
Tailoring the electrochemical activity of magnesium chromium oxide towards Mg batteries through control of size and crystal structure†
Linhua
Hu‡
 ab,
Ian D.
Johnson‡
c,
Soojeong
Kim
bd,
Gene M.
Nolis
ab,
John W.
Freeland
e,
Hyun Deog
Yoo
f,
Timothy T.
Fister
bd,
Liam
McCafferty
c,
Thomas E.
Ashton
c,
Jawwad A.
Darr
*c and
Jordi
Cabana
*ab
ab,
Ian D.
Johnson‡
c,
Soojeong
Kim
bd,
Gene M.
Nolis
ab,
John W.
Freeland
e,
Hyun Deog
Yoo
f,
Timothy T.
Fister
bd,
Liam
McCafferty
c,
Thomas E.
Ashton
c,
Jawwad A.
Darr
*c and
Jordi
Cabana
*ab
aDepartment of Chemistry, University of Illinois at Chicago, Chicago, IL 60607, USA. E-mail: jcabana@uic.edu
bJoint Center for Energy Storage Research, Argonne National Laboratory, Argonne, IL 60439, USA
cDepartment of Chemistry, University College London, 20 Gower Street, London, WC1H 0AJ, UK. E-mail: j.a.darr@ucl.ac.uk
dChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, IL 60439, USA
eX-ray Sciences Division, Argonne National Laboratory, Argonne, IL 60439, USA
fDepartment of Chemistry and Chemical Institute for Functional Materials, Pusan National University, Busan 46241, Republic of Korea
First published on 3rd December 2018
Abstract
Chromium oxides with the spinel structure have been predicted to be promising high voltage cathode materials in magnesium batteries. Perennial challenges involving the mobility of Mg2+ and reaction kinetics can be circumvented by nano-sizing the materials in order to reduce diffusion distances, and by using elevated temperatures to overcome activation energy barriers. Herein, ordered 7 nm crystals of spinel-type MgCr2O4 were synthesized by a conventional batch hydrothermal method. In comparison, the relatively underexplored Continuous Hydrothermal Flow Synthesis (CHFS) method was used to make highly defective sub-5 nm MgCr2O4 crystals. When these materials were made into electrodes, they were shown to possess markedly different electrochemical behavior in a Mg2+ ionic liquid electrolyte, at moderate temperature (110 °C). The anodic activity of the ordered nanocrystals was attributed to surface reactions, most likely involving the electrolyte. In contrast, evidence was gathered regarding the reversible bulk deintercalation of Mg2+ from the nanocrystals made by CHFS. This work highlights the impact on electrochemical behavior of a precise control of size and crystal structure of MgCr2O4. It advances the understanding and design of new cathode materials for Mg-based batteries.
Introduction
Mg batteries have been predicted to have higher energy densities than Li-ion batteries, and have received significant attention in recent years as a potential alternative to meet looming energy storage demands for grid and transportation technologies.1 Since a redox reaction involving a Mg2+ cation is a two-electron process, it could, theoretically possess double the charge storage of an intercalation host, compared to single-electron processes involving Li+. Moreover, Mg metal can be more safely used directly as the anode material, offering higher energy densities in comparison to Li-ion graphite anodes, because unlike Li metal, Mg is not prone to form metallic dendrites upon repeated cycling.1,2 The first stable Mg cell was demonstrated by Aurbach in 2000, using a Mg anode and an intercalation-based MgxMo6S8 cathode.2 However, its overall energy density was well short of the target for modern batteries (77 vs. a target of 908 W h kg−1),1,2 and, therefore, that system did not offer an alternative to Li-ion technology.3 Investigation of cathode materials leading to higher energy density has been limited by the low operating voltages of candidate compounds,2,4,5 their incompatibility with electrolytes,6 poor power capabilities,4,7 and propensity for degradation processes that have resulted in poor cycle life.6,8 Therefore, there is a need to discover novel phases that can act as high-voltage cathode materials in a Mg battery by meeting performance metrics while maintaining stability during charge/discharge processes.Recently, a computational study by Liu et al. proposed that spinel-type MgM2O4 (where M is a transition metal ion) materials have potential for use as high-voltage intercalation hosts for Mg-ion batteries.9 In the normal spinel structure, Mg2+ cations occupy available tetrahedral sites and should diffuse throughout the structure within a 3D network of tunnels.10 Reversible Mg2+ intercalation within spinel structures has only been experimentally demonstrated within Mn2O4, made electrochemically by delithiating LiMn2O4.11 Previous efforts to reversibly remove Mg from spinel-type Mg0.5Mn2.5O4 revealed reversible structural rearrangements between the spinel and a layered framework upon cycling.12 Furthermore, the potential use of MgMn2O4 spinels may be hampered by high degrees of inversion (i.e. Mg on octahedral sites, and Mn on tetrahedral sites), which could block Mg diffusion channels.13 MgCr2O4 has been predicted in silico to potentially overcome these issues,9 because MgCr2O4 possesses virtually no degree of inversion due to the crystal field stabilisation of Cr3+ as a d3 ion, which reduces the likelihood of diffusion channel blockages.14 Furthermore, out of all considered spinel compounds, MgCr2O4 possesses one of the highest predicted energy densities (∼800 W h kg−1), and lower predicted Mg2+ diffusion activation energy barriers, compared to MgMn2O4 (650 vs. 800 meV). Therefore, MgCr2O4 is clearly of interest as a potential cathode material.
Beyond seeking materials with minimized hopping barriers, designing them at small particle sizes would shorten bulk diffusion lengths and maximize sites for cathode/electrolyte transfer of Mg2+, thereby further facilitating electrochemical performance. However, the reproducible and scalable fabrication of multimetallic metal oxides in the sub 10 nm regime, is challenging using conventional synthesis methods. Typically, laborious and energy-intensive solid-state processes are used to synthesize materials such as MgCr2O4 from the parent oxides of MgO and Cr2O3, which are a few microns in size.15 In contrast, solution-based techniques have successfully produced more regular and nano-sized MgCr2O4 materials; Li et al. used a citrate sol–gel process to generate 20 nm particles,16 and Morozova et al. used mixed Mg and Cr hydroxide precursors (with heat-treatment) to synthesise nanocrystalline (range 10 to 40 nm) MgCr2O4; using XRD, they proposed that during heat-treatment, the onset of crystallisation to form the spinel phase, began at ca. 500 °C (and was complete by 600 °C), with particle sizes growing from 10 to 40 nm in the temperature range 500 to 1000 °C.17 Durrani et al. produced ca. 40 nm MgCr2O4 crystallites in a similar manner and suggested that the reaction proceeded via initial formation of MgO and Cr2O3 that underwent further reactions during heat-treatment.18
Continuous Hydrothermal Flow Synthesis (CHFS) is regarded as a highly versatile, scalable synthesis approach to more conventional batch hydrothermal synthesis methods.19 In CHFS, rapid nucleation of nanoparticles is achieved by in-flow mixing of supercritical water (typically at 450 °C) with an ambient temperature aqueous precursor solution in a well-defined mixing arrangement;20 particle nucleation and growth can be effectively controlled to achieve small particles using highly supersaturating conditions and a relatively short residence time (total reaction time of a few seconds).19 Therefore, CHFS synthesis can generally access significantly smaller particle sizes in comparison to batch hydrothermal techniques; the former method has successfully been used to generate a wide variety of nano-sized electrode materials for Li-ion batteries and supercapacitors, including olivine materials LiMPO4 (M = Fe or Mn),21–23 TiO2,24 Nb2O5,25 and VO2,26 among others.19 In this report, the authors compared the crystal chemistry and electrochemical behaviour of MgCr2O4 synthesized by both a batch hydrothermal and a continuous hydrothermal flow synthesis method. It was found that the two products possessed subtly different crystal structures, resulting in distinctly different electrochemical behaviours. These results suggest highly defective MgCr2O4 can reversibly remove/intercalate Mg2+ and as such, is an exciting new candidate cathode for Mg batteries.
Experimental
Synthesis
Magnesium chromium oxide nanocrystals were synthesized by two different methods.(a) Continuous Hydrothermal Flow Synthesis (CHFS).19 The as-prepared MCO(CHFS) sample was synthesized using a laboratory-scale CHFS process incorporating a Confined Jet Mixer (CJM), which has been described in detail in previous publications,20 and a diagram of which is provided in Fig. S1a and S1b,† respectively. The synthesis process can be described as follows; two precursor solutions were prepared in which the first precursor solution (fed in via pump P2) consisted of the dissolved Cr and Mg precursors: 0.1 M Cr(NO3)3·9H2O (99%, Acros Organics, Loughborough, UK) and 0.05 M Mg(NO3)2·6H2O (99%, Sigma Aldrich, Steinheim, Germany) in D.I. water, respectively. The second solution (fed in via pump P3), was a molar excess of base, in the range 0.4 to 0.5 M KOH (86%, Fisher Scientific, Loughborough, UK), in D.I. water to ensure an overall reaction pH of 14. Each solution was pumped into a 1/4′′ stainless steel T-piece mixer at a flow rate of 40 mL min−1 each. The combined mixture of solution 1 and 2 flowed at 80 mL min−1 into the side arms of the CJM, where it rapidly combined with 80 mL min−1 of supercritical water at 450 °C and 24.1 MPa (delivered via pump P1 and an inline heater), which emerged from the inner tube of the CJM as a turbulent jet, resulting in extremely rapid mixing between the precursor mixture and the sc-water feed (mixing temperature of 335 °C,27 and in a turbulent regime due to a Reynolds number of >6900).20,27 The nanoparticles of MgCr2O4−x(OH)2x rapidly formed, (residence time of ca. 7 s) before cooling to near ambient temperature through a pipe-in-pipe countercurrent heat exchanger. The aqueous product slurry was collected in a beaker at a particle production rate of ca. 30 g h−1. The slurry was allowed to settle (1 h) and the supernatant siphoned off. Repeated centrifugation (3 × 4500 rpm for 5 minutes) and washing with D.I. water (after each centrifugation) was performed until the conductivity of the supernatant was below 60 μS m−1. The cleaned, wet product paste was freeze-dried by slowly heating from −60 °C to 25 °C over 24 h under a vacuum of <13 Pa using a VirTis Genesis 35 XL (SP Scientific, New York, U.S.) to yield a poorly-crystalline initial product. Crystalline MgCr2O4 was generated from a flash heat-treatment of the as-prepared material (Fig. S2†), which is described further in the ESI;† this essentially involved the introduction of the as-prepared CHFS powder into a pre-heated (500 °C) tube furnace for 10 minutes, before being removed. The heat-treated product of as-prepared MCO(CHFS) was designated as MCO(ΔCHFS).
(b) Standard Batch Hydrothermal Synthesis (BHS): As-prepared MCO (BHS) sample was synthesized using a 125 mL batch autoclave reactor with a PTFE liner in a stainless steel vessel (4878 model, Parr Instrument Company). 1 mmol Mg(ac)2·4H2O (99%, Alfa Aesar) and 2 mmol Cr3(ac)6·H2O (99%, Alfa Aesar) were dissolved into 70 mL D.I. water, and 1 M NaOH solution was added dropwise (99%, Thermo Fisher Scientific) until pH ∼9. The resulting brown solution was loaded into an autoclave and heated to 160 °C for a total reaction time of 12 h. The precipitation was washed with D.I. water several times, dried at 60 °C and finally heat-treated at 500 °C in air for 3 h. The heat-treated product of as-prepared MCO (BHS) was designated as MCO (ΔBHS).
Physical–chemical characterization of the pristine powders
Powder X-ray diffraction (XRD) patterns were collected on a Bruker D8 Advance X-ray Diffractometer (X-ray wavelength = 1.5406 Å). High-resolution synchrotron X-ray powder diffraction were performed at beamline 11-ID-B of Advanced Photon Source, Argonne National Laboratory (wavelength = 0.2114 Å). Pawley refinements were performed using GSAS II.Transmission electron microscopy (TEM) and Energy-dispersive X-ray (EDX) was performed using JEOL-3010 microscope operated at 300 kV. The surface area of the samples was measured using a Micromeritics TriStar II PLUS. Simultaneous differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) was carried out using a Netzsch STA 1500 using a heating rate of 5 °C min−1 in the temperature range 25 to 1000 °C in air. Samples were run within an alumina crucible and under a constant flow of air. A background scan was conducted with an empty crucible in the same temperature range and heating rate to subtract from the raw data, which eliminated buoyancy effects.
Hard X-ray Absorption Spectroscopy (XAS) measurements at the Cr K-edge were performed at the MRCAT bending magnet beamline at the Advanced Photon Source, Argonne National Laboratory.28 X-ray absorption spectra were collected in transmission mode through the MgCr2O4 laminates. A Si(111) water-cooled double-crystal monochromator was used to scan incident X-ray and scanned energies were detuned by 50 percent. The beam profile was collimated to 3 × 0.75 mm. A Cr reference foil was measured simultaneously with every sample for energy calibration (Cr K-edge set to 5989 eV). All the data analysis was completed by IFEFFIT software package.29,30 Background removal was completed with AUTOBK30 using the ATHENA graphical interface.30
Soft X-ray Absorption Spectroscopy (XAS) measurements were also performed across the Cr L-edges at beamline 4-ID-C at the Advanced Photon Source, Argonne National Laboratory. Absorption spectra were obtained at a resolution of ∼0.2 eV in total electron yield (TEY). Energy scales were aligned using known Mn and Mg reference samples for an accurate comparison.
Electrochemical measurements
The working electrodes were prepared by mixing the MgCr2O4 nanocrystal samples, carbon black (Denka), and polyvinylidene difluoride (PVDF) (Kynar) in N-methylpyrrolidone (NMP) (Sigma-Aldrich) (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:20 (wt%)), which were then cast on an electrochemical-grade stainless steel 316 mesh using a doctor blade, followed by drying in vacuo at 110 °C overnight. Dried electrodes, with a loading level of ∼3 mg cm−2, were punched with a diameter of 1/2′′ and assembled into 2032 coin-type cells in an Ar-filled glovebox (water and oxygen, ≤0.1 ppm). Electrochemical cells contained an activated carbon mesh as a counter electrode, a glass fiber separator (VWR, grade 691, 28297-289) and 0.5 M Mg[N(SO2)2(CF3)2]2-(C9H20N)N(SO2)2(CF3)2 (abbreviated as MgTFSI2-PY14TFSI) with low H2O content (∼43 ppm) as electrolyte. For 2-electrode cells, the potential of cathode was calibrated by considering the AC anode's potential, which is originally 2.2 V vs. Mg/Mg2+ and linearly proportional to the state-of-charge (SoC). Electrochemistry was carried out at 110 °C in the potential range 0.1–3.5 V vs. Mg/Mg2+. The charge/discharge rate (C/50) was galvanostatically controlled by a Bio-Logic VMP3 potentiostat. After oxidation or reduction of the MgCr2O4 electrodes, they were recovered and rinsed in acetonitrile three times, and dried at room temperature under vacuum for 1 minute before characterization.
:20:20 (wt%)), which were then cast on an electrochemical-grade stainless steel 316 mesh using a doctor blade, followed by drying in vacuo at 110 °C overnight. Dried electrodes, with a loading level of ∼3 mg cm−2, were punched with a diameter of 1/2′′ and assembled into 2032 coin-type cells in an Ar-filled glovebox (water and oxygen, ≤0.1 ppm). Electrochemical cells contained an activated carbon mesh as a counter electrode, a glass fiber separator (VWR, grade 691, 28297-289) and 0.5 M Mg[N(SO2)2(CF3)2]2-(C9H20N)N(SO2)2(CF3)2 (abbreviated as MgTFSI2-PY14TFSI) with low H2O content (∼43 ppm) as electrolyte. For 2-electrode cells, the potential of cathode was calibrated by considering the AC anode's potential, which is originally 2.2 V vs. Mg/Mg2+ and linearly proportional to the state-of-charge (SoC). Electrochemistry was carried out at 110 °C in the potential range 0.1–3.5 V vs. Mg/Mg2+. The charge/discharge rate (C/50) was galvanostatically controlled by a Bio-Logic VMP3 potentiostat. After oxidation or reduction of the MgCr2O4 electrodes, they were recovered and rinsed in acetonitrile three times, and dried at room temperature under vacuum for 1 minute before characterization.
Results and discussion
The initial products of batch hydrothermal synthesis and continuous hydrothermal synthesis were poorly-crystalline powders and were designated MCO(BHS) and MCO(CHFS), respectively. Subsequent heat-treatment of these powders was required to crystallize MgCr2O4, as detailed in the Experimental section, generating samples MCO(ΔBHS) and MCO(ΔCHFS), respectively.To understand the formation of MgCr2O4, MCO(CHFS) was analyzed using TGA-DSC (Fig. S3†). As the mass yield of the as-prepared MCO(CHFS) powder was ∼41% greater than that expected for pure MgCr2O4, an initial chemical composition of MgCr2O4−x(OH)2x is suggested, reflecting the possible partial decomposition to oxide from the hydroxide during hydrothermal reaction. TGA-DSC analysis in the temperature range 25 to 1000 °C, revealed a mass loss of ca. 32.5%, which resulted in an overall mass yield of 95% of MgCr2O4 from a combination of CHFS and subsequent heat-treatment. In the same figure, DSC measurements identified two endothermic events that occurred below 400 °C (at 80 and 200 °C, respectively). These two events were assigned to the loss of surface-adsorbed water (80 °C) and the conversion of a hydroxide to oxide (200 °C), respectively. Significant mass loss ended by 500 °C and suggested the formation of the oxide was effectively complete by this temperature. This endotherm was consistent with similar endothermic processes observed in batch hydrothermally prepared samples (attributed to hydroxyl conversion to oxide).18 In the temperature range 200 to 1000 °C, a large exotherm was observed (maximum at ∼600 °C), suggesting crystallization and particle growth of the MgCr2O4 phase occurred smoothly across this temperature range. Therefore, these results suggest that the crystallinity and particle size of MgCr2O4 can be altered by careful selection of annealing temperature and time.
The powder X-ray diffraction pattern for MCO(ΔBHS) nanocrystals was a good match to the standard reference pattern of spinel-type MgCr2O4 (Fig. 1a, PDF No. 00-010-351), with a Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and a cubic structure. However, with regards to the powder X-ray diffraction data of MCO(CHFS) in comparison to the reference pattern, the peak intensity ratios were different and the (220) and (511) Bragg reflections were not evident. These differences suggested that MCO(CHFS) possessed a highly defective cubic spinel structure. The unit cell parameters, calculated by Pawley refinement using the Fdm space group, were a = 8.30411 Å and 8.32942 Å for MCO(CHFS) and MCO(BHS) samples, respectively (Fig. S4a, 4b, Table S1†). The a parameter of MCO(BHS) samples agreed well with literature values (a = 8.3378(3) Å, using standard reference pattern PDF No. 00-010-351), whereas the defective nature of the MCO(ΔCHFS) sample resulted in the slightly smaller a value. The evaluation of morphology and size of the as-synthesized materials, was carried out using electron microscopy. This confirmed that MCO(ΔCHFS) nanoparticles had a distorted-sphere shape, with an average particle size of 3.7 ± 2.4 nm (300 particles sampled), whereas MCO(ΔBHS) nanoparticles were cube shaped with a particle size of 7.2 ± 2.0 nm (100 particles sampled) (Fig. 1c). To the authors’ knowledge, the particle size of MCO(ΔCHFS) is the smallest ever reported for this phase, with an extremely high specific surface area of 331 m2 g−1, as measured from experiments of N2 adsorption (Fig. S5†). In contrast, MCO(ΔBHS) possessed a significantly smaller specific surface area of 76 m2 g−1.
m space group and a cubic structure. However, with regards to the powder X-ray diffraction data of MCO(CHFS) in comparison to the reference pattern, the peak intensity ratios were different and the (220) and (511) Bragg reflections were not evident. These differences suggested that MCO(CHFS) possessed a highly defective cubic spinel structure. The unit cell parameters, calculated by Pawley refinement using the Fdm space group, were a = 8.30411 Å and 8.32942 Å for MCO(CHFS) and MCO(BHS) samples, respectively (Fig. S4a, 4b, Table S1†). The a parameter of MCO(BHS) samples agreed well with literature values (a = 8.3378(3) Å, using standard reference pattern PDF No. 00-010-351), whereas the defective nature of the MCO(ΔCHFS) sample resulted in the slightly smaller a value. The evaluation of morphology and size of the as-synthesized materials, was carried out using electron microscopy. This confirmed that MCO(ΔCHFS) nanoparticles had a distorted-sphere shape, with an average particle size of 3.7 ± 2.4 nm (300 particles sampled), whereas MCO(ΔBHS) nanoparticles were cube shaped with a particle size of 7.2 ± 2.0 nm (100 particles sampled) (Fig. 1c). To the authors’ knowledge, the particle size of MCO(ΔCHFS) is the smallest ever reported for this phase, with an extremely high specific surface area of 331 m2 g−1, as measured from experiments of N2 adsorption (Fig. S5†). In contrast, MCO(ΔBHS) possessed a significantly smaller specific surface area of 76 m2 g−1.
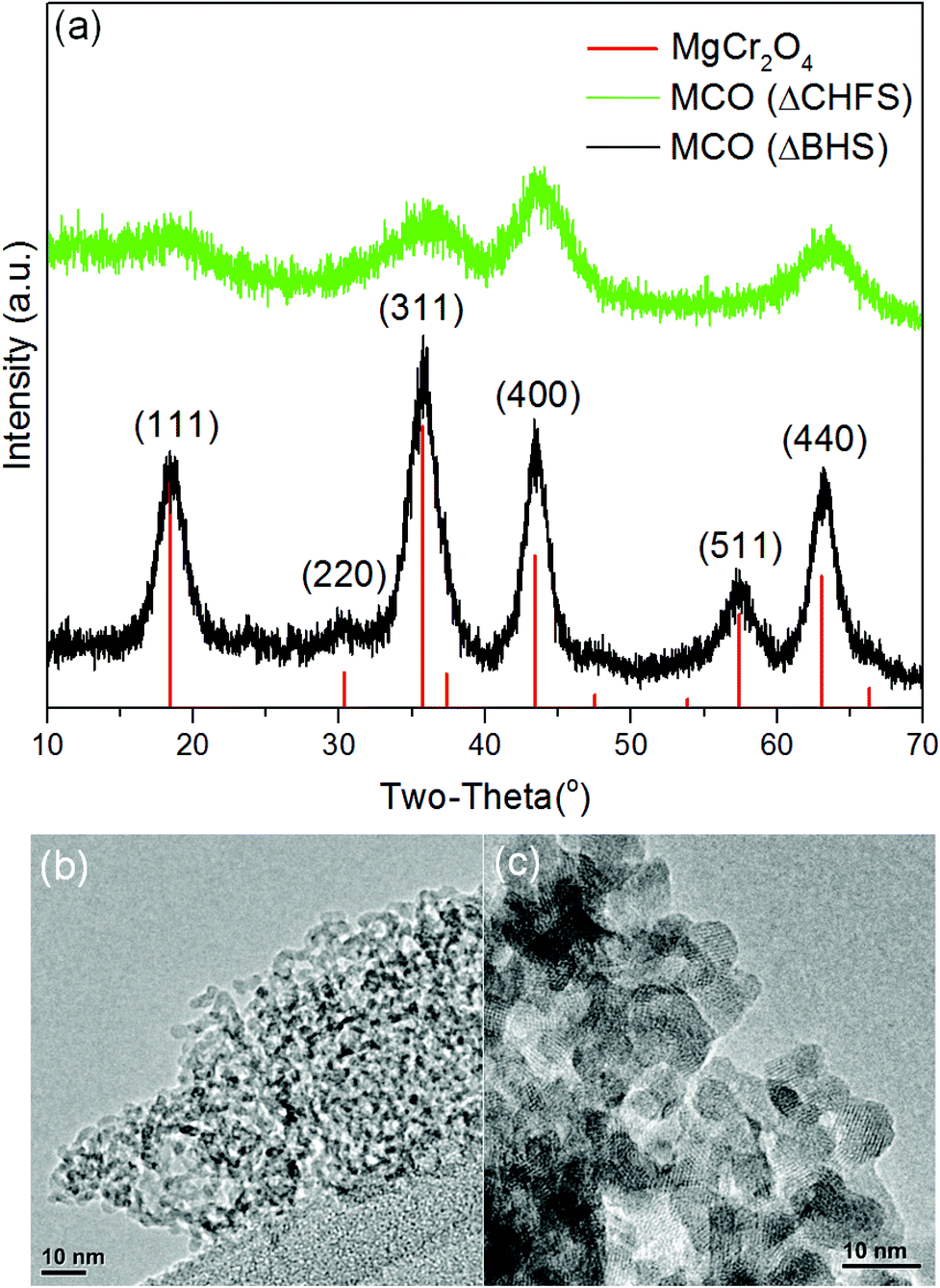 | ||
| Fig. 1 (a) Powder X-ray diffraction (λ = 1.5406 Å) patterns of the heat-treated magnesium chromium oxide (MCO) nanocrystals, including a MgCr2O4 spinel standard XRD pattern (red line, PDF No. 00-010-351), the batch hydrothermal sample MCO(ΔBHS) (black line), and the Continuous Hydrothermal Flow Synthesis sample MCO(ΔCHFS) (green line). MCO(ΔBHS) matched the main diffraction peaks of the reference at (111), (220), (311), (400), (511), and (440) Miller indices. MCO(ΔCHFS) matched the main diffraction peaks of the reference at (111), (311), (400), and (440). (b) Representative TEM image of MCO (ΔCHFS) nanocrystals. (c) Representative TEM image of MCO(ΔBHS) nanocrystals. | ||
All electrochemical experiments were conducted using the magnesium chromium oxide as the cathode and activated carbon (AC) as the anode. The electrolyte used was an ionic liquid (IL) composed of Mg[N(SO2)2(CF3)2]2-(C9H20N)N(SO2)2(CF3)2 (abbreviated as MgTFSI2-PY14TFSI). This electrolyte had a very low H2O content (∼43 ppm).31 Also, this electrolyte has high anodic and thermal stability.32 MgTFSI2-PY14TFSI was used to prevent convolution of electrochemical signals from competing reactions. In turn, the incompatibility of the IL with Mg metal, required the use of AC as the counter electrode. The charge profile of the cell with MCO(ΔCHFS), Fig. 2a, exhibited an increase in potential from 2.7 to 3.5 V vs. Mg/Mg2+ where a charge capacity of 120 mA h g−1 was observed. The total charge capacity was 44% of the theoretical capacity (∼270 mA h g−1), assuming partial demagnesiation of Mg2+ from the host and partial oxidation of Cr3+ to Cr4+. The discharge profile of MCO(ΔCHFS) in Fig. 2b presented a progressive voltage drop from 2.7 to 0.1 V vs. Mg/Mg2+ (270 mA h g−1). The charge profile of the cell with MCO(ΔBHS), Fig. 2c, demonstrated a faster increase in potential than MCO(ΔCHFS) before 3.3 V vs. Mg/Mg2+ (30 mA h g−1) and a slower increase from 3.3 to 3.4 V (110 mA h g−1). The corresponding discharge profile of MCO(ΔBHS) (Fig. 2d) showed a faster drop in potential than MCO(ΔCHFS) from 2.7 to 0.9 V vs. carbon (30 mA h g−1) and a slower drop from 0.9 to 0.6 V vs. carbon (270 mA h g−1).
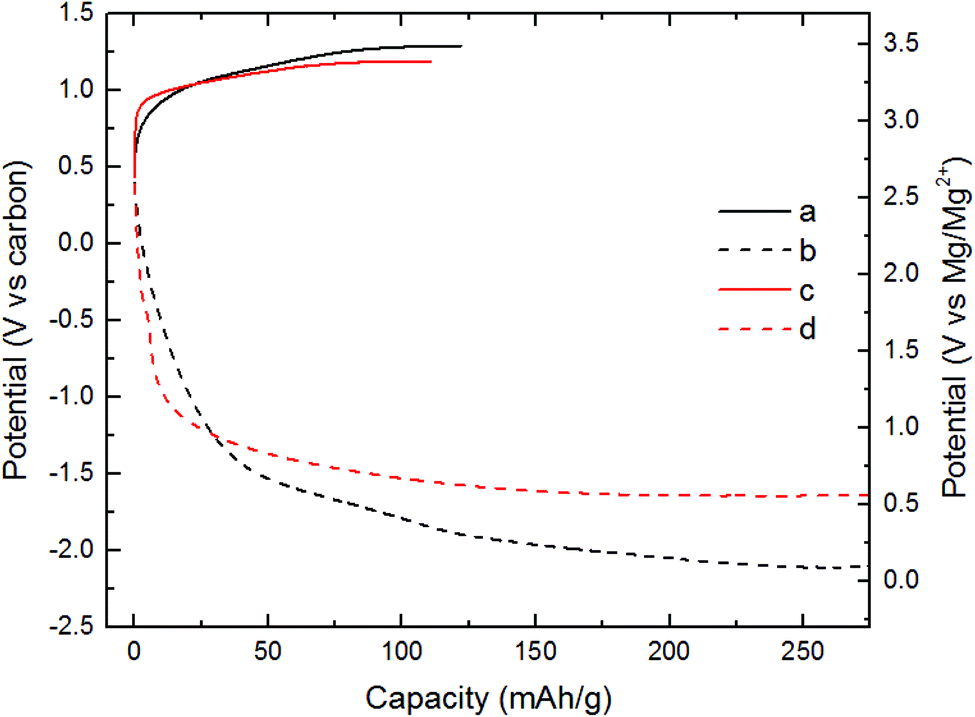 | ||
| Fig. 2 Electrochemical evaluation of nanocrystals made by different synthesis methods. Specific capacity vs. potential profile of (a) MCO(ΔCHFS) charged, (b) MCO(ΔCHFS) discharged, (c) MCO(ΔBHS) charged, and (d) MCO(ΔBHS) discharged. Electrodes were charged and discharged in a Mg2+ electrolyte, at 110 °C. | ||
To further track possible electrochemically-induced phase transformations, high-resolution powder XRD was collected for pristine MCO(ΔCHFS) and MCO(ΔBHS), as well as powders from electrodes harvested from charged electrochemical cells. After charging, the recovered powder for MCO(ΔCHFS) showed a clear shift to higher 2θ values for the (111) peak (Fig. 3), consistent with a contraction in the unit cell that was assumed to be due to the deintercalation of Mg2+ from the structure. The unit cell parameters, calculated by Pawley refinement using the Fd3m space group, were a = 8.30591 and 8.29574 Å for the MCO(ΔCHFS) pristine and charged samples, respectively (Fig. S4c, S4d, Table S1†). The demagnesiation after charge was implied in the bulk phase, with the decrease of unit cell volume. A concurrent ∼45% decrease in the Mg:Cr ratio was found from Transmission Electron Microscopy-Energy-Dispersive X-ray Spectroscopy (TEM-EDX) analysis of the particles (Fig. S6†). In contrast, XRD analysis for the pristine and charged MCO(ΔBHS) powder samples, showed no clear differences. However, EDX still demonstrated a ∼40% decrease in the Mg:Cr ratio (Fig. S7†). As there was no bulk Mg removal/insertion observed, this suggested that electrochemical oxidation decomposed the MgCr2O4 structure with selective dissolution of Mg2+, especially at the surface, a process that likely involved the electrolyte.
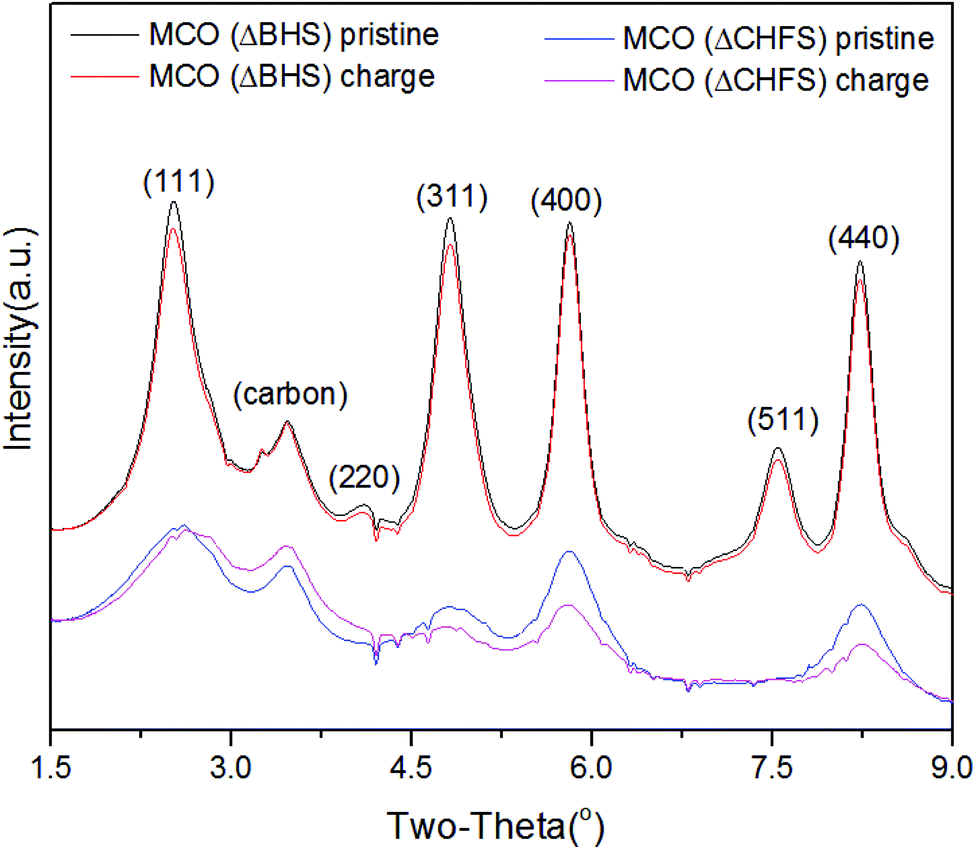 | ||
| Fig. 3 High resolution XRD diffraction data in the narrow 2 theta range 1.5 to 9 degrees for magnesium chromium oxide electrodes in pristine and charged states. The carbon peak arises from the conductive additive within the electrode. Wavelength = 0.2114 Å. | ||
Cr K-edge X-ray Absorption Near Edge Structure (XANES) in transmission mode, was used to probe the redox states in the bulk ensemble of the MgCr2O4 materials after charge/discharge. The changes in intensity of the pre-edge feature of MCO(ΔCHFS) (region A, Fig. 4a), indicated Cr oxidation and reduction took place during charge and discharge, respectively. The corresponding derivative profile of the Cr K-edge in Fig. S8a† was used to follow changes in position of the absorption threshold, for instance, through the inflection point (local maximum in the derivative) around 6000 eV. It demonstrated that the absorption energy shifted to higher and lower energies with charge and discharge, respectively, further confirming Cr redox activity during cycling; the changes would be consistent with at least partial oxidation/reduction reactions between Cr3+ and Cr4+.33 Fully demagnesiated Cr2O4 (Cr4+ only) has been predicted computationally to be unstable and, therefore, potentially not electrochemically attainable.34 The peak intensity in region A, showed a subtle, yet significant increase upon charge, indicative of changes in the centro-symmetry of the coordination environment of Cr. The changes were only partly reversed by discharging the electrode, as revealed, for instance, by the higher intensity of region A compared to the pristine state. In the case of MCO(ΔBHS), the difference in peak positions between pristine, charge and discharge, was negligible (Fig. 4b), which was further confirmed by the derivative profile (Fig. S8b†). Therefore, little or no redox activity occurred in the BHS ensemble during charge/discharge processes. This strongly suggests the highly defective nature of MCO(ΔCHFS) allowed some reversible Cr3+–Cr4+ redox chemistry to be accessed, in contrast to MCO(ΔBHS).
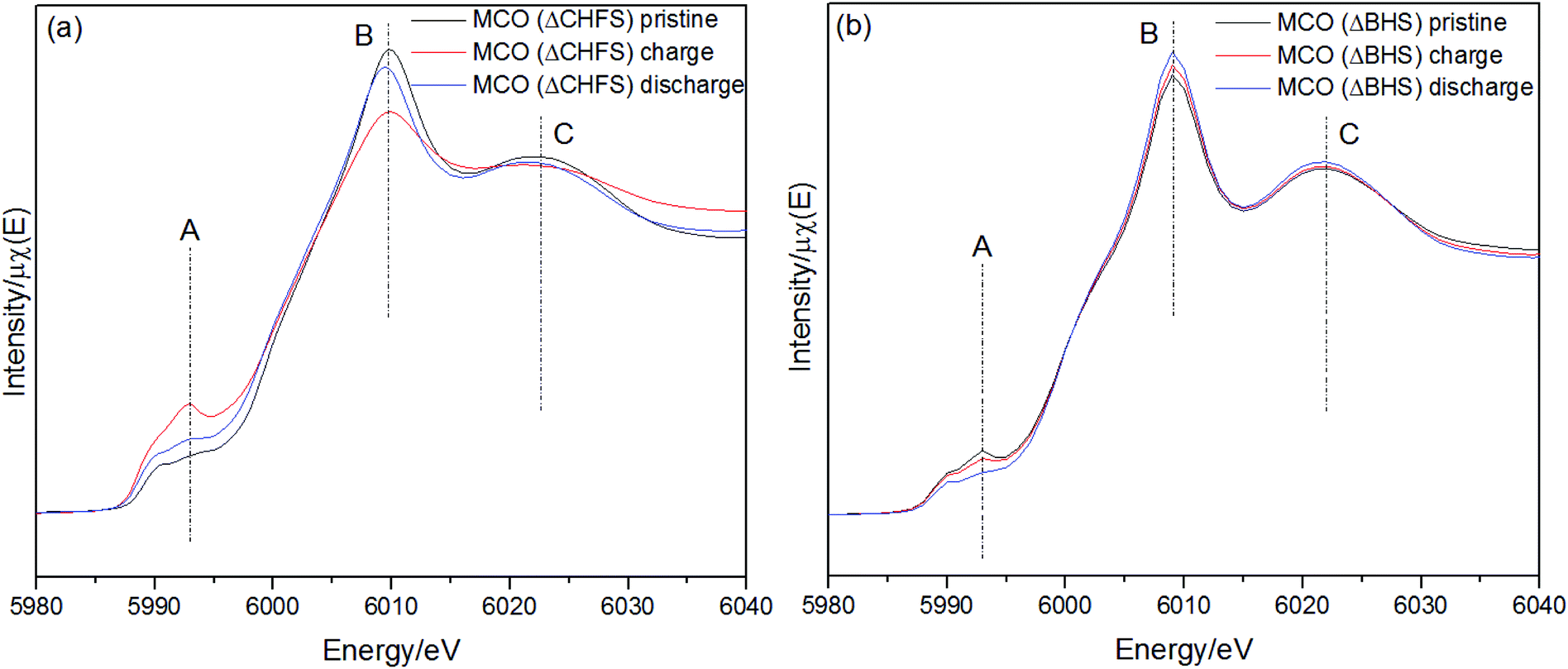 | ||
| Fig. 4 Cr K-edge XANES data (transmission mode) collected for the pristine, charge, and discharged states (a) MCO(ΔCHFS) and (b) MCO(ΔBHS) electrodes. | ||
Extended X-ray Absorption Fine Structure (EXAFS) analysis provided local structure information of the Cr in MCO(ΔCHFS) nanoparticles (Fig. 5a). The Fourier transform data revealed average Cr–O and Cr–Cr bond lengths (before phase shift) of ∼1.5 and ∼2.5 Å, respectively, in the pristine compound. In the material that was analyzed after charging, the Cr–O bond lengths were reduced and the amplitude of the Cr–O signal decreased. These results indicate that charging resulted in oxidation of Cr3+ and a contraction of the Cr–O bond as a result of Mg2+ deintercalation, consistent with the increase in pre-edge intensity (Fig. 4a). The Cr–O bond lengths increased again for the discharged MCO(ΔCHFS) material, suggesting redox activity was, at least, partially reversible. A subtly narrower spread of Cr–Cr bond lengths was also observed, suggesting small changes in the defect structure of the material. Unfortunately, this issue could not be further assessed with cycling due to the anodic instability of the electrolyte, which prevented the collection of multiple cycles. It is suggested that this is an important issue for future study subsequent to the development of more stable electrolytes. In comparison, no significant change was observed in the MCO(ΔBHS) sample (Fig. 5b). This result was consistent with the XRD and XANES analyses, suggesting that Mg2+ was not removed from or inserted into MCO(ΔBHS) during charge/discharge processes.
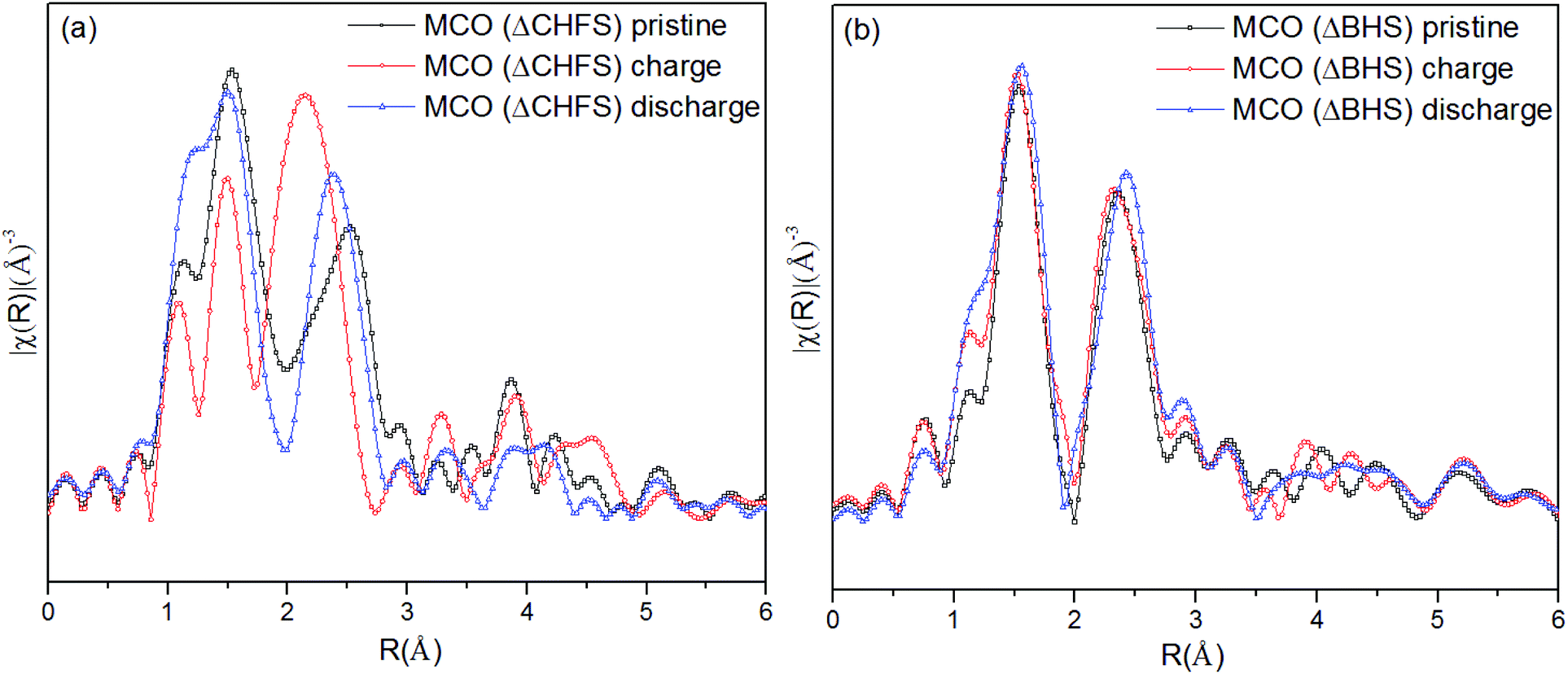 | ||
| Fig. 5 The R-weighted Fourier transform of the Cr K-edge X-ray Absorption Spectroscopy data of (a) MCO(ΔCHFS) and (b) MCO(ΔBHS) electrodes collected for pristine, charged, and discharged materials. | ||
X-ray Absorption Spectroscopy (XAS) at the Cr L2,3-edge using a TEY detector, further provided insight into the possible changes in formal redox state of the transition metal at the surface of the electrodes. These edges directly probe unfilled 3d states, through promotion of 2p electrons. The pristine electrode of sample MCO(ΔCHFS) showed the L3 absorption centered at ∼576.3 eV and the L2 absorption at ∼584.9 eV, as indicted in Fig. 6a. The complex spectra could be broadly divided in four fine absorption lines, numbered 1 to 4. These features subtly shifted to higher energies after charging of MCO(ΔCHFS), consistent with Cr3+ oxidizing to Cr4+ (Fig. S9a†).35 This change was reversed after discharge, further supporting evidence of reversible Cr redox activity with Mg deintercalation/intercalation (Fig. S9b†). In turn, no peak shift was observed with charge or discharge for the MCO(ΔBHS) electrode material (Fig. 6b), but rather a slight peak intensity decrease (line 4) after charge, which increased again with discharge. This indicated that the Cr oxidation state was unchanged, but there may have been some changes in local coordination around the Cr atoms.
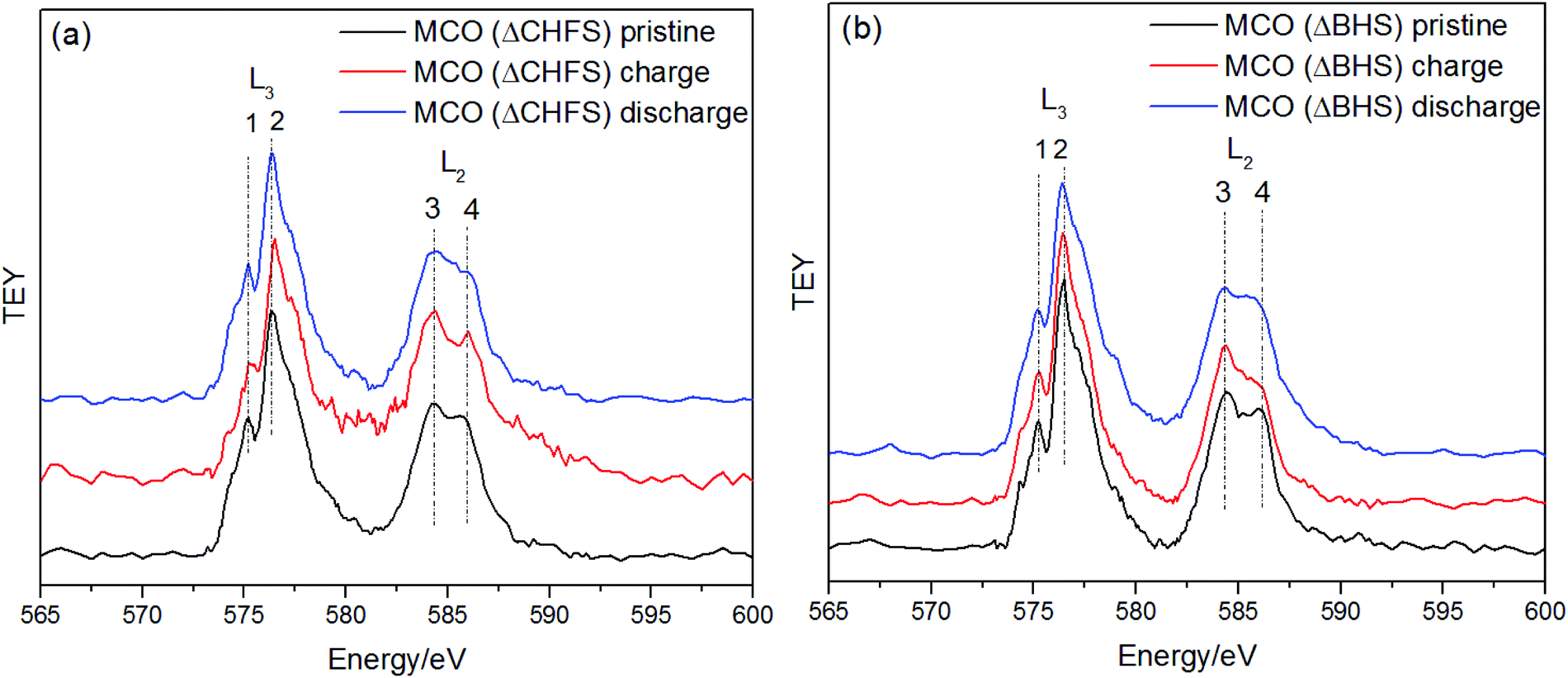 | ||
| Fig. 6 Cr L2,3-edge X-ray Absorption Spectroscopy of (a) MCO (ΔCHFS) and (b) MCO (ΔBHS) electrodes, in TEY mode collected in the pristine, charged, and discharged states. | ||
Overall, the results of XRD, XANES, EXAFS and EDX analysis on charged and discharged electrodes of MCO(ΔCHFS) were consistent with bulk Mg2+ removal and insertion, with accompanying Cr redox activity. In contrast, there was no evidence of this in MCO(ΔBHS), which seemingly only displayed decomposition reactions, likely in concert with the electrolyte at the surface. It is conceivable that MCO(ΔCHFS) allowed reversible Mg2+ bulk removal by the exploitation of diffusion pathways of lower activation energy, unlocked by its highly defective structure compared to the ordered spinel structure of MCO(ΔBHS), which only allowed surface reactions despite the small particle size. The high specific surface area of the powders of MCO(ΔCHFS), may further enhance Mg2+ removal and insertion kinetics, but it is worth noting that, ultimately, the electrochemically active surface area was similar in both spinel electrodes, as probed by the similar values of specific capacity (Fig. 2). The difference between the two was instead rooted in the specific reactions leading to the measured capacities.
Conclusion
Batch Hydrothermal Synthesis (BHS) and a Continuous Hydrothermal Flow Synthesis (CHFS) plus a flash heat-treatment method, were used to successfully manufacture crystalline <10 nm MgCr2O4 particles with an ordered or a highly defective cubic spinel structure, respectively. These crystal sizes were significantly smaller than previously reported in the literature, especially compared to those made using more conventional solid-state techniques. Electrochemical evaluation in cells with non-aqueous Mg2+ electrolytes, at 110 °C, suggested that Mg could be successfully removed and inserted from the bulk structure of highly-defective MgCr2O4 (made via CHFS and flash-heating step), as verified by a combination of EDX, XRD and XAS. In comparison, an ordered cubic MgCr2O4 (made via the BHS method) displayed loss of Mg without accompanying structural or redox changes, suggesting different mechanisms of reaction in the two cells. This suggested the defective cubic spinel structure (made via CHFS) may have facilitated new diffusion pathways for Mg2+ ions with a lower activation energy barrier compared to the ordered crystalline cubic phase, modulated by microstructural effects from different particle size and powder surface area. Experiments with other electrolyte systems are required to explore the inherent nature of the electrochemical activity of the CHFS nanocrystals. These results will enrich our understanding of Mg2+ intercalation reactions in oxides, and hint at strategies to overcome existing bottlenecks. In the future, it is suggested that the development of distorted/defective spinel structures should be investigated further as potential cathode materials.Author contributions
LH and IDJ jointly wrote the manuscript and synthesized samples (assistance from LM for CHFS, and TEA who designed and built the flash heating apparatus). LH characterized materials by XRD, TEM, Cr LII, III-edge XAS and electrochemical measurements. JWF and GN provided assistance with the measurement and analysis of Cr LII, III-edge XAS. SK and TTF collected and analyzed Cr K-edge XAS data. HDY designed the electrochemical test methods. JC leads the Mg battery research team at the University of Illinois at Chicago. JAD is academic lead of the synthesis team at University College London, developed the CHFS process, and is a coinventor of the CJM mixer that was used in this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported as part of the Joint Center for Energy Storage Research (JCESR), an Energy Innovation Hub funded by the US Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under contract DE-AC02-05CH11231. IDJ, TEA, LM and JAD would like to thank the EPSRC for funding the JUICED Energy Hub (EP/R023662/1) and the ELEVATE (Electrochemical Vehicle Advanced Technology; P/M009394/1) project. IDJ would also like to thank the Materials Modelling and Molecular Doctoral Training Centre (EP/G036675/1), and the STFC for providing funding support for travel within the collaboration (STFC/MDC Futures Early Career Award, ST/N002385/1).References
- P. Canepa, G. S. Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- D. Eroglu, S. Ha and K. G. Gallagher, J. Power Sources, 2014, 267, 14–19 CrossRef CAS.
- E. Levi, Y. Gofer and D. Aurbach, Chem. Mater., 2010, 22, 860–868 CrossRef CAS.
- D. Aurbach, I. Weissman, Y. Gofer and E. Levi, Chem. Rec., 2003, 3, 61–73 CrossRef CAS PubMed.
- M. E. Spahr, P. Novak, O. Haas and R. Nesper, J. Power Sources, 1995, 54, 346–351 CrossRef CAS.
- Z. G. Wang, Q. L. Su and H. Q. Deng, Phys. Chem. Chem. Phys., 2013, 15, 8705–8709 RSC.
- W. Kaveevivitchai and A. J. Jacobson, Chem. Mater., 2016, 28, 4593–4601 CrossRef CAS.
- M. Liu, Z. Q. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder and K. A. Persson, Energy Environ. Sci., 2015, 8, 964–974 RSC.
- M. M. Thackeray, P. J. Johnson, L. A. Depicciotto, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1984, 19, 179–187 CrossRef CAS.
- C. Kim, P. J. Phillips, B. Key, T. H. Yi, D. Nordlund, Y. S. Yu, R. D. Bayliss, S. D. Han, M. N. He, Z. C. Zhang, A. K. Burrell, R. F. Klie and J. Cabana, Adv. Mater., 2015, 27, 3377–3384 CrossRef CAS PubMed.
- C. Kim, A. A. Adil, R. D. Bayliss, T. L. Kinnibrugh, S. H. Lapidus, G. M. Nolis, J. W. Freeland, P. J. Phillips, T. H. Yi, H. D. Yoo, B. J. Kwon, Y. S. Yu, R. Klie, P. J. Chupas, K. W. Chapman and J. Cabana, Chem. Mater., 2018, 30, 1496–1504 CrossRef CAS.
- G. S. Gautam, P. Canepa, A. Urban, S. H. Bo and G. Ceder, Chem. Mater., 2017, 29, 7918–7930 CrossRef.
- H. S. Oneill and W. A. Dollase, Phys. Chem. Miner., 1994, 20, 541–555 CrossRef CAS.
- K. Nagata, R. Nishiwaki, Y. Nakamura and T. Maruyama, Solid State Ionics, 1991, 49, 161–166 CrossRef CAS.
- S. P. Li, X. L. Jia and Y. F. Qi, Adv. Mater. Res., 2011, 284–286, 730–733 CAS.
- L. V. Morozova and V. P. Popov, Glass Phys. Chem., 2010, 36, 86–91 CrossRef CAS.
- S. K. Durrani, S. Naz, M. Nadeem and A. A. Khan, J. Therm. Anal. Calorim., 2014, 116, 309–320 CrossRef CAS.
- J. A. Darr, J. Y. Zhang, N. M. Makwana and X. L. Weng, Chem. Rev., 2017, 117, 11125–11238 CrossRef CAS PubMed.
- R. I. Gruar, C. J. Tighe and J. A. Darr, Ind. Eng. Chem. Res., 2013, 52, 5270–5281 CrossRef CAS.
- I. D. Johnson, M. Lubke, O. Y. Wu, N. M. Makwana, G. J. Smales, H. U. Islam, R. Y. Dedigama, R. I. Gruar, C. J. Tighe, D. O. Scanlon, F. Cora, D. J. L. Brett, P. R. Shearing and J. A. Darr, J. Power Sources, 2016, 302, 410–418 CrossRef CAS.
- I. D. Johnson, E. Blagovidova, P. A. Dingwall, D. J. L. Brett, P. R. Shearing and J. A. Darr, J. Power Sources, 2016, 326, 476–481 CrossRef CAS.
- I. D. Johnson, M. Loveridge, R. Bhagat and J. A. Darr, ACS Comb. Sci., 2016, 18, 665–672 CrossRef CAS PubMed.
- M. Lubke, J. H. Shin, P. Marchand, D. Brett, P. Shearing, Z. L. Liu and J. A. Darr, J. Mater. Chem. A, 2015, 3, 22908–22914 RSC.
- J. Chouler, G. A. Padgett, P. J. Cameron, K. Preuss, M. M. Titirici, I. Ieropoulos and M. Di Lorenzo, Electrochim. Acta, 2016, 192, 89–98 CrossRef CAS.
- M. Luebke, N. Ding, M. J. Powell, D. J. L. Brett, P. R. Shearing, Z. L. Liu and J. A. Darr, Electrochem. Commun., 2016, 64, 56–60 CrossRef.
- W. Wagner and A. Pruss, J. Phys. Chem. Ref. Data, 2002, 31, 387–535 CrossRef CAS.
- A. J. Kropf, J. Katsoudas, S. Chattopadhyay, T. Shibata, E. A. Lang, V. N. Zyryanov, B. Ravel, K. McIvor, K. M. Kemner, K. G. Scheckel, S. R. Bare, J. Terry, S. D. Kelly, B. A. Bunker and C. U. Segre, AIP Conf. Proc., 2010, 1234, 299 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 3 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- G. Vardar, A. E. S. Sleightholme, J. Naruse, H. Hiramatsu, D. J. Siegel and C. W. Monroe, ACS Appl. Mater. Interfaces, 2014, 6, 18033–18039 CrossRef CAS PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- A. Pantelouris, H. Modrow, M. Pantelouris, J. Hormes and D. Reinen, Chem. Phys., 2004, 300, 13–22 CrossRef CAS.
- T. N. Chen, G. S. Gautam, W. X. Huang and G. Ceder, Chem. Mater., 2018, 30, 153–162 CrossRef CAS.
- Y. S. Dedkov, A. S. Vinogradov, M. Fonin, C. Konig, D. V. Vyalikh, A. B. Preobrajenski, S. A. Krasnikov, E. Y. Kleimenov, M. A. Nesterov, U. Rudiger, S. L. Molodtsov and G. Guntherodt, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 4 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr08347a |
| ‡ The contribution of LH and IDJ to this work was equal. |
| This journal is © The Royal Society of Chemistry 2019 |