.7%. The extraction equilibrium isotherms showed that two-stage counter current extraction was required.The cobalt extraction process was controlled by chemical reactions. The activation energy calculated by the Arrhenius equation was 44.98 kJ mol−1, and it can be found that the kinetics mechanism result of Co(II) is basically consistent with the theoretical kinetics.
MTOAC extraction works relatively well in recovery cobalt from spent LIBs. Meanwhile, a large amount of Cl− was pulled in the aqueous phase, which should be considered to remove.
Conflicts of interest
No potential conflict of interest was reported by the authors.Acknowledgements
This work was supported by Jiangsu Government Scholarship for Overseas Studies Project (2017) and the Key University Natural Science Research Project of Jiangsu Province of China (15KJA610001), National Natural Science Foundation of China (21607101).Notes and references
- X. L. Zeng, J. H. Li and Y. S. Ren, Prediction of various discarded lithium batteries in China, IEEE International Symposium on Sustainable Systems and Technology, 2012, 1–4 Search PubMed.
- L. Sun and K. Qiu, Vacuum pyrolysis and hydrometallurgical process for the recovery of valuable metals from spent lithium-ion batteries, J. Hazard. Mater., 2011, 194, 378–384 CrossRef CAS PubMed.
- L. Li, J. B. Dunn, X. X. Zhang, L. Gaines, R. J. Chen, F. Wu and K. Amine, Recovery of metals from spent lithium-ion batteries with organic acids as leaching reagents and environmental assessment, J. Power Sources, 2013, 233, 180–189 CrossRef CAS.
- X. Zeng, J. Li and N. Singh, Recycling of spent lithium-ion battery: a critical review, Crit. Rev. Environ. Sci. Technol., 2014a, 44, 1129–1165 Search PubMed.
- S. P. Barik, G. Prabaharan and B. Kumar, An innovative approach to recover the metal values from spent lithium-ion batteries, Waste Manage., 2016, 51, 222–226 CrossRef CAS PubMed.
- L. Li, J. Ge, F. Wu, R. Chen, S. Chen and B. Wu, Recovery of cobalt and lithium from spent lithium ion batteries using organic citric acid as leachant, J. Hazard. Mater., 2010b, 176, 288–293 Search PubMed.
- J. Dewulf, D. V. G. Van, K. Denturck, H. Langenhovea, W. Ghyootb and K. Vande-putte, Recycling rechargeable lithium-ion batteries: critical analysis of natural resource savings, Resour., Conserv. Recycl., 2010, 54, 229–234 CrossRef.
- X. Chen, H. Ma, C. Luo and T. Zhu, Recovery of valuable metals from waste cathode materials of spent lithium-ion batteries using mild phosphoric acid, J. Hazard. Mater., 2017, 326, 77–86 CrossRef CAS PubMed.
- L. Sun and K. Qiu, Organic oxalate as leachant and precipitant for the recovery of valuable metals from spent lithium-ion batteries, Waste Manage., 2012, 32, 1575–1582 CrossRef CAS PubMed.
- A. A. Nayl, M. M. Hamed and S. E. Rizk, Selective extraction and separation of metal values from leach liquor of mixed spent Li-ion batteries, J. Taiwan Inst. Chem. Eng., 2015, 55, 119–125 CrossRef CAS.
- G. P. Nayaka, J. Manjanna, K. V. Pai, R. Vadavi, S. J. Keny and V. S. Tripathi, Recovery of valuable metal ions from the spent lithium-ion battery using aqueous mixture of mild organic acids as alternative to mineral acids, Hydrometallurgy, 2015, 151, 73–77 CrossRef CAS.
- G. P. Nayaka, K. V. Pai, J. Manjanna and S. J. Keny, Use of mild organic acid reagents to recover the Co and Li from spent Li-ion batteries, Waste Manage., 2016a, 51, 234–238 Search PubMed.
- G. P. Nayaka, K. V. Pai, G. Santhosh and J. Manjanna, Dissolution of cathode active material of spent Li-ion batteries using tartaric acid and ascorbic acid mixture to recover Co, Hydrometallurgy, 2016b, 161, 54–57 Search PubMed.
- X. Chen, C. Luo, J. Zhang, J. Kong and T. Zhou, Sustainable recovery of metals from spent lithium-ion batteries: a green process, ACS Sustainable Chem. Eng., 2015b, 3(12), 3104–3113 Search PubMed.
- X. Zeng, J. Li and B. Shen, Novel approach to recover cobalt and lithium from spent lithium-ion battery using oxalic acid, J. Hazard. Mater., 2015, 295, 112–118 CrossRef CAS PubMed.
- A. Chagnes and B. Pospiech, A brief review on hydrometallurgical technologies for recycling spent lithium-ion batteries, J. Chem. Technol. Biotechnol., 2013, 88, 1191–1199 CrossRef CAS.
- J. Kang, G. Senanayake, J. Sohn and S. M. Shin, Recovery of cobalt sulfate from spent lithium ion batteries by reductive leaching and solvent extraction with Cyanex 272, Hydrometallurgy, 2010, 100, 168–171 CrossRef CAS.
- M. T. Coll, A. Fortuny, C. S. Kedari and A. M. Sastre, Studies on the extraction of Co(II) and Ni(II) from aqueous chloride solutions using Primene JMT-Cyanex272 ionic liquid extractant, Hydrometallurgy, 2012, 125–126, 24–28 CrossRef CAS.
- X. Zhang, H. Cao, Y. Xie, P. Ning, H. An, H. You and F. Nawaz, A closed-loop process for recycling LiNi1/3Co1/3Mn1/3O2 from the cathode scraps of lithium-ion batteries: process optimization and kinetics analysis, Sep. Purif. Technol., 2015, 150, 186–195 CrossRef CAS.
- S. Zhang, X. Huang and D. Wang, Review on Comprehensive Recovery of Valuable Metals from Spent Electrode Materials of Nickel-Hydrogen Batteries, Rare Met. Mater. Eng., 2015, 44(1), 73–78 CrossRef CAS.
- L. Chen, X. Tang, Y. Zhang, L. Li, Z. Zeng and Y. Zhang, Process for the recovery of cobalt oxalate from spent lithium-ion batteries, Hydrometallurgy, 2011, 108, 80–86 CrossRef CAS.
- X. Chen, Y. Chen, T. Zhou, D. Liu, H. Hu and S. Fan, Hydrometallurgical recovery of metal values from sulfuric acid leaching liquor of spent lithium-ion batteries, Waste Manage., 2015a, 38, 349–356 Search PubMed.
- G. Granata, F. Pagnanelli, E. Moscardini, Z. Takacova, T. Havlik and L. Toro, Simultaneous recycling of nickel metal hydride, lithium ion and primary lithium batteries: Accomplishment of European Guidelines by optimizing mechanical pre-treatment and solvent extraction operations, J. Power Sources, 2012, 212, 205–211 CrossRef CAS.
- R. Torkaman, M. Asadollahzadeh, M. Torab-Mostaedi and M. G. Maragheh, Recovery of cobalt from spent lithium ion batteries by using acidic and basic extractants in solvent extraction process, Sep. Purif. Technol., 2017, 186, 318–325 CrossRef CAS.
- L. I. Xiao-hui, H. Song-tao, J.-k. Wen and G.-y. Zhou, Research progress of synergistic solvent extraction of cobalt and nickel, Chin. J. of Nonferrous Met., 2013, 23(7), 2026–2032 Search PubMed.
- P. Francesca, M. Emanuela, A. Pietro, A. T. Abo and L. Toro, Cobalt products from real waste fractions of end of life lithium ion batteries, Waste Manage., 2016, 51, 214–221 CrossRef PubMed.
- A. Nogueira Carlos, C. Oliveira Paula and M. Pedrosa Fátima, Separation of Cadmium, Cobalt, and Nickel by Solvent Extraction using the Nickel Salts of the Extractants, Solvent Extr. Ion Exch., 2009, 27(2), 295–311 CrossRef.
- Y. Yang, S. Xu and Y. He, Lithium recycling and cathode material regeneration from acid leach liquor of spent lithium-ion battery via facile co-extraction and coprecipitation processes, Waste Manage., 2017, 64, 219–227 CrossRef CAS PubMed.
- C. Bourget, B. Jakovljevic and D. Nucciarone, CYANEXR 301 binary extractant systems in cobalt/nickel recovery from acidic sulphate solutions, Hydrometallurgy, 2005, 77, 203–218 CrossRef CAS.
- A. A. Nayl, Extraction and separation of Co(II) and Ni(II) from acidic sulfate solutions using Aliquat 336, J. Hazard. Mater., 2010, 173, 223–230 CrossRef CAS PubMed.
- A. Fernandes, J. C. Afonso and A. J. B. Dutra, Separation of nickel(II), cobalt(II) and lanthanides from spent Ni-MH batteries by hydrochloric acid leaching, solvent extraction and precipitation, Hydrometallurgy, 2013, 133, 37–43 CrossRef CAS.
- V. Innocenzi, N. M. Ippolito, I. D. Michelis, M. Prisciandaro, F. Medici and F. Vegliò, A review of the processes and lab-scale techniques for the treatment of spent rechargeable NiMH batteries, J. Power Sources, 2017, 362, 202–218 CrossRef CAS.
- M. K. Jha, A. Kumari, R. Panda, J. R. Kumar, K. Yoo and J. Y. Lee, Review on hydrometallurgical recovery of rare earth metals, Hydrometallurgy, 2016, 165, 2–26 CrossRef CAS.
- C. K. Gupta and N. Krishnamurthy, Extractive Metallurgy of Rare Earths, CRC press, NY, USA, 2005 Search PubMed.
- T. Hirai and K. Isao, Mechanism of Extraction of Cobalt from Hydrochloric Acid by Tri-n-Octylmethylammonium Chloride, J. Chem. Eng. Jpn., 1991, 24(1), 58–62 CrossRef CAS.
- D. E. Horner, J. C. Mallen, J. R. Jr Coggins and R. G. Yates, Inter phase Transfer Kinetics of Thorium between Nitric Acid and Tributyl Phosphate Solutions Using the Single Drop and the Lewis Cell Techniques, Ind. Eng. Chem. Fundam., 1980, 19(3), 287–291 CrossRef CAS.
- Y. Chen and J. H. Cheng, Copper separation from spent lithium ion battery by hydroxyoxime extractants, Chin. J. Process Eng., 2017, 17(6), 1170–1175 Search PubMed.
- D. S. Flett, Cobalt–nickel separation in hydrometallurgy: a review, Chem. Sustainable Dev., 2004, 12, 81–91 Search PubMed.
- D. Wassink and H. J. Dreisinger, Solvent extraction separation of zinc and cadmium from nickel and cobalt using Aliquat 336, a strong base anion exchanger, in the chloride and thiocyanate forms, Hydrometallurgy, 2000, 57, 235–252 CrossRef.
- Z. Zhu, W. Zhang, Y. Pranolo and C. Y. Cheng, Separation and recovery of copper, nickel, cobalt and zinc in chloride solutions by synergistic solvent extraction, Hydrometallurgy, 2012, 127–128, 1–7 CrossRef CAS.
- R. Sattar, S. Ilyas, H. N. Bhatti and A. Ghaffar, Resource recovery of critically-rare metals by hydrometallurgical recycling of spent lithium ion batteries, Sep. Purif. Technol., 2019, 209, 725–773 CrossRef CAS.
- B. Swain, J. Jeong, J. Lee and G. H. Lee, Development of process flow sheet for recovery of high pure cobalt from sulfate leach liquor of LIB industry waste: A mathematical model correlation to predict optimum operational conditions, Sep. Purif. Technol., 2008, 63, 360–369 CrossRef CAS.
- N. E. El-Hefny, Kinetics and mechanism of extraction of Cu(II) by CYANEX 302 from nitrate medium and oxidative stripping of Cu(I) using Lewis cell technique, Chem. Eng. Process., 2010, 49, 84–90 CrossRef CAS.
- P. R. Danesi, R. Chiarizia and C. F. Coleman, The kinetics of metal solvent extraction, CRC Crit. Rev. Anal. Chem., 1980, 10, 1–126 CrossRef.
- Y. Xiong, Z. Lou, S. Yue, J. Song, W. Shan and G. Han, Kinetics and mechanism of Re(VII) extraction with mixtures of tri-alkylamine and tri-n-butylphosphate, Hydrometallurgy, 2010, 100, 110–115 CrossRef CAS.
- Z. Takacova, T. Havlik, F. Kukurugya and D. Orac, Cobalt and lithium recovery from active mass of spent Li-ion batteries: Theoretical and experimental approach, Hydrometallurgy, 2016, 163, 9–17 CrossRef CAS.
- S. Yin, S. Li, B. Zhang, J. Peng and L. Zhang, Extraction kinetics of neodymium(III) from chloride medium in the presence of two complexing agents by D2EHPA using a constant interfacial area cell with laminar flow, Hydrometallurgy, 2016, 161, 160–165 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |