
Direct visualization of the ouzo zone through aggregation-induced dye emission for the synthesis of highly monodispersed polymeric nanoparticles†
Eshu
Middha
 ,
Purnima Naresh
Manghnani
,
Denise Zi Ling
Ng
,
Huan
Chen
,
Saif A.
Khan
and
Bin
Liu
*
,
Purnima Naresh
Manghnani
,
Denise Zi Ling
Ng
,
Huan
Chen
,
Saif A.
Khan
and
Bin
Liu
*
Department of Chemical and Biomolecular Engineering, Faculty of Engineering, National University of Singapore, Engineering Drive 4, Singapore 117585. E-mail: cheliub@nus.edu.sg; Tel: +65-65168049
First published on 29th April 2019
Abstract
Polymeric nanoparticles (NPs) have drawn significant attention for use in optoelectronic devices and biomedical applications. Among their physiochemical properties, the size of NPs is considered to be one of their most crucial parameters. Taking the encapsulation of hydrophobic drug or dye molecules into biocompatible polymers as an example, the solvent displacement method (also known as nanoprecipitation) offers good control over the mixing process to synthesize NPs with sizes ranging from 25 to 300 nm. However, nanoprecipitation produces large aggregates above a certain solvent fraction and polymer concentration, which results in the synthesis of highly polydisperse particles with uncontrolled sizes. Therefore, for the systematic and controlled synthesis of monodispersed NPs, we have plotted the ouzo zones of two polymers, PLGA and DSPE–mPEG, in a novel and simple way by taking advantage of the unique properties of aggregation-induced emission (AIE) dyes, which show different fluorescence emissions in different states. Furthermore, we have developed a new process, the enhanced solvent displacement method (ESDM), for the production of highly monodispersed NPs with ultra-low PDI values (0.05 to 0.1) and sizes ranging from 25 to 200 nm by increasing the miscibility between the anti-solvent and solvent with pre-mixing of the solvent (tetrahydrofuran) into the anti-solvent (water).
Introduction
Over the past few decades, nanoprecipitation has revolutionized the synthesis of lipid and polymer-based nanoparticles (NPs).1–9 It is considered to be a fast, straightforward and reproducible method to synthesize small and monodisperse NPs with size ranges of 20 to 300 nm and polydispersity indices (PDI) of less than 0.2.9–11 To date, the nanoprecipitation method has been used for the synthesis of many drug-loaded NPs and dye-loaded NPs composed of biocompatible and biodegradable polymers, such as poly(lactide-co-glycolic acid) (PLGA), poly(lactide-co-glycolide)-b-poly(ethylene glycol) (PLGA–PEG), poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG-b-PCL) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine–polyethylene glycol (DSPE–mPEG).7,12–17 For biomedical applications, the size of NPs plays an important role; it directly impacts the pharmacokinetics and pharmacodynamics of NPs post intravenous administration.18,19 NP samples with broad size distributions or high PDI contain many large and small NPs; additionally, their mean average size affects their performance during biological applications, especially biodistribution. It is much more difficult to achieve a highly monodispersed distribution (PDI < 0.1) for small size ranges of NPs (25 to 100 nm) than for large NPs (>100 nm). Therefore, an advanced process and strategy is required which exhibits ultra-high uniformity during mixing to synthesize highly monodispersed NPs for all size ranges.Nanoprecipitation is a solvent displacement method (SDM) in which precursors (polymers, dyes and drugs) are dissolved in a water-miscible organic solvent, such as acetone, ethanol or tetrahydrofuran (THF).7,9 One of the major limitations of SDM is the formation of large aggregates above certain solvent fractions and polymer concentrations, which results in the synthesis of highly polydisperse particles.20 According to Vitale and Katz, there is a metastable region in the ternary phase diagram of a solute/solvent/anti-solvent system, known as the “ouzo region”, where local supersaturation of solute results in the synthesis of small, stable NPs.21 In this respect, the introduction of the “ouzo region” in ternary phase diagrams enables more systematic preparation of stable polymeric NPs at desired solute concentrations and solvent compositions.2,6,20 To date, the ouzo diagram of PLGA has served as a meaningful resource for the synthesis of small and uniform NPs.22,23 However, the complete construction of an ouzo diagram for any polymer is a tedious task which requires filtration, particle size analysis, and zeta potential analysis for many data points with different solute and solvent compositions.20–23
To achieve high uniformity of NPs, efforts have been made to enhance the mixing of the two liquid phases by varying several parameters, such as the Reynolds number (Re), water-to-solvent ratio, properties of solvents, and amount of surfactant in the anti-solvent, using numerous mixing devices. In the past few years, microfluidic 2D and 3D hydrodynamic devices,24 the Co-axial Turbulent Jet-Mixer,25 the Multi Inlet Vortex Mixer (MIVM) and the Confined Impinging Jets Mixer (CIJM)17,26,27 have been used to synthesize polymeric NPs with precise size control and uniform size distribution through SDM. Despite rapid mixing, due to either high Re or high surface area-volume interactions, the above mixing devices were not able to achieve the same level of monodispersity for small NPs (25 to 100 nm) as for large NPs (>100 nm).28 Moreover, they could only achieve PDI values greater than 0.15 for NPs smaller than 100 nm.
In this paper, firstly, we present a new NP synthesis method called the enhanced solvent displacement method (ESDM) for the production of highly monodispersed NPs (PDI ≤ 0.1) with a broad size range of 25 to 200 nm (Fig. 1) by enhancing the miscibility between the solvent and antisolvent in the ouzo zone. Compared to the conventional solvent displacement method (SDM), a significant decrease in PDI is achieved through ESDM at different Reynolds numbers (Re) for all size ranges of NPs. Secondly, a novel and facile method to construct the ouzo diagrams of polymers, namely DSPE–mPEG and PLGA, has been introduced by taking advantage of the properties of aggregation-induced emission (AIE) dyes,13,29–31 which show different fluorescence emissions in different aggregate states, to achieve the systematic and controlled synthesis of highly monodispersed NPs through ESDM. To demonstrate how the ouzo zone can be used to guide NP synthesis, an organic AIE dye molecule, 4,7-bis{4-(1,2,2-triphenylvinyl)phenyl}benzo-2,1,3-thiadiazole (BTPEBT), and an anti-cancer drug, tamoxifen (TAM), were selected to fabricate organic NPs using DSPE–mPEG and PLGA as the matrices.32,33
 | ||
| Fig. 1 Schematic of the microfluidic glass capillary mixer for the synthesis of monodisperse BTPEBT-loaded DSPE–mPEG2000 NPs. Case A represents the synthesis of NPs through conventional SDM with 100% water (x = 0) from the outlet stream (pump A). Case B depicts the synthesis through ESDM with a mixture of THF (x%) and water (100 − x%) in the outlet stream for the preparation of highly monodisperse NPs (PDI values as low as 0.05). | ||
Results and discussion
Synthesis of NPs through the enhanced solvent displacement method (ESDM)
We fabricated a microfluidic glass capillary mixer with coaxial flow for the synthesis of monodisperse NPs. Fig. 1 presents a schematic of the microfluidic glass capillary mixer with two inlets for the synthesis of BTPEBT-loaded DSPE–mPEG NPs through the solvent displacement method (SDM) and enhanced solvent displacement method (ESDM). BTPEBT is a dye with aggregation-induced emission (AIE) properties; it shows strong fluorescence in the aggregate state.31 Encapsulating BTPEBT molecules through conventional SDM is a widely applicable technique to produce bright NPs for cell tracking, vascular imaging and organelle-specific imaging.13,30,31,34 In the mixer, each inlet is connected with a syringe controlled by a programmable syringe pump. Syringe pump A is used to control the flow of the outer fluid (anti-solvent), which contains water during SDM and a mixture of water and x% THF during ESDM (where x represents the % volume of THF in the anti-solvent). Meanwhile, syringe pump B controls the flow rate of the inner fluid, an organic solvent (100% THF) which contains BTPEBT and DSPE–mPEG2000. The volumetric flow rate ratio of outer to inner fluid is maintained at 10, and the percentage of THF in the outer fluid (x%) is varied from 0% to 80% (Table S1, ESI†). The solvent stream comprises BTPEBT and DSPE–mPEG2000 with a polymer-to-dye ratio (PDR) of 0.5; this was chosen based on optimized dye-to-polymer loading and minimum dye loss (Fig. S1, ESI†). The solvent flowing through the inner capillary (inner fluid) is mixed with the anti-solvent (water) flowing from the outer capillary in the mixing zone. After emerging from the tip of the inner capillary, the solvent rapidly mixes with the anti-solvent in the coaxial direction, which results in the formation of NPs through a self-assembly process; then, the mixture is maintained for solvent evaporation (Fig. S2, ESI†).Rapid mixing of organic solvent into the anti-solvent (aqueous phase) causes spontaneous formation of NPs. As reported by Lince et al.,35 supersaturation has been described as the main driving force for the synthesis of nanoparticles. High and uniform supersaturation throughout the mixing zone affords small NPs with high homogeneity. In the mixing zone, the ESDM process occurs with higher miscibility of the solvent and antisolvent than the SDM process, and we could synthesize monodispersed NPs of different sizes by varying the total flow rate from 1.1 to 16.5 mL min−1 (Re 23.33 to 350, eqn (S4) and Table S2, ESI†).
For comparison between the conventional solvent displacement method (SDM) and enhanced solvent displacement method (ESDM), we synthesized NPs in the microfluidic glass capillary mixer at different Re (23.33 to 350) and a total solute (BTPEBT + DSPE–mPEG) concentration of 2 mg mL−1 in an organic solvent, THF (Fig. 2). With increasing Re from 23.33 to 350 through conventional SDM (x = 0), a decrease in the size of the BTPEBT-loaded DSPE–mPEG2000 NPs from 95 nm to 32 nm was noted, with PDI values in the range of 0.14 to 0.16. Meanwhile, in the ESDM process, with increasing x from 5% to 15%, a slight decrease in the size of the NPs was noted for all Re, with a substantial decrease in the PDI of the NPs compared to conventional SDM (from 0.16 to 0.08 in the case of Re 350) (Fig. 2b). However, on further increasing x from 15% to 30%, the sizes of the NPs increased for all Re, with increases in homogeneity. For all Re, significant increases in the uniformity of the NPs were noted with increasing x from 0% to 30%. At ESDM 30% THF (x = 30), the effects of mixing and the control over the size of the NPs decreased, with no significant changes in the sizes of the NPs (∼170 nm) with varying Re (Fig. 2a and b). The sizes of the particles entered the micron range upon further increasing the amount of THF in the anti-solvent (data not shown). The number average diameter of NPs obtained from DLS analysis (Fig. 2a) is in good agreement with the TEM images (Fig. 2e) and shows very high uniformity for the NPs synthesized by ESDM with 20% THF in the anti-solvent at Re 23.33 and 350.
 | ||
| Fig. 2 Effects of varying the composition of THF (x) in the anti-solvent during the synthesis of NPs in the microfluidic glass capillary mixer. (a) Variation in the size of NPs obtained using different processes with Re from 23.33 to 350. (b) Effects of the ESDM process on the uniformity (PDI) of NPs synthesized by varying x from 0% to 30% and Re from 23.33 to 350. (c) Variation of the interaction parameters between the solvent and anti-solvent (s–as) by changing the amount of THF in the anti-solvent. (d) Changes in Tmix between the solvent and anti-solvent for different processes at different Re. (e) TEM images of BTPEBT-loaded DSPE–mPEG NPs with homogeneous size distributions synthesized using ESDM with 20% THF at Re 23.3 and 350. | ||
The process of solvent displacement occurs due to mixing between the solvent and anti-solvent. As the solvent mixes with the anti-solvent, it creates local supersaturation for the solute dissolved in the solvent, which results in spontaneous formation of NPs.6,8,9 The mixing of the solvent and anti-solvent can be altered by changing their miscibility. The miscibilities of different solvents in water were proved to have pronounced effects on the sizes of NPs.12 In this work, we changed the properties of the anti-solvent by pre-mixing a certain composition of THF (x) in water to enhance the miscibility of the solvent and anti-solvent. The miscibility of a solvent in an anti-solvent can be determined by comparing the solvent and anti-solvent interaction parameters (χs–as) for different values of x as per eqn (1) (Table S3 and eqn (S5), ESI†). A lower value of χs–as signifies a high affinity between the solvent and anti-solvent at that particular THF composition.12,22
 | (1) |
With increasing THF (x) content from 0% to 30% in the anti-solvent, the solubility parameter of the anti-solvent (δas) decreases constantly, which results in a significant decrease of the interaction parameter χs–as from 28.02 to 13.73 (Fig. 2c and Table S3, ESI†). Therefore, the miscibility of the solvent in the anti-solvent increases with increasing amount of THF in the anti-solvent, which also generates uniform and fast mixing in ESDM compared to the conventional SDM process for the same Re.
We used computational fluid dynamics (CFD) to analyze the mixing of the solvent and anti-solvent by estimating the mixing lengths (Lmix) and mixing times (Tmix) for SDM and ESDM (Fig. S2, ESI†). Lmix is taken as the distance between the inner glass capillary hole and the point where the mass fraction of THF achieves saturation along the axial direction (Fig. S3, ESI†). As per CFD simulation, at the same Re, Lmix and Tmix decrease 1.5 to 2 times with increasing THF composition from 0% to 30% in anti-solvent (Fig. 2d and Fig. S3, ESI†). The benefits of achieving low Tmix compared to SDM at the same Re enables ESDM to achieve highly monodispersed BTPEBT-loaded DSPE–mPEG NPs with a PDI of less than 0.1. The continuous decrease in PDI of BTPEBT-loaded DSPE–mPEG2000 NPs with increasing x from 0% to 30% in the anti-solvent can be explained by the increase in the miscibility of the solvent and anti-solvent due to the decrease in Tmix. However, the size of the NPs first decreases and then increases with increasing THF composition in the anti-solvent (Fig. 2a). This phenomenon occurs due to a decrease in the level of supersaturation with increasing THF composition, which is the main driving force in the synthesis of NPs. As the total composition of THF in the mixture increases, the solute experiences a low level of supersaturation and the nucleation rate decreases significantly. Because the nucleates are far apart, they grow by aggregating with nearby isolated solute molecules until they collect all the available solute molecules, resulting in larger NPs.5,20
To check the reliability of our enhanced solvent displacement method (ESDM), we synthesized BTPEBT-loaded DSPE–mPEG2000 NPs through ESDM with 20% THF at Re 350 for 5 experimental runs (Fig. S4, ESI†). The number average mean diameters of the NPs were reproducible, with particle sizes of 48 ± 2 nm in different runs; this indicates high reproducibility of the process in terms of size. For all the runs, particles were synthesized with highly uniform size distributions and an average PDI of 0.081 ± 0.002. In addition, we performed stability analyses of the synthesized NPs from one of the above runs for 2 weeks (Fig. S12, ESI†). It was found that the size and polydispersity (PDI) of the NPs did not show any obvious changes, even after 2 weeks. Due to this high degree of stability and reproducibility in terms of NP size and distribution, the enhanced solvent displacement method (ESDM) is a simple, reproducible and reliable method for synthesizing highly monodispersed polymeric NPs.
Construction of the ouzo diagram of DSPE–mPEG with and without AIE dye
The level of supersaturation experienced by the solute decreases with the addition of THF, which results in the formation of large particles above a certain composition of THF in the anti-solvent. As the concentration of the solute increases, the probability of collision between existing particles rises due to the formation of large numbers of nuclei. Random collisions between the particles result in the synthesis of large, non-uniform aggregates through a diffusion-limited cluster–cluster aggregation mechanism.6 At a high concentration of solute or a high THF composition, the control over the size and PDI of the NPs decreases. Several investigations have been performed on the ternary phase diagrams of solute/solvent/water systems in order to identify the zone where only NPs are formed with good control over the size. As per Vitale and Katz, there is a small metastable region between the binodal and spinodal curves of the ternary phase diagram of a solute/solvent/water system, known as the ouzo zone, which affords stable, small NPs with a narrow distribution through the process of solvent displacement (Fig. 3).21,36 This region enables methodical synthesis of stable polymeric NPs with defined size characteristics. As per Monte-Carlo numerical analysis, the reason for the stability of NPs synthesized in the ouzo zone is the minimum total energy achieved by the system, which prevents further coalescence of the NPs.37 | ||
| Fig. 3 (a) Ternary phase diagram of a solute/solvent/water system containing the ouzo zone, non-ouzo zone and binodal curve. (b) Magnified small portion of the ternary phase diagram obtained by plotting the solute mass fraction on the x-axis and the solvent mass fraction on the y-axis with the ouzo zone, non-ouzo zone and binodal curve. | ||
We have focused on the magnified part of the ternary phase diagram with a right triangle ouzo diagram (Fig. 3b). The ouzo zone of DSPE–mPEG2000 was constructed via two methods. We started with the traditional method using a microfluidic glass capillary, where the ouzo zone was plotted by varying the composition of THF in the anti-solvent (x) from 0% to 80% at a particular concentration of solute for the synthesis of particles and analysis of their sizes without THF evaporation through dynamic light scattering (DLS). The total composition of THF in the final mixture (y%) also changes linearly with increasing THF content in the anti-solvent (x%) (Fig. 1 and Table S1, ESI†), which results in the synthesis of particles with different mass fractions of THF (fTHF) and water (fwater) in the final mixture (eqn (S12), ESI†). To construct the ouzo zone in the ternary phase diagram, the mass fraction of each component in the final mixture is taken as a reference. The mass fraction of DSPE–mPEG2000 (fsolute) in the mixture during synthesis is plotted on the x-axis with a logarithm scale due to the low mass fraction (eqn (S10), ESI†). The y axis signifies the mass fraction of solvent (fTHF) in the final mixture, which varies linearly with the % volume of THF (x%) added to the anti-solvent during the pre-mixing step (eqn (S8) and (S12), ESI†). The mass fraction of the third component (water) can be determined by the difference (fwater = 1 − fsolute − fTHF).
With a concentration of 2 mg mL−1 of DSPE–mPEG2000 in solvent or fsolute 1.8 × 10−4 in the final mixture (eqn (S7) and (S10), ESI†), the sizes of the produced particles are in the range of 100 to 250 nm when the mass fraction of THF (fTHF) is 0.34 in the final mixture (Fig. 4a). When fTHF reaches 0.38, the mixture enters the non-ouzo region to yield large aggregates of micron size. As fTHF increases further, the particles remain in the non-ouzo zone until the mass fraction of THF reaches 0.66. Above fTHF 0.66, there is a sudden decrease in the size of the particles to a few nanometers, which signifies that the mixture crosses the miscibility limit (binodal curve). For DSPE–mPEG2000 at a mass fraction (fsolute) of 1.8 × 10−4, all three zones of DSPE–mPEG2000, namely ouzo, non-ouzo and binodal, were determined using ESDM through varying fTHF from 0.08 to 0.80 in the final mixture (or the % volume of THF from 0% to 80% in the anti-solvent).
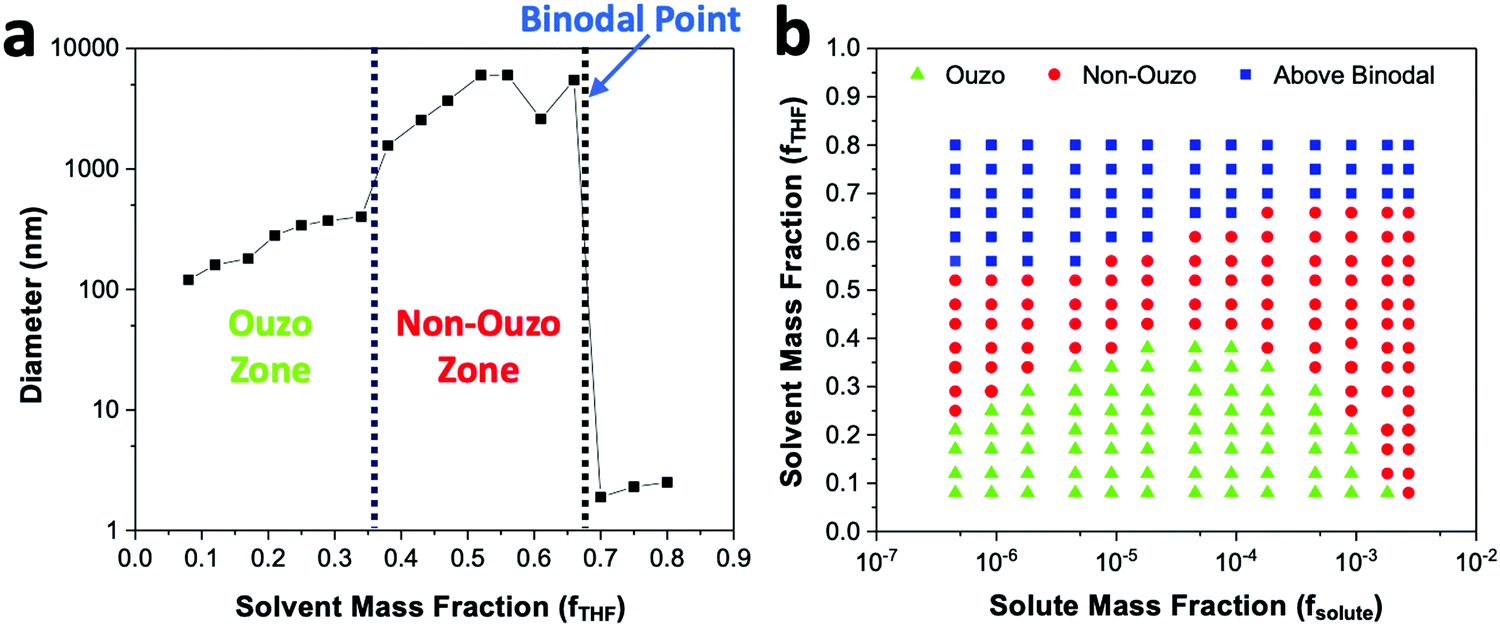 | ||
| Fig. 4 (a) Changes in the sizes of particles formed by blank DSPE–mPEG2000 with a mass fraction of 1.8 × 10−4 at various fTHF to determine the ouzo zone, non-ouzo zone and binodal point. (b) Ternary phase diagram of the DSPE–mPEG2000/THF/water combination containing the ouzo zone, non-ouzo zone and binodal curve by varying fTHF from 0.08 to 0.80 and fsolute from 4.5 × 10−7 to 2.7 × 10−3. | ||
In our approach, we analyzed the sizes of the samples without THF evaporation and constructed ouzo and binodal curves simultaneously. However, as per Aubry et al., the size of a sample should be analyzed after evaporating THF from it. As the solvent evaporates, the mixture with high THF content (above binodal curve) will shift towards the non-ouzo zone and form large aggregates below a certain THF content (Fig. S11c, ESI†); thus, it is difficult to plot the binodal curve through this approach. We acquired a TEM image of DSPE–PEG synthesized above the binodal zone (Fig. S11, ESI†). The image shows the formation of large aggregations due to shifting of the composition from binodal to non-ouzo after solvent evaporation. Therefore, we used a size analysis approach without THF evaporation to construct the ouzo zone and binodal curve one at a time by varying the total amount of THF in the samples. With a concentration of 2 mg mL−1 of DSPE–mPEG2000 in the solvent, the transition between the ouzo and non-ouzo zones is similar for both methods (with and without THF evaporation, Fig. S10, ESI†).
We varied fsolute from 4.5 × 10−7 to 2.7 × 10−3 and fTHF from 0.08 to 0.80 to construct the small region of the DSPE–mPEG/THF/water ternary phase diagram containing the ouzo, non-ouzo and binodal regions using the microfluidic glass capillary mixer (Fig. 4b and Fig. S5, ESI†). Under the ouzo boundary, the blank particles formed are small (<300 nm) with a uniform size distribution (Fig. S11a, ESI†). The level of supersaturation rises with the formation of large numbers of nuclei due to the increasing solute concentration.
The ouzo zone also disappears with decreasing solute concentration or increasing fTHF (Fig. 4b). At very low solute concentrations or high THF compositions, only a few solute molecules experience supersaturation and form nuclei. Instead of encountering each other, these nuclei grow by accumulating all nearby isolated solute molecules and synthesize large aggregates through the nucleation-growth mechanism (Fig. S11b, ESI†). Overall, there is an optimum region in terms of water, THF and solute fraction, known as the ouzo zone, to yield small, stable NPs.
For biological applications, the empty DSPE–mPEG NPs must be loaded with drugs or dyes. Therefore, we repeated the construction of the ternary phase diagram with the solute consisting of a combination of AIE dye BTPEBT and polymer DSPE–mPEG2000 at a polymer-to-dye mass ratio of 0.5 using a microfluidic mixer. Similar to the method discussed above, the ouzo region of BTPEBT-loaded DSPE–mPEG2000 was constructed by varying fTHF from 0.08 to 0.80 at different mass fractions of solute for the synthesis of the particles. We varied the mass fraction of the solute (fsolute), BTPEBT + DSPE–mPEG2000, from 4.5 × 10−7 to 2.7 × 10−3 to construct the small region of the BTPEBT + DSPE–mPEG/THF/water ternary phase diagram containing the ouzo, non-ouzo and binodal regions (Fig. 5a and Fig. S6, ESI†).
 | ||
| Fig. 5 (a) Ternary phase diagram of the BTPEBT + DSPE–mPEG2000/THF/water combination containing the ouzo zone, non-ouzo zone and binodal curve by varying fTHF from 0.08 to 0.80 and fsolute from 4.5 × 10−7 to 2.7 × 10−3. (b) SEM images of BTPEBT–DSPE–mPEG particles synthesized in the ouzo and non-ouzo regions at 1.8 × 10−4 mass fraction of solute. | ||
The ouzo zone of the BTPEBT + DSPE–mPEG2000/THF/water combination is similar to that of DSPE–mPEG2000/THF/water, with a minor shift of the ouzo boundary to a lower mass fraction of THF (Fig. 5a). The NPs synthesized in the ouzo zone ranged between 50 and 300 nm in size (Fig. S6, ESI†). Outside the ouzo region, the polymer loses its capability to encapsulate BTPEBT due to its weak self-assembly, which results in the formation of large aggregates of BTPEBT.38 The particles synthesized at a 1.8 × 10−4 mass fraction of solute in the ouzo and non-ouzo zones were visualized using SEM (Fig. 5b). In the ouzo zone, all particles were spherical in shape with sizes less than 200 nm and a uniform size distribution. Meanwhile, large aggregates with sizes greater than 1 μm and a non-uniform size distribution were observed in the non-ouzo region (Fig. 5b).
The ouzo diagram of BTPEBT + DSPE–mPEG can be used as a resourceful tool for the synthesis of monodisperse NPs with defined size distributions. For example, with fsolute of 1.8 × 10−4 (i.e. the initial concentration of BTPEBT and polymer in the organic solvent is 2 mg mL−1) (eqn (S7) and (S10), ESI†), the ouzo boundary region lies under fTHF 0.34 (Fig. 5a). Using this information, the maximum amount of solvent to add to the anti-solvent in the pre-mixing step is determined for the synthesis of highly monodisperse NPs through ESDM. With fTHF 0.34 in the final mixture, the amount of solvent to mix with the anti-solvent (x%) during ESDM is 30% (Table S4, ESI†). At the ouzo boundary (with fTHF 0.34 in final mixture or 30% THF in anti-solvent), the process did not show significant changes in the size of the NPs by varying Re from 23.33 to 350 (Fig. 2a). This signifies that the process starts losing its sensitivity to the mixing conditions near the ouzo boundary. Meanwhile, in the ouzo region, with less THF in the anti-solvent, significant changes in the NP size with varying Re are noted (Fig. 2a and 5). Therefore, under the guidance of the ouzo zone, the sizes of NPs can be optimized, with defined size characteristics and uniform size distributions (PDI as low as 0.05), through ESDM.
Aggregation-induced emission (AIE) dyes generally have rotor-like structures; they are known for their varying fluorescence emissions in different aggregate states due to different levels of restriction in their intramolecular rotation.29–31 In this work, we chose an ultra-bright AIE dye, BTPEBT, which emits intense fluorescence in its nanoaggregate state relative to its dissolved state in dilute solvent.13,29 This special characteristic of the AIE dye enables direct visualization of the ouzo, non-ouzo and binodal zones for the BTPEBT + DSPE–mPEG/THF/water combination in a unique manner (Fig. 6a). In the ouzo zone, the nanoaggregates of BTPEBT showed very high fluorescence. Meanwhile, the emission band of the BTPEBT particles blue shifted with increasing amount of THF and when entering the non-ouzo zone. Upon further increasing the amount of THF, the mixture moved above the binodal curve, with the same colour of emission as the dye molecules in pure THF solution (Fig. 6a). The binodal curve corresponds to the miscibility limit of the solute, above which the solute exists as a monophasic system with no sharp interface.38,39 Therefore, the unique characteristics of AIE, showing different fluorescence emissions in different states, can be used to plot the ouzo zones of polymers for the systematic and controlled synthesis of NPs.
 | ||
| Fig. 6 (a) Different fluorescence emissions of the AIE dye BTPEBT in combination with DSPE–mPEG in the ouzo, non-ouzo and binodal zones by varying fTHF from 0.08 to 0.80 at different fsolute. (b) Absorbance and (c) PL intensities of BTPEBT–DSPE–mPEG particles synthesized in different regions at a solute mass fraction of 9.0 × 10−5. | ||
The BTPEBT particles synthesized in different regions at a solute mass fraction of 9.0 × 10−5 were analysed by evaluating their absorbance and photoluminescence12 values (Fig. 6b and c). In the ouzo zone, the mixture shows the maximum PL intensity, with a peak at 547 nm.13 On shifting from ouzo to non-ouzo, the maximum PL intensity peak is blue shifted to 514 nm, with a significant decrease in fluorescence. On further addition of THF and upon crossing the binodal curve, the PL intensity peak is red shifted to 556 nm, with a lower emission than in the ouzo zone. The trend of the changes in emission of BTPEBT in different states is in good agreement with the DLS analysis to predict the ouzo, non-ouzo and binodal regions (Fig. 5a and 6a). Hence, the production of polymeric particles inside or outside the ouzo zone can be easily adjudged with direct visualization through the AIE dye properties instead of by filtration, particle size analysis and zeta potential analysis for numerous data points at different solute and solvent compositions.
Construction of the ouzo diagram of PLGA using AIE dye as an indicator
To validate the reliability of using the AIE properties of BTPEBT for the construction of ouzo zones, we used PLGA in combination with BTPEBT as the solute to plot the ouzo diagram and compared it with the ouzo zone of PLGA constructed in the literature through filtration and particle size analysis (Fig. 7a).22 We varied the mass fraction of solute (fsolute) from 9.0 × 10−5 to 4.5 × 10−3 and the mass fraction of solvent (fTHF) from 0.08 to 0.80 using the microfluidic mixer to construct the BTPEBT + PLGA/THF/water ternary phase diagram containing the ouzo, non-ouzo and binodal regions (Fig. 7a). The ouzo diagram of BTPEBT in combination with PLGA is very similar to the ouzo diagram of PLGA plotted through particle size analysis (Fig. 7b).22 In the ouzo zone, BTPEBT encapsulated into PLGA experiences maximum restriction of intramolecular rotation, which leads to high fluorescence emission. Meanwhile, outside the ouzo zone, the polymer loses its capability to encapsulate the dye tightly due to its weak self-assembly,38 which results in the formation of large aggregates of BTPEBT with low fluorescence emission.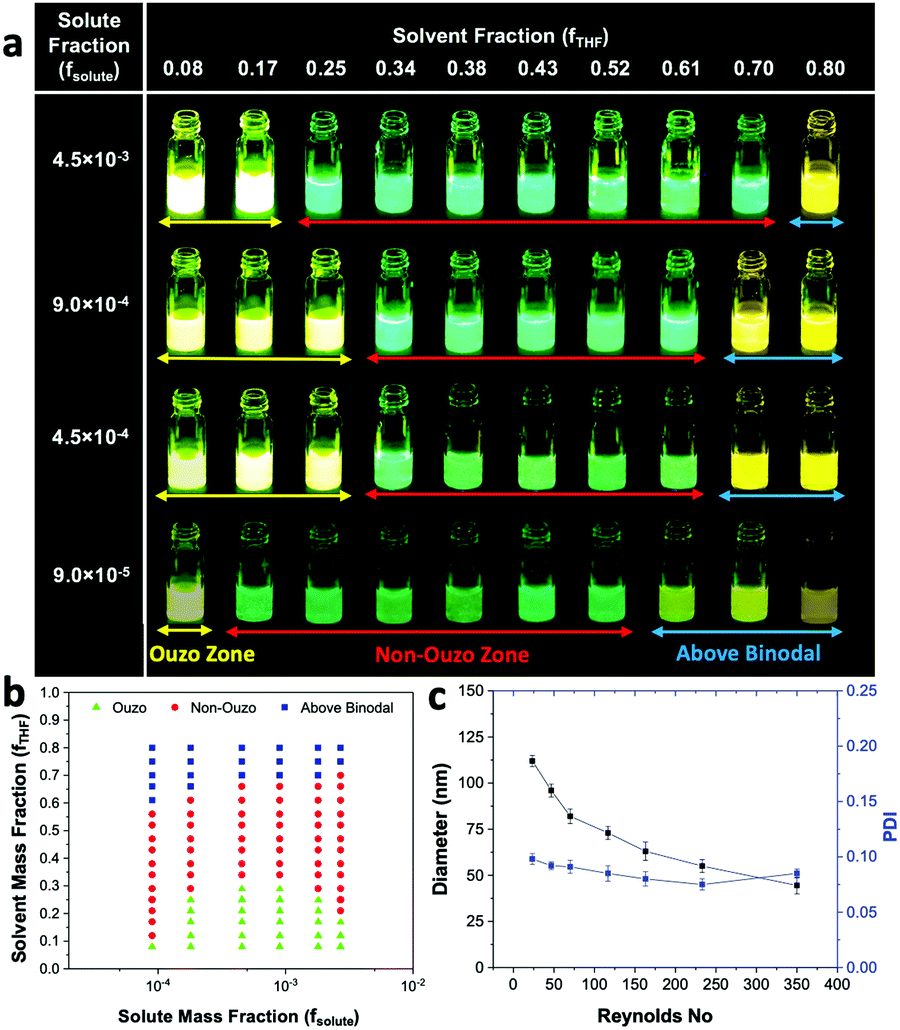 | ||
| Fig. 7 (a) Different fluorescence emissions of the AIE dye BTPEBT in combination with PLGA in the ouzo, non-ouzo and binodal zones by varying fTHF from 0.08 to 0.80 and fsolute from 9.0 × 10−5 to 4.5 × 10−3. (b) Ternary phase diagram of the BTPEBT + PLGA/THF/water combination containing the ouzo zone, non-ouzo zone and binodal curve by varying fTHF from 0.08 to 0.80 and fsolute from 9.0 × 10−5 to 4.5 × 10−3. (c) Variation in the size of BTPEBT-loaded PLGA NPs using ESDM with 10% THF in the anti-solvent in the ouzo zone with varying Re from 23.33 to 350. | ||
As above, the ouzo diagram of BTPEBT + PLGA can be used as guidance for the synthesis of highly monodispersed NPs with defined size distributions through ESDM. In the ouzo zone, at fsolute 1.8 × 10−4 (initial concentration of 2 mg mL−1 of solute in THF) (eqn (S7) and (S10), ESI†), we synthesized BTPEBT-loaded PLGA NPs at different Re through ESDM with 10% THF in the anti-solvent (or fTHF 0.17 in the final mixture) (Fig. 7c). With increasing Re from 23.33 to 350, a decrease in the size of NPs from 112 nm to 44 nm was noted, with highly monodispersed size distributions (PDI as low as 0.075). Therefore, under the guidance of the ouzo zone, we can synthesize highly monodispersed polymeric NPs of different sizes in a systematic and controlled manner using the ESDM process.
Versatility of the enhanced solvent displacement method (ESDM)
To check the versatility of the ESDM, we synthesized tamoxifen (TAM)-loaded DSPE–mPEG2000 NPs and TAM-loaded PLGA NPs using ESDM with 10% THF in the anti-solvent in the ouzo zone (Fig. 8). With increasing Re from 23.33 to 350, the average diameter of TAM-loaded DSPE–mPEG NPs decreases from 268 to 126 nm, with PDI values in the range of 0.078 to 0.098 (Fig. 8a). In the case of TAM-loaded PLGA NPs, the size of the NPs decreases from 242 to 89 nm with increasing Re from 23.33 to 350 and PDI values in the range of 0.075 to 0.095 (Fig. 8b). Using ESDM with 10% THF in the anti-solvent, the drug loading capacity (LC) and encapsulation efficiency (EE) obtained for TAM-loaded PLGA NPs synthesized at Re 350 were 8.1% and 88%, respectively (Fig. S9, ESI†). Meanwhile, in the case of TAM-loaded DSPE–mPEG NPs, the LC and EE achieved at Re 350 were 7.4% and 81%, respectively. Compared to previously reported bulk synthesis and conventional SDM, the ESDM produces smaller and more narrowly distributed TAM-PLGA NPs.33 | ||
| Fig. 8 (a) Effects of varying Re from 23.33 to 350 on the size and uniformity (PDI) of TAM-loaded DSPE–mPEG NPs. (b) Variation in the size and uniformity (PDI) of TAM-loaded PLGA NPs by varying Re from 23.33 to 350. | ||
Conclusions
We have developed a new process, the enhanced solvent displacement method (ESDM), for the synthesis of highly monodispersed NPs (PDI ≤ 0.1) with precise control over the size range of 25 to 200 nm, by enhancing the miscibility of the solvent and anti-solvent through pre-mixing of THF in water. During NP preparation, particles can flocculate above certain solute or solvent compositions. Therefore, to achieve systematic and controlled synthesis of NPs through ESDM, we constructed the ouzo zone of DSPE–mPEG2000 with and without an AIE dye to determine the maximum amount of THF to add to the anti-solvent. Additionally, we propose a novel approach to plot the ouzo zone of a polymer in a unique and facile way by taking advantage of the characteristics of AIE dyes, which show different fluorescence emissions in different states. In summary, location of the ouzo zone through AIE dye can be used to synthesize highly monodispersed dye or drug-loaded polymeric NPs in a controllable and systematic manner through the ESDM process.Experimental section
Materials
The lipid-polymer, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy(polyethylene glycol)-2000) (DSPE–PEG2000), was purchased from Avanti Polar Lipids, Inc. The PLGA, Resomer RG 502H (acid-terminated, average molecular weight (Mw) 7000 to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), the anti-cancer drug tamoxifen and the solvent tetrahydrofuran (THF) were purchased from Sigma Aldrich. Milli-Q water was supplied by an Ultra-Pure water system (Fisher Scientific, Singapore). The AIE dye, 4,7-bis(4-(1,2,2-triphenylvinyl)phenyl)benzo-2,1,3-thiadiazole (BTPEBT), was synthesized according to the literature.13 Round glass capillaries with inner diameters (ID) of 1 mm and 0.7 mm, respectively, were purchased from Arte Glass Associates Co. Ltd, Japan. Teflon tubing with an ID of 1.6 mm was purchased from Spectra Teknik Pte Ltd.
000), the anti-cancer drug tamoxifen and the solvent tetrahydrofuran (THF) were purchased from Sigma Aldrich. Milli-Q water was supplied by an Ultra-Pure water system (Fisher Scientific, Singapore). The AIE dye, 4,7-bis(4-(1,2,2-triphenylvinyl)phenyl)benzo-2,1,3-thiadiazole (BTPEBT), was synthesized according to the literature.13 Round glass capillaries with inner diameters (ID) of 1 mm and 0.7 mm, respectively, were purchased from Arte Glass Associates Co. Ltd, Japan. Teflon tubing with an ID of 1.6 mm was purchased from Spectra Teknik Pte Ltd.
Fabrication of the microfluidic glass capillary mixer
The coaxial flow microfluidic glass capillary mixer was assembled using two round glass capillaries with diameters of 1 mm (outer capillary) and 0.7 mm (inner capillary), Teflon tubing and fittings from Chemikalie Pte Ltd. The surface of the outer glass capillary was rendered hydrophilic by treating it with oxygen plasma at 18 W for 120 seconds. The inner round capillary was pulled using a micropipette puller (Sutter Instruments, P-97) to produce a tapered end. The tapered end was then enlarged to 150 μm using abrasive paper. Teflon tubing was used to connect the glass capillary device to the respective syringes controlled by syringe pumps. The inner glass capillary was inserted into the outer glass capillary through a side hole in the Teflon tubing. The outer glass capillary was connected with a plastic monoject syringe through Teflon tubing and fittings for the supply of water. The dye (BTPEBT), drug (TAM) and polymer (DSPE–mPEG) dissolved in tetrahydrofuran (THF) were injected into the mixer through the inner glass capillary connected with a Hamilton Gastight syringe via Teflon tubing. All the flows were controlled by syringe pumps. The pump used for the water supply was a Chemyx Nexus-3000 pump, and the organic phase was injected using a Chemyx Fusion-200 pump.Preparation of drug or dye-loaded DSPE–mPEG2000 NPs
The NPs were synthesized using the microfluidic glass capillary with two inlets, one for organic solvent with drug, dye or polymer dissolved in it and the other for anti-solvent (100 − x% water + x% THF) linked with a syringe driven by a programmable syringe pump. The total flow rate of water and organic solvent was varied from 1.1 mL min−1 to 16.5 mL min−1 (Re from 23.33 to 350) by maintaining the outer (anti-solvent) to inner (solvent) ratio at 10. BTPEBT dye and polymer (DSPE–mPEG or PLGA) were dissolved in THF with a total concentration of 2 mg mL−1 and a polymer-to-dye mass ratio of 0.5. The synthesized NPs were characterized using dynamic light scattering (DLS) at 90-degree scattering (ZetaSizer Nano-ZS, Malvern Instruments, UK) after solvent evaporation. Each measurement was repeated 3 times. For the synthesis of TAM-loaded DSPE–mPEG NPs, the polymer-to-drug ratio was maintained at 10 while maintaining a total NP precursor concentration of 2 mg mL−1. Same as above, the sizes of the TAM-loaded DSPE–mPEG NPs were analyzed using DLS.Preparation of TAM-loaded PLGA NPs
The TAM-loaded PLGA NPs were prepared through ESDM with 10% THF in the anti-solvent to check the versatility of the process. The NPs were synthesized using the microfluidic glass capillary by maintaining the outer (anti-solvent) to inner fluid (organic solvent) flow ratio of 10. The solvent stream comprised TAM and PLGA with a polymer-to-drug mass ratio of 10 while maintaining the total NP precursor concentration at 2 mg mL−1.33 To determine the encapsulation efficiency and drug loading capacity, we filtered the solution with a 0.44 μm filter to remove any non-encapsulated TAM, while the TA-loaded NPs solution was collected. The filtered TAM NPs were resuspended in THF solution, and the percentage of TAM encapsulated was determined using a standard UV absorbance calibration curve measured at 300 nm (Fig. S8 and S9, ESI†).Construction of ouzo diagrams
As a map of composition, a right triangle, three-component phase diagram (“ouzo diagram”) was chosen, where the compositions were plotted according to the mass fraction of a solute (fsolute) on the abscissa and the mass fraction of a solvent (fTHF) on the ordinate. Due to the low polymer mass fractions (≤10−1), a logarithmic scale was adopted for the abscissa. The mass fraction of the third component (water) can be determined by the difference (fwater = 1 − fsolute − fTHF). The ouzo zones of DSPE–mPEG2000 with or without AIE dye were constructed by varying the mass fraction of the solute (fsolute) from 4.5 × 10−7 to 2.7 × 10−3 and fTHF from 0.08 to 0.80 in the final mixture to plot a small region of the solute/THF/water ternary phase diagram containing the ouzo, non-ouzo and binodal regions. In the ouzo zone, supersaturation allows the formation of suspensions of small, stable nanoparticles. The ouzo zone of PLGA in combination with BTPEBT was constructed by varying the mass fraction of the solute (fsolute) from 9.0 × 10−5 to 4.5 × 10−3 and fTHF from 0.08 to 0.80 in the final mixture by exploiting the unique characteristics of AIE dyes, which show different fluorescence emissions in different aggregate states.Characterization of NPs
The sizes of the NPs were measured using (a) a dynamic light scattering (DLS) instrument (ZetaSizer Nano-ZS, Malvern Instruments, UK) and (b) field-emission transmission electron microscopy (FETEM). UV-vis absorption spectra were attained on a Shimadzu Model UV-1700 spectrometer. Photoluminescence12 spectra were measured on a Perkin- Elmer LS 55 spectrofluorometer. All UV and PL spectra were collected at 24 ± 1 °C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Research Foundation Investigatorship (R279-000-444-281 and R279-000-483-281) and the National University of Singapore (R279-000-482-133). The authors acknowledge helpful discussions regarding the molecular dynamics of ouzo and non-ouzo zones with Ramakrishna Krishnan and computational fluid dynamics (CFD) with Vishvak Kannan.Notes and references
- Z. Wang, B. Guo, E. Middha, Z. Huang, Q. Hu, Z. Fu and B. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 11167–11176 CrossRef CAS PubMed.
- B. Guo, E. Middha and B. Liu, ACS Nano, 2019, 13, 2675–2680 CrossRef CAS PubMed.
- W. Huang and C. Zhang, Biotechnol. J., 2018, 13, 1700203 CrossRef PubMed.
- Z. Wang, J. Huang, W. Huang, H. Yamamoto, S. Kawaguchi and M. Nagai, Colloid Polym. Sci., 2018, 297, 69–76 CrossRef.
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Nano Lett., 2008, 8, 2906–2912 CrossRef CAS PubMed.
- E. Lepeltier, C. Bourgaux and P. Couvreur, Adv. Drug Delivery Rev., 2014, 71, 86–97 CrossRef CAS PubMed.
- K. Miladi, S. Sfar, H. Fessi and A. Elaissari, in Polymer Nanoparticles for Nanomedicines: A Guide for their Design, Preparation and Development, ed. C. Vauthier and G. Ponchel, Springer International Publishing, Cham, 2016, ch. 2, pp. 17–53 DOI:10.1007/978-3-319-41421-8_2.
- C. J. Martínez Rivas, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari, S. A. Galindo Rodríguez, R. Á. Román, H. Fessi and A. Elaissari, Int. J. Pharm., 2017, 532, 66–81 CrossRef PubMed.
- S. Schubert, J. J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581–1588 RSC.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS.
- B. K. Johnson and R. K. Prud’homme, Phys. Rev. Lett., 2003, 91, 118302 CrossRef PubMed.
- J. Cheng, B. A. Teply, I. Sherifi, J. Sung, G. Luther, F. X. Gu, E. Levy-Nissenbaum, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Biomaterials, 2007, 28, 869–876 CrossRef CAS PubMed.
- G. Feng, C. Y. Tay, Q. X. Chui, R. Liu, N. Tomczak, J. Liu, B. Z. Tang, D. T. Leong and B. Liu, Biomaterials, 2014, 35, 8669–8677 CrossRef CAS PubMed.
- S. Pieper and K. Langer, Mater. Today: Proc., 2017, 4, S188–S192 Search PubMed.
- H. Deng, J. Liu, X. Zhao, Y. Zhang, J. Liu, S. Xu, L. Deng, A. Dong and J. Zhang, Biomacromolecules, 2014, 15, 4281–4292 CrossRef CAS PubMed.
- I. Barwal, A. Sood, M. Sharma, B. Singh and S. C. Yadav, Colloids Surf., B, 2013, 101, 510–516 CrossRef CAS PubMed.
- R. H. Fang, K. N. Chen, S. Aryal, C. M. Hu, K. Zhang and L. Zhang, Langmuir, 2012, 28, 13824–13829 CrossRef CAS PubMed.
- B. L. Banik, P. Fattahi and J. L. Brown, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 271–299 Search PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- J. Aubry, F. Ganachaud, J.-P. C. Addad and B. Cabane, Langmuir, 2009, 25, 1970–1979 CrossRef CAS PubMed.
- S. A. Vitale and J. L. Katz, Langmuir, 2003, 4105–4110, DOI:10.1021/la026842o.
- M. Beck-Broichsitter, J. Nicolas and P. Couvreur, Nanoscale, 2015, 7, 9215–9221 RSC.
- M. Beck-Broichsitter, Int. J. Pharm., 2016, 511, 262–266 CrossRef CAS PubMed.
- M. Lu, A. Ozcelik, C. L. Grigsby, Y. Zhao, F. Guo, K. W. Leong and T. J. Huang, Nano Today, 2016, 11, 778–792 CrossRef CAS PubMed.
- J.-M. Lim, A. Swami, L. M. Gilson, S. Chopra, S. Choi, J. Wu, R. Langer, R. Karnik and O. C. Farokhzad, ACS Nano, 2014, 8, 6056–6065 CrossRef CAS PubMed.
- Y. Zhang and A. R. Clapp, RSC Adv., 2014, 4, 48399–48410 RSC.
- C. Zhang, V. J. Pansare, R. K. Prud’homme and R. D. Priestley, Soft Matter, 2012, 8, 86–93 RSC.
- A. Albisa, E. Piacentini, V. Sebastian, M. Arruebo, J. Santamaria and L. Giorno, Pharm. Res., 2017, 34, 1296–1308 CrossRef CAS PubMed.
- Y. F. Wang, T. Zhang and X. J. Liang, Small, 2016, 12, 6451–6477 CrossRef CAS PubMed.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed.
- D. Ding, C. C. Goh, G. Feng, Z. Zhao, J. Liu, R. Liu, N. Tomczak, J. Geng, B. Z. Tang, L. G. Ng and B. Liu, Adv. Mater., 2013, 25, 6083–6088 CrossRef CAS PubMed.
- J. Ma, S. M. Lee, C. Yi and C. W. Li, Lab Chip, 2017, 17, 209–226 RSC.
- J. Xu, S. Zhang, A. Machado, S. Lecommandoux, O. Sandre, F. Gu and A. Colin, Sci. Rep., 2017, 7, 4794 CrossRef PubMed.
- X. Gu, E. Zhao, J. W. Lam, Q. Peng, Y. Xie, Y. Zhang, K. S. Wong, H. H. Sung, I. D. Williams and B. Z. Tang, Adv. Mater., 2015, 27, 7093–7100 CrossRef CAS PubMed.
- F. Lince, D. L. Marchisio and A. A. Barresi, J. Colloid Interface Sci., 2008, 322, 505–515 CrossRef CAS PubMed.
- G. Francois and J. L. Katz, ChemPhysChem, 2005, 6, 209–216 CrossRef PubMed.
- R. Botet, J. Phys.: Conf. Ser., 2012, 352, 012047 CrossRef.
- T. N. Zemb, M. Klossek, T. Lopian, J. Marcus, S. Schöettl, D. Horinek, S. F. Prevost, D. Touraud, O. Diat, S. Marčelja and W. Kunz, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4260–4265 CrossRef CAS PubMed.
- S. Shimizu and N. Matubayasi, Phys. Chem. Chem. Phys., 2017, 19, 26734–26742 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation of THF composition in mixture, Reynolds no in mixer, solubility interaction parameter and mass fraction of solute and solvent in final mixture; construction of ouzo diagram of DSPE–mPEG with and without AIE dye; effect of polymer to dye ratio on intensity size distribution; reproducibility of ESDM for the synthesis of NPs and drug loading and encapsulation efficiency. See DOI: 10.1039/c9qm00020h |
| This journal is © the Partner Organisations 2019 |