
Tunable circularly polarized luminescence from molecular assemblies of chiral AIEgens†
Fengyan
Song
ab,
Yanhua
Cheng
 *c,
Qiuming
Liu
b,
Zijie
Qiu
ab,
Jacky W. Y.
Lam
ab,
Liangbin
Lin
d,
Fafu
Yang
*d and
Ben Zhong
Tang
*abe
*c,
Qiuming
Liu
b,
Zijie
Qiu
ab,
Jacky W. Y.
Lam
ab,
Liangbin
Lin
d,
Fafu
Yang
*d and
Ben Zhong
Tang
*abe
aDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, Institute for Advanced Study, Department of Chemical and Biological Engineering and Division of Life Science, The Hong Kong University of Science and Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
bHKUST-Shenzhen Research Institute, No. 9 Yuexing 1st RD, South Area, Hi-tech Park, Nanshan, Shenzhen 518057, China
cState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, China. E-mail: cyh@dhu.edu.cn
dCollege of Chemistry and Materials Science, Fujian Normal University, Fuzhou 350007, P. R. China. E-mail: yangfafu@fjnu.edu.cn
eNSFC Center for Luminescence from Molecular Aggregates, SCUT-HKUST Joint Research Institute, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
First published on 10th July 2019
Abstract
Circularly polarized luminescence (CPL) is important to chiral photonic technologies. In molecular systems, besides their intrinsic chemical structures, architectures of molecular assemblies at the mesoscopic scale also account for the final macroscopic CPL properties. Herein, tunable CPL responses can be induced through architectural regulation of these molecular assemblies in suspension and solid states. A liquid crystalline assembled system of DPCE-ECh exhibiting a smectic C* phase with a high dissymmetry factor (gCD = −0.20 and glum = +0.38) is reported. The intense and apparent CD and CPL of the film stem from the intrinsic helical structure of the molecular assembles with a weak contribution of Bragg reflection, where the helical axis is perpendicular to the optical axis and parallel to the direction of the glass substrate. To the best of our knowledge, this large glum factor is very rare for organic compounds even in the assembled state formed by annealing at the smectic liquid crystalline temperature. Interestingly, a strong CPL signal with a glum value of +0.18 is still recorded when DPCE-ECh is annealed in a chiral isotropic liquid (Iso*) state. On the other hand, DPCE-ACh can form two coexisting phases of chiral hexagonal and smectic liquid-crystalline phases due to intermolecular hydrogen bonding. The non-periodic molecular orientations of DPCE-ACh break its helical structure to give a weak negtive CPL signal of the order of 10−3. This work thus provides a new insight for developing efficient chiroptical materials in the aggregate state that have profound implications for high-performance CPL-based devices.
Introduction
Development of circularly polarized luminescent (CPL) materials has gained increasing interest owing to their potential applications in stereoscopic optical information storage and processing,1 optical recognition sensors,2–5 quantum computing,6 and circularly polarized electroluminescence for 3D displays.7–20 The CPL response of a molecular system is quantified by the dissymmetry factor (glum),21–27 where glum = 2(IL − IR)/(IL + IR) and IL and IR denote the emission intensitiy of left- and right-CPL, respectively. The common strategy to achieve CPL is to synthesise molecules with a specific chiral configuration.28–32 However, the CPL response of synthetic advanced materials not only relies on chiral functions on the molecular level, but also depends on the mesoscopic architectures of the molecular assemblies. Through a self-assembly approach, nanostructured chiral materials are able to transfer and amplify the molecular functions to an amplified CPL property at a specific length scale.33–41 Therefore, an investigation on the relationship between the hierarchical structure of molecular assembles and their corresponding CPL properties is still an important issue to achieve efficient CPL materials.Normally, the luminescence normalized dissymmetry factor of organic systems ranges between 10−4 to 10−2.3,5,16–27 In rare cases, extremely high g-values exceeding 0.2 or even up to 1 have been reported for polyfluorene thin films8–10,42–44 or cholesteric organic systems.45–51 In a polyfluorene system, the circular polarization is largely determined by the anisotropy of the cholesteric dielectric medium. The glum value is thickness dependent and a strong CPL effect originates from the selective CP reflection due to the long-range cholesteric ordering (Bragg reflection).10,44 The helical axis of this system is perpendicular to the direction of the substrate. In cholesteric films, hierarchical chiral mesoscopic structures were found. A strong CPL response can be generated from the sum of two main contributions, including the inherent chiral supramolecular structure and birefringence pattern (Bragg reflection). However, these doped cholesteric systems often suffer from the problems of incompatibility and instability. Thus the pursuance of a strong chiroptical signal from pure organic compounds remains challenging. Akagi's group reports a glum of +0.29 in a chiral bithiophene-phenylene copolymer film annealed in a chiral nematic state.45 They also report a high glum of −0.23 in chiral disubstituted polyacetylene without no chiral dopant.46
Recently, chiral molecular assemblies with an aggregation-induced emission (AIE) effect have become the focus of attention.52–55 Benefiting from the enhanced emission intensity upon aggregation of AIEgens, an efficient CPL response can be generated in the solid state to realize their applications in devices.17 Although significant progress has been achieved to access efficient glum value increase, approaches to controlling the mesoscopic structure and the ensuing CPL properties are still limited.41–47 Therefore, the CPL properties of chiral luminogens in the condensed matter state might have profound implications for high performance CPL-based devices at the macroscopic scale. Herein, two rod-like aggregation-induced emission luminogens with a rigid core containing an ester or amide linkage and a cholesterol moiety at one end and long aliphatic chains at the other end, namely, DPCE-ECh and DPCE-ACh, are presented and illustrated in Fig. 1A. In the solid state, DPCE-ECh self-assembles into a supramolecular liquid-crystalline smectic C* (SC*) phase and shows an impressive highly positive CPL response with a glum of +0.380 ± 0.011 and a gCD of −0.20. The intense and apparent CD and CPL of the film stem from the intrinsic helical structure of the molecular assemblies with a small contribution of Bragg reflection, where the helical axis is parallel to the direction of the glass substrate. To the best of our knowledge, this large glum factor is very rare for organic compounds even in the assembled state formed by annealing at the smectic liquid crystalline temperature. On the other hand, DPCE-ACh can form two coexisting phases of hexagonal and smectic liquid-crystalline phases with a weak negative CPL response. The glum falls in the range of −0.61 × 10−3 to −5.96 × 10−3. Such non-periodic molecular orientations give a weak CPL signal of the order of 10−3. The large difference in |glum| is attributed to the amplified artifact induced by the birefringent domains of the thick film.
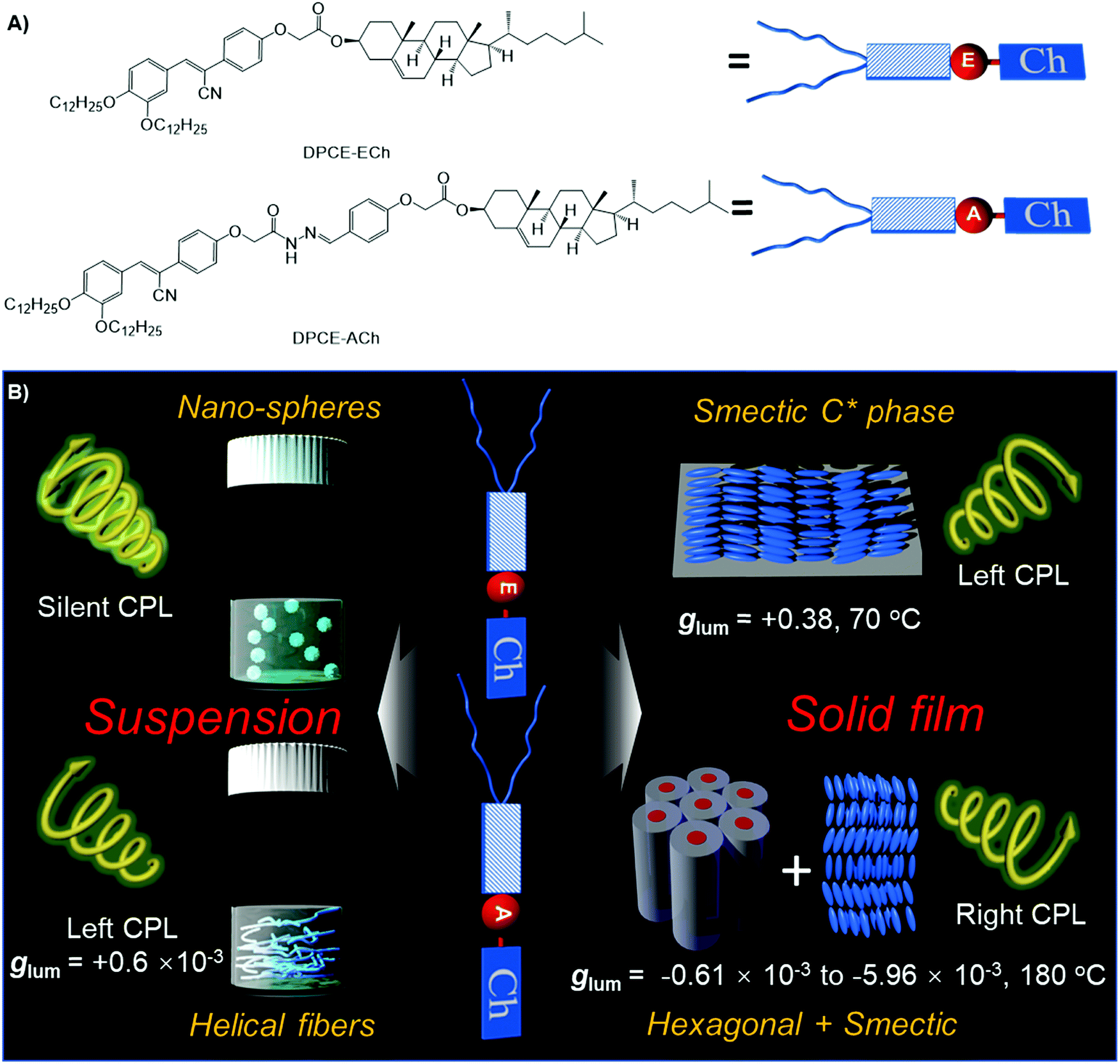 | ||
| Fig. 1 (A) Molecular structures of DPCE-ECh and DPCE-ACh. (B) Schematic illustration of chiral supramolecular assemblies in solution and liquid crystalline states and their corresponding glum values. | ||
Results and discussion
Synthesis and characterization
The synthetic procedures of DPCE-ECh and DPCE-ACh are outlined in Scheme S1 (ESI†). Their structures were confirmed by NMR and high resolution mass spectroscopies (Fig. S1–S15, ESI†). Thermogravimetric analysis (TGA) revealed that the two compounds have high decomposition temperatures (Td) which could go up to 300 °C (Fig. S16, ESI†), suggesting that they are thermally stable. In dilute THF solution, DPCE-ECh and DPCE-ACh show an absorption band centered at 364 and 360 nm, respectively (Fig. S17A, ESI†). DPCE-ECh and DPCE-ACh exhibit similar fluorescence spectra with a peak maximum centered at 430 nm (Fig. S17B, ESI†). As shown in Table S1 (ESI†), the two compounds are weakly emissive in THF solutions with quantum yield ΦF, soln. of 0.005 and 0.003 and emissive in solid powders with ΦF, solid of 0.12 and 0.114. Their αAIE (ΦF, solid/ΦF, soln.) values were calculated to be 24 and 38, suggesting a typical AIE feature for these compounds.Chiroptical properties in solution and suspension
The weakly-emissive THF solutions of DPCE-ECh and DPCE-ACh become progressively emissive upon addition of water (Fig. 2A and Fig. S18, S19, ESI†), demonstrating an AIE phenomenon. Such chiral AIEgens with cholesterol moieties are promising candidates for chiral induction, which might be capable of displaying supramolecular helicity with the assistance of the long alkoxy chains. The chiroptical properties of the aggregates generated in THF/water mixtures with different H2O fractions (fw) were then investigated (Fig. 2B–E and Fig. S20–S35, ESI†). The aggregates of DPCE-ECh with an ester linkage are CD silent regardless of water fraction variation (Fig. S20–S28, ESI†). In contrast, at fw ≥ 40%, aggregates of DPCE-ACh with an amide linkage exhibit obvious CD signals with negative and positive Cotton effects at wavelengths between 300 nm and 400 nm (Fig. 2B). It is noted that at fw = 60%, an induced positive split-type Cotton effect consisting of a positive Cotton effect at 375 nm and a negative Cotton effect at 338 nm was observed in DPCE-ACh aggregates, suggesting the formation of organized helical superstructures in solution.56–58 The maximum absorption anisotropy factor (gCD) reaches 2.78 × 10−3 at 375 nm (Fig. 2C). In addition, the UV-Vis absorption intensities in the long wavelength region began to increase substantially when fw = 60% (Fig. S25, ESI†); this could be attributed to the production of large aggregates with strong light scattering.59 The above CD and UV data further proved the formation of large-sized helical aggregates with the addition H2O into THF solvent (fw = 60%). However, the continuous increment of fw to 90% leads to a dramatic decrease in gCD by an order of magnitude (+2.3 × 10−4 at 375 nm, Fig. 2C), suggesting the dissociation of the chiral helical aggregates. Analogous to CD spectroscopy, CPL reflects the chiroptical properties of the luminescent materials upon excitation. Consistent with those of CD results, the isolated species in THF solution and aggregates in the THF/H2O mixture of DPCE-ECh are all CPL silent (Fig. S29–S35, ESI†). However, the DPCE-ACh aggregates suspended in the THF/H2O mixture show a positive CPL signal at fw = 40% with a glum of 2.0 × 10−4 (Fig. 2D). The detailed glum values of DPCE-ACh aggregates in the THF/H2O mixture are depicted in Fig. 2E. The maximum glum reaches ∼6.0 × 10−4 at fw between 50% and 70% (Fig. 2E).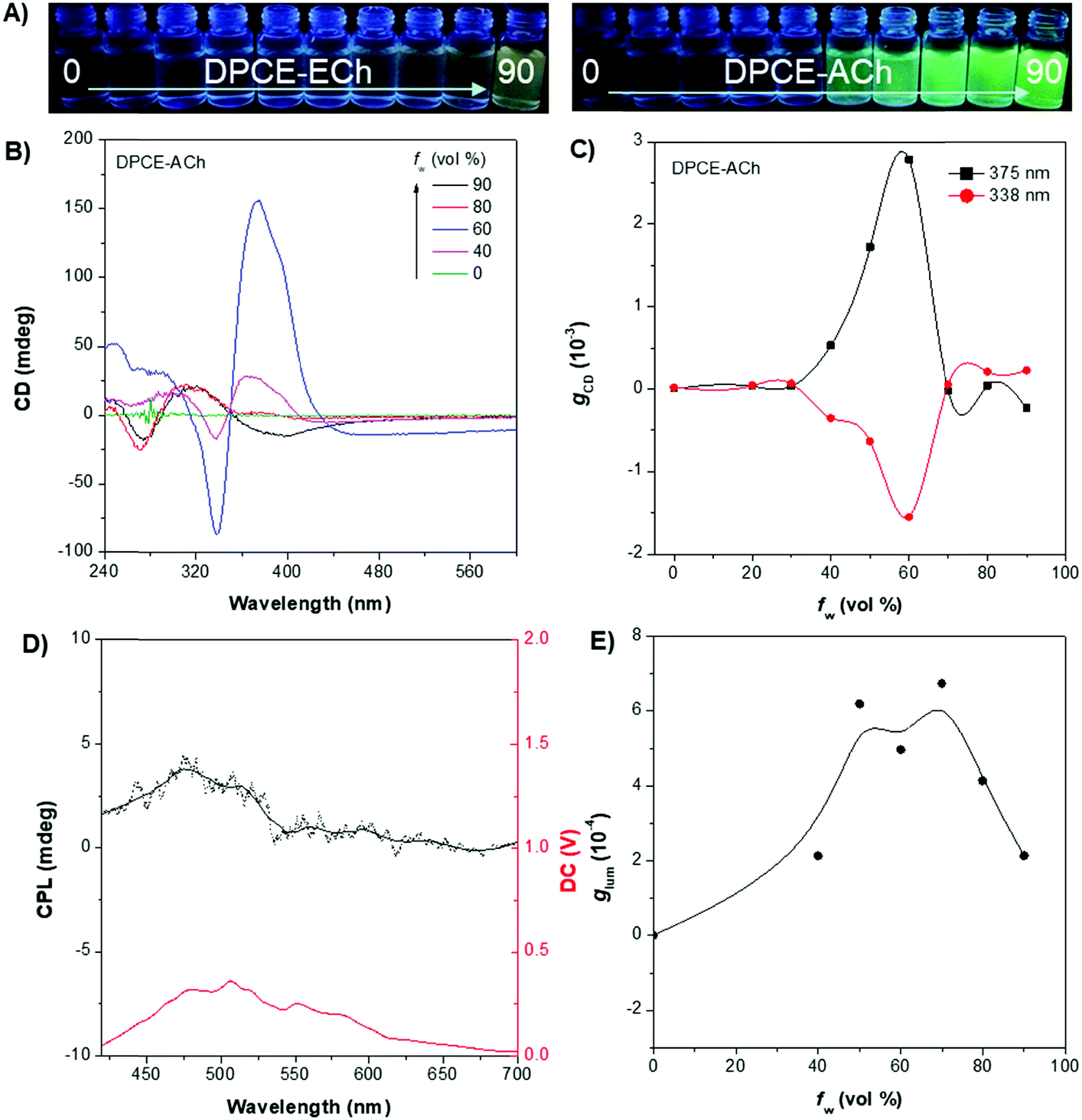 | ||
| Fig. 2 (A) Fluorescence photographs of DPCE-ECh and DPCE-ACh in THF/H2O mixtures with different water fractions (fw). (B) CD spectra and corresponding (C) gabs factor of DPCE-ACh in THF/H2O mixtures with different fw. (D) CPL spectra of DPCE-ACh in THF/H2O mixtures at fw = 60%. (E) glum factor of DPCE-ACh in THF/H2O mixtures with fw from 40% to 90%. Solution concentration: 5 × 10−5 M. | ||
To better understand the origins of the chiroptical properties of DPCE-ECh and DPCE-ACh aggregates, scanning electron microscopy (SEM) was employed to study their assembled structures in THF/H2O mixtures with fw varying from 40%–90% (Fig. S36–S40, ESI†). For DPCE-ECh, its aggregates maintain the spherical structure regardless of the water fraction variation (Fig. S36, ESI†). Such a symmetrical morphology leads to a silent CPL response. For DPCE-ACh, its aggregates show an obvious morphological evolution from an intertwined network (fw = 40%, Fig. S37, ESI†) to left-handed helical nanofibers (fw = 60%, Fig. 3A and B). Benefiting from the helical fibrous morphology, DPCE-ACh aggregates show higher dissymmetry factors compared to those of the spherical aggregates. On the other hand, M-helices and P-supra-helices are found in DPCE-ACh suspension (Fig. 3A and B). According to Michael D. Barnes’ report60 they thought that the measured g value in the disperse phase represents a weighted average of all possible orientations and interactions with the host. As for DPCE-ACh in suspension, the |glum| value detected in this system can be attributed to cancellation effects in ensemble measurements of a randomly oriented (M-helices and P-suprahelices) bulk sample. As seen in Fig. 3A, M-helices account for most of the proportion, thus leading to a positive CPL signal. A careful examination of the chemical structures of the AIEgens suggested that intermolecular hydrogen bonding between the amide linkages and the chiral nature of the cholesterol moiety of DPCE-ACh may serve as the external driving forces for the formation of the helical self-assembled structure to generate CD and CPL signals. To gain further insight into the dynamic nature of hydrogen bonding within the induced helical fibrous structure,56 temperature-dependent CD spectra are monitored on the DPCE-ACh-based aggregates (fw = 60%, Fig. 3C, D and Fig. S41, ESI†). At a low temperature of 5 °C, the CD spectrum of DPCE-ACh aggregates shows an obvious positive Cotton effect with an absorption peak at 375 nm and gives a corresponding gCD value of +2.58 × 10−3. The peak at 375 nm is ascribed to the achiral aromatic rigid core of DPCE-ACh, which is induced by the molecular helical arrangement. It is found that the CD signals of helical aggregates are sensitive to the temperature. The CD signals gradually decrease and completely disappear upon heating to 49 °C (Fig. 3C). Such temperature-dependent gabs factor variations are summarized and plotted in Fig. 3D. It is noted that the gCD of 375 nm was only +2.5 × 10−4 at the high temperature of 49 °C, indicating that the hydrogen bonding became very weak and thus led to the dissociation of the helical assembled structure of DPCE-ACh.
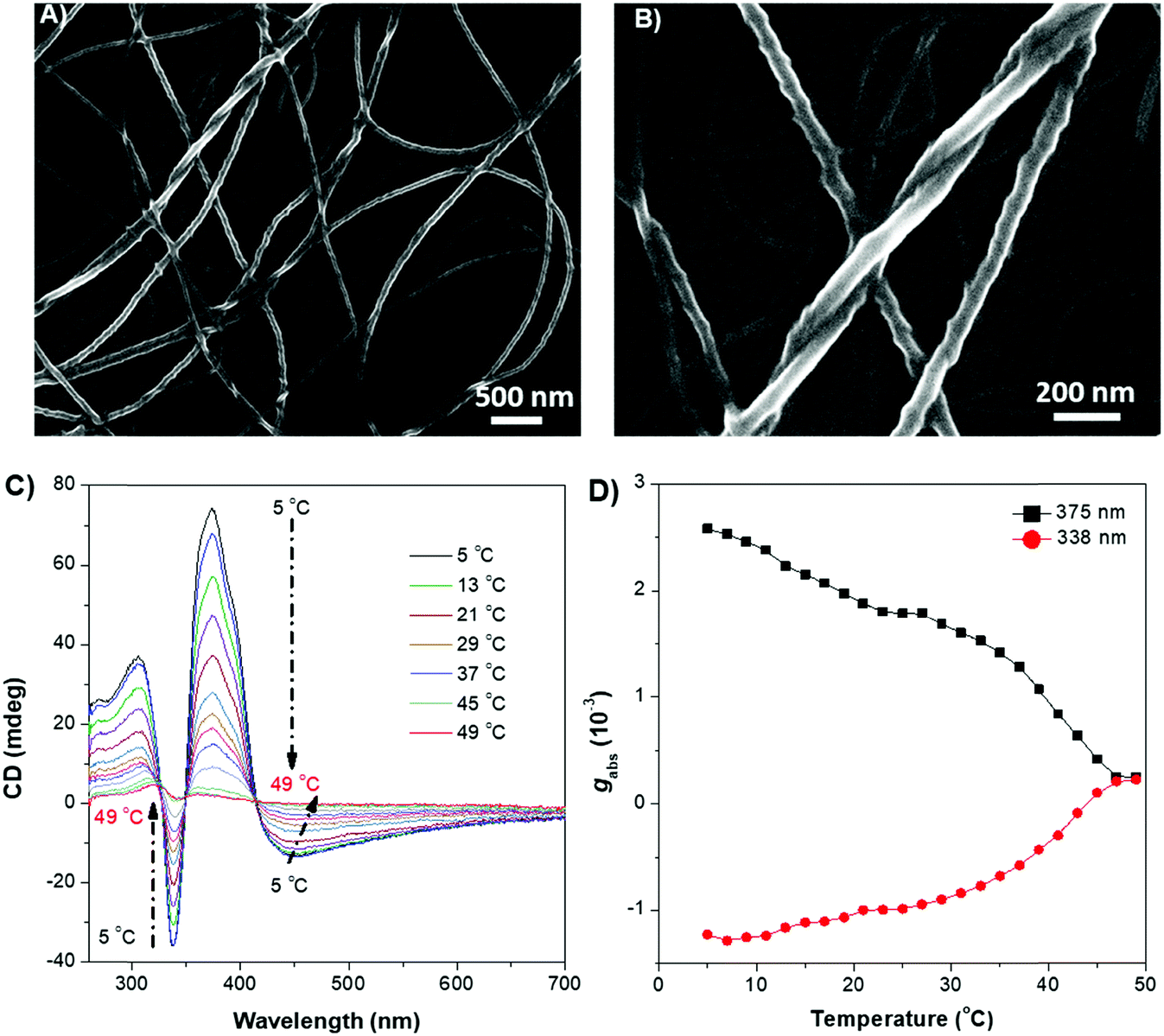 | ||
| Fig. 3 SEM images of DPCE-ACh with M-helical nanofibers obtained from a THF/H2O mixture at fw = 60% in (A) 500 nm, (B) 200 nm. Solution concentration: 1 × 10−4 M. Temperature-dependent CD (C) and temperature-dependent gabs factor changes (D) for DPCE-ACh in THF/H2O when fw = 60%. Solution concentration: 1 × 10−5 M. | ||
Chiroptical properties in the condensed phase
The rod-like molecular structure of the two AIEgens with a cholesterol moiety and flexible tails at two ends makes them promising for the formation of a liquid-crystalline phase in a chiral fashion. DPCE-ACh was first explored considering its capability to form helical fibers mentioned above. The phase transition temperatures of DPCE-ACh in solid films are shown graphically in Fig. 4A (top, Fig. S42, ESI†). Upon cooling the isotropic liquid of DPCE-ACh to 210 °C, a smectic liquid-crystalline phase with a fan-shaped texture followed by a columnar liquid-crystalline phase with a mosaic texture is observed (Fig. 4A, bottom; Fig. S43, ESI†). The molecular orientations in the liquid-crystalline phases are revealed by 1D wide angle X-ray diffraction (1D WAXD, Fig. S44–S46, ESI†). The 1D WAXD pattern at 210 °C shows a sharp peak at 2θ = 2.97° and a high-order diffraction peak at 2θ = 5.66°. These two diffraction peaks are associated with a smectic phase structure61,62 with a layer thickness of 3.05 nm (Fig. 4B and Fig. S46, ESI†). The 1D WAXD diagrams at 200 °C, 170 °C and 150 °C show the appearance of three new peaks with scattering vectors of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :√3:2, which are the typical reflection mode for the (100), (110) and (200) planes of the hexagonal columnar liquid crystals34,37,63,64 (Fig. 4B and Fig. S46, ESI†). Analysis of small-angle X-ray scattering (SAXS) confirms the lamellar and columnar organization of DPCE-ACh with a lamellar thickness of 3.05 nm and a columnar diameter of 4.33 nm, respectively (Fig. S46, ESI†). Electron density reconstruction was calculated according to the method of a previously published paper.60 The electron density map (Fig. 4C) of the phase based on the XRD result shows that the high electron density (red) is concentrated at the center of columns and low-density areas (green and blue) are located at the column periphery associated with the alkyl chains and the cholesterol side chains, respectively. On the other hand, as DPCE-ACh is a hexagonal columnar mesophase, the average number (n) of molecules per slice of the column could be obtained by the following formula.65
:√3:2, which are the typical reflection mode for the (100), (110) and (200) planes of the hexagonal columnar liquid crystals34,37,63,64 (Fig. 4B and Fig. S46, ESI†). Analysis of small-angle X-ray scattering (SAXS) confirms the lamellar and columnar organization of DPCE-ACh with a lamellar thickness of 3.05 nm and a columnar diameter of 4.33 nm, respectively (Fig. S46, ESI†). Electron density reconstruction was calculated according to the method of a previously published paper.60 The electron density map (Fig. 4C) of the phase based on the XRD result shows that the high electron density (red) is concentrated at the center of columns and low-density areas (green and blue) are located at the column periphery associated with the alkyl chains and the cholesterol side chains, respectively. On the other hand, as DPCE-ACh is a hexagonal columnar mesophase, the average number (n) of molecules per slice of the column could be obtained by the following formula.65| n = (a2)(√3/2)(hρNA/M) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. These results certainly support the fact that the hydrogen bond plays the crucial role in inducing the columnar mesophase of the asymmetrical diphenylacrylonitrile derivatives.
O groups. These results certainly support the fact that the hydrogen bond plays the crucial role in inducing the columnar mesophase of the asymmetrical diphenylacrylonitrile derivatives.
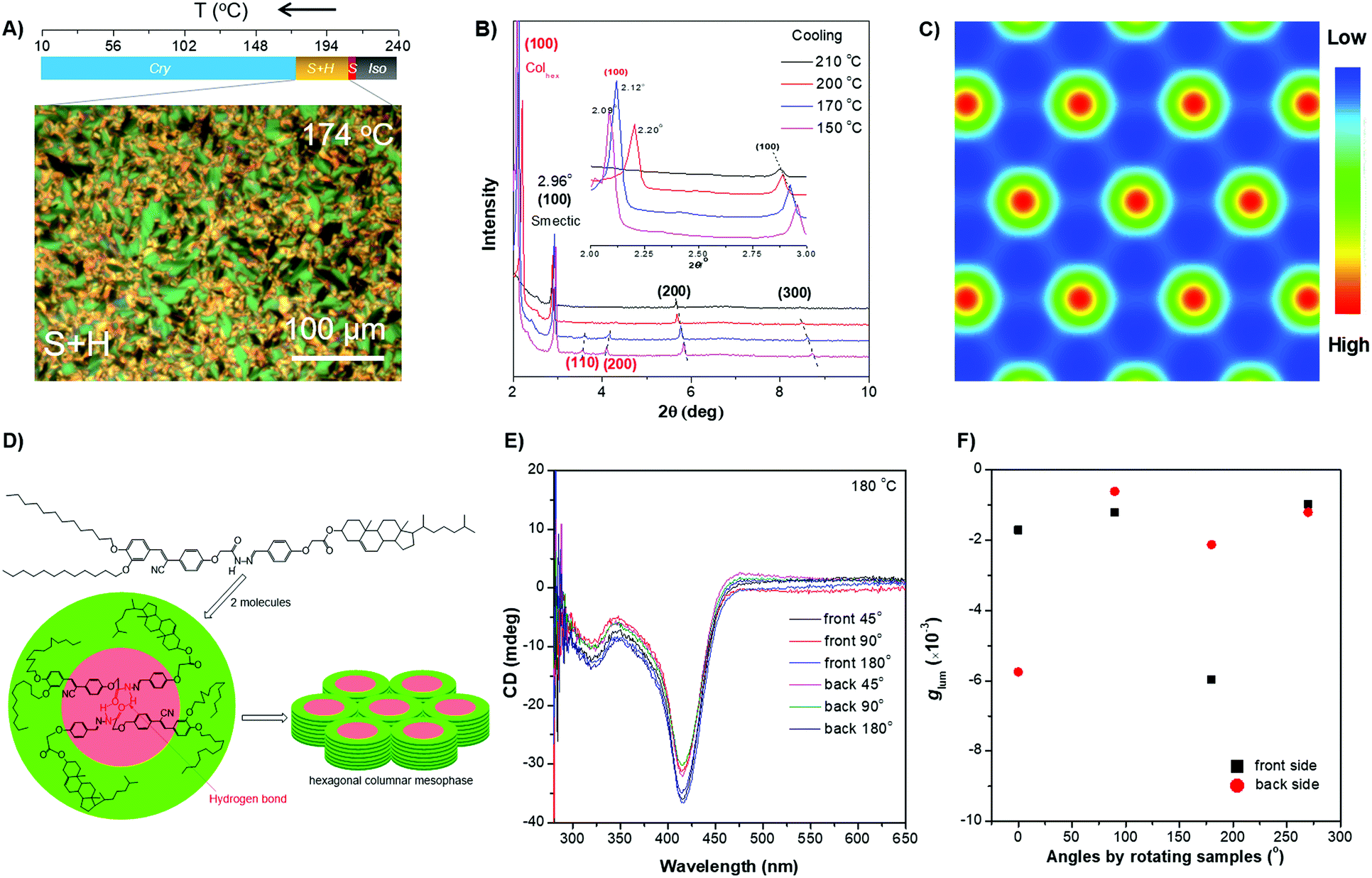 | ||
| Fig. 4 (A) (Top) Phase transition of DPCE-ACh recorded during the first cooling scan at a rate of 5 °C min−1. Abbreviation: Cry = solid crystal; S + H: smectic (S) + hexagonal (H) columnar phase; Iso: isotropic liquid. (Bottom) POM texture of DPCE-ACh at 174 °C. (B) 1D WAXD pattern of DPCE-ACh recorded during the first cooling process at a temperature of 150–210 °C (2θ = 2°–10°; Inset: Low-angle diffractions). (C) Reconstructed electron density map of the columnar phase generated from Fig. S46 (ESI†). Color scale is shown in the right column. (D) The proposed molecular stacking of hexagonal columns for DPCE-ACh. (E) CD spectra of front and back sides of DPCE-ACh films annealed at 180 °C for 45 min at different rotation angles perpendicular to the light axis. The film thickness is 50 nm for CD detection. (F) glum (500 nm) of front and back sides of DPCE-ACh films annealed at 180 °C for 45 min at different rotation angles perpendicular to the light axis. | ||
As illustrated in Fig. 4E, the film exhibits a negative signal at 415 nm with a gCD of −(1.92 ± 0.063) × 10−3 after annealing at 180 °C. A moderate profile change in the CD spectrum was obtained by rotating the sample at different angles around the optical axis (Fig. 4F and Fig. S47–S49, ESI†), suggesting that the LDLB effect35 (birefringent phenomenon) contributes the final CD. Because DPCE-ACh forms two coexisting hexagonal and smectic phases, the non-periodic molecular orientations break its helical structure to give a weak CD signal of the order of 10−3. On the other hand, we also investigated the CPL spectra at different angles on both sides in a 7 μm liquid crystal cell. However, a large difference in |glum| was observed at different angles on both sides and the |glum| falls in the range from −0.61 × 10−3 to −5.96 × 10−3 (Fig. 4F and Fig. S50, S51, ESI†). This phenomenon is attributed to the amplified artifact (Bragg reflection) induced by the birefringent domains of the thick film (7 μm).
The ester linkage of DPCE-ECh offers only a weak intermolecular interaction and the relationship between the molecular orientations and the chiroptical properties are also investigated. The DSC trace of DPCE-ECh recorded during the first cooling cycle shows three exothermic transitions at around 121, 86 and 30 °C upon cooling from 180 °C (Fig. 5A). The POM image shows oily-streak textures that are typical of the sematic phase in the liquid crystalline state at 85 °C (Fig. S52, ESI†). 1D WAXD measurements were then carried out to monitor the structural evolution. It is found that a sharp peak at 2θ = 2.35° appears at 80 °C, indicating the formation of an ordered structure. In addition, a high-order diffraction peak at 2θ = 4.78° is also observed. The ratio of the scattering vectors of the two peaks is approximately 1:2, indicating the formation of a smectic structure (Fig. 5B and Fig. S53, S54, ESI†). Such an ordered structure was retained in the temperature range of 86–33 °C. The transition recorded by DSC at 86 °C corresponds to the clearing point (Fig. 5A). In the temperature range of 86–121 °C (DSC), a broader and weaker peak compared with that of the smectic phase was observed (Fig. S55, ESI†) and we identified it as the isotropic liquid phase. To further prove the smectic structure of DPCE-ECh, 2D SAXS and wide-angle X-ray scattering (WAXS) were carried out (Fig. 5C and D). The oriented sample for the measurements was prepared by mechanically shearing the melted film at 85 °C. As shown in the illustration in Fig. S56 (ESI†), the point-focused X-ray beam was aligned perpendicular to the shear direction. It is noted that the diffuse peaks at smaller (Fig. 5C) and larger angles (Fig. 5D, marked with a dashed line) are not orthogonal to each other. This suggests that the director makes a tilt angle with respect to the smectic layer and the angle rotates from layer to layer to form a smectic C phase. CD experiments were conducted to analyse the chiroptical activities of the DPCE-ECh solid film on glass slides. Irrespective of rotating or flipping the samples, strong and consitent CD signals were obtained (Fig. 5E and Fig. S57, ESI†). This implied that the long helical molecular stacking axis was perpendicular rather than parallel to the optical axis (Fig. S58A, ESI†). In addition, the in-plane molecular orientation was supposed to be aligned randomly. In this sense, the LDLB effect in such a solid film could be neglected and genuine chiroptical signals from the chiral supramolecular structure resulted. A gCD value of −0.20 at 404 nm was achieved (Fig. 5F). Similarly, we also fabricated a film in a 7 μm thick liquid crystal cell for CPL measurement. CPL spectra were also obtained by rotating the sample at different angles on both sides (Fig. 5G). Strong CPL responses with positive signals were observed in this annealled film with a maximum glum value (average, 70 °C) of +0.380 ± 0.011. However, different glum values ranging from 0.342 to 0.438 were observed with varied angles and sides. The strong CPL response is generated from the chiral supramolecular structure in which the helical axis is perpendicular to the optical axis and parallel to the direction of the glass substrate. The large difference in glum (about 0.1) at different angles on both sides is attributed to the birefringence pattern. Because the film thickness is 7 μm for CPL detection which is much thicker than the film for CD detection (50 nm), the artifact induced by the birefringent domains is amplified in such a thick film. To the best of our knowledge, such a large glum value (+0.380 ± 0.011) with a weak contribution of Bragg reflection is very rare for organic compounds.
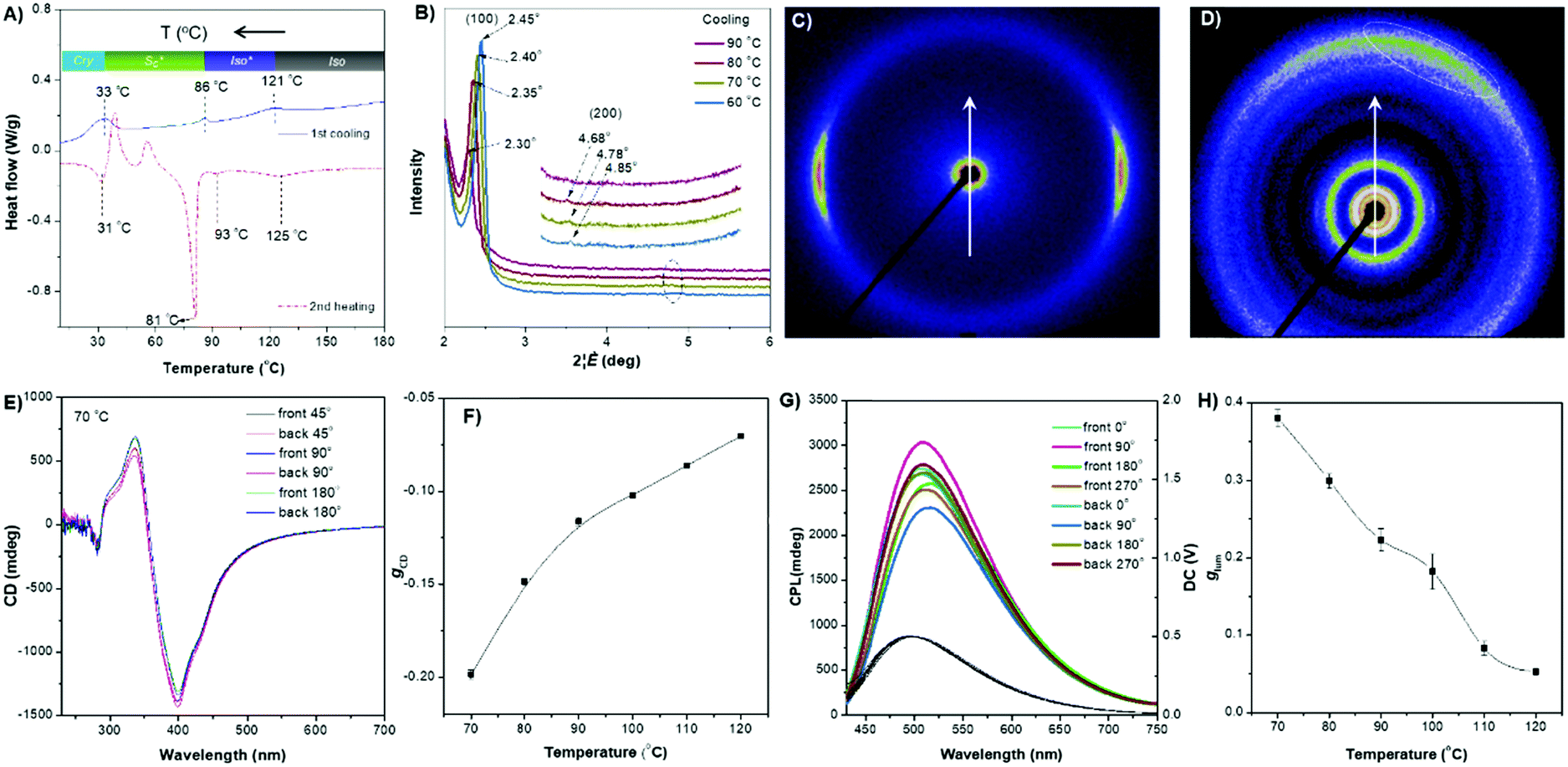 | ||
| Fig. 5 (A) DSC thermograms of DPCE-ECh recorded at the second heating and first cooling scans (5 °C min−1). Inset: Phase transition of DPCE-ECh determined from the first cooling DSC scans at a rate of 5 °C min−1. (B) 1D WAXD pattern of DPCE-ECh recorded in the first cooling process at a temperature of 60–90 °C. (2θ = 2°–6°, inset: high-order diffractions). (C) 2D SAXS and (D) 2D WAXS patterns of the mechanically aligned DPCE-ECh with X-ray beam perpendicular to the shear direction. (E) CD spectra (404 nm) of front and back sides of DPCE-ECh films annealed at 70 °C recorded for 45 min at different rotation angles perpendicular to the light axis. The film thickness is 50 nm for CD detection. (G) glum (510 nm) of front and back sides of DPCE-ECh films annealed at a temperature of 70 °C for 45 min at different rotation angles perpendicular to the light axis. The film thickness is 7 μm for CPL detection. (F and H) gCD and glum of DPCE-ECh at different temperatures. | ||
With increasing annealing temperature (>70 °C), the dissymmetry factor (gCD and glum) of the thin film decreases (Fig. 5F and H), indicating the dissociation of the smectic C phase. Interestingly, when the isotropic liquid of DPCE-ECh was annealed in the temperature range of 90–120 °C followed by CPL measurement, a strong CPL signal with a gCD value of −0.11 and a glum value of +0.18 was still recorded (Fig. S59–S71, ESI†). The CPL response of the isotropoic state of DPCE-ECh indicates that a twisted organization is still retained in aggregates and such a twisting is still sufficient for CPL induction. Thus, we identify this chiral isotropic state as a chiral isotropic liquid (Iso*)66–68 in Fig. S55 (ESI†) (inset), which was recently discovered as a new phase.
Normally, the measured g value in a disperse phase represents a weighted average of all possible orientations.60 In THF solution, DPCE-ECh and DPCE-ACh (10−5 mol L−1) are soluble and dispersed isolatedly. Therefore, the measured g value in dilute solution comes from a single molecule itself. For a single molecule, the chiral function is mainly focused on the cholesterol unit and the luminescent function is mainly focused on the diphenylacrylonitrile unit. Hence, no CPL signal is observed in these single molecules. On the other hand, the aggregates of DPCE-ECh keep the spherical structure regardless of the water fraction variation. Such a symmetrical morphology leads to a silent CPL response. Meanwhile, in the solid state, combining the X-ray results and chiroptical activity of DPCE-ECh, a semctic C* phase was identified finally. Moreover, SEM textures of the fracture plane of DPCE-ECh with layered and arched structures further support our hypothesis (Fig. S72, ESI†). Such a smectic C* state leads to a giant CPL response. For DPCE-ACh, a positive CPL signal is observed due to M-helical nanofiber formation in suspension. A negative CPL signal is observed due to the complex liquid crystalline (H + S) orientations in the solid state. These findings demonstrate that the CPL response (intensity and orientation) of synthetic advanced materials not only relies on chiral functions on the molecular level, but also depends on the mesoscopic architectures of the molecular assemblies.
Conclusions
In summary, two AIEgens with rigid cores containing different linkages are developed. These chiral AIEgens show a silent CPL response when existing as species in THF solution. In contrast, a tunable CPL response is achieved through regulating their aggregated structures in solution and solid states. Driven by the intermolecular hydrogen bonding in DPCE-ACh, opposite CPL responses with glum of the order of 10−3 are obtained from a M-helical nanofibrous structure and complex liquid crystalline (H + S) orientations. Meanwhile, DPCE-ECh exhibits a liquid crystalline assembled system (smectic C*) with a high dissymmetry factor (gCD = −0.20 and glum = +0.38). The intense and apparent CD and CPL of the film stem from the intrinsic helical structure of the molecular assemblies with a weak contribution of Bragg reflection, where the lone helical molecular stacking axis is perpendicular to the optical axis and parallel to the direction of the glass substrate. To the best of our knowledge, this large glum factor is very rare for organic compounds even in the assembled state formed by annealing at the smectic liquid crystalline temperature. This path opens new capabilities for structural control of molecular assemblies to generate versatile CPL responses that are inaccessible from isolated AIEgens. These findings demonstrate that the CPL response (intensity and orientation) of synthetic advanced materials not only relies on chiral functions on the molecular level, but also depends on the mesoscopic architectures of the molecular assemblies. We hope that the present strategy for constructing CPL-active materials in the condensed matter state will open numerous opportunities for applications in photonic devices.Experimental section
Chemicals and methods
All chemicals were purchased from Sigma-Aldrich and J&K Chemical Co. and used as received without further purification unless otherwise specified. Anhydrous THF and CH3CN were used for fluorescence property investigation. Deionized water was used throughout this study. Pre-coated glass plates were used for TLC analysis. Column chromatography was carried out by using silica gel (200–300 mesh) as the adsorbent.1H and 13C NMR spectra were measured on a Bruker ARX 400 NMR spectrometer and reported as parts per million (ppm) referenced to the internal standard TMS. High-resolution mass spectra (HR-MS) were obtained on a Finnigan MAT TSQ 7000 Mass Spectrometer System operated in a MALDI-TOF mode. Thermogravimetric analysis (TGA) was performed on a TA TGA Q5000 under nitrogen at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) analysis was performed on a TA Instruments DSC Q1000 at a heating rate of 5 °C min−1. The sample size was about 2 mg and encapsulated in hermetically sealed aluminum pans, and the pan weights were kept constant. The temperature and heat flow were calibrated using standard materials such as indium and benzoic acid. Polarized optical microscopy (POM) was carried out to observe the liquid crystalline textures of the samples on a Leitz Laborlux 12 microscope with a Leitz 350 hot stage.
The morphological structures of the aggregates were investigated by a HITACHI-SU8010 scanning electron microscope (SEM) at accelerating voltages of 200 and 5 kV. Stock solutions of DPCE-ECh and DPCE-ACh in THF (10−3 mol L−1) were prepared. A certain volume (30 μL) of such stock solutions was transferred to small glass vials (5 mL). After addition of appropriate amounts of THF, distilled water was added dropwise under vigorous stirring to afford 5 × 10−5 mol L−1 of DPCE-ECh and DPCE-ACh solutions. The mixtures were dropped on silicon wafers, the solvents were removed under reduced pressure at room temperature, and the SEM images of the aggregates on silicon wafers were taken.
To identify the liquid crystalline structure of DPCE-ECh and DPCE-ACh, 1D XRD experiments were performed on a Philips X’Pert Pro diffractometer equipped with a 3 kW ceramic tube as the X-ray source (Cu Kα), an X’celerator detector, and a temperature control unit of Paar Physica TCU 100. The sample stage was set horizontally. The diffraction peak positions of the 1D XRD were calibrated with silicon powder for the wide-angle region and silver behenate for the small-angle region, respectively. The data were collected using a Mar165 detector and calibrated with CeO2 powder. The sample temperature was controlled using a Linkman THMSE600 hot stage. The heating and cooling rates in the experiments were 5 °C min−1. The data were collected using an exposure time of 120 s. The 2D SAXS and WAXS data of DPCE-ECh were collected on a Xeuss 2.0 (Xenocs, France), and the measurement details are listed in Table S2 (ESI†).
Absorption spectra were measured on a Milton Roy Spectronic 3000 Array spectrophotometer. Steady-state photoluminescence (PL) spectra were measured on a PerkinElmer spectrofluorometer LS 55. The lifetime and the absolute luminescence quantum yield were measured on an Edinburgh FLSP 920 fluorescence spectrophotometer equipped with an integrating sphere (0.1 nm step size, 0.3 second integration time, 5 repeats).
Circular dichroism (CD) spectra were recorded with a Chirascan spectrometer (Applied Photophysics, England). Circularly polarized photoluminescence (CPPL) spectra of the films and solution were recorded at a scan speed of 50 nm min−1 with a commercialized instrument JASCO CPL-300 at room temperature with a resolution of 15 nm. The film samples for CD and CPL measurements were prepared by drop-casting on the quartz substrate from the CHCl3 solution (5 mg mL−1) of DPCE-ECh and DPCE-ACh, and subsequently by volatilization of CHCl3 solvent at room temperature. Samples were subsequently thermally annealed for 45 min at the indicated temperatures. Preparation took place under an inert atmosphere in a nitrogen filled glove box. To freeze temporarily the phase of the DPCE-ECh and DPCE-ACh, the film sample was quenched from the indicated temperatures to liquid nitrogen temperature. The CD and CPL response of the quenched sample was recorded over the same time interval (per 3 min) at room temperature. The magnitude of circular polarisation in the excited state is defined as glum = 2 (IL – IR)/(IL + IR), where IL and IR indicate the output signals for the left and right circularly polarized luminescence, respectively. Experimentally, the value of glum is defined as ΔI/I = [ellipticity/(32980/ln10)]/(unpolarized PL intensity) at the CPL extremum.69
Electron density reconstruction was calculated according to the method of a previously published paper.64 The diffraction peaks were indexed on the basis of their peak positions, and the lattice parameters and the space groups were subsequently determined. Once the diffraction intensities are measured and the corresponding space group determined, three dimensional (3D) electron density maps can be reconstructed, on the basis of the general formula
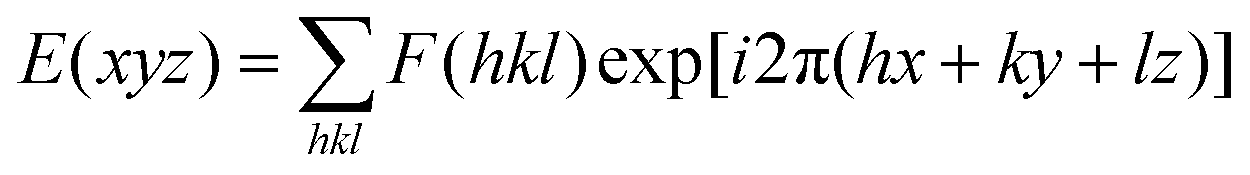 | (1) |
| I(hkl) = KF(hkl)F × (hkl) = K|F(hkl)|2 | (2) |
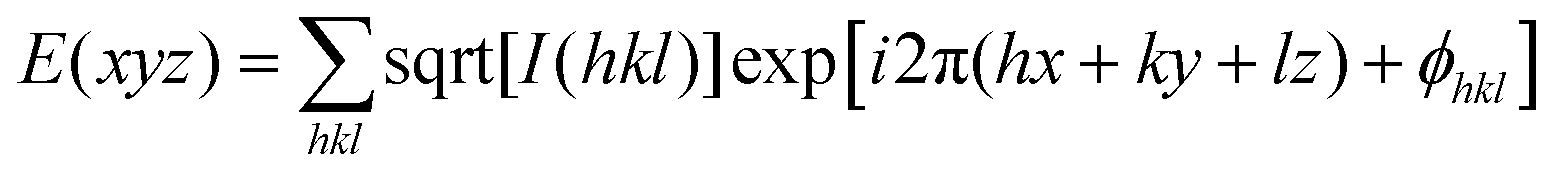 | (3) |
For 2D structures I(hk) and eqn (4) were used:
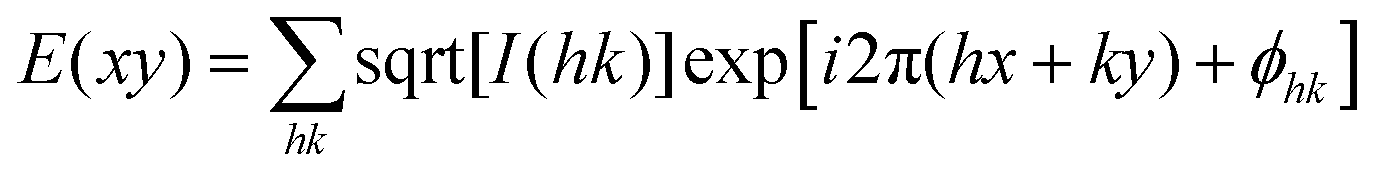 | (4) |
This makes it possible for a trial-and-error approach, where candidate electron density maps are reconstructed for all possible phase combinations, and the “correct” phase combination is then selected on the merit of the maps, helped by prior physical and chemical knowledge of the system. This is in particular useful for the study of nanostructures, where normally only a limited number of diffraction peaks are observed.
Synthesis of 3-(3,4-bis(dodecyloxy)phenyl)-2-(4-hydroxyphenyl)acrylonitrile (2)
A mixture of 4-hydroxyphenylacetonitrile (1.62 g, 12.0 mmol), compound 1 (5.7 g, 12.0 mmol) and NaOH (0.96 g, 24.0 mmol) in a mixed solution of 60 mL of EtOH and 30 mL of THF was refluxed for 24 h. After cooling to room temperature, 24 mL of HCl solution (1 M) was poured into the reaction mixture, then the solvent of C2H5OH was removed by a rotary evaporator. And water (60 mL) was added. The mixture was extracted with ethyl acetate (3 × 50 mL). The combined organic layers were dried with anhydrous Na2SO4 and evaporated under reduced pressure to obtain the crude product. The residue was purified by silica-gel column chromatography using hexane/ethyl acetate (15:1) as an eluent. Compound 2 was obtained as a light brown powder with 35% yield (2.48 g, 4.2 mmol). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.62 (s, 1H), 7.52 (d, J = 8.4 Hz, 2H), 7.34–7.31 (m, 2H), 6.94–6.90 (m, 3H), 4.63 (s, 1H), 4.10–4.04 (m, 4H), 1.86–1.79 (m, 4H), 1.58–1.43 (m, 4H), 1.34–1.27 (m, 32H), 0.89 (t, J = 6.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ (ppm): 155.9, 150.4, 148.7, 139.8, 126.6, 126.2, 123.2, 119.2, 118.2, 115.3, 112.4, 112.2, 107.4, 68.7, 68.5, 64.8, 31.3, 29.09, 29.06, 29.03, 28.83, 28.80, 28.77, 28.55, 28.48, 25.4, 22.1, 13.5. MALDI-TOF-MS (C39H59NO3) calcd for m/z = 589.9050, found: m/z = 589.4465 (M + H+).
Synthesis of ethyl-2-(4-(2-(3,4-bis(dodecyloxy)phenyl)-1-cyanovinyl)phenoxy)acetate (3)
A mixture of compound 2 (2.09 g, 3.55 mmol), excess ethyl choroacetate (0.80 mL, 7.10 mmol), and K2CO3 (2.45 g, 17.75 mmol) was stirred and refluxed in 60 mL of MeCN and 30 mL of THF for 12 h at 90 °C. After cooling the room temperature, the solvent was removed by a rotary evaporator. And then water (60 mL) was added. The mixture was extracted with CH2Cl2 (3 × 40 mL). The combined organic layers were dried with anhydrous Na2SO4 and evaporated under reduced pressure to obtain the crude product. The residue was purified by silica-gel column chromatography (hexane/ethyl acetate = 20:1) to yield 2.01 g (84%) of the product as a yellow powder after removal of the solvent. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.63 (s, 1H), 7.59 (d, J = 9.2 Hz, 2H), 7.34–7.32 (m, 2H), 6.97 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.4 Hz, 1H), 4.67 (s, 2H), 4.30 (q, J = 6.8 Hz, 2H), 4.10–4.05 (m, 4H), 1.89–1.82 (m, 4H), 1.59–1.45 (m, 4H), 1.39–1.27 (m, 35H), 0.89 (t, J = 6.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.0, 157.6, 150.5, 148.4, 140.3, 127.9, 126.5, 126.1, 123.3, 118.1, 114.5, 112.4, 112.2, 107.1, 68.6, 68.4, 64.8, 60.9, 31.3, 29.1, 29.06, 29.01, 28.82, 28.79, 28.77, 28.55, 28.49, 25.44, 25.38, 22.1, 13.56, 13.52. MALDI-TOF-MS (C43H65NO5) calcd for m/z = 675.9950, found: m/z = 675.4874 (M+).
Synthesis of 2-(4-(2-(3,4-bis(dodecyloxy)phenyl)-1-cyanovinyl)phenoxy)acetohydrazide (4)
A mixture of excess NH2NH2 (0.49 mL, 10.0 mmol) and compound 3 (0.66 g, 1.0 mmol) in 40 mL of EtOH solution was stirred for 10 h at room temperature. The reaction was examined by the TLC technique, which suggested the disappearance of the starting materials. After the reaction, the precipitate was formed and filtered. The obtained precipitate was purified by recrystallization. After drying, compound 4 was collected as a dark yellow solid in yield of 77%. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.77 (s, 1H, NH), 7.64–7.60 (m, 3H), 7.33–7.32 (m, 2H), 6.97 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz, 1H), 4.62 (s, 2H), 4.06 (t, J = 8.2 Hz, 4H), 3.97 (br, 2H, NH2), 1.87–1.84 (m, 4H), 1.50–1.27 (m, 36), 0.89 (t, J = 6.4 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ (ppm): 167.5, 156.7, 150.7, 148.4, 140.7, 128.6, 126.7, 125.9, 123.4, 118.0, 114.4, 112.4, 112. 2, 106.7, 68.7, 68.4, 66.4, 31.3, 29.08, 29.05, 29.01, 28.81, 28.78, 28.75, 28.56, 28.49, 25.43, 25.37, 22.1, 13.5. MALDI-TOF-MS: (C41H63N3O4) calcd for m/z = 662.0, found: m/z = 662.3 (M), m/z = 685.2 (M + Na+), m/z = 701.3 (M + K+).Synthesis of DPCE-ECh ((3S,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-((R)-6-methylheptan-2-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl 2-(4-(2-(3,4-bis(dodecyloxy)phenyl)-1-cyanovinyl)phenoxy)acetate)
Under a N2 atmosphere, a mixture of compound 2 (0.3 g, 0.5 mmol), compound 5 (0.23 mL, 0.5 mmol), and K2CO3 (0.28 g, 2.0 mmol) was stirred and refluxed in 30 mL of dry MeCN for 24 h at 90 °C. The reaction was monitored by the TLC technique, implying the disappearance of reactants. After the reaction, the mixture was treated with 40 mL of HCl (1 M) and extracted with 50 mL of CHCl3. The CHCl3 layer was partitioned, washed with 20 × 3 mL of distilled water, dried over anhydrous MgSO4, and then concentrated. The crude product was purified by recrystalliztion in CH2Cl2/MeOH (1:5, v/v). After drying, compound 6 was collected as a dark yellow solid in yield of 90%. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.64 (s, 1H), 7.60 (d, J = 8.0 Hz, 2H), 7.35 (m, 2H), 6.97 (d, J = 8.0 Hz, 2H), 6.92 (d, J = 8.0 Hz, 1H), 5.40 (br, 1H), 4.77 (br, 1H), 4.65 (s, 2H), 4.04–4.11 (m, 4H), 0.69–2.41 (m, 89H); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.0, 158.3, 151.1, 149.0, 140.8, 139.2, 130.5, 128.4, 127.0, 126.7, 124.0, 123.1, 118.6, 115.0, 112.9, 107.6, 75.4, 69.7, 69.1, 65.6, 56.6, 56.2, 50.0, 39.5, 36.2, 31.9, 29.7, 29.4, 28.0, 24.0, 22.7, 19.3, 18.7, 14.1, 11.9. MALDI-TOF-MS: calcd for m/z = 1054.7, found: m/z = 1055.9 (M + K+). HR-MS (ESI) (C68H105NO5) [M + K]+: calcd: 1054.7624. Found: 1154.7697(M + K)+.
Synthesis of DPCE-ACh ((3S,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-((R)-6-methylheptan-2-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl 2-(4-((2-(2-(4-(2-(3,4-bis(dodecyloxy)phenyl)-1-cyanovinyl)phenoxy)acetyl)hydrazono)methyl)phenoxy)acetate)
Under a N2 atmosphere, a mixture of compound 4 (0.17 g, 0.25 mmol) and compound 6 (0.25 mmol) was stirred and refluxed in 30 mL of CH2Cl2–MeOH solution (3:1, v/v). Several drops of glacial acetic acid were added as the catalyst. The reaction was examined by the TLC technique, implying the disappearance of reactants. After the reaction, most of the solvents were distilled under reduced pressure at room temperature. The obtained residue was further purified by recrystalliztion in CH2Cl2/MeOH (1:5, v/v) three times. DPCE-ACh was collected as a yellow solid in yields of 88%. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.45 (br, 1H), 8.17 (s, 1H), 6.90–7.78 (m, 12H), 5.40 (br, 1H), 5.20 (br, 1H), 4.72 (br, 2H), 4.65 (s, 2H), 4.08 (br, 4H), 0.68–2.42 (m, 89H); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.0, 163.7, 160.0, 159.7, 158.7, 149.2, 149.0, 144.9, 141.3, 140.7, 139.1, 129.6, 128.9, 127.4, 127.1, 126.6, 124.0, 123.2, 115.2, 114.9, 113.0, 112.7, 107.8, 107.2, 75.5, 69.2, 69.1, 67.2, 65.2, 56.8, 56.0, 55.4, 50.0, 42.3, 39.4, 38.0, 36.9, 36.6, 36.1, 35.8, 32.0, 29.7, 29.4, 27.9, 27.8, 26.1, 24.1, 23.7, 22.6, 21.0, 19.2, 18.5, 14.1, 11.7. MALDI-TOF-MS: calcd for m/z = 1192.7, found: m/z = 1194.0 (M + H+). HR-MS (ESI) (C77H113N3O7) [M + H]+: calcd: 1192.8651. Found: 1192.8643 (M + H)+.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Science Foundation of China (21788102, 21406036, 51508242 and 51603035), the Research Grants Council of Hong Kong (16308016, A-HKUST605/16 and C6009-17G), the Innovation and Technology Commission (ITC-CNERC14SC01 and ITS/254/17), the Shenzhen Science and Technology Program (JCYJ20160229205601482), the China Postdoctoral Science Foundation (2016M602532 and 2017T100646), and Science and Technology Commission of Shanghai Municipality (17ZR1446300). We thank Prof. Shuang Yang at Peking University for the simulation work.References
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC.
- F. Song, G. Wei, X. Jiang, F. Li, C. Zhu and Y. Cheng, Chem. Commun., 2013, 49, 5772–5774 RSC.
- Y. Imai, Y. Nakano, T. Kawai and J. Yuasa, Angew. Chem., Int. Ed., 2018, 57, 8973–8978 CrossRef CAS PubMed.
- S. Shuvaev, M. A. Fox and D. Parker, Angew. Chem., Int. Ed., 2018, 57, 7488–7492 CrossRef CAS PubMed.
- K. Takaishi, M. Yasui and T. Ema, J. Am. Chem. Soc., 2018, 140, 5334–5338 CrossRef CAS PubMed.
- C. Wagenknecht, C.-M. Li, A. Reingruber, X.-H. Bao, A. Goebel, Y.-A. Chen, Q. Zhang, K. Chen and J.-W. Pan, Nat. Photonics, 2010, 4, 549–552 CrossRef CAS.
- E. Peeters, M. P. T. Christiaans, R. A. J. Janssen, H. F. M. Schoo, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1997, 119, 9909–9910 CrossRef CAS.
- M. Oda, H.-G. Nothofer, G. Lieser, U. Scherf, S. C. J. Meskers and D. Neher, Adv. Mater., 2000, 12, 362–365 CrossRef CAS.
- Y. Geng, A. Trajkovska, S. W. Culligan, J. J. Ou, H. M. P. Chen, D. Katsis and S. H. Chen, J. Am. Chem. Soc., 2003, 125, 14032–14038 CrossRef CAS PubMed.
- D. Di Nuzzo, C. Kulkarni, B. Zhao, E. Smolinsky, F. Tassinari, S. C. J. Meskers, R. Naaman, E. W. Meijer and R. H. Friend, ACS Nano, 2017, 11, 12713–12722 CrossRef CAS PubMed.
- Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS PubMed.
- F. Zinna, U. Giovanella and L. D. Bari, Adv. Mater., 2015, 27, 1791–1795 CrossRef CAS PubMed.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed.
- F. Zinna, M. Pasini, F. Galeotti, C. Botta, L. Di Bari and U. Giovanella, Adv. Funct. Mater., 2017, 27, 1603719 CrossRef.
- D.-M. Lee, J.-W. Song, Y.-J. Lee, C.-J. Yu and J.-H. Kim, Adv. Mater., 2017, 29, 1700907 CrossRef PubMed.
- J. Han, S. Guo, J. Wang, L. Wei, Y. Zhuang, S. Liu, Q. Zhao, X. Zhang and W. Huang, Adv. Opt. Mater., 2017, 5, 1700359 CrossRef.
- F. Song, Z. Xu, Q. Zhang, Z. Zhao, H. Zhang, W. Zhao, Z. Qiu, C. Qi, H. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, Z. Zhao, A. Qin, D. Ma and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1800051 CrossRef.
- M. Li, S.-H. Li, D. Zhang, M. Cai, L. Duan, M.-K. Fung and C.-F. Chen, Angew. Chem., Int. Ed., 2018, 57, 2889–2893 CrossRef CAS PubMed.
- L. Yang, Y. Zhang, X. Zhang, N. Li, Y. Quan and Y. Cheng, Chem. Commun., 2018, 54, 9663–9666 RSC.
- X. Zhang, Y. Zhang, H. Zhang, Y. Quan, Y. Li, Y. Cheng and S. Ye, Org. Lett., 2019, 21, 439–443 CrossRef CAS PubMed.
- F. Zinna and L. Di Bari, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. – Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452 CrossRef CAS PubMed.
- J. Roose, B. Z. Tang and K. S. Wong, Small, 2016, 12, 6495–6512 CrossRef CAS PubMed.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef.
- M. Li, W.-B. Lin, L. Fang and C.-F. Chen, Acta Chim. Sin., 2017, 75, 1150–1163 CrossRef.
- Y. Inoue, D. Sakamaki, Y. Tsutsui, M. Gon, Y. Chujo and S. Seki, J. Am. Chem. Soc., 2018, 140, 7152–7158 CrossRef CAS PubMed.
- C. M. Cruz, S. Castro-Fernández, E. Maçôas, J. M. Cuerva and A. G. Campaña, Angew. Chem., Int. Ed., 2018, 57, 14782–14786 CrossRef CAS PubMed.
- J. Sturala, M. K. Etherington, A. N. Bismillah, H. F. Higginb, S. Hirata and M. Vacha, J. Phys. Chem. Lett., 2016, 7, 1539–1545 CrossRef PubMed.
- N. Hellou, M. Srebro-Hooper, L. Favereau, F. Zinna, E. Caytan, L. Toupet, V. Dorcet, M. Jean, N. Vanthuyne, J. A. G. Williams, L. Di Bari, J. Autschbach and J. Crassous, Angew. Chem., Int. Ed., 2017, 56, 8236–8239 CrossRef CAS PubMed.
- J.-B. Xiong, H.-T. Feng, J.-P. Sun, W.-Z. Xie, D. Yang, M. Liu and Y.-S. Zheng, J. Am. Chem. Soc., 2016, 138, 11469–11472 CrossRef CAS PubMed.
- Y. Kim, B. Yeom, O. Arteaga, S. Jo Yoo, S.-G. Lee, J.-G. Kim and N. A. Kotov, Nat. Mater., 2016, 15, 461 CrossRef CAS PubMed.
- C. Roche, H.-J. Sun, P. Leowanawat, F. Araoka, B. E. Partridge, M. Peterca, D. A. Wilson, M. E. Prendergast, P. A. Heiney, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and V. Percec, Nat. Chem., 2015, 8, 80 CrossRef PubMed.
- G. Albano, M. Lissia, G. Pescitelli, L. A. Aronica and L. Di Bari, Mater. Chem. Front., 2017, 1, 2047–2056 RSC.
- R. Aoki, R. Toyoda, J. F. Kögel, R. Sakamoto, J. Kumar, Y. Kitagawa, K. Harano, T. Kawai and H. Nishihara, J. Am. Chem. Soc., 2017, 139, 16024–16027 CrossRef CAS PubMed.
- D. Sahoo, M. R. Imam, M. Peterca, B. E. Partridge, D. A. Wilson, X. Zeng, G. Ungar, P. A. Heiney and V. Percec, J. Am. Chem. Soc., 2018, 140, 13478–13487 CrossRef CAS PubMed.
- D. A. Wilson, K. A. Andreopoulou, M. Petera, P. Leowanawat, D. Sahoo, B. E. Partridge, Q. Xiao, N. Huang, P. A. Heiney and V. Percec, J. Am. Chem. Soc., 2019, 141, 6162–6166 CrossRef CAS PubMed.
- M. Li, H.-Y. Lu, C. Zhang, L. Shi, Z. Tang and C.-F. Chen, Chem. Commun., 2016, 52, 9921–9924 RSC.
- D.-Q. He, H.-Y. Lu, M. Li and C.-F. Chen, Chem. Commun., 2017, 53, 6093–6096 RSC.
- M. Li, C. Zhang, L. Fang, L. Shi, Z. Tang, H.-Y. Lu and C.-F. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 8225–8230 CrossRef CAS PubMed.
- S. H. Chen, D. Katsis, A. W. Schmid, J. C. Mastrangelo, T. Tsutsui and T. N. Blanton, Nature, 1999, 397, 506 CrossRef CAS.
- Y. Geng, A. Trajkovska, D. Katsis, J. J. Ou, S. W. Culligan and S. H. Chen, J. Am. Chem. Soc., 2002, 124, 8337–8347 CrossRef CAS PubMed.
- C. Kulkarni, M. H. C. van Son, D. Di Nuzzo, S. C. J. Meskers, A. R. A. Palmans and E. W. Meijer, Chem. Mater., 2019 DOI:10.1021/acs.chemmater.9b00601.
- K. Watanabe, I. Osaka, S. Yorozuya and K. Akagi, Chem. Mater., 2012, 24, 1011–1024 CrossRef CAS.
- B. A. San Jose, S. Matsushita and K. Akagi, J. Am. Chem. Soc., 2012, 134, 19795–19807 CrossRef CAS.
- J. Yan, F. Ota, B. A. San Jose and K. Akagi, Adv. Funct. Mater., 2017, 27, 1604529 CrossRef.
- B. A. San Jose, J. Yan and K. Akagi, Angew. Chem., Int. Ed., 2014, 53, 10641–10644 CrossRef CAS PubMed.
- X. Li, Q. Li, Y. Wang, Y. Quan, D. Chen and Y. Cheng, Chem. – Eur. J., 2018, 24, 12607–12612 CrossRef CAS PubMed.
- X. Yang, J. Han, Y. Wang and P. Duan, Chem. Sci., 2019, 10, 172–178 RSC.
- S. Yamakawa, K. Wada, M. Hidaka, T. Hanasaki and K. Akagi, Adv. Funct. Mater., 2019, 29, 1806592 CrossRef.
- J. Liu, H. Su, L. Meng, Y. Zhao, C. Deng, J. C. Y. Ng, P. Lu, M. Faisal, J. W. Y. Lam, X. Huang, H. Wu, K. S. Wong and B. Z. Tang, Chem. Sci., 2012, 3, 2737–2747 RSC.
- D. Yang, P. Duan, L. Zhang and M. Liu, Nat. Commun., 2017, 8, 15727 CrossRef CAS PubMed.
- L. Ji, Y. Sang, G. Ouyang, D. Yang, P. Duan, Y. Jiang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 844–848 CrossRef CAS PubMed.
- J. Han, J. You, X. Li, P. Duan and M. Liu, Adv. Mater., 2017, 29, 1606503 CrossRef PubMed.
- G. Liu, J. Sheng, H. Wu, C. Yang, G. Yang, Y. Li, R. Ganguly, L. Zhu and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 6467–6473 CrossRef CAS PubMed.
- Q. Liu, Q. Xia, S. Wang, B. S. Li and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 4807–4816 RSC.
- F. Li, Y. Li, G. Wei, Y. Wang, S. Li and Y. Cheng, Chem. – Eur. J., 2016, 22, 12910–12915 CrossRef CAS PubMed.
- L. Shi, L. Zhu, J. Guo, L. Zhang, Y. Shi, Y. Zhang, K. Hou, Y. Zheng, Y. Zhu, J. Lv, S. Liu and Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 15397–15401 CrossRef CAS PubMed.
- R. Hassey, E. J. Swain, N. I. Hammer, D. Venkataraman and M. D. Barnes, Science, 2006, 314, 1437–1439 CrossRef CAS PubMed.
- Y.-H. Cheng, W.-P. Chen, Z. Shen, X.-H. Fan, M.-F. Zhu and Q.-F. Zhou, Macromolecules, 2011, 44, 1429–1437 CrossRef CAS.
- S. Dai, Z. Cai, Z. Peng, Z. Wang, B. Tong, J. Shi, S. Gan, Q. He, W. Chen and Y. Dong, Mater. Chem. Front., 2019, 3, 1105–1112 RSC.
- Q.-H. Zhou, J.-K. Zheng, Z. Shen, X.-H. Fan, X.-F. Chen and Q.-F. Zhou, Macromolecules, 2010, 43, 5637–5646 CrossRef CAS.
- J. Wang, X.-Q. Liu, X.-K. Ren, B. Zhang, S. Yang, Y. Cao, F. Liu, B. Lotz and E.-Q. Chen, Chem. – Asian J., 2016, 11, 2387–2391 CrossRef CAS PubMed.
- M. Zhu, H. Guo, F. Yang and Z. Wang, RSC Adv., 2017, 7, 4320–4328 RSC.
- H. Lu, X. Zeng, G. Ungar, C. Dressel and C. Tschierske, Angew. Chem., Int. Ed., 2018, 57, 2835–2840 CrossRef CAS PubMed.
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355–4371 CrossRef CAS PubMed.
- C. Dressel, T. Reppe, M. Prehm, M. Brautzsch and C. Tschierske, Nat. Chem., 2014, 6, 971 CrossRef CAS PubMed.
- Z. Shen, T. Wang, L. Shi, Z. Tang and M. Liu, Chem. Sci., 2015, 6, 4267–4272 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00332k |
| This journal is © the Partner Organisations 2019 |