
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe roles of Lewis acidic additives in organotransition metal catalysis
Joseph
Becica
and
Graham E.
Dobereiner
 *
*
Department of Chemistry, Temple University, Philadelphia, Pennsylvania 19122, USA. E-mail: dob@temple.edu
First published on 22nd January 2019
Abstract
We describe recent examples of prominent reactions in organic synthesis that involve transition metal and Lewis acid cocatalysts. Introducing Lewis acid additives to transition metal catalysis can enable new reactivity or improve activity and selectivity of an existing process. Several studies are highlighted to illustrate the possible roles of Lewis acids in catalytic mechanisms. The uses of Lewis acid additives in bond-breaking catalysis (C–C activation, C–H activation, and hydrogenolysis reactions) and bond-forming catalysis (Au-catalysed alkyne functionalisation, Pd-catalysed C–C and C–N bond formation) are reviewed.
 Joseph Becica | Joseph was born in Camden, New Jersey in 1990. He is currently pursuing a PhD in Inorganic Chemistry at Temple University (Philadelphia, USA) under the direction of Prof. Graham E. Dobereiner and is a visiting researcher at GlaxoSmithKline (Upper Providence, USA). Previously, he studied organometallic chemistry in the laboratories of Nathan M. West at University of the Sciences (Philadelphia, USA) and Prof. Ola F. Wendt at Lund University (Lund, Sweden). His current research interests include organometallic synthesis and homogeneous catalysis. |
 Graham E. Dobereiner | Graham received his B.S. from Brandeis University working with Jin-Quan Yu and completed his Ph.D. at Yale University with Robert H. Crabtree and Scott J. Miller. After a postdoctoral appointment with Richard R. Schrock (MIT) he began his independent career at Temple University in 2014. His research group investigates how weak or transient interactions influence catalysis and chemistry of the organotransition metals. |
1. Introduction
In pursuit of more efficient organotransition metal catalysis, many chemists have boosted activity and/or selectivity using Lewis acid additives.1 Significant recent contributions have advanced the mechanistic understanding of transition metal-Lewis acid synergy, and therefore it is timely to provide a concise review of highlights of this area. Out of a broad array of examples in which Lewis acidic additives are used in organometallic catalysis, we present here four classes of Lewis acid effects in catalytic reactions, chosen to highlight a few important ways in which Lewis acids may influence organometallic reactions. This Review is intended as a guide for practitioners who seek to improve catalytic reactions by the addition of additives and to understand the role of additives from a mechanistic perspective. We focus on mechanistic effects of Lewis acids throughout catalytic cycles, including alkylaluminums, alkyl and arylboranes, alkylzincs, and metal halides and triflates (trifluoromethanesulfonate, or OTf) of various cations. These classes of Lewis acids play a role in the cooperative activation of strong bonds, such as in the oxidative addition of C–C and C–H bonds to transition metals, or the hydrogenolysis of C–O and C–N bonds. Lewis acids can also facilitate bond-forming reactions, such as Au-catalysed reactions of alkynes (and related reactions), as well as Pd-catalysed C–C and C–N bond forming reactions.Lewis acids can play myriad roles within a given transition metal-catalysed reaction (Fig. 1). Some examples in this Review will involve tandem catalysis,2 in which two discrete catalytic cycles occur simultaneously in a single reaction mixture, where M1 is a transition metal catalyst and M2 is an acid catalyst. More complex mechanisms see the involvement of a Lewis acid cocatalyst (LA) upon a single catalytic cycle mediated by a transition metal catalyst (M). In a simplified mechanistic model, the Lewis acid can affect the overall cycle by forming Lewis acid–base adducts with different components of the catalytic reaction.
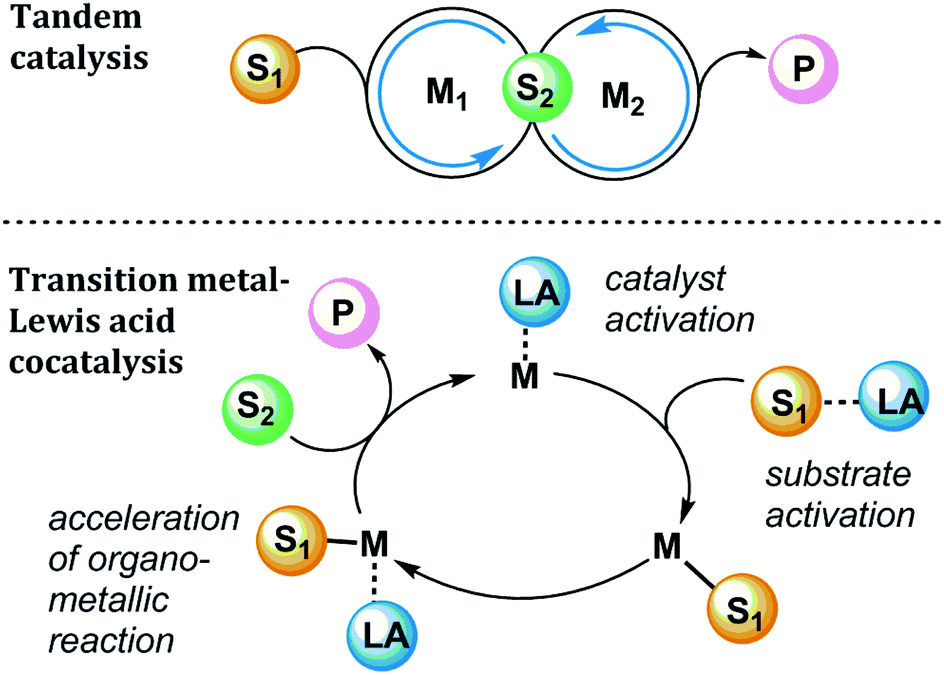 | ||
| Fig. 1 Generalized mechanism for tandem catalysis (top) and transition metal and Lewis acid cocatalysis (bottom). S1, S2 = substrates; M = metal complex; LA = Lewis Acid; P = product. | ||
For example, cooperative substrate-Lewis acid interactions (S1-LA) or catalyst-Lewis acid interactions (M-LA) can promote the turnover-limiting step of catalysis or direct the reaction pathway in a selectivity-determining step. In this Review, we will encounter examples of reactions in which the Lewis acid is revealed to take several possible roles, such as activating substrate toward a transition metal centre, accelerating a key elementary organometallic step (i.e., migratory insertion, transmetalation, or reductive elimination), affecting the catalyst activation or deactivation pathways, or promoting the controlled release of a Brønsted acid in situ. Here we highlight recent examples that elucidate the numerous possible sources of Lewis acid effects in organometallic catalysis and identify the modes of action of the Lewis acid in different reaction contexts.
One challenge is understanding why a particular Lewis acid is more effective than another in a given reaction. Electrophilic species can be found across the periodic table, and the methods to systematically categorize Lewis acids or measure Lewis acidity are limited.3 In select cases, the behaviour of the Lewis acids can be differentiated. For example, Organ and coworkers4 suggest that the different susceptibility of boron Lewis acids to autoxidation can result in different reaction outcomes in Pd and boron-catalysed C–N bond forming reactions. Another example is provided by Marks and coworkers,5 who show that the capacity of Pd and metal triflate cocatalysts to affect the hydrogenolysis of ethers is dependent on the effective charge density of the metal triflate cation employed.
In each section, we highlight synthetic methods enabled by cocatalytic systems, as well as insights into potential modes of cooperativity. There are several methods for rationalising Lewis acid effects observed in catalytic reactions. Density functional theory calculations enable the assessment of the role of Lewis acid–base adducts of substrates and key catalyst intermediates on catalytic reaction pathways, as well as parametrisation of Lewis acids. Experimental insight into catalyst-Lewis acid cooperativity can be ascertained through the isolation of catalytic intermediates or model complexes thereof and assessing their reactivity towards Brønsted or Lewis acids. Lastly, monitoring of reaction rates can confirm an empirical Lewis acid effect and provide insight into possible reaction pathways. Here we piece together these clues to unveil numerous modes of catalyst-Lewis acid interactions in productive catalysis.
2. Cooperative catalyst systems for C–C and C–H bond activation
A representative example of transition metal/Lewis acid cooperative bond activation is DuPont's Adiponitrile Process (Scheme 1),6 which is thought to proceed via oxidative addition of HCN, Ni–H insertion of diene, and reversible elimination to generate alkylnitrile. In the presence of a Lewis acid cocatalyst (typically, BPh3), the Ni/phosphine-catalysed hydrocyanation of 1,3-butadiene (1) shows increased rate and selectivity for the desired linear adiponitrile (2) versus branched isomers. Since Lewis acids can promote the oxidative addition of a C–CN bond to Ni(0) – i.e., the microscopic reverse of alkylnitrile-forming reductive elimination – branched nitriles can be thus re-engaged by the catalyst, and again eliminated to form linear 2. Jones7,8 revealed that a cooperative Ni/LA system reacts preferentially with C–CN bonds over activated C–H bonds, e.g. in allylcyanide. Together, these early works in cooperative bond activation are the basis for a wide variety of synthetic methods in C–C and C–H bond activation discussed in this section.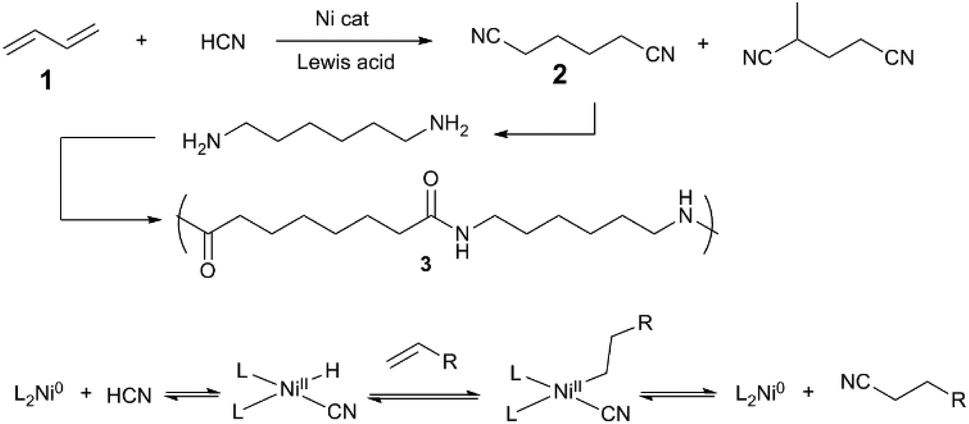 | ||
| Scheme 1 Hydrocyanation of butadiene en route to 6,6-nylon (3)6 (top) and the mechanism of Ni-catalysed alkene hydrocyanation (bottom). | ||
2.1 Synthetic methods that apply Ni and Lewis acid cocatalysts
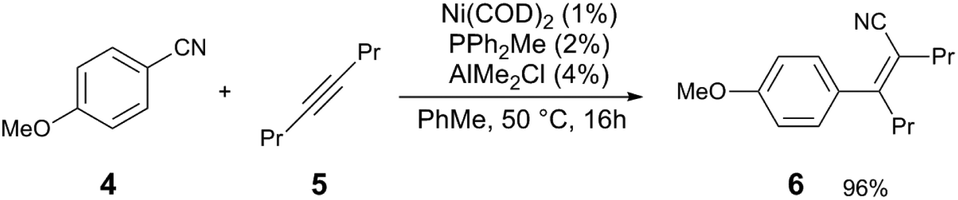 | ||
| Scheme 2 Ni and Al-catalysed carbocyanation of alkynes with benzonitriles.11 | ||
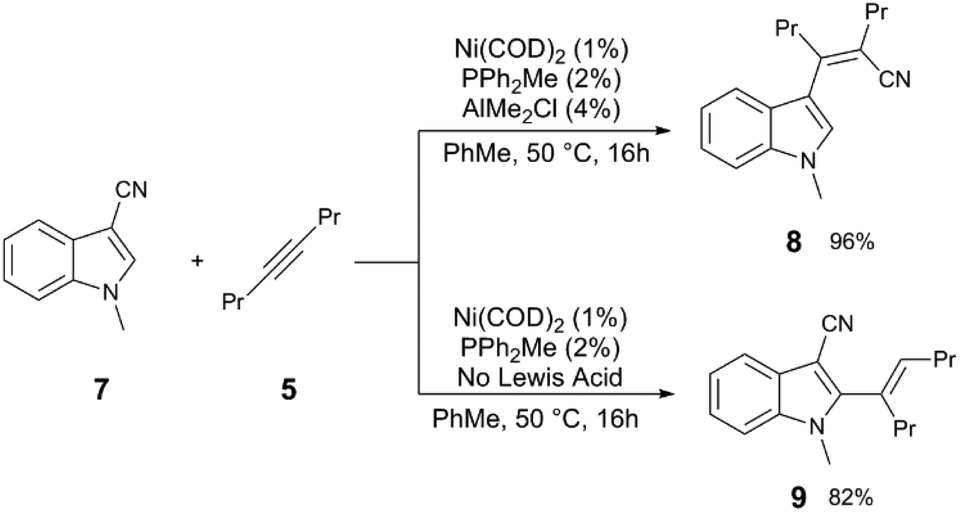 | ||
| Scheme 3 Divergent reactivity in the Ni and Al-catalysed carbocyanation of alkynes with 3-cyanoindoles.11 | ||
Other nitriles, such as allylnitriles,12 benzylnitriles,13 and alkynylnitriles14 can be employed in similar catalytic scaffolds. Intramolecular versions in which alkenes are used as the coupling partner also have been reported,15,16 including variants which include Pd catalysts.17–20 The reaction of cyanoester 10, a more activated electrophile, with 5 to give alkenylnitrile 11 uses an electron-poor phosphine PArF3 (ArF = 3,5-C6H3(CF3)2) in combination with Ni(COD)2 and B(C6F5)3 as the Lewis acid cocatalyst (Scheme 4).21 Other C–C bonds can be activated such as in anhydrides,22 cyclopropanes,23 and esters.24 In the latter example, the reaction of arylnitrile 12 with 2-butyne (13) and catalytic Ni(COD)2/P(CH2Ph)3/MAD (MAD = methylaluminum bis(2,6-di-t-butyl-4-methylphenoxide)) results in sequential C–C bond activation events resulting in formation of coumarin 14 and elimination of arylnitrile 15.
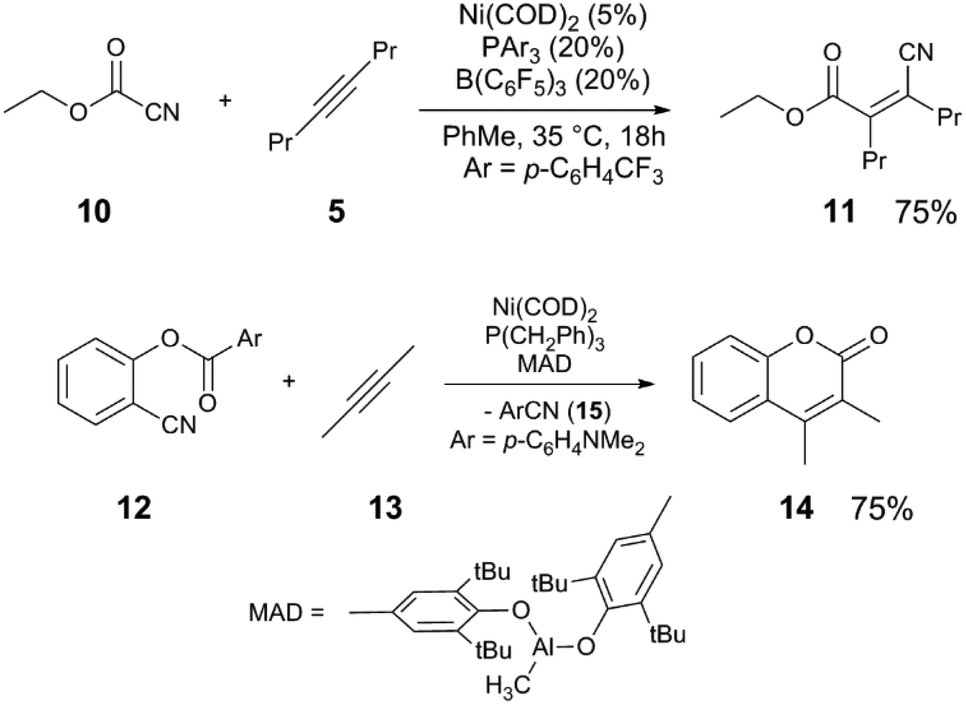 | ||
| Scheme 4 Alkynylation of cyanoesters21 (top) and sequential C–CN and C–CO bond activation to form coumarins24 (bottom). | ||
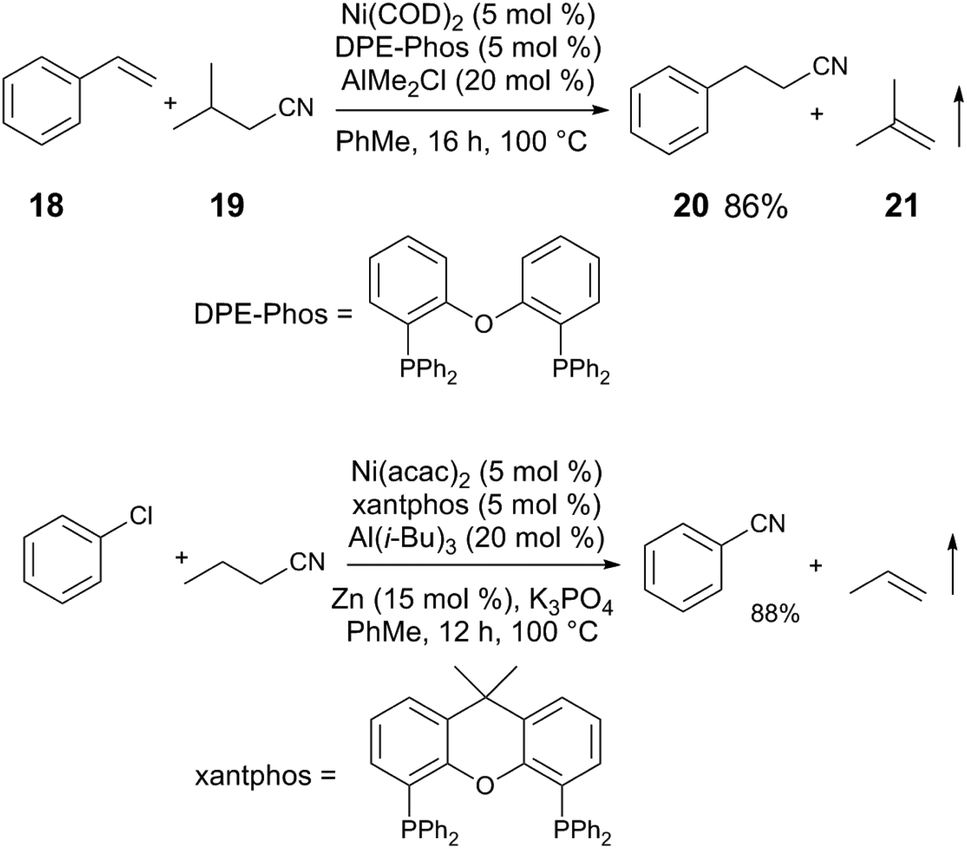
The activation of C(sp3)–CN bonds in unactivated aliphatic nitriles is more challenging, due to lower C–CN reactivity and competing β-hydride elimination of alkylnickel intermediates. These challenges can be overcome using a directing group on the nitrile to kinetically stabilize the resulting alkylnickel intermediate, and/or using a sterically-demanding ligand to reduce the rate of β-H elimination.25 The pyrrolidine-substituted nitrile 16 and 5 react to give alkenylnitrile 17 in 86% yield using a Ni(COD)2/SPhos/AlMe3 catalyst mixture (Scheme 5). A clever manifestation of cooperative C(sp3)–CN bond activation is realized by the Morandi group, in which alkylnitriles are used as HCN group transfer reagents (Scheme 6),26,27 utilizing the reversibility of the steps in the general alkene hydrocyanation mechanism to drive formation of the desired product without resorting to the use of hazardous HCN. For example, styrene (18) and isobutyrylnitrile (19) are converted to alkylnitrile 20 and isobutylene (21), using Ni(COD)2/DPE-Phos/AlMe2Cl; isobutyrynitrile is selected as HCN transfer agent due to removal of gaseous isobutylene, driving selective formation of the desired product. A subsequent publication from the Morandi group uses a similar method to synthesize arylnitriles.28
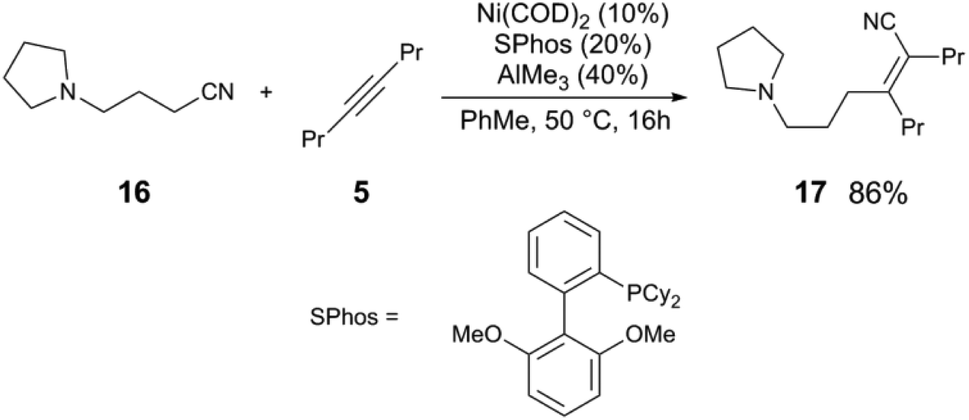 | ||
| Scheme 5 Activation of alkylnitriles using a directing group.24 | ||
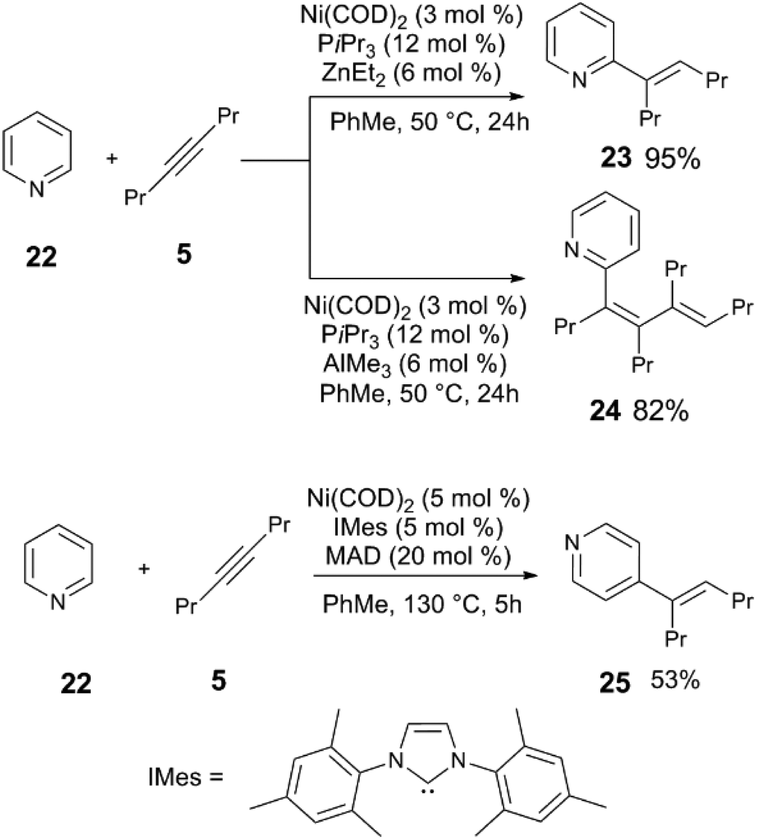 | ||
| Scheme 7 Divergent reactivity in the C–H alkynylation of heterocycles.29,30 | ||
2.2. The role of the Lewis acid in cyanofunctionalisation and C–H functionalisation
In these reactions, the Lewis acid influences selectivity of C–C and C–H bond activation.37–41 Sakaki and coworkers37 investigated the mechanism of the aforementioned dual C–C cleavage reaction of 12 to form the coumarin 14 by computation. An abridged mechanism determined by DFT calculations is depicted in Fig. 2 to reflect key elementary steps of the reaction; the mechanism proceeds by oxidative addition of the arylcyanide to Ni(0), followed insertion of the alkyne into an arylnickel bond, followed by C–C bond formation and β-aryl elimination, yielding 14 and benzonitrile byproduct 15. The calculations show that binding of the Lewis acid (which has been simplified from MAD to Al(OMe)3) at both the cyano and ester groups of the substrate are essential to achieving the two C–C cleavage steps of the reaction. In the presence of two equivalents of Lewis acid, both N-bound and O-bound, the transition state energies for both C–C bond cleavage events are significant reduced, and moderate activation barriers are found: for C–CN activation, the ΔG°‡ is +14.4 kcal mol−1, and for C–CO activation the ΔG°‡ is +17.3 kcal mol−1, whereas in the absence of any Lewis acid, the barriers are ΔG°‡ = +44.8 kcal mol−1 and ΔG°‡ = +23.8 kcal mol−1, respectively. The significant reduction in barrier of C–CN cleavage is attributed to charge transfer from the Ni 3dπ to the C–CN σ* + π* molecular orbital and increasing population of the LUMO of antibonding character is crucial to C–CN bond cleavage. The computations support the experimental result that no conversion of 12 is observed in the absence of Lewis acid.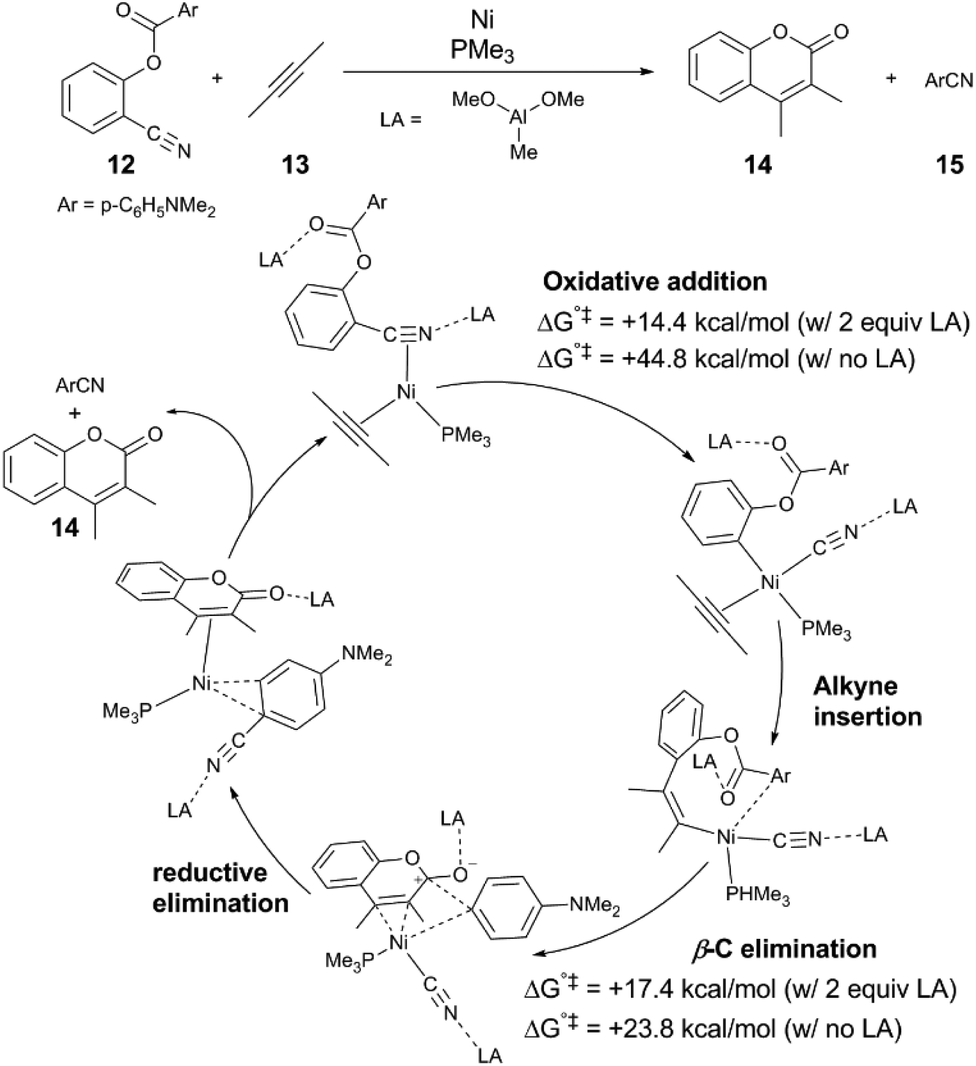 | ||
| Fig. 2 A proposed mechanism of Ni and Lewis acid-cocatalysed C–CN and C–CO bond activation informed by DFT calculations.37 For the purpose of clarity, isomerization and ligand exchange steps are omitted from the figure. | ||
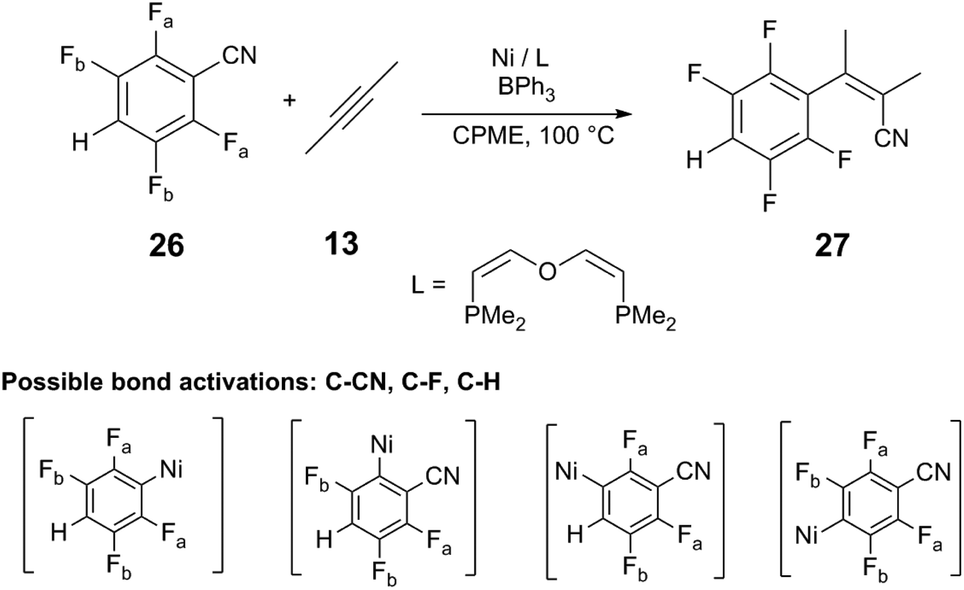
Another computational study performed by Guan and coworkers38 provides further insight into the regioselectivity observed by Ni/LA catalyst systems. The reaction of fluorinated benzonitrile 26 with 2-butyne (13) is investigated using a simplified Ni/DPE-Phos catalyst with BPh3 cocatalyst. The selectivity of rate-limiting C–CN bond cleavage to yield the alkenylnitrile 27 is evaluated in comparison to other competitive bond cleavages (C–H, C–F) in both the presence and absence of BPh3. In the absence of BPh3, the energy barriers of C–CN and C–H bond cleavage are similar (ΔG°‡ = +22.0 kcal mol−1 and ΔG°‡ = +21.2 kcal mol−1, respectively; Table 1). Upon binding of BPh3 to 26, the barrier of C–CN cleavage is reduced to +17.4 kcal mol−1, whereas the barrier of C–H cleavage increases to +28.8 kcal mol−1, confirming that the Lewis acid can increase the kinetic preference for C–CN cleavage versus other reactive bonds. The BDEs of the C–H and C–F bonds in the 26:BPh3 Lewis acid–base adduct are determined to be similar in free 26; however, the BDE of the C–CN bond is significantly reduced (ΔH = +136.2 kcal mol−1, free 26; ΔH = +112.7 kcal mol−126:BPh3) upon adduct formation. The results affirm findings in other studies,39 where Lewis acids reduce activation barriers of oxidative addition of cyanoester and alkylnitrile to Ni(0). Lewis acids can also reduce the barrier to migratory insertion of the alkyne into Ni–C bonds.
|
|
||||
|---|---|---|---|---|
| Bond activation | ΔG°‡ (kcal mol−1) | ΔG°‡ w/BPh3 (kcal mol−1) | BDE (kcal mol−1) | BDE w/BPh3 (kcal mol−1) |
| C–CN | +22.0 | +17.4 | +136.2 | +112.7 |
| C–Fa | +27.8 | +33.1 | +125.8 | +125.8 |
| C–Fb | +32.8 | +43.0 | +125.2 | +125.0 |
| C–H | +21.2 | +28.8 | +122.5 | +122.8 |
To summarize, advances in C–C and C–H bond activation have been enabled by cooperative Ni and Lewis acid catalysis, and numerous novel synthetic methods have resulted. A survey of examples indicates dependence of the electron-donating capacity of the ancillary ligand and the electron-withdrawing capacity of the Lewis acid in these catalytic systems strongly dictates the catalyst activity and selectivity. To generalize, the most obvious role of the Lewis acid in this context is to effect substrate activation, though effects of chemo- and regioselectivity are also identified. In the following sections, the origins of catalyst-Lewis acid cooperativity will be more ambiguous.
3. Hydrogenation of carboxylic acids, esters, amides, and ethers
We now turn our attention to the hydrogenation of C–O and C–N bonds, where Lewis acid additives influence activity and selectivity chiefly by acting as source of Brønsted acid. To contextualise the role of Lewis acids as potential sources of Brønsted acids, our discussion begins by exploring the influence of acid additives upon Klankermeyer and Leitner's levulinic acid (28) hydrogenation,42,43 catalysed by a Ru(acac)3/1,1,1-Tris(diphenylphosphinomethyl)ethane (triphos) catalyst (Scheme 8). This example represents an advance in hydrogenation of challenging carboxylic acid derivative substrates through the use of a multifunctional catalyst system. The reaction sequence for the conversion of 28 to γ-valerolactone (29), 1,4-pentanediol (30), and 2-methyltetrahydrofuran (MeTHF, 31) can be formulated using distinct Ru-catalysed hydrogenation steps and acid-catalysed dehydration steps. The overall performance of the reaction (i.e., activity and selectivity) is highly dependent on both the Ru catalyst (ligand, and counteranion) and pKa of the reaction medium. Mechanistic evidence also points to acidic additives influencing catalyst activation and deactivation modes during Ru-mediated reaction steps.43,44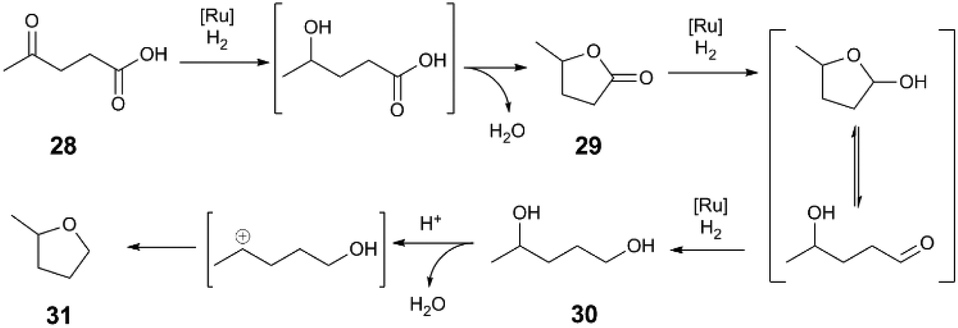 | ||
| Scheme 8 Conversion of levulinic acid to 2-methyltetrahydrofuran.42 | ||
3.1. Ru-Catalysed hydrogenation and Brønsted acid cocatalysis
By modifying Klankermeyer and Leitner's catalyst and conditions, the activity and selectivity of the reaction can be dramatically affected.42 Ru(acac)3, tri(octyl)phosphine, and NH4PF6 selectively converts 28 to γ-valerolactone (29) in >99% yield (Scheme 9), without further reduction of the lactone. In contrast, the use of Ru(acac)3, triphos, along with a mixed acid system (using a sulfonic acid (aIL) and NH4PF6) provides excellent selectivity for the complete reduction of 28 to MeTHF (31) in 92% yield.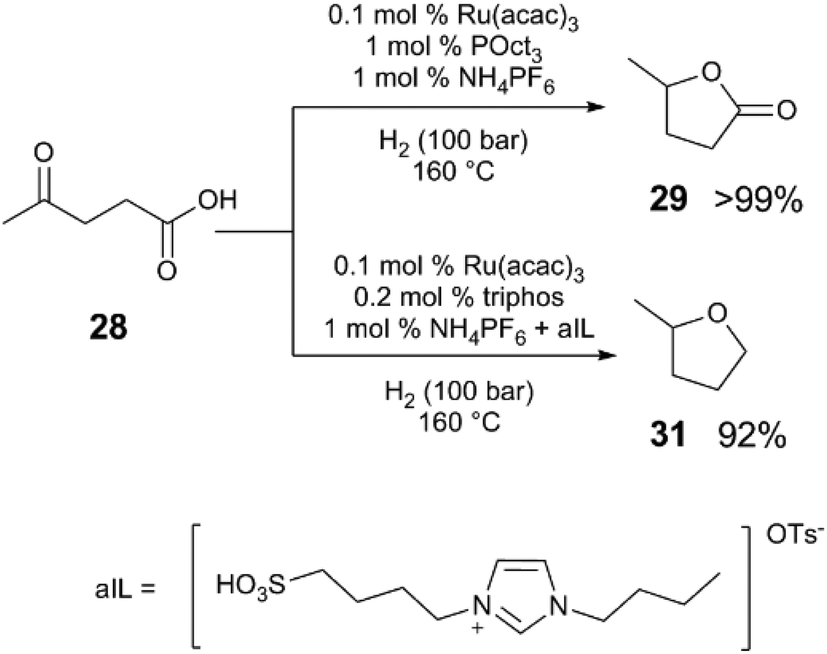 | ||
| Scheme 9 Acid-dependent selectivity in the hydrogenative cyclisation of levulinic acid.42 | ||
In the latter reaction, an off-cycle catalyst resting state (triphos)Ru(CO)(H)2 (32) was isolated from the reaction mixture.43 The carbonyl ligand is most likely derived from decarbonylation of aldehyde formed in situ. As precatalyst, 32 has lower catalytic activity than the combination of Ru(acac)3 and triphos, but can be reactivated by adding substrate and/or acid (Fig. 3). The ability of an acid to restore activity is dependent on pKa, conjugate base identity, and Ru/acid ratio. Acid could react with neutral [Ru(H)2] to form the non-classical hydride [Ru(H2)H+] (33). Subsequent dihydrogen dissociation could then open up a site for substrate binding.
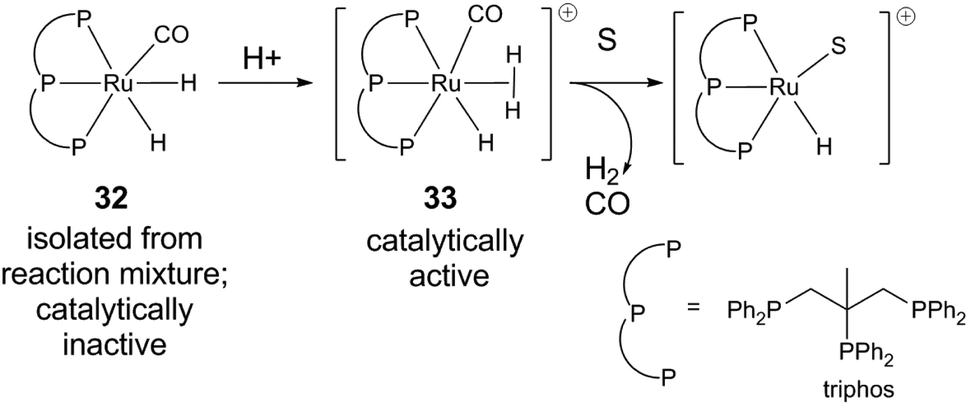 | ||
| Fig. 3 Catalyst resting state in levulinic acid hydrogenation.43 | ||
In a related amide hydrogenation, activity of (triphos)Ru(OAc)(H) (34), also depends on the Ru/acid ratio.44 Stoichiometric reactions of 34 and methanesulfonic acid (MsOH) form a mixture of species depending on equivalents of acid added (Fig. 4), suggesting acid influences catalyst activity by controlling speciation of Ru intermediates. The acidity of the reaction medium influences both metal-mediated and acid-promoted steps in these reaction sequences.
 | ||
| Fig. 4 Reactivity of a Ru(triphos) complex with methanesulfonic acid.44 | ||
3.2. Hydrogenation reactions enabled by Lewis acid cocatalysis
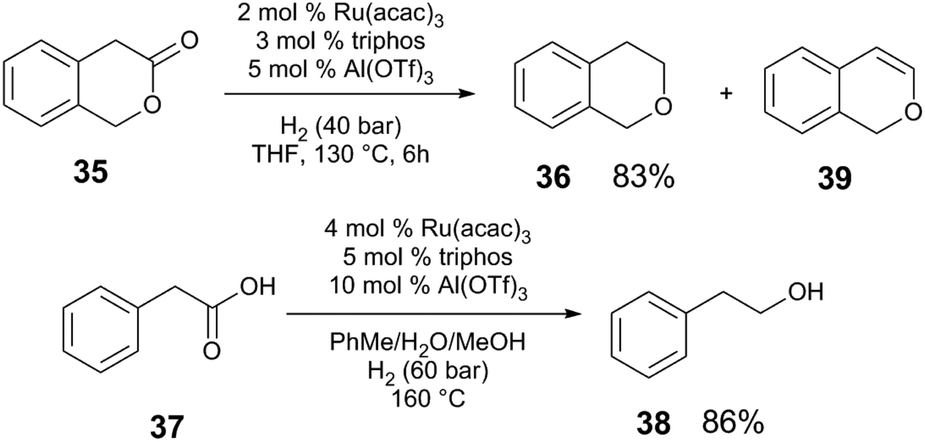
Under lower pressures of H2, metal triflate cocatalysts greatly outperform Brønsted acids. The hydrogenation of 37 (Scheme 10) does not proceed in the absence of cocatalyst, or with Brønsted acids (MsOH, diphenylphosphate), whereas when Al(OTf)3 is employed, >99% conversion of 35 is observed with 83% yield of the desired ether product 36. Although triflic acid (TfOH) also promotes conversion of 35, poor selectivity of the reaction is observed; a competing elimination from the intermediate acetal provides the enol ether 39 as the major product in 45% yield. Al(OTf)3 provides much better performance than other Al compounds. Qualitative mechanistic experiments suggest that an off-cycle catalyst resting state is reactivated in the presence of relatively acidic protons, resembling the reactivity of Ru and Brønsted acid cocatalytic systems (section 3.1). The presence of H2O is important for efficient catalysis; the reaction is suppressed when molecular sieves are added, or when non-polar solvents are used. The authors propose that Al(OTf)3 is important to generate Brønsted acid in situ upon combination with H2O or alcohols. Like the system reported by Klankermeyer and Leitner, the acid could act to generate the active cationic [RuH+] species. Additionally, the weakly-coordinating OTf counteranion of the M(OTf)3 Lewis acid may play an important role in the reaction, as chloride anions inhibit catalysis.
The Ru(acac)3/triphos/M(OTf)3 system also mediates the hydrogenation of secondary amides to amines (Scheme 11) at low pressures of H2 (5–15 bar).47 A screen of acidic additives reveals that in the hydrogenation of amide 40, Yb(OTf)3·H2O cocatalyst provides the highest yield of amine 41 and that again, Lewis acidic metal triflate salts outperform Brønsted acidic cocatalysts under these reaction conditions. The reaction proceeds by initial hydrogenolysis of the amide to benzyl alcohol 42 and aniline 43, which are observable intermediates. A subsequent condensation reaction between 42 and 43 yields the benzylamine 41. Control experiments show that both the individual Ru and Yb catalysts are necessary for the condensation reaction to occur, and that H2, while not necessary for condensation, improves the efficiency of this step.
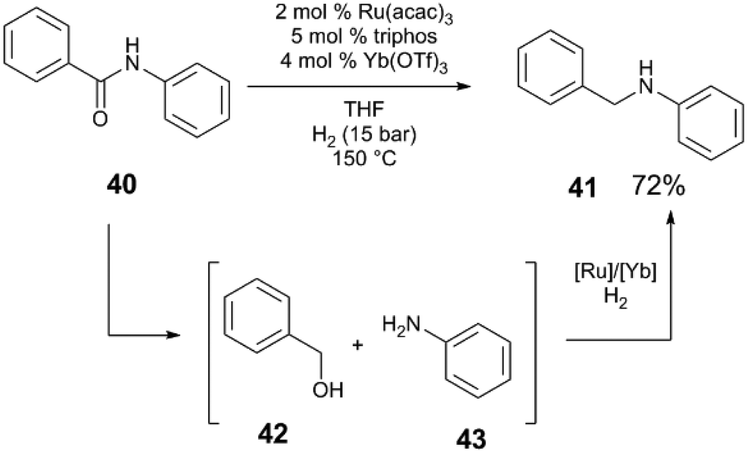 | ||
| Scheme 11 Hydrogenation of secondary amides.47 | ||
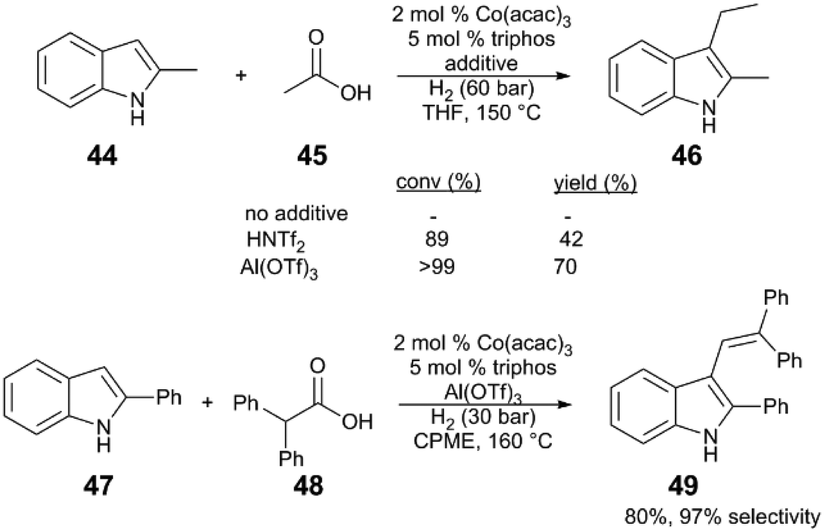 | ||
| Scheme 12 Alkylation of indoles via carboxylic acid hydrogenation.48 | ||
A plausible reaction sequence (Fig. 5) starts with Co/Al(OTf)3-catalysed hydrogenation of acetic acid (45) to the respective acetaldehyde (50), followed by nucleophilic attack of the indole 44 to 51. Acid-catalysed dehydration of the resulting functionalized indole intermediate 51 generates alkenylated indole 52; subsequent Co and Al(OTf)3-catalysed hydrogenation of the alkene yields the desired alkylindole product 46. Evidence for an alkenylindole intermediate comes from the reaction of α,α-disubstituted carboxylic acid (48). Here the major product is alkenylindole 49, probably because the trisubstituted alkene is unreactive reactive towards successive hydrogenation.
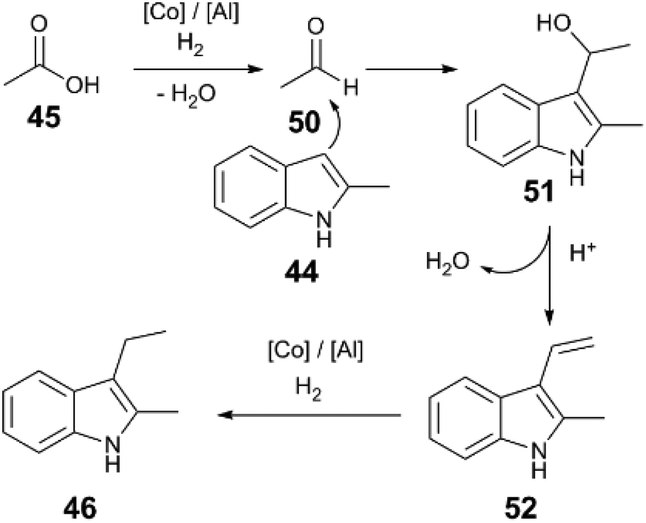 | ||
| Fig. 5 Proposed mechanism of acetic acid hydrogenation and indole alkylation.48 | ||
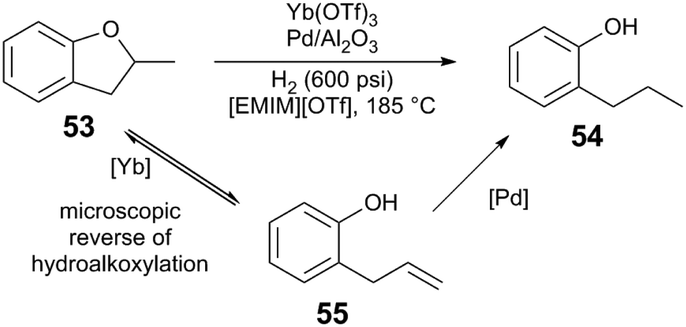 | ||
| Scheme 13 Hydrogenolysis of ethers.49 | ||
The competency for metal triflates to mediate the key acid-catalyzed C–O bond cleavage step indicates the transition metal Lewis acids are particularly suitable for ether cleavage (Table 2). In the absence of H2 pressure, the conversion of 1,8-cineole (56) is greatest when reacted with 0.5 mol% Zr(OTf)4 or Hf(OTf)4 and Pd/C (which has been pre-treated with H2).51 Computationally-determined (B3LYP) effective charge density (ρ) of metal triflate Lewis acids tracks with catalytic performance, as determined by conversion and turnover numbers. DFT exploration of the reaction's potential energy surface indicates that C–O bond cleavage of 2-methyltetrahydropyran via Yb(OTf)3 has a higher activation barrier (ΔG‡ = +32.4 kcal mol−1), than when Hf(OTf)4 is used (ΔG‡ = +16.8 kcal mol−1) – in agreement with experimental observation. The authors propose that the Lewis acid may bind and increase acidity of intermediate alcohols, potentially mitigating side reactions, such as carbocation rearrangement.
|
|
|||
|---|---|---|---|
| Acid | Time (min) | Conv. (%) | ρ (eff. charge density) |
| — | 90 | — | — |
| La(OTf)3 | 30 | 0.2 | 2.60 |
| Yb(OTf)3 | 30 | 0.2 | 2.81 |
| Ce(OTf)3 | 10 | 5.7 | 3.44 |
| Sc(OTf)3 | 10 | 12.4 | 3.23 |
| Fe(OTf)3 | 10 | 13.6 | 3.71 |
| Al(OTf)3 | 10 | 23.7 | 3.87 |
| Zr(OTf)4 | 10 | 33.5 | 4.29 |
| Hf(OTf)4 | 10 | 46.0 | 4.37 |
| HOTf | 10 | 10.4 | — |
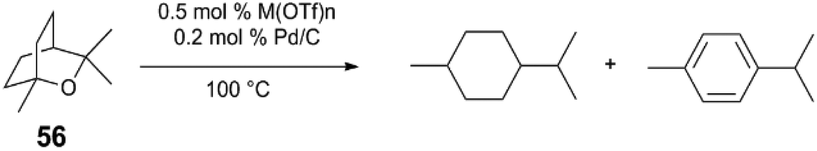
Metal triflate Lewis acids are competent cocatalysts to mediate challenging, multi-step hydrogenation reaction sequences catalysed by a variety of different transition metals. In some cases, it is evident that a Lewis acid can promote the formation of a Brønsted acid in situ, which may be important to catalyse dehydration steps or other acid-catalysed reactions. It is also evident that Brønsted acids activate catalytic intermediates. As is demonstrated by Pd and metal triflate mediated ether hydrogenolysis, the coupling of two catalyst systems can “leverage” demanding endothermic reactions.
4. Tandem Au(I) catalysis: Lewis acids as Brønsted acids, catalyst activators and reactivators
The ability of Lewis and Brønsted acids to form highly-active cationic Au(I) catalysts was noted in the seminal 1998 report by Teles53 on Au-mediated alkyne hydroalkoxylation, a reaction of immense subsequent interest.54 Au halide catalyst precursors are typically combined with Ag salts, which are thought to generate catalytically-active cationic Au species. Although the non-innocent role of Ag in such reactions is recognized,55 less attention has been afforded to the role of Brønsted or Lewis acids in Au-catalysed reactions.56 In similar fashion to the analogy of Ru/Brønsted acid and Ru/Lewis acid cocatalytic systems (sections 3.1 and 3.2), it is useful to consider the potential role of Lewis acids to effect the generation of Brønsted acid in situ within the context of Au catalysis (section 4.2). We note that although several Au/Brønsted acid catalysed reactions are known,57–61 we present evidence that the action of Lewis acids in some cocatalytic systems are distinct from that of Brønsted acids. Here we summarize recent efforts to understand the mechanism of such cooperative catalytic systems.4.1. Au and Lewis acid tandem catalysis
First, examples are discussed in which Au and the Lewis acid act synergistically in tandem catalysis. To the best of our knowledge, the first evidence of metal triflate Lewis acids to serve as a catalyst activator in Au-catalysed reactions is the report of Shi,62 in which (PMe3)AuCl and Yb(OTf)3 catalyse the rearrangement of epoxy alkynes to carbocycles (Scheme 14). In addition to Yb(OTf)3 acting as an apparent halide abstractor in this reaction, the catalyst is more active when Yb(OTf)3 is employed rather than when AgOTf is employed. The first step of the reaction involves the ring-opening of epoxy alkyne 57 to the enol alkyne 58, which is catalysed independently by Yb(OTf)3 without activation of the alkyne, and the Au-catalysed rearrangement of 57 to the carbocycle 59 occurs in much greater yield with Yb(OTf)3. The combination of (PMe3)AuCl and Yb(OTf)3 generates (PMe3)Au(OTf) in situ – presumably the active form of the catalyst. The authors claim that the mixture of (PMe3)AuCl and Yb(OTf)3 provides improved catalytic activity than when (PMe3)AuCl and AgOTf are used, as well as when pre-formed (PMe3)Au(OTf) is used under the standard reaction conditions.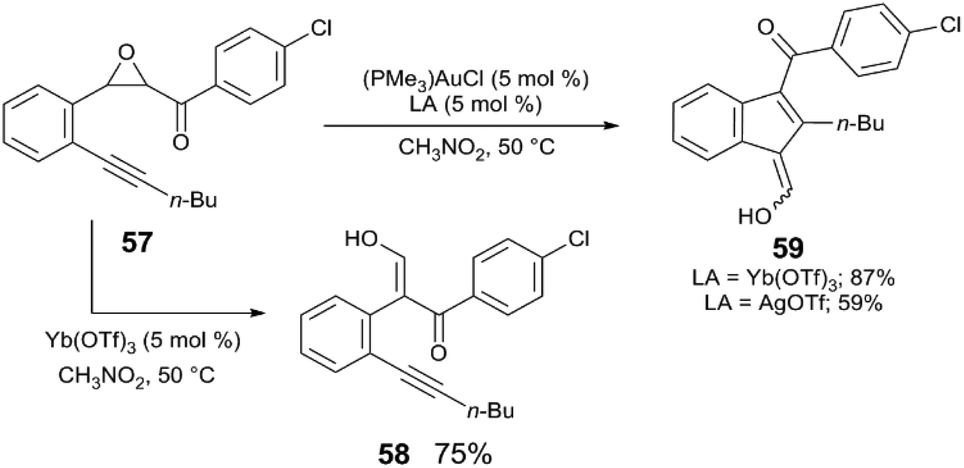 | ||
| Scheme 14 Tandem epoxide ring-opening and cyclisation.62 | ||
Several other synthetic applications have more recently emerged, particularly in cascade reactions where Lewis acids promote steps not catalysed by Au. The groups of Jia and Xu report several synergistic Au and Lewis acid reactions in the synthesis of spirocyclic63–65 and fused bicyclic compounds.66,67 For example, bicyclic aminal 60 is synthesized via a Au-catalysed intramolecular alkyne hydroamination followed by an Ga(OTf)3-catalysed inverse-electron-demand hetero-Diels–Alder cyclisation (Scheme 15).66 Analogous Au and Lewis acid tandem reactions are reported to selectively favour spiroaminals63 and spiro-heterocycles64 over bicyclic cyclisation patterns. Spiroketals65 and fused tricyclic compounds67 are also accessible from similar alkyne hydrofunctionalisation/cycloaddition sequences, where the Lewis acid also acts to generate ortho-quinonemethides from 2-(hydromethyl)phenol derivatives. Au and Lewis acid-catalysed alkyne hydroamination has also been applied to the synthesis of N-heterocycles such as a pyrroles68 and indoles.69
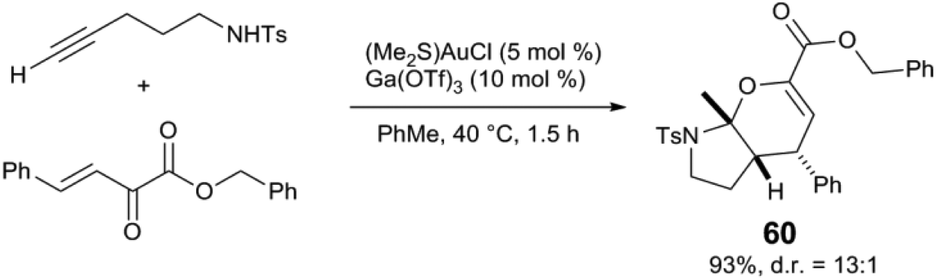 | ||
| Scheme 15 Tandem intramolecular alkyne hydroamination and inverse electron demand Diels–Alder reaction.66 | ||
Lewis acids can also affect Au-mediated steps of the catalytic reaction. In a Au/Ga-cocatalysed intermolecular Nakamura reaction, nucleophilic attack of 1,3-diketone 61 upon phenylacetylene (62) yields the alkenylated 1,3-diketone 63 (Scheme 16).70 The authors find that (XPhos)Au(TA)(OTf) (TA = 1H-benzo[1,2,3]triazole) is only a competent precatalyst in the presence of Ga(OTf)3; other Lewis acids, such as Zn(OTf)2, or other cocatalysts such as AgOTf or HOTf, do not cocatalyse the reaction. The authors propose that Ga(OTf)3 may bind, and therefore, activate the 1,3-dicarbonyl substrate, but may also be critical for the formation of catalytic Brønsted acid.
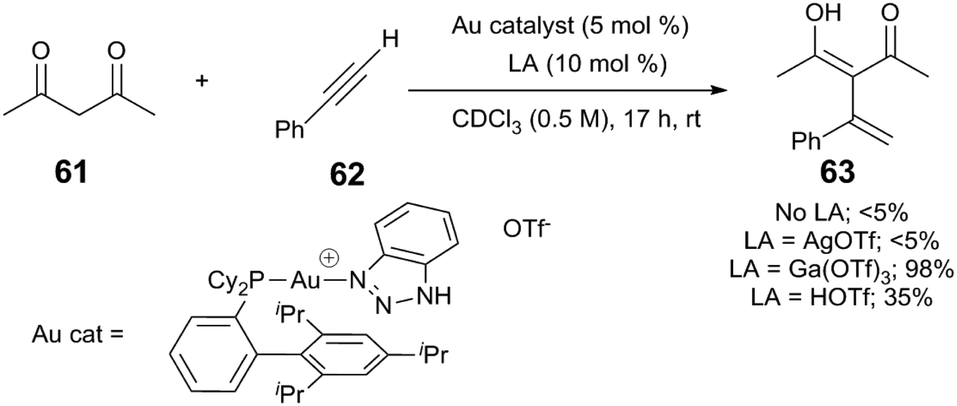 | ||
| Scheme 16 Alkylation of phenylacetylenes.70 | ||
4.2. Distinguishing between Ag, Brønsted and Lewis acid effects in Au catalysis
Hammond and Xu have comprehensively investigated Brønsted and Lewis acid effects in Au-catalysed reactions.71–73 In one study, the effect of cocatalytic additives on Au-catalysed alkyne and allene hydrofunctionalisation reactions was explored.71 Au catalysis at extremely low catalyst loadings (0.02–0.5 mol%) are strongly influenced by impurities such as halides anions, or basic anions (OH− or CO32−). The authors explore a series of reactions catalysed under base- and halide-free conditions using precatalysts (JohnPhos)AuOTf (JohnPhos = (2-biphenyl)di-tert-butylphosphine) and (PPh3)AuOTf. In a low-catalyst loading hydration of alkyne 64 (Fig. 6), no catalytic activity is observed in the reaction without co-catalyst; in the reaction with cocatalytic (0.6 mol%) Ga(OTf)3 or HOTf, the reaction proceeds to high conversion in 12 hours. Ga(OTf)3 may act as a source of Brønsted acid (HOTf) in this case. But in other reactions, such as the cycloisomerisation of enyne 65 (Fig. 7), cocatalytic HOTf is ineffective, whereas cycloisomerisation proceeds to quantitative yield in 20 min in the presence of In(OTf)3. On the other hand, in the hydrocarboxylation of hexynoic acid, the reaction is promoted by HOTf, AgOTf, and In(OTf)3 at an approximately similar rate; here, Ga(OTf)3 is ineffective.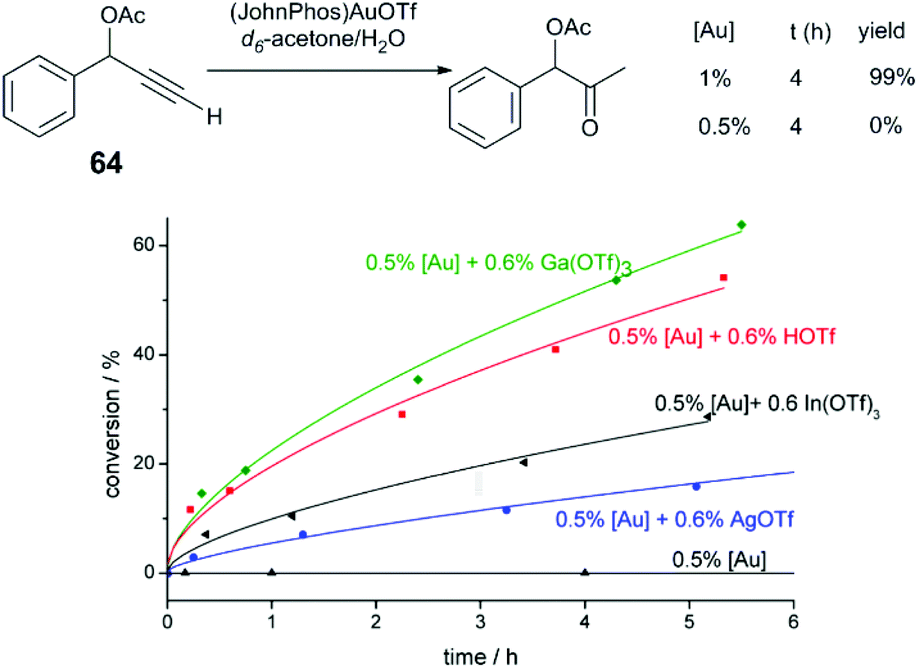 | ||
| Fig. 6 Effect of acidic additives on the reaction profile of alkyne hydration.72 Reprinted with permission. Copyright 2014 American Chemical Society. | ||
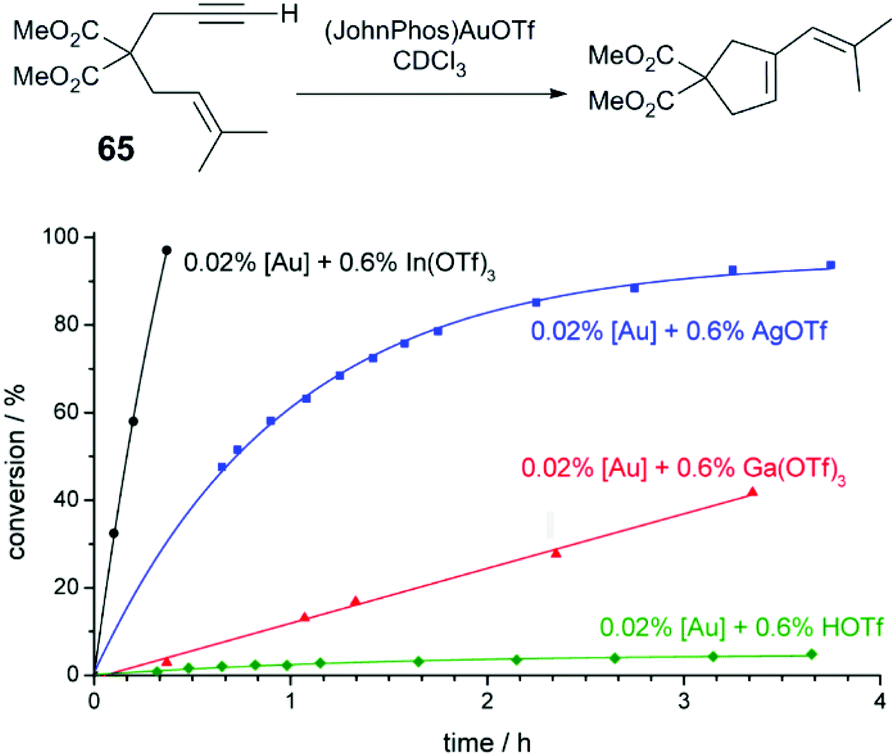 | ||
| Fig. 7 Effect of acidic additives on the reaction profile of enyne cyclisation.72 Reprinted with permission. Copyright 2014 American Chemical Society. | ||
These examples demonstrate the complexity of catalyst-Lewis acid interactions and their dramatic effect on catalyst performance. An acidic cocatalyst may regenerate catalytically-active cationic Au deactivated by base or halide impurities. A Brønsted acid-mediated mechanism for catalyst reactivation may proceed by protonolysis of a basic Au species, whereas Lewis acid-mediated catalyst reactivation may involve either anion abstraction or in situ generation of HOTf or other Brønsted acids. The authors demonstrate that basic impurities can come from filtration agents or desiccants, such as Celite or 4 Å molecular sieves.
Acid acceleration in Au reactions presumably influence the rate-limiting step in catalysis, but the rate-limiting step in Au catalysis is not always clear.71 In certain cases protodeauration is believed to be rate-limiting;74,75 strong Au–C bonds are generally robust to protonolysis and likely present in on-cycle and off-cycle pathways. Hammond75 and Yu76 find that protonolysis of heteroarylgold complexes is first order in gold complex and acid, and that the rate depends on the acid pKa. In protonolysis of heteroarygold complex 66 the rate is evidently dependent on the acid strength: TfOH yields 100% conversion of 66 in 5 min of reaction, whereas the weaker acid TsOH requires a longer duration (80%, 1 h); HOAc produces no reaction (Scheme 17).75 Accelerating turnover-limiting protodeauration is therefore a plausible role for Lewis acids, in addition to decreasing the rate of catalyst deactivation.
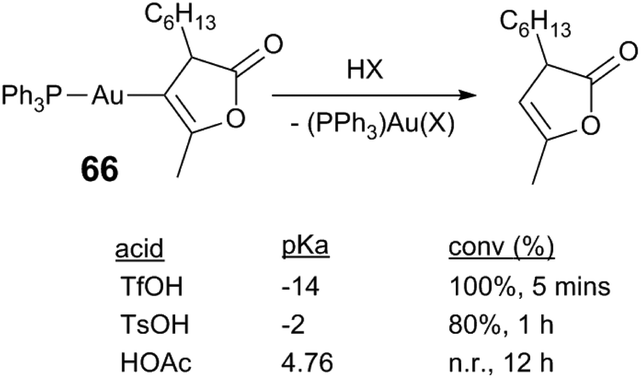 | ||
| Scheme 17 Protonolysis of an arylgold complex.75 | ||
As noted previously, Lewis acids can also affect the activation of Au precatalysts by abstracting a coordinating anion. The use of Lewis acids as activators of Au halide precursors has been systematically studied by Gandon and coworkers,77–79 who sought alternatives to Ag activation. Ag is thought to form inactive bimetallic Au–Ag80 or dimeric Au species,81 and increase rates of catalyst decomposition; fast, irreversible anion abstraction can actually slow catalysis via favouring formation of Au(0) or inactive gold species (such as [L2Au][X]).77 Decomposition is of special concern in reactions of reducing substrates (alkynes or alkenes) or in reactions requiring elevated temperatures.75
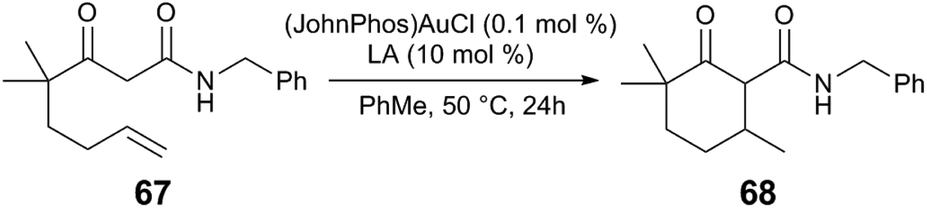
Alternative halide abstractors, such as Cu, Zn, In, and Bi Lewis acids, are effective in Au-catalysed hydroalkylation reactions, enyne cyclisations, and other Au catalysis.77,78 Unusually low Au loadings are effective in C–C bond forming reactions with Cu cocatalysts. The Au/Cu cocatalyst system also offers the advantage of scalability; for example, the ene-β-ketoamide 67 is transformed into the cyclised product 68 using (L)AuCl and Cu(OTf)2 (80% yield, dr 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, L = JohnPhos or PPh3) and notably, the reaction only proceeds to <30% conversion when AgOTf is used instead of Cu(OTf)2. Following the speciation of the (PPh3)Au(Cl) precatalyst by 31P NMR reveals slow generation of [(PPh3)2Au][OTf] upon combination with Cu(OTf)2, presumably via unobserved (PPh3)Au(OTf). During catalytic conditions, Cu(OTf)2 may slowly and reversibly abstract the chloride anion from the Au precatalyst, minimising catalyst decomposition by maintaining low concentration of [LAu][X]. In a separate report, a screen of Lewis acids at similar reaction conditions (Table 3) indicates that In(OTf)3 and Bi(OTf)3 also promote the hydroalkylation, whereas Sc(OTf)3, Yb(OTf)3, and TfOH are not effective. The Au/LA catalyst system is also useful in Au-catalysed alkyne hydroarylation, enyne cyclisation, and alkyne hydrofunctionalisation reactions.
:5, L = JohnPhos or PPh3) and notably, the reaction only proceeds to <30% conversion when AgOTf is used instead of Cu(OTf)2. Following the speciation of the (PPh3)Au(Cl) precatalyst by 31P NMR reveals slow generation of [(PPh3)2Au][OTf] upon combination with Cu(OTf)2, presumably via unobserved (PPh3)Au(OTf). During catalytic conditions, Cu(OTf)2 may slowly and reversibly abstract the chloride anion from the Au precatalyst, minimising catalyst decomposition by maintaining low concentration of [LAu][X]. In a separate report, a screen of Lewis acids at similar reaction conditions (Table 3) indicates that In(OTf)3 and Bi(OTf)3 also promote the hydroalkylation, whereas Sc(OTf)3, Yb(OTf)3, and TfOH are not effective. The Au/LA catalyst system is also useful in Au-catalysed alkyne hydroarylation, enyne cyclisation, and alkyne hydrofunctionalisation reactions.
|
|
||
|---|---|---|
| Lewis acid | Conv. (%) | Conv. (w/o Au, %) |
| Sc(OTf)3 | 0 | 0 |
| Yb(OTf)3 | 0 | 0 |
| Zn(OTf)2 | 83 | 0 |
| Al(OTf)3 | 26 | 16 |
| AlCl3 | 0 | 0 |
| Ga(OTf)3 | Trace | 12 |
| GaCl3 | 0 | 0 |
| In(OTf)3 | 100 | 77 |
| Bi(OTf)3 | 100 | 38 |
| HOTf | 0 | 0 |
A Lewis acid can take on numerous possible roles in Au catalysis. The most obvious role is anion abstractor, and sometimes Lewis acids provide improved activity vs. the more common Ag activators. The Lewis acid can also generate Brønsted acid in situ – thereby affecting Au-centred activation/deactivation pathways, accelerating the turnover-limiting protonolysis of key Au–C intermediates, or initiating acid-catalysed reaction sequences. We note that control experiments are critical when evaluating acid effects in Au catalysis. Several Lewis acids, such as Ga or In compounds, are also π-acids and can sometimes independently catalyse alkyne functionalisation reactions.82 Furthermore alkyne functionalisation reactions can also be independently catalysed by Brønsted acids.83
5. Enabling transmetalation and reductive elimination with Lewis acids in Pd catalysis
In this section, we will present several examples of Lewis acid additives and their effects in C–C and C–N coupling reactions. Pd coupling reactions are almost always run in the presence of alkali bases, and boron, zinc, magnesium, and copper nucleophiles are common alkyl and aryl group transfer agents. The Lewis acidity of these reagents and their byproducts are seldom considered when exploring reaction mechanisms.The influence of reagent metal cations is evident in Liebeskind's coupling of aromatic thioethers with organometallic nucleophiles.84 The coupling reaction proceeds in the presence of strongly nucleophilic alkylmagnesium or alkylzinc reagents. For boronic acids, a separate Lewis acid additive must be used. For example, addition of Zn(OAc)2 enables the coupling heteroaromatic thioether 69 and boronic acid 70 using the Pd2(dba)3/P(fur)3/CuTC catalyst yields biaryl 71 (76% yield) (Scheme 18). The thiophilic Zn Lewis acid is thought to bind the thiolate intermediate formed upon oxidative addition of the C–S bond to Pd(0), facilitating the difficult transmetalation step.
 | ||
| Scheme 18 Pd-Catalyed coupling of arylthioethers and boronic acids.84 | ||
This work inspired others to consider potential roles of Lewis acids in C–N bond-forming reactions. Hartwig and Shen isolated arylpalladium(II) amido complexes – proposed intermediates in amine N-arylation – and explored their reactivity in the presence of Lewis acidic species (Scheme 19).85 Upon heating a solution of complex 72 or 73 in PhMe in the absence of Lewis acid, the predominant reaction pathway is protonolysis of the amido ligand, liberating the free amine or sulphonamide. With added BPh3 or BEt3, on-cycle reductive elimination of the N-heteroarylamine occurs in 90% yield. The heteroaryl ligand of 72 acts as a Lewis acid “docking site,” making the Pd centre more electron deficient and accelerating reductive elimination versus competing protonolysis. BEt3 and complex 73, which lacks a heteroatom for Lewis acid binding, instead forms products of ethyl group transfer to the arene as well as amido protonolysis. The stoichiometric Pd-Lewis acid interaction may be catalytically relevant: coupling of heteroarene 74 with amide 75 using the Pd(dba)2/xantphos yields N-arylamide 76 in 84% yield in the presence of BEt3, but only in 17% yield without Lewis acid (Scheme 20). The control over the metal electronic character via remote binding of a Lewis acid resembles effects reported by Nolan and Moloy,86 and Bergman and Tilley.87
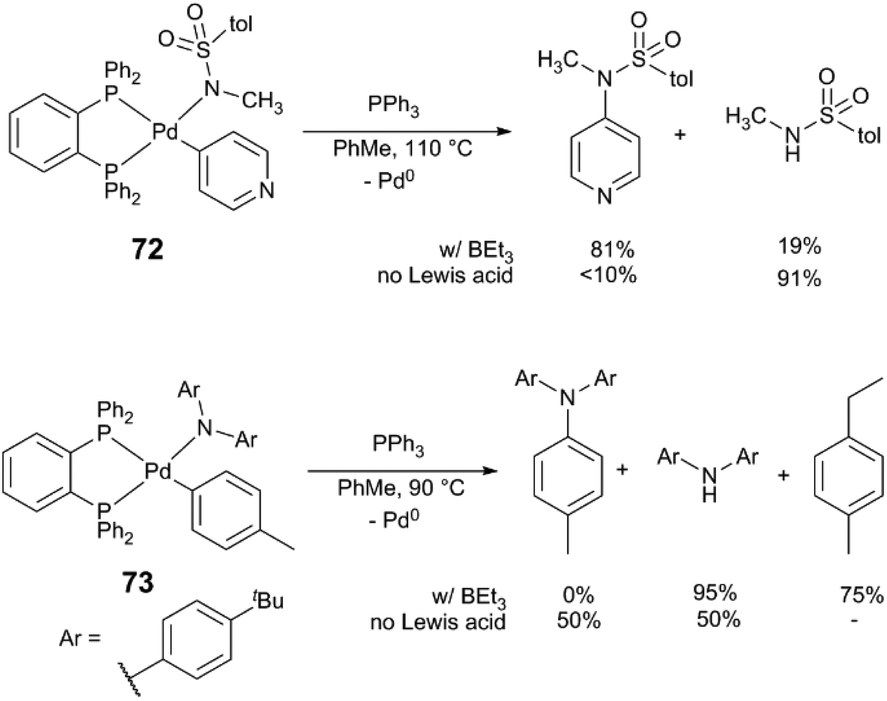 | ||
| Scheme 19 Reactivity of amidopalladium complexes.85 | ||
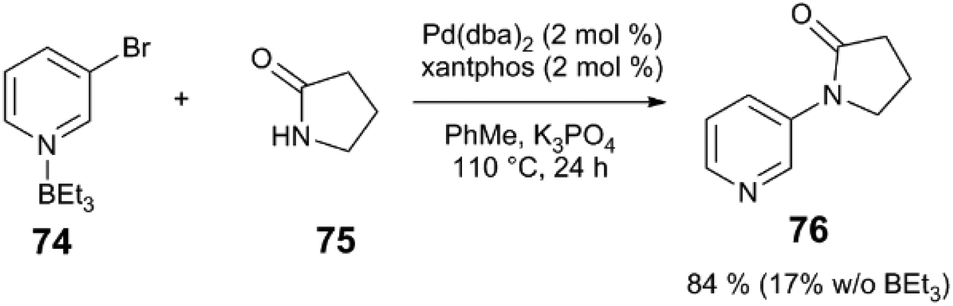 | ||
| Scheme 20 Cross coupling of heteroaryl halides and lactams.85 | ||
Further examples demonstrate that Lewis acid cocatalysts can enable challenging C–N coupling reactions. Researchers at Bristol-Myers Squibb serendipitously discovered that Zn salts enable the Pd-catalysed formation of a N-heteroarylazaindole 77.88 The azaindole precursor 78 is prepared from the Pd-catalysed cyanation of bromoarene 79 with Zn(CN)2, and the subsequent N-arylation step exhibited different performance depending on whether 78 was isolated by recrystallisation (38% yield) or chromatography (11% yield) (Scheme 21). The varying reaction performance was attributed to the presence of residual Zn present in recrystallized 78. Indeed, the reaction of heteroaryl chloride 80 with 78 catalysed by Pd2(dba)3/xantphos yields 78 in only 3% yield. The reaction instead returns >98% yield when Zn(OAc)2 or Zn(OPiv)2 is employed as a cocatalyst (60 mol%).
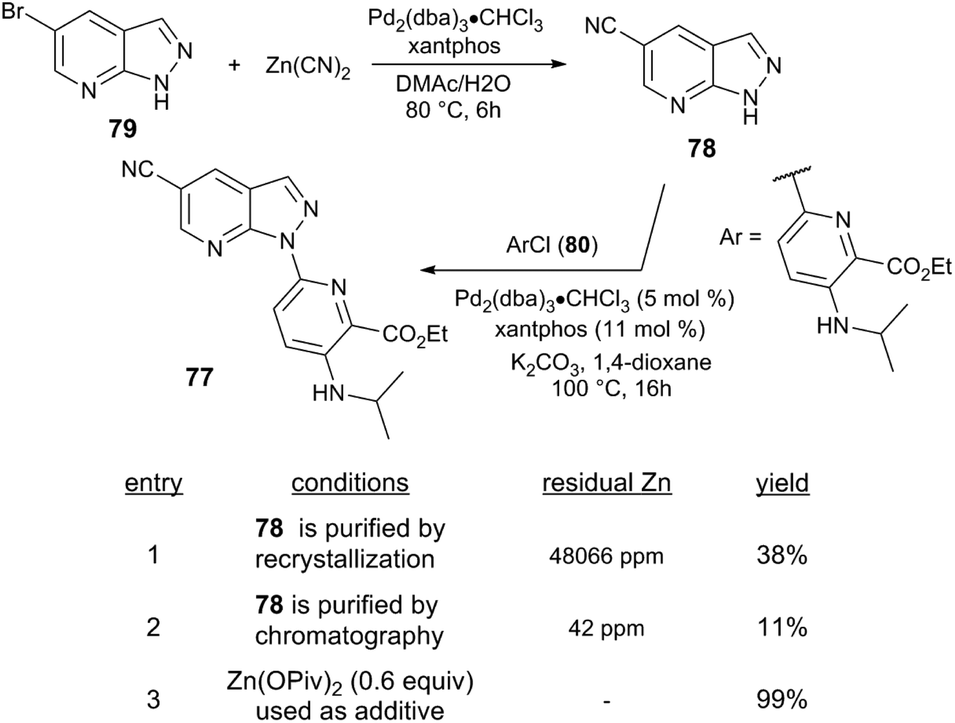 | ||
| Scheme 21 Route to an N-heteroarylazaindole by bromoarene cyanation and azaindole N-arylation.88 | ||
Although the mechanistic proposal by Hartwig and Shen invokes the binding of Lewis acid to a heteroarylpalladium moiety, this catalyst-Lewis acid interaction is not a requirement for a Lewis acid effect to be observed in C–N coupling reactions. Our group has observed that metal triflate Lewis acids are suitable cocatalysts in Pd catalysis, improving the yield of various N-arylamide products.89 In a representative example (Scheme 22), the reaction of bromoarene 81 with N-methylacetamide (82) via Pd(dba)2/xantphos/Al(OTf)3 yields the tertiary amide 83 in 89% yield, whereas only 53% yield of 83 is observed in the absence of an Al salt. Based on mechanistic insight and kinetic data, the Lewis acid appears to accelerate the rate-limiting transmetalation step of catalysis. However, deconvolution of key steps of transmetalation is difficult. At the very least, this step includes various reversible pre-equilibria as represented in Fig. 8: (a) halide dissociation from the oxidative addition complex I, (b) amide coordination to II, (c) dissolution of heterogenous base (N-arylation reactions in the presence of insoluble, inorganic bases can be mass-transfer limited),90 and (d) amide deprotonation to yield intermediate III.
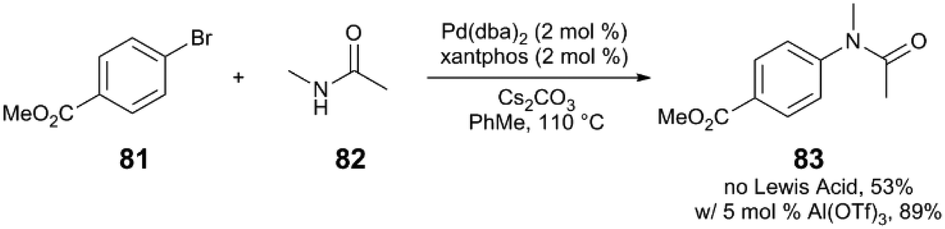 | ||
| Scheme 22 N-Arylation of a secondary acyclic amide.89 | ||
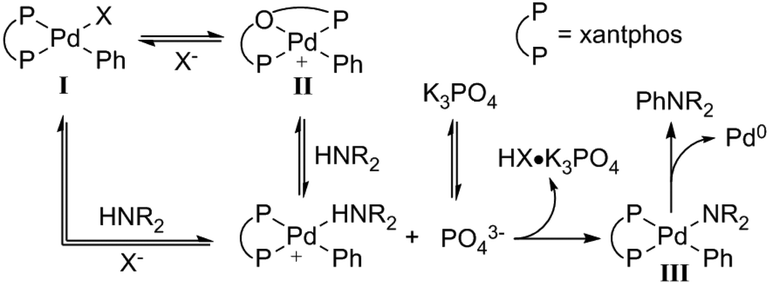 | ||
| Fig. 8 Proposed mechanism of dehydrohalogenation and amide coordination in Pd-catalysed amide N-arylation.89 Reprinted with permission. Copyright 2017 American Chemical Society. | ||
Measurement of the initial rates of catalysis of the coupling of bromobenzene (84) and pyrrolidin-2-one (78) indicate that an approximately three-fold acceleration is observed in the presence of Yb(OTf)3 (1:1 Yb:Pd), and that inhibition of catalysis is observed in the presence of exogenous bromide salts (NBu4Br) (Fig. 9). Our group proposed that the Lewis acid acts to mitigate competitive binding of bromide (a reaction byproduct) and the weakly nucleophilic amide by catalytic halide abstraction from the key Pd intermediate I. Indeed, by 31P NMR, evidence for the generation of the cationic Pd intermediate [(xantphos)Pd(Ph)]+ from the interaction of the oxidative addition complex, (xantphos)Pd(Ph)(Br), with Yb(OTf)3, suggesting that Lewis acids can promote the dissociative pathway in transmetalation represented in Fig. 8.
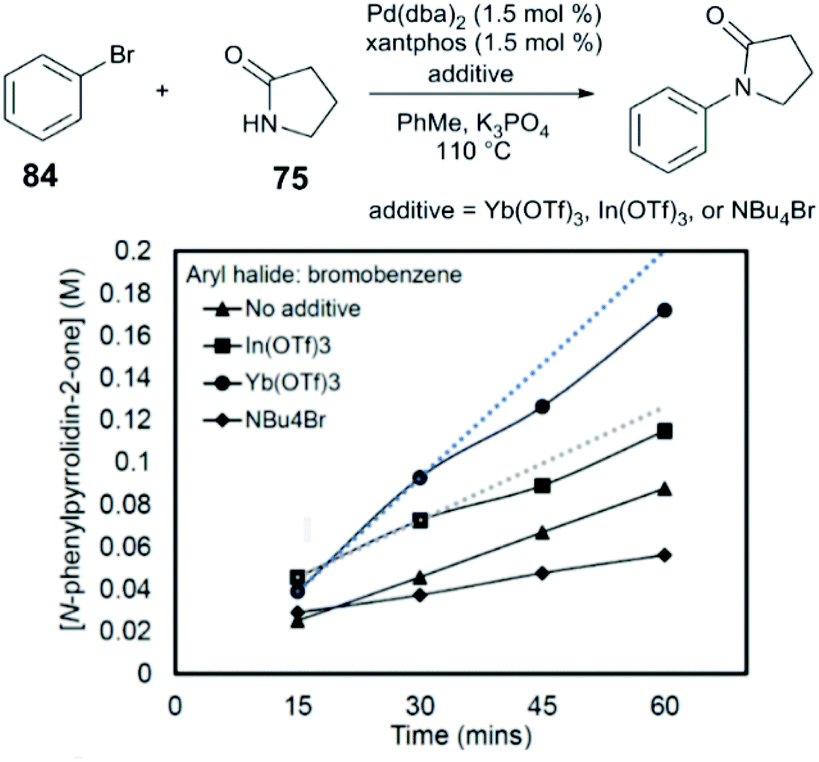 | ||
| Fig. 9 Effect of additives on the initial rate of amide N-arylation.89 Reprinted with permission. Copyright 2017 American Chemical Society. | ||
Another example in which a dramatic Lewis acid effect is observed comes from the group of Organ,4 where a sterically hindered NHC ligand enables the coupling of aryl halides and primary amides when borane cocatalysts are employed. Unactivated aryl chloride 85 and benzamide 86 react with a PEPPSI-Pd catalyst to yield the secondary amide 87 in quantitative yield with various borane additives (BEt3, tri(sec-butyl)borane, B(C6F5)3), while the product is only obtained in 21% yield in the absence of a borane (Scheme 23). The authors observe different behaviour between the three borane reagents. Whereas alkylboranes undergo autoxidation readily, B(C6F5)3 has stronger B–C bonds and therefore does not undergo immediate reaction with O2. It is observed that atmospheric contaminants and/or added radical traps affect the catalysis differently depending on which borane is used; for example, reactions that employ BEt3 do not yield product when the borane is first exposed to air. The authors provide spectroscopic evidence for the formation of borane-amide adducts (Scheme 24), which may be responsible for increasing the nucleophilicity of the amide and thereby facilitating transmetalation.
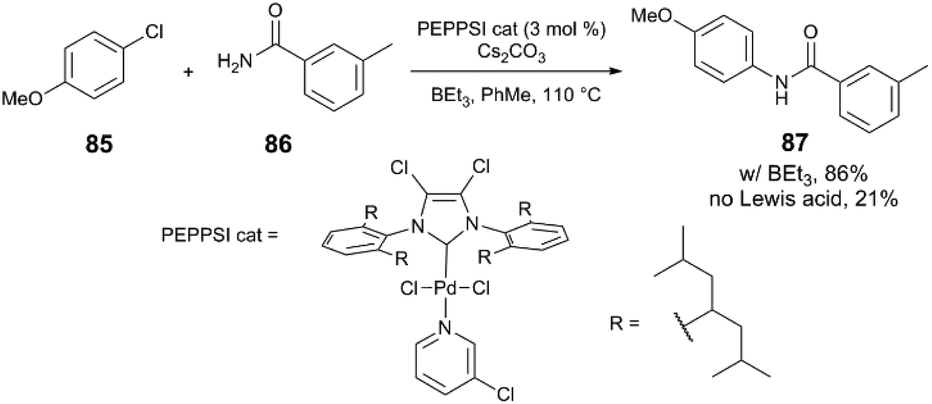 | ||
| Scheme 23 N-Arylation of a primary amide.4 | ||
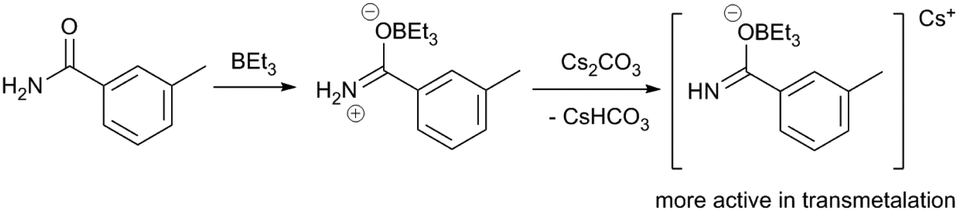 | ||
| Scheme 24 Formation of an amidonium boronate complex.4 | ||
In these Pd-catalysed reactions, various evidence points to the Lewis acid modulating the reactivity of the key Pd oxidative addition intermediates by catalyst-Lewis acid interactions, though Lewis acids of different character are employed (alkylborane, arylborane, zinc salts, or metal triflates, such as Al(OTf)3 or Yb(OTf)3) and therefore different possible modes of action of the Lewis acid may be operative. Based on the stoichiometric study by Shen and Hartwig, alkylboranes may increase the rate of reductive elimination versus the rate of amido protonolysis. Based on the works of our group and Organ, where delivery of the nitrogen nucleophile to Pd appears to be turnover-limiting, binding of amide may require sequestration of halide, or the generation of a more nucleophilic boron-amidate complex.
6. Conclusions
Lewis acid cocatalysts influence a diverse array of transition metal-catalysed reactions. Through mechanistic insight from a variety of studies, it appears that the predominant roles of the Lewis acid in the described transformations are as follows: (1) to form a Lewis acid base-adduct of the substrate to enable a challenging bond-cleavage event or to provide a thermodynamic driving force for a bond-cleavage event, (2) to modulate the pKa of the reaction medium, effect the controlled release of a Brønsted acid in situ, or facilitate proton transfer by some means, (3) to affect the coordination sphere of catalyst precursors or key catalyst intermediates by anion abstraction, and in doing so promote catalyst activation, reactivate off-cycle catalyst species, or accelerate slow organometallic elementary steps. In some cases, parametrisation or spectroscopic study has enabled insight into the behaviour of Lewis acidic species. Further mechanistic insight and novel experimental means of Lewis acid parameterisation are crucial for further application and rational design of cocatalytic systems.Lewis acid additives can provide more active or selective catalysts for existing processes, or enable orthogonal reactivity of catalytic reactions by the Lewis acid-mediated interception of key intermediates. Thus, further development in this area will impact new and existing chemistries alike, and we expect Lewis acids will figure prominently in many further catalytic applications. Additional work in understanding and controlling Lewis acid/organometallic interactions will be instrumental in offering new opportunities in homogeneous catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. CHE-1565721.References
- C. Wang and Z. Xi, Chem. Soc. Rev., 2007, 36, 1395–1406 RSC
.
- T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS PubMed
- H. Böhrer, N. Trapp, D. Himmel, M. Schleep and I. Krossing, Dalton Trans., 2015, 44, 7749–7499 RSC
- S. Sharif, J. Day, H. N. Hunter, Y. Lu, D. Mitchell, M. J. Rodriguez and M. G. Organ, J. Am. Chem. Soc., 2017, 139, 18436–18439 CrossRef CAS PubMed
- Z. Li, R. S. Assary, A. C. Atesin, L. A. Curtiss and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 104–107 CrossRef CAS PubMed
- C. A. Tolman, W. C. Seidel, J. D. Druliner and P. J. Domaille, Organometallics, 1984, 3, 33–38 CrossRef CAS
- N. M. Brunkan, D. M. Brestensky and W. D. Jones, J. Am. Chem. Soc., 2004, 126, 3627–3641 CrossRef CAS PubMed
- J. J. García, A. Arévalo, N. M. Brunkan and W. D. Jones, Organometallics, 2004, 23, 3997–4002 CrossRef
- Y. Nakao, Chem. Rec., 2011, 11, 242–251 CrossRef CAS PubMed
- W. Guan, G. Zeng, H. Kameo, Y. Nakao and S. Sakaki, Chem. Rec., 2016, 16, 2405–2425 CrossRef CAS PubMed
- Y. Nakao, A. Yada, S. Ebata and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 2428–2429 CrossRef CAS PubMed
- Y. Hirata, T. Yukawa, N. Kashihara, Y. Nakao and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 10964–10973 CrossRef CAS PubMed
- A. Yada, T. Yakawa, Y. Nakao and T. Hiyama, Chem. Commun., 2009, 3931–3933 RSC
- Y. Nakao, Y. Hirata, M. Tanaka and T. Hiyama, Angew. Chem., Int. Ed., 2008, 47, 385–387 CrossRef CAS PubMed
- Y. Nakao, S. Ebata, A. Yada, T. Hiyama, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2008, 130, 12874–12875 CrossRef CAS PubMed
- M. P. Watson and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 12594–12595 CrossRef CAS PubMed
- D. C. Koester, M. Kobayashi, D. B. Werz and Y. Nakao, J. Am. Chem. Soc., 2012, 134, 6544–6547 CrossRef CAS PubMed
- Y. Miyazaki, N. Ohta, K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 3732–3735 CrossRef CAS PubMed
- G. B. Frost, N. A. Serratore, J. M. Ogilvie and C. J. Douglas, J. Org. Chem., 2017, 82, 3721–3726 CrossRef CAS PubMed
- Z. Pan, S. Wang, J. T. Brethorst and C. J. Douglas, J. Am. Chem. Soc., 2018, 140, 3331–3338 CrossRef CAS PubMed
- Y. Hirata, A. Yada, E. Morita, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2010, 132, 10070–10077 CrossRef CAS PubMed
- Y. Kajita, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2008, 130, 17226–17227 CrossRef CAS PubMed
- T. Tamaki, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2011, 50, 12067–12070 CrossRef CAS PubMed
- K. Nakai, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2011, 133, 11066–11068 CrossRef CAS PubMed
- Y. Nakao, A. Yada and T. Hiyama, J. Am. Chem. Soc., 2010, 133, 10024–10026 CrossRef PubMed
- X. Feng, P. Yu and B. Morandi, Science, 2016, 351, 832–836 CrossRef PubMed
- X. Feng, P. Yu, G. P. Cerai and B. Morandi, Chem. – Eur. J., 2016, 22, 15629–15633 CrossRef PubMed
- P. Yu and B. Morandi, Angew. Chem., Int. Ed., 2017, 56, 15693–15697 CrossRef CAS PubMed
- Y. Nakao, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 2448–2449 CrossRef CAS PubMed
- Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666–13668 CrossRef CAS PubMed
- Y. Nakao, H. Idei, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 15996–15997 CrossRef CAS PubMed
- S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed
- S. Okumura and Y. Nakao, Org. Lett., 2017, 19, 584–587 CrossRef CAS PubMed
- M. Ohashi, H. Saijo, T. Arai and S. Ogoshi, Organometallics, 2010, 29, 6534–6540 CrossRef CAS
- Y. Nakao, H. Idei, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 5070–5071 CrossRef CAS PubMed
- Y. Nakao, E. Morita, H. Idei and T. Hiyama, J. Am. Chem. Soc., 2011, 133, 3264–3267 CrossRef CAS PubMed
- W. Guan, S. Sakaki, T. Kurahashi and S. Matsubara, ACS Catal., 2015, 5, 1–10 CrossRef CAS
- H. Ren, G.-F. Du, B. Zhu, G.-C. Yang, L.-S. Yao, W. Guan and Z.-M. Su, Organometallics, 2018, 37, 2594–2601 CrossRef CAS
- B. Zhu, G. F. Du, H. Ren, L.-K. Yan, W. Guan and Z.-M. Su, Organometallics, 2017, 36, 4713–4720 CrossRef CAS
- M. Anand and R. B. Sunoj, Org. Lett., 2012, 14, 4584–4587 CrossRef CAS PubMed
- S.-F. Ni, T.-L. Yang and L. Dang, Organometallics, 2017, 36, 2746–2754 CrossRef CAS
- F. M. A. Geilin, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermeyer and W. Leitner, Angew. Chem., Int. Ed., 2010, 122, 5642–5646 CrossRef
- F. M. A. Geilin, B. Engendahl, M. Hölscher, J. Klankermeyer and W. Leitner, J. Am. Chem. Soc., 2011, 133, 14349–14358 CrossRef PubMed
- J. Coetzee, D. L. Dodds, J. Klankermeyer, S. Brosinski, W. Leitner, A. M. Z. Slawin and D. J. Cole-Hamilton, Chem. – Eur. J., 2013, 19, 11039–11050 CrossRef CAS PubMed
- Y. Li, C. Topf, X. Cui, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 5196–5200 CrossRef CAS PubMed
- X. Cui, Y. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10596–10599 CrossRef CAS PubMed
- J. R. Cabrero-Antonino, E. Alberico, K. Junge, H. Junge and M. Beller, Chem. Sci., 2016, 7, 3432–3442 RSC
- J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Chem. Sci., 2017, 8, 6439–6450 RSC
- C. Atesin, N. A. Ray, P. C. Stair and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 14682–14685 CrossRef PubMed
- R. S. Assary, A. C. Atesin, Z. Li, L. A. Curtiss and T. J. Marks, ACS Catal., 2013, 3, 1908–1914 CrossRef CAS
- Z. Li, R. S. Assary, C. Atesin, L. A. Curtiss and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 104–107 CrossRef CAS PubMed
- T. L. Lohr, Z. Li, R. S. Assary, L. A. Curtiss and T. J. Marks, ACS Catal., 2015, 5, 3675–3679 CrossRef CAS
- J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415–1418 CrossRef CAS PubMed
-
A. S. K. Hashmi and F. D. Toste, Modern Gold Catalyzed Synthesis, Wiley-VCH, Weinheim, 2012 Search PubMed
- D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed
- S. Zhang, F. Wei, C. Song, J. Jia and Z. Xu, Chin. J. Chem., 2014, 32, 937–956 CrossRef CAS
- Z.-Y. Han, H. Xiao, X.-H. Chen and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 9182–9183 CrossRef CAS PubMed
- D. Qian and J. Zhang, Chem. – Eur. J., 2013, 19, 6984–6988 CrossRef CAS PubMed
- X. Wu, M.-L. Li and P.-S. Wang, J. Org. Chem., 2014, 79, 419–425 CrossRef CAS PubMed
- S. Dhiman and S. S. V. Ramasastry, Org. Lett., 2015, 17, 5116–5119 CrossRef CAS PubMed
- S. Dhiman and S. S. V. Ramasastry, Chem. Commun., 2015, 51, 557–560 RSC
- L.-Z. Dai and M. Shi, Chem. – Eur. J., 2010, 16, 2496–2502 CrossRef CAS PubMed
- S. Zhang, Z. Xu, J. Jia, C.-H. Tung and Z. Xu, Chem. Commun., 2014, 50, 12084–12087 RSC
- B. Wang, M. Liang, J. Tang, Y. Deng, J. Zhao, H. Sun, C.-H. Tung, J. Jia and Z. Xu, Org. Lett., 2016, 18, 4614–4617 CrossRef CAS PubMed
- M. Liang, S. Zhang, J. Jia, C.-H. Tung, J. Wang and Z. Xu, Org. Lett., 2017, 19, 2526–2529 CrossRef CAS PubMed
- X. Wang, Z. Yao, S. Dong, F. Wei, H. Wang and Z. Xu, Org. Lett., 2013, 15, 2234–2237 CrossRef CAS PubMed
- P. Fernández, P. Alonso, F. J. Fañanás and F. Rodríguez, Eur. J. Org. Chem., 2018, 3957–3964 CrossRef
- A. S. Demir, M. Emrullahoğlu and K. Buran, Chem. Commun., 2010, 46, 8032–8034 RSC
- Y. Wang, L. Liu and L. Zhang, Chem.
Sci., 2013, 4, 739–746 RSC
- Y. Xi, D. Wang, X. Ye, N. G. Akhmedov, J. L. Petersen and X. Shi, Org. Lett., 2014, 16, 306–309 CrossRef CAS PubMed
- J. Han, N. Shimizu, Z. Lu, H. Amii, G. B. Hammond and B. Xu, Org. Lett., 2014, 16, 3500–3503 CrossRef CAS PubMed
- M. Kumar, G. B. Hammond and B. Xu, Org. Lett., 2014, 16, 3452–3455 CrossRef CAS PubMed
- P. Barrio, M. Kumar, Z. Lu, J. Han, B. Xu and G. B. Hammond, Chem. – Eur. J., 2016, 22, 16410–16414 CrossRef CAS PubMed
- C. Obradors and A. M. Echavarren, Chem. Commun., 2014, 50, 16–28 RSC
- W. Wang, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697–5705 CrossRef CAS PubMed
- Y. Zhu and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 8329–8332 CrossRef CAS PubMed
- A. Guérinot, W. Fang, M. Sircoglou, C. Bour, S. Bezzenine-Lafollée and V. Gandon, Angew. Chem., Int. Ed., 2013, 52, 5848–5852 CrossRef PubMed
- W. Fang, M. Presset, A. Guérinot, C. Bour, S. Bezzenine-Lafollée and V. Gandon, Chem. – Eur. J., 2014, 10, 5439–5446 CrossRef PubMed
- W. Fang, M. Presset, A. Guérinot, C. Bour, S. Bezzenine-Lafollée and V. Gandon, Org. Chem. Front., 2014, 1, 608–613 RSC
- D. Weber and M. Gagne, Org. Lett., 2009, 11, 4962–4965 CrossRef CAS PubMed
- A. Homs, I. Escofet and A. M. Echavarren, Org. Lett., 2013, 15, 5782–5785 CrossRef CAS PubMed
- C. Bour and V. Gandon, Synlett, 2015, 26, 1427–1436 CrossRef CAS
- T. Tsuchimoto, T. Joya, E. Shirakawa and Y. Kawakami, Synlett, 2000, 1777–1778 CAS
- L. S. Liebeskind and J. Srogl, Org. Lett., 2002, 4, 979–981 CrossRef CAS PubMed
- Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7734–7735 CrossRef CAS PubMed
- J. K. Huang, C. M. Haar, S. P. Nolan, J. E. Marcone and K. G. Moloy, Organometallics, 1999, 18, 297–299 CrossRef CAS
- A. L. Liberman-Martin, R. G. Bergman and T. D. Tilley, J. Am. Chem. Soc., 2013, 135, 9612–9615 CrossRef CAS PubMed
- R. Ayothiraman, S. Rangwaswamy, P. Maity, E. M. Simmons, G. L. Beutner, J. Janey, D. S. Treitler, M. D. Eastgate and R. Vaidyanathan, J. Org. Chem., 2017, 82, 7420–7427 CrossRef CAS PubMed
- J. Becica and G. E. Dobereiner, ACS Catal., 2017, 7, 5862–5870 CrossRef CAS
- J. M. Dennis, N. A. White, R. Y. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 4721–4725 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2019 |