
Insights into the adsorption mechanism and dynamic behavior of tetracycline antibiotics on reduced graphene oxide (RGO) and graphene oxide (GO) materials†
Yuejie
Ai
 *a,
Yang
Liu
a,
Yingzhong
Huo
a,
Chaofeng
Zhao
a,
Lu
Sun
b,
Bing
Han
a,
Xinrui
Cao
c and
Xiangke
Wang
a
*a,
Yang
Liu
a,
Yingzhong
Huo
a,
Chaofeng
Zhao
a,
Lu
Sun
b,
Bing
Han
a,
Xinrui
Cao
c and
Xiangke
Wang
a
aMOE Key Laboratory of Resources and Environmental System Optimization, College of Environmental Science and Engineering, North China Electric Power University, Beijing 102206, P. R. China. E-mail: aiyuejie@ncepu.edu.cn; Fax: +86 10 61772890; Tel: +86 10 61772890
bInstitute of Modern Optics, Nankai University, Tianjin, 300350, P.R. China
cDepartment of Physics and Collaborative Innovation Center for Optoelectronic Semiconductors and Efficient Devices, Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, Xiamen University, Xiamen 361005, P.R. China
First published on 16th September 2019
Abstract
The widespread use of antibiotics from both agricultural and human sources has led to their environmental dissemination which is now recognized as a potential hazard to human health and aquatic ecosystems. Along with the extensive academic and social concerns on the impact of this new kind of emerging pollutant, the knowledge on their effective removal from the environment is increasing. However, the complex interactions between antibiotics and sorbents make the experimental studies difficult at the molecular level. To provide an insight into the adsorption mechanism and dynamic behavior of antibiotics, in this work, three tetracycline molecules, namely tetracycline (TTC), oxytetracycline (OTC), and chlortetracycline (CTC), have been chosen as the representative antibiotics to present a theoretical study on their adsorption properties by reduced graphene oxide (RGO) and graphene oxide (GO). The density functional theory (DFT) method and molecular dynamics (MD) simulations were used to address a number of key issues, such as the effects of distinct adsorption sites, pH, and solvent on the adsorption capacity. A closer look at the adsorption configuration and binding energy showed that the π–π interaction was the driving force when TCs adsorbed on GO (or RGO), and hydrogen bonds played a significant role in the GO_TC systems. The computed results showed that the tetracycline adsorption affinity for the graphene-based materials followed the order CTC > TTC > OTC and TTC > CTC > OTC in the GO and RGO systems, respectively. The comparison of binding energies at different pH values and solvents proposed that low pH and less polar solvent environments were beneficial to the adsorption efficiency of TCs on GO and RGO. In addition, molecular dynamics simulations have been used to assign the dynamic behavior of the TCs, analyzing the competitive adsorption process, and the intermolecular accumulation was verified to be involved in the adsorption behavior of TCs. The CTC molecule appeared to exhibit the strongest competitiveness. Our work gives a deep insight into the interactions between the graphene-based materials and TCs, and provides a theoretical basis for the further design of adsorbents used for the removal of tetracycline antibiotics in the environment.
Environmental significanceThe environmental risks of antibiotics have attracted increasing attention due to their potential adverse effects on the environment. The complex interactions and especially the dynamic behaviors such as the competitive adsorption process make the experimental studies difficult at the molecular level. This work investigated the adsorption behavior of three kinds of representative tetracycline antibiotics (tetracycline, oxytetracycline, and chlortetracycline) on GO (or RGO) via the density functional theory (DFT) method and molecular dynamics (MD) simulations. This work provides a theoretical basis for the removal of tetracycline antibiotics in the environment. |
1. Introduction
In recent years, antibiotics have been widely used in human therapy, livestock industries, modern husbandry and aquaculture.1,2 However, due to the poor metabolism of antibiotics in organisms, a significant percentage (about 60–90%) of antibiotics were excreted into the ecosystem in the form of either a parent compound or metabolite via a variety of sources,3–5 including expired prescriptions, animal excretion,6,7 agricultural waste storage and disposal in manufacturing processes.8,9 The environmental dissemination of various antibiotics has led to an increasing academic and social concern on their adverse effects on human health and epigeic and aquatic ecosystems. Tetracyclines (TCs), as one kind of great antibiotics,10 including tetracycline (TTC), oxytetracycline (OTC), chlortetracycline (CTC) and their derivatives, are toxic and antimicrobial resistant.11,12 TCs are composed of a parent naphthalene ring as shown in Scheme 1. Since they have already been detected in agricultural fields, surface waters and even drinking water,13 the TCs can reach the human body through the food chain or drinking water, inducing damage to the human liver system and interfering with the physiological function of human bodies.14 As a result, over the years, researchers have devoted effort to studying the distribution, fate, metabolism and transport of TCs in the environment. Moreover, developing and applying efficient technologies to remove the TCs from the environment have emerged as an expanding research topic.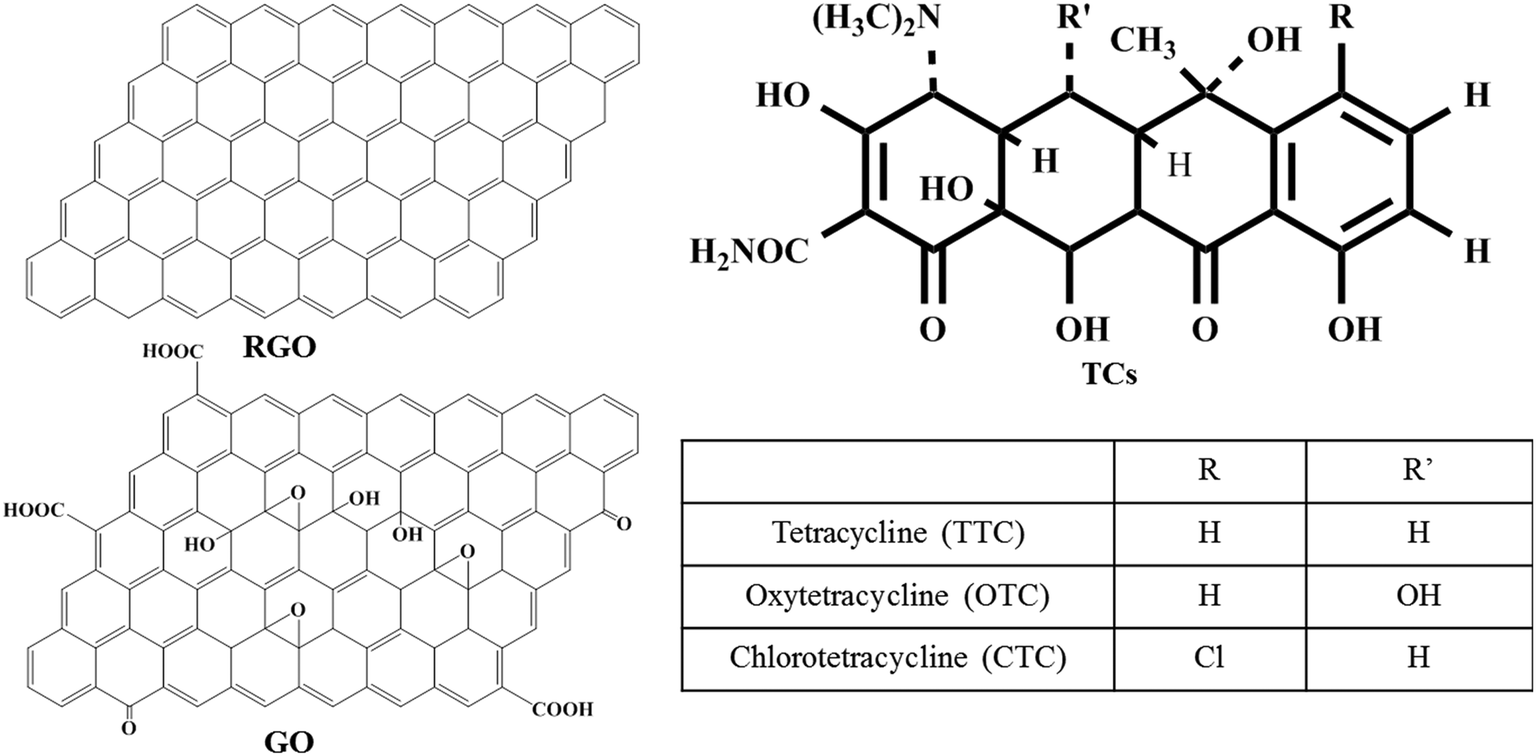 | ||
| Scheme 1 Schematic presentation of RGO, GO and TC molecules. | ||
At present, many methods have been applied to the treatment of tetracycline antibiotics in the environment such as adsorption,15 photodegradation,16 microbial degradation17 and so on. Among these methods, sorption18–27 is widely practiced as a low cost, high efficiency technology for the removal of organic contaminants in wastewater treatment. Recently, graphene-based materials such as graphene oxide (GO) and reduced graphene oxide (RGO) (the schematic presentations are shown in Scheme 1) have attracted special interest in the removal of contaminants due to their unique structure and physicochemical characteristics;28–32 graphene oxide (GO) and reduced graphene oxide (RGO) have also been applied to remove the antibiotics in the environment and provided remarkable sorption performance then. For instance, Yadav's group33 used GO as a potential effective adsorbent for the elimination of fluoroquinolones (ciprofloxacin, norfloxacin, ofloxacin) and they found out that the pH of the solution was the most crucial parameter in the adsorption process. Three typical antibiotics have been efficiently removed by graphene-based adsorbents in Zhang's studies,34 which also showed that the optimal adsorption pH values for NOR, SDZ, and TC were 6.2, 4.0, and 4.0, respectively. Particularly, for TCs, Ghadim and coworkers10 achieved a high adsorption capacity of 323 mg g−1 towards tetracycline by GO from an aqueous solution. Song's group35 prepared RGO for the removal of tetracycline, and the maximum adsorption capacity they obtained was 219.10 mg g−1 under the condition of pH = 6. Gao et al.19 investigated the removal of tetracyclines using GO by batch experiments, and the maximum adsorption capacity of GO towards tetracycline and oxytetracycline was 313 mg g−1 and 212 mg g−1, respectively. They also found out that the adsorption capacity decreased with the increase of the pH value. In addition, Li et al.20 reported a novel magnetic graphene oxide (NDMGO) which exhibited excellent adsorption properties (356 mg g−1) towards tetracycline at 298 K. The former experimental studies10,19,20,34–36 presented the adsorption performance of graphene-based materials toward antibiotics and also deduced the adsorption mechanism from the experimental perspective, such as π–π interaction, cation–π bonding, etc.
Overall, although efficient adsorption capacities of GO and RGO for TCs have been extensively investigated experimentally, a significant gap still exists in the understanding of the interaction mechanism between tetracycline antibiotics and graphene-based materials. Obtaining such an underlying mechanism is difficult, which makes the experimental studies problematic, and it has sparked theoretical research that focused on the adsorption behavior of TCs on graphene-based materials. Peng et al.37 investigated the adsorption of TTC on a graphene surface based on first principles calculations, showing the importance of π–π interactions in the adsorption process. Another DFT calculation study by Zhang et al.34 explored the adsorption of TTC on GO and RGO and they proposed that the TTC molecule showed strong affinity with sp3 and sp2 regions. Besides, a specific configuration of TTC has been proposed by Song and coworkers35 in their theoretical investigation about RGOs. However, with the exception of a few research studies, there is a paucity of data on several key issues in the adsorption process of TCs onto graphene-based materials, such as the detailed geometric and electronic nature of different tetracyclines (TTC, OTC, and CTC) when they bind onto GO (or RGO). Meanwhile, since experimental results have shown that the pH value was the most crucial parameter among the other experimental conditions in the adsorption of TCs, the internal mechanism of this phenomenon is not clear and needs further elucidation. On the other hand, although the occurrence, fate and even removal of TCs have been extensively studied in the literature, their coexistence or competitive behavior is less publicly well understood when multiple pollutants coexist. In the natural environment, the coexistence of pollutants is very common and limited competitive adsorption experiments have only focused on the apparent adsorption phenomena, such as a decrease of adsorption capacity caused by competitive adsorption, or an increase caused by synergistic effect. Therefore, to sum up, it is crucial to use the means of theoretical methods and develop a thorough and comprehensive study to address major questions and concerns discussed above.
In this article, the density functional theory (DFT) method and molecular dynamics were carried out to investigate the geometric and electronic characters of tetracyclines (TCs) on graphene oxide (GO) and reduced graphene oxide (RGO). This study aims to (1) find out the most stable adsorption configuration of TCs on RGO and GO, and compare the adsorption performance of two adsorbents (RGO and GO) towards TCs; (2) explore the influence of low pH values and solvent on the adsorption process; (3) understand the competitive adsorption behavior of multiple tetracycline molecules on graphene materials (GO and RGO) by molecular dynamics calculations. This study can provide a theoretical basis for the application of graphene-based materials in the removal of tetracycline antibiotics.
2. Computational details
DFT calculations
To understand the binding characteristics between GO (or RGO) and TCs, a 7 × 6 supercell of graphene with a lattice constant of 1.402 Å was chosen as the computational model of RGO. In addition, to save computational costs, the same supercell modified with an epoxy group was taken as the representative model of GO. The electronic structures of adsorption complexes were obtained using the projector augmented-wave (PAW)38 potentials of DFT calculations that were implemented in Vienna ab-initio simulation package (VASP).39 The Perdew–Burke–Ernzerhof (PBE) version of the generalized gradient approximation (GGA) was employed to describe the exchange correlation potential and energy.40 In addition, the dispersive interaction was considered using the DFT-D2 method of Grimme.41 The cutoff radius for pair interactions and the global scaling factors S6 were 50 Å and 0.75, respectively. The C6 parameters were set to 1.75, 0.14, 0.70, 1.23, and 5.07 for C, O, H, N and Cl atoms, and the R0 parameters for C, O, H, N and Cl atoms were 1.452, 1.001, 1.342, 1.397 and 1.639, respectively. Besides, the convergence criterion for the total energy was set to 10−5 eV and the cut-off energy was chosen to be 400 eV. Meanwhile, all of the structures were optimized until the Hellmann–Feynman force tolerance was smaller than 0.02 eV Å−1 and the Brillouin-zone integrations were sampled with 3 × 3 × 1 Monkhorst–Pack special k-points. The adsorption energies (Ead) of the adsorbents (GO or RGO) towards TCs (TTC, OTC or CTC) were calculated according to the following formula:| Ead = E(adsorbents) + E(TCs) − E(adsorbents_TCs) |
Meanwhile, the solvation effect was calculated using the joint density functional theory framework as implemented in the VASP by Mathew and Hennig.42
Classical MD simulations
According to the above DFT calculations, the GO model used for the MD simulation contains epoxy groups with a size of 4.90 × 4.97 nm2 which is shown in Fig. S1.† For each kind of tetracycline antibiotic, eight molecules were inserted into a 5.10 × 5.04 × 5.00 nm3 simulation box that contains 3323 water molecules. Water molecules were simulated using the simple point charge (SPC) model.43 The optimized potentials for liquid simulations-all atoms (OPLS-AA)44 force field was used to parameterize the GO and TCs. Periodic boundary conditions were selected in all three directions of the box to eliminate adverse effects caused by boundary effects and other factors.45 The cutoff radius for the vdW interaction was set to 1.2 nm. After annealing and equilibration, the systems were run for 100 ns in an NPT ensemble which used a Berendsen thermostat and an isotropic Parrinello–Rahman barostat to control the constant temperature (T = 298 K) and pressure (P = 1 bar), respectively.46,47 The time-step was 1 fs and the trajectories were recorded every 10 ps. All the simulations were performed with the GROMACS software package.483. Results and discussion
3.1. Electrostatic potential (ESP) of adsorbents and TCs
The molecular electrostatic potential was usually used to describe the surface electron density and electron activity49,50 of a molecule. It has considerable fundamental significance in the study of molecular reactivity and intermolecular interaction, especially in non-covalent interactions. In order to investigate the possible active sites inside the TCs during the sorption, the electrostatic potentials have been plotted as shown in Fig. 1. The red region represented positive electrostatic potential and showed the electrophilic property, while the blue negative region was more nucleophilic.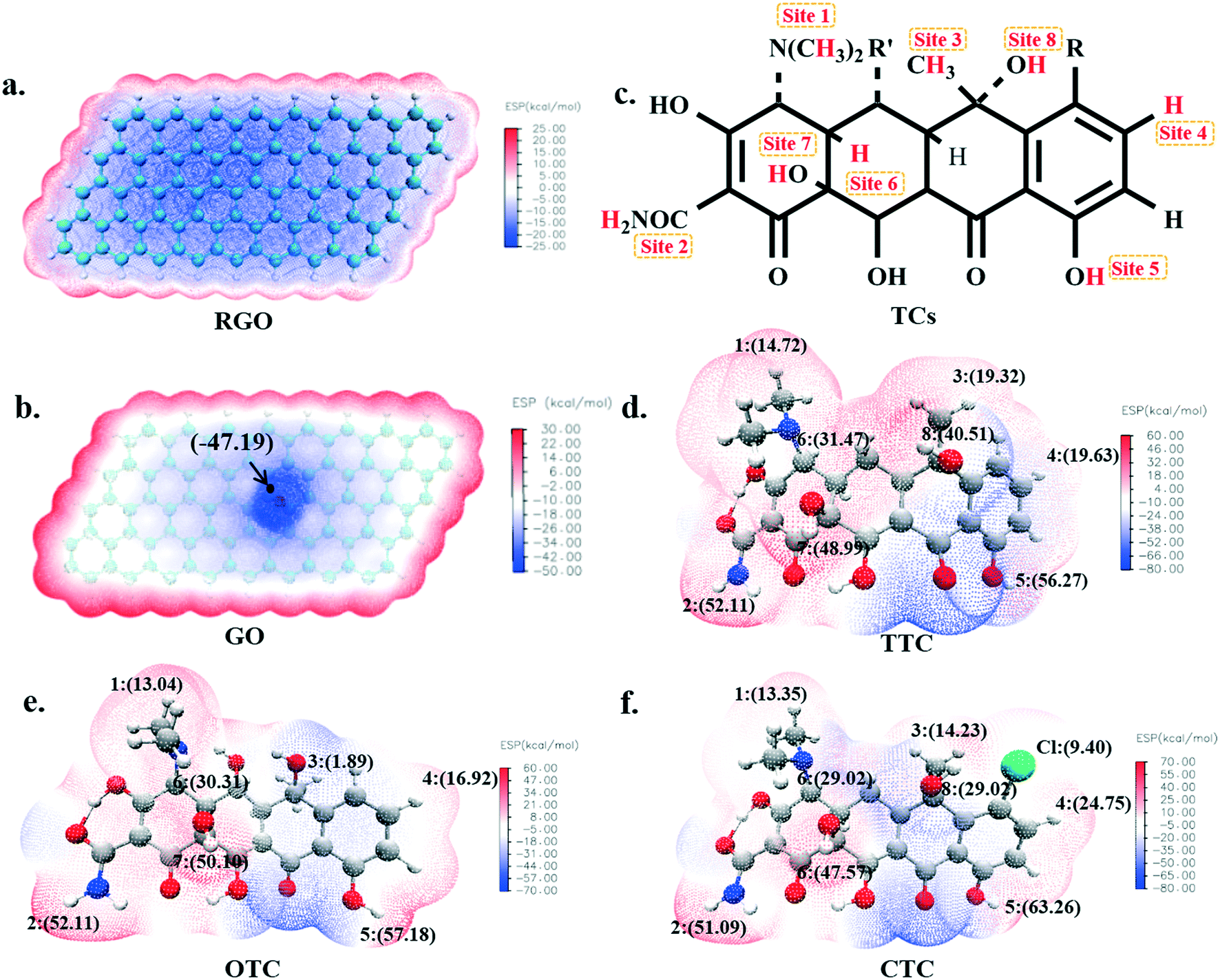 | ||
| Fig. 1 The electrostatic potential (ESP) distribution of RGO (a), GO (b), TTC (d), OTC (e) and CTC (f); the possible adsorption sites (positive ESP regions) on the surface of tetracycline molecules (c). | ||
The electrostatic potential map of RGO is shown in Fig. 1a, where homogeneous electron density with an average value of −24.15 kcal mol−1 was found, and abundant π electrons were present. It was proposed that the π–π electron-donor–acceptor (EDA) interactions and van der Waals forces were most likely to occur between the RGO and TCs due to their large ring structure;51 while for the GO, although the presence of oxygen-containing functional groups weakened the π-electron activity because of the high fraction of sp3 C atoms, it may also introduce possible hydrogen bonds with TC molecules. As seen in Fig. 1b, the maximum negative electrostatic potential (−47.19 kcal mol−1) appeared around the O atom which was an active site when GO interacted with other substances.
As shown in Fig. 1c, there are nine positive electrostatic potential regions on the surface of the tetracycline molecules namely, H1, H2, ..., H8 and Cl which can serve as the binding sites with a negative O atom on the GO surface. The specific values of these active sites on TCs are shown in Fig. 1 and Table S1.† It's worth mentioning that the electrostatic potential value of each H5 atom (56.27 kcal mol−1 for TTC, 57.18 kcal mol−1 for OTC and 63.26 kcal mol−1 for CTC) was the largest among all of the possible active sites. Therefore, theoretically, the H5 site is supposed to most likely interact with the GO surface and we will discuss this in the next section.
3.2. Binding configuration
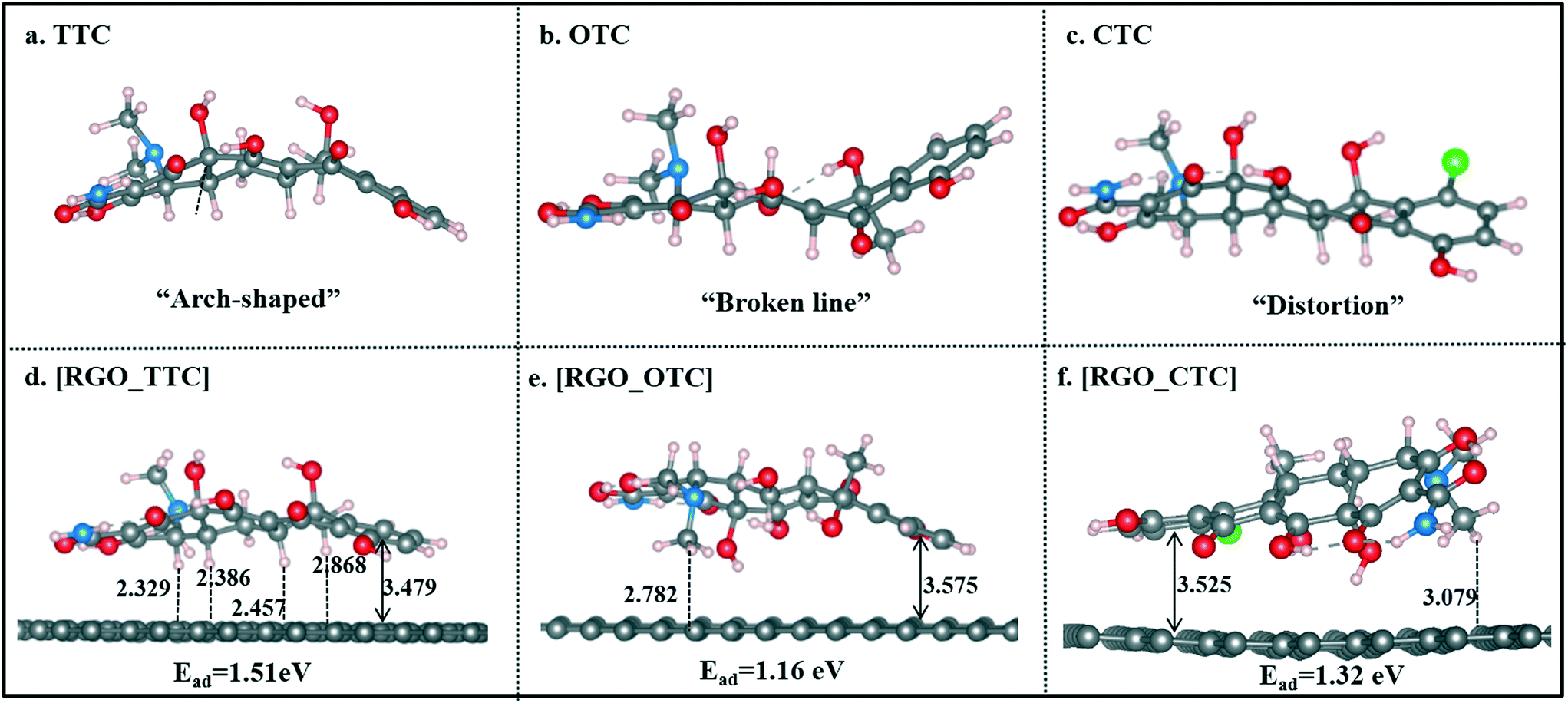 | ||
| Fig. 2 The optimized geometries of TC molecules and RGO_TC systems, bonds are in Å (the vertical distance refers to the distance between the centroid of benzene of TCs to the carbon plane of RGO). | ||
Single TC molecules present quite different configurations as shown in Fig. 2a–c. TTC was in “arch” shape, and OTC showed a “broken line” shape, while the CTC molecule was distorted. The different configurations of these three TC molecules lead to the difference in interaction strength when they adsorbed on RGO. As shown in Fig. 2d–f, TCs lie flat on the surface and the vertical distance between the aromatic ring (denoted as Dv) and the RGO surface is 3.479 Å, 3.575 Å, and 3.525 Å for TTC, OTC, and CTC, respectively. The distance followed the order [RGO_TTC] < [RGO_CTC] < [RGO_OTC]. This demonstrated that the adsorption ability of RGO towards the three kinds of TC molecules was in the order TTC > CTC > OTC. We can draw the same conclusion from the adsorption energy results. Meanwhile, the distance between the H atoms of TCs and the carbon plane was also labeled in Fig. 2. The C–H⋯π interaction may also contribute to the adsorption mechanism.
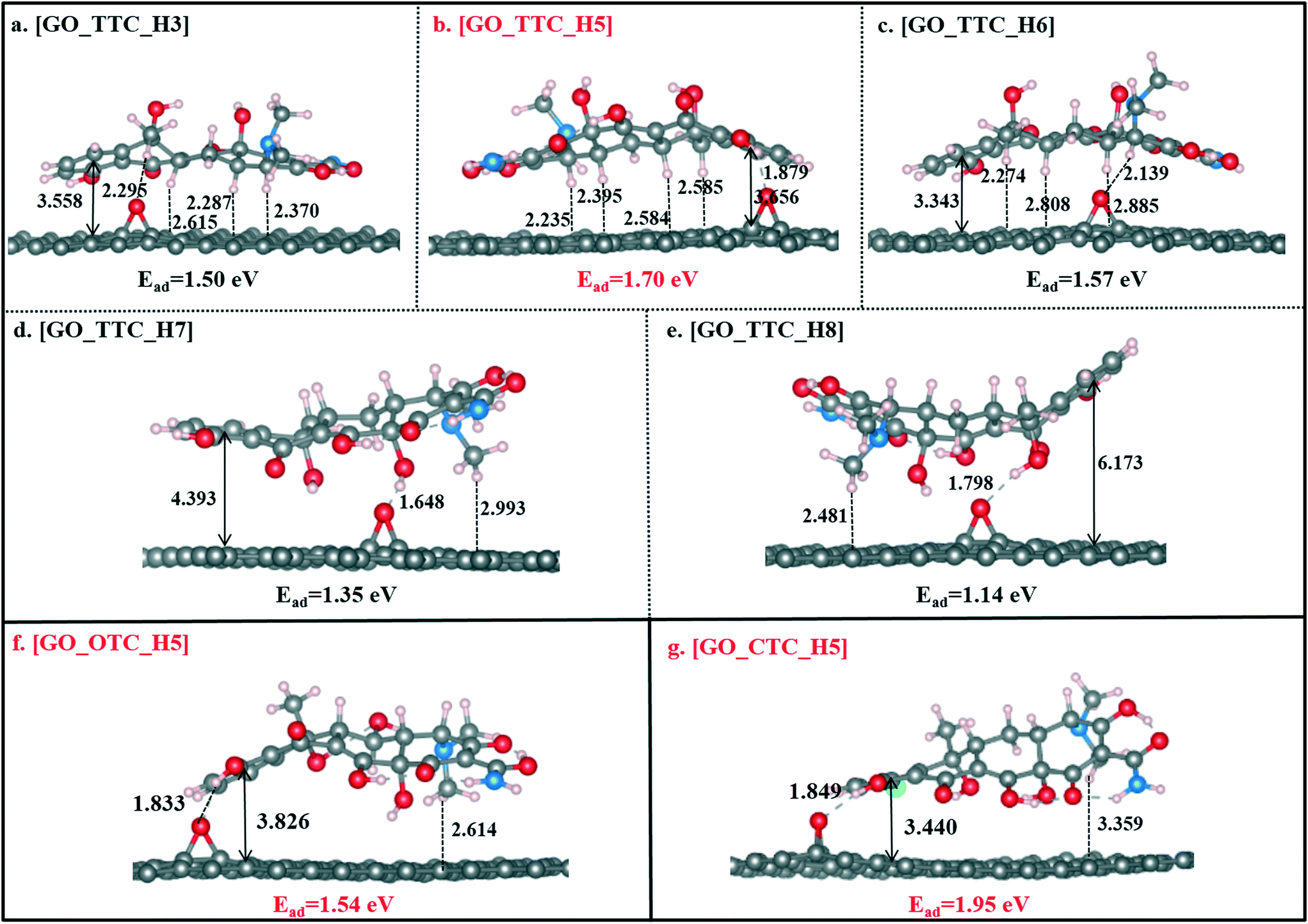 | ||
| Fig. 3 The optimized geometries of GO_TTC, GO_OTC, and GO_CTC complexes, bond lengths are in Å (the vertical distance refers to the distance between the centroid of benzene of TCs to the basal plane of GO). | ||
As shown in Fig. 3b, [GO_TTC_H5] exhibited the highest adsorption energy of 1.70 eV in all the complexes, which was attributed to the strong π–π EDA interaction and hydrogen bond effect. In detail, for [GO_TTC_H5], the distance between H5 and the O atom (denoted as OGO) on the surface of GO was 1.879 Å, and the vertical distance between the aromatic ring of TTC and the GO surface was 3.656 Å. In Fig. 3a and c, the OOG–H3 and OOG–H6 bond lengths were 2.295 Å and 2.139 Å, respectively, and the weaker hydrogen bond led to smaller adsorption energies as compared with [GO_TTC_H5]. In addition to the hydrogen bonds, the π–π interaction was also important and decisive which can be deduced from the comparison between [GO_TTC_H5] and [GO_TTC_H7] (or [GO_TTC_H8]). The vertical distance between the aromatic ring of TTC and the GO surface in [GO_TTC_H7] was 4.393 Å, and it was 6.173 Å in [GO_TTC_H8]. The arched structure of the TTC molecule largely weakened the π–π EDA interaction between the TTC molecule and GO. Although strong hydrogen bonds existed in [GO_TTC_H7] and [GO_TTC_H8], the Ead for these two complexes was much smaller than that for [GO_TTC_H5]. Thus, based on the above discussion, we can conclude that the interaction between GO and TTC relied on the hydrogen bonds and π–π interactions, while the latter one should be the decisive effect. On the other hand, the favorable binding site of TTC was H5, which was consistent with the above ESP results.
Fig. 3g and S4† show the different optimized structures of GO_CTC complexes with H and Cl atoms. As shown in Fig. 3g, the Dv of [GO_CTC_H5] was only 3.440 Å, indicating a strong π–π interaction between GO and CTC. The binding energy of [GO_CTC_H5] was then up to 1.95 eV, which was higher than those of [GO_TTC_H5] and [GO_OTC_H5] systems. Thus, the overall consideration of the binding configuration and adsorption energy makes us conclude that the adsorption ability of GO towards TCs followed the order CTC > OTC > OTC. Specifically, here we also optimized the GO_OTC structure with the binding site of CTC being the Cl atom and the Ead was calculated to be 1.35 eV (seen in Fig. S4g†). However, due to the repulsion between the electron-rich GO and Cl atom, the adsorption of the CTC molecule in this situation also mainly depended on the π–π interaction and hydrogen bonds.
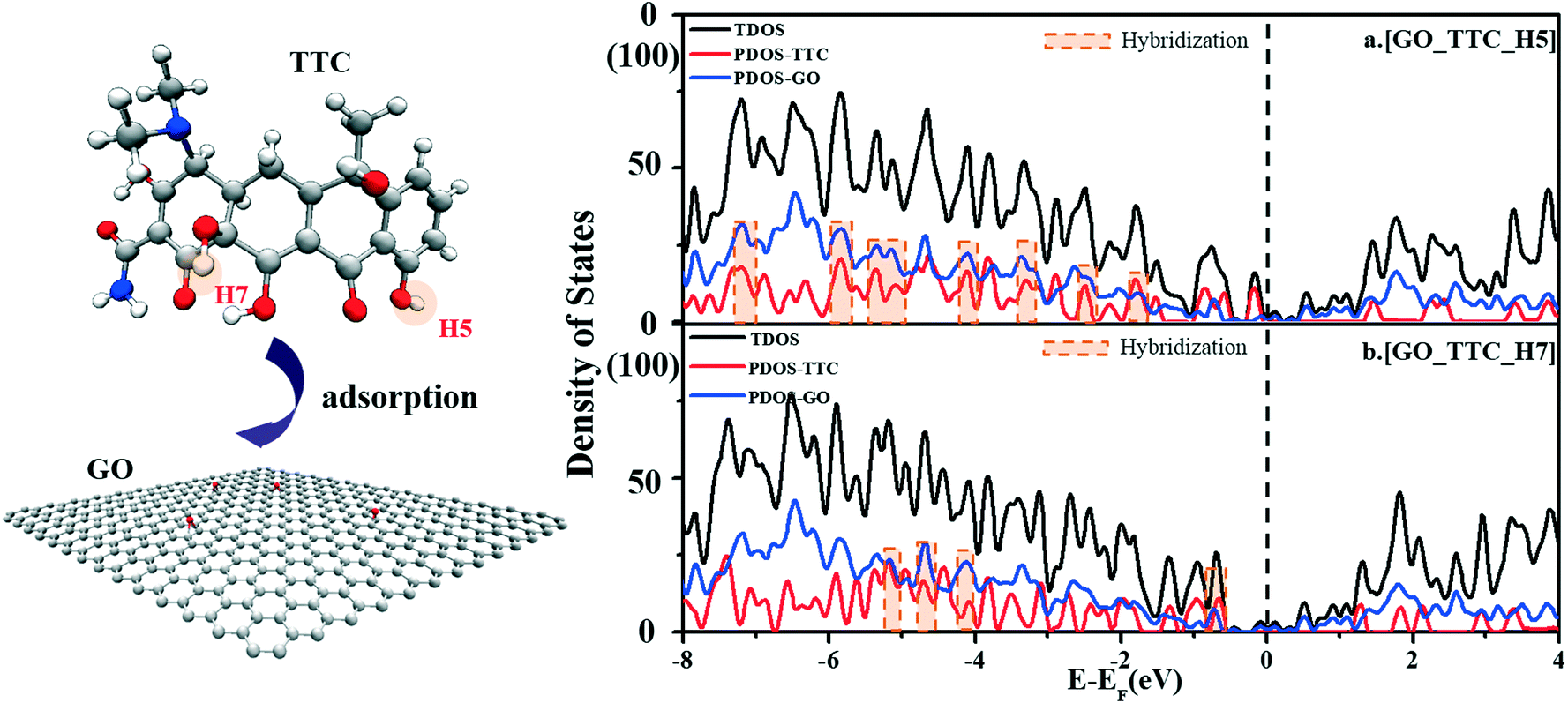 | ||
| Fig. 4 The density of states of the GO_TTC systems as the active sites of TTC was H5(a) and H7(b), respectively. The black lines represent the TDOS of GO_TTC; the red and blue ones represent the PDOS of TTC and GO in these complexes, respectively. | ||
3.3. The effect of the pH value on the adsorption process
Former experimental studies stressed that the pH value of the solution was the most important factor in the adsorption process of TTC onto graphene-based materials and low pH conditions are preferred.19 Thus, in this section, we took the TTC molecule for instance to investigate its adsorption process under low pH conditions covering a range of 2–7. According to the experimental results, the TTC molecule existed in the fully protonated form when the pH value was less than 4, while it was in the form of zwitterion (TTC0) when the pH was in the range of 4–7 as seen in Fig. 5.10,19 The zwitterion forms included three possible patterns: TTC0(1) (the model we have discussed above), TTC0(2) and TTC0(3).52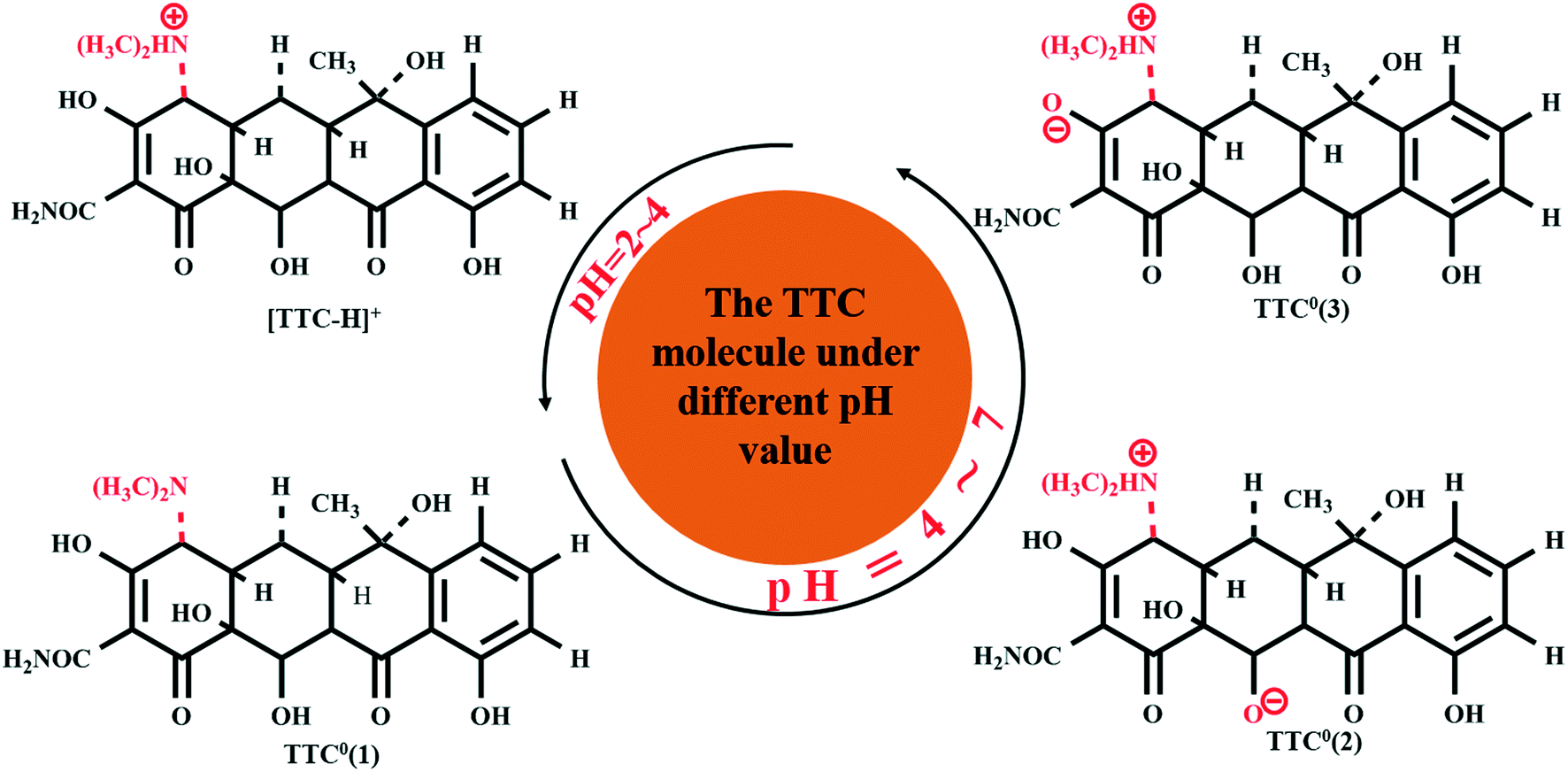 | ||
| Fig. 5 The possible forms of the TTC molecule under different pH conditions. | ||
The optimized structures of TTC0(2), TTC0(3) and [TTC-H]+ as well as their complexes with RGO are shown in Fig. 6. The remarkable change in the geometry of TTC0 molecules after adsorption lay in the molecular planarity from the initial arch-shaped molecule to parallel π–π interactions with the RGO surface. This can be reflected in the short Dv values (3.498 Å in TTC0(2) and 3.506 Å in TTC0(3)), and the adsorption energies of [RGO_TTC0(2)] and [RGO_TTC0(3)] were 1.38 eV and 1.43 eV, respectively. On the other hand, as shown in Fig. 6f, when the pH of the solution was less than 4, the tetracycline molecule was in the fully protonated state, and the vertical distance was only 3.324 Å which was much shorter than that of the [RGO_TTC0] systems (3.479–3.506 Å). The configurational change was accompanied by the enhancement of the adsorption energy which was 3.27 eV for [RGO_TTC_H]+, indicating that the acidic environment was favorable for the adsorption process. The significant improvement of the adsorption energy may be due to not only the reinforcement of π–π interaction, but also the cation–π bonding occurring between the protonated amino group of TTC and the π-electron-rich aromatic structure of GO. Besides, there was an obvious charge transfer between RGO and TTC molecules in the charge density difference plots as shown in Fig. 6g–i, which further elucidated the interaction between RGO and TTC molecules.
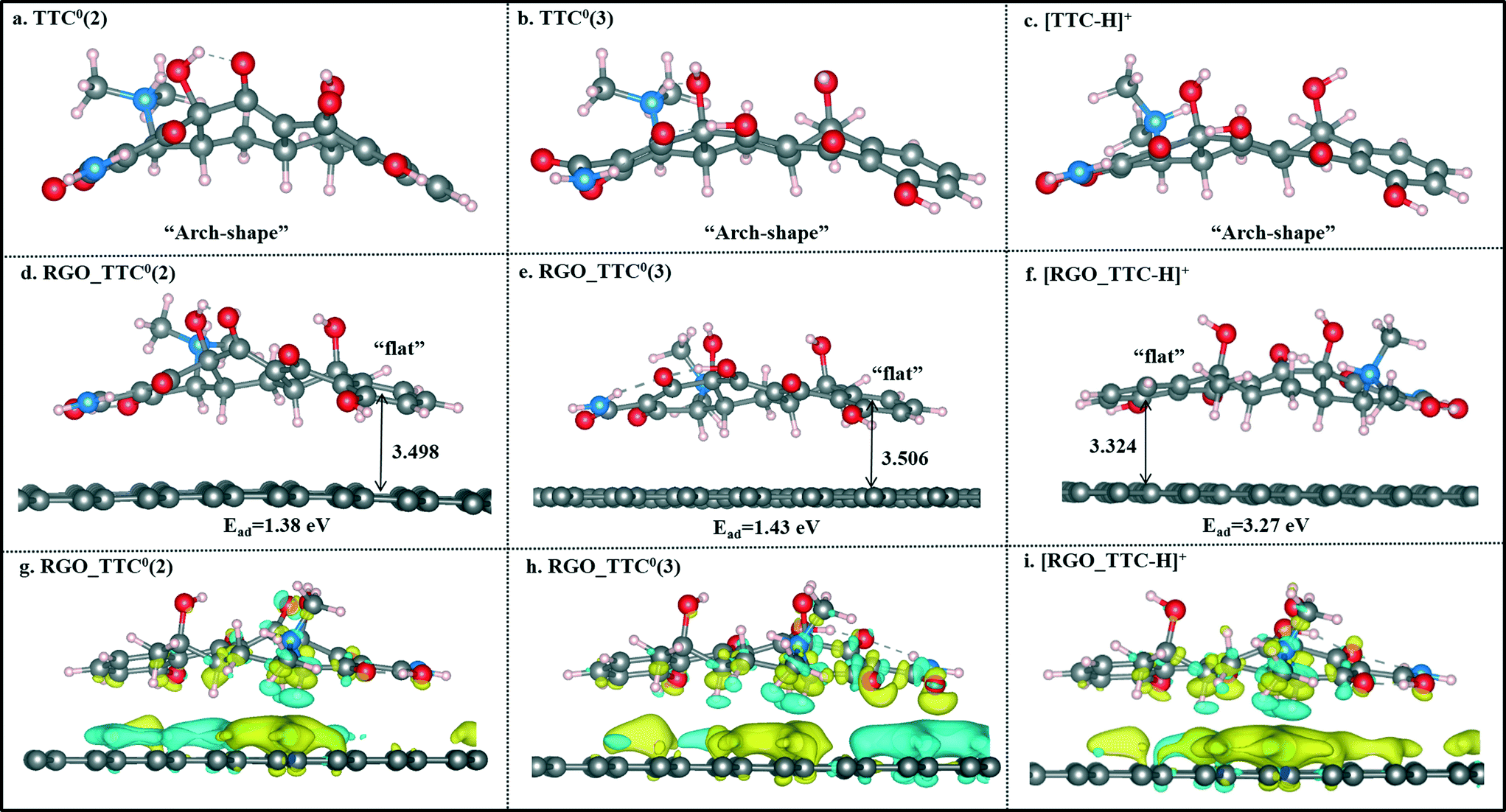 | ||
| Fig. 6 The optimized geometries of TTC0(2) (a), TTC0(3) (b), [TTC-H]+ (c), [RGO_TTC0(2)] (d), [RGO_TTC0(3)] (e), [RGO_TTC-H]+ (f), and the charge density difference plots of [RGO_TTC0(2)] (g), [RGO_TTC0(3)] (h), [RGO_TTC-H]+ (i) systems. | ||
We also investigated the effects of the pH value on the adsorption of TTC molecules on the GO surface. According to the discussion above, we can conclude that the H5 site was the active site of tetracyclines. Thus, for comparison, we also selected the H5 site as the active site of the TTC molecule in the model of protonation and the other two zwitterion forms. As seen in Fig. 7, compared with the RGO results above, there has been a slight increase in the binding energies. Such an enhancement could be explained by the hydrogen bonds between the GO surface and the TTC molecules. Besides, the protonation states [TTC-H]+ still exhibit superior capability in adsorption compared to the zwitterion forms, as reflected in the Ead values shown in Fig. 7. This phenomenon was apparently present in the [GO-H_TTC-H]+ system where both the TTC and GO were protonated. It has the highest Ead value of 3.86 eV and remarkable charge transfer as seen in Fig. 7g and h. Consequently, the low pH value is beneficial to the protonation of TTC molecules and will induce the cation–π interaction between the adsorbates and sorbents.
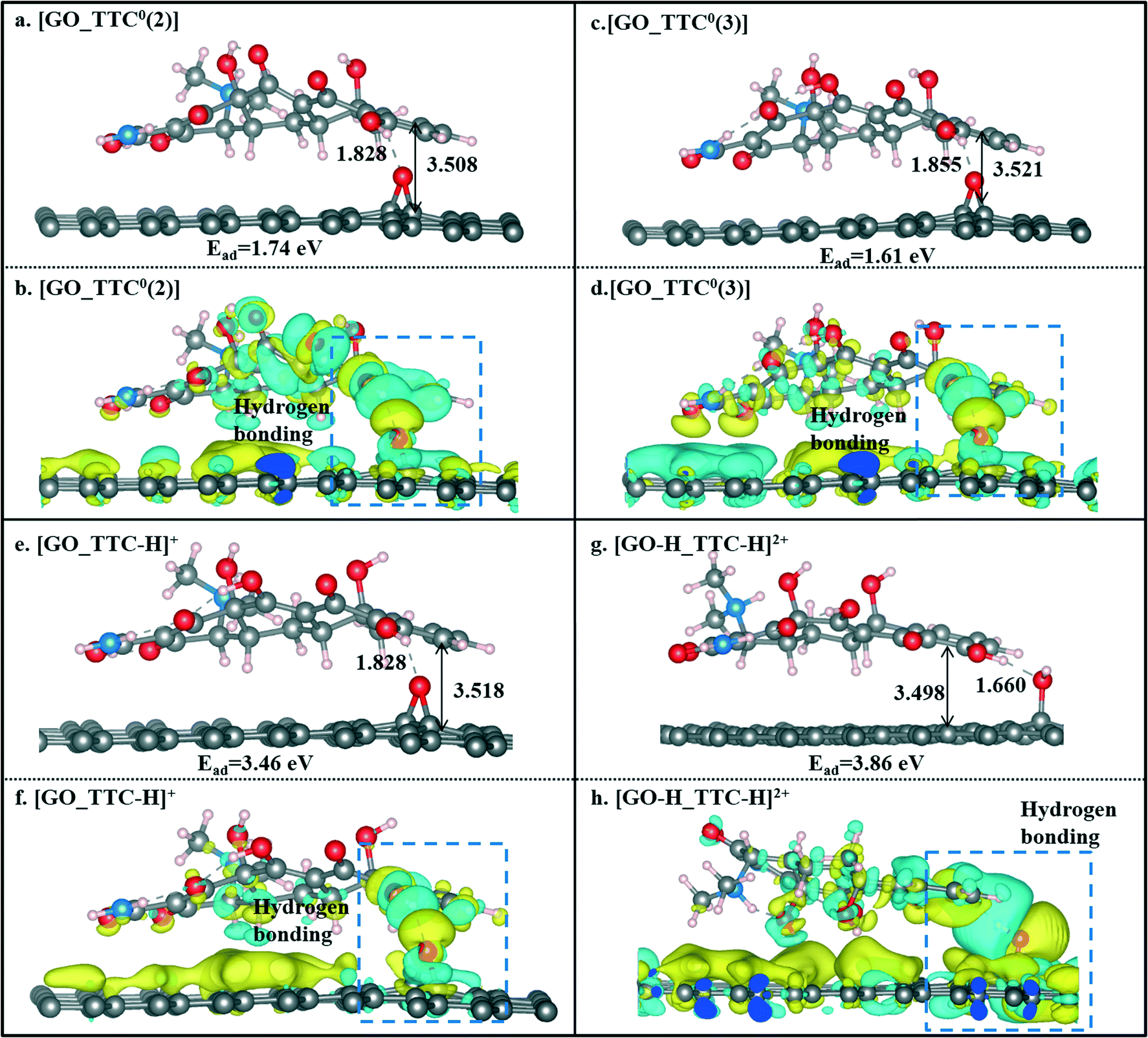 | ||
| Fig. 7 The optimized geometries of [GO_TTC0(2)] (a), [GO_TTC0(3)] (c), [GO_TTC-H]+ (e), and [GO-H_TTC-H]2+ (g) systems and their charge density difference plots (b, d, f and h). | ||
3.4. Effect of the solvents
In this work, we also investigated the influence of the solvents on the adsorption capacity. Three types of solvents with different relative permittivity values were chosen here, such as ethanol (ε = 24.3), acetonitrile (ε = 37.5), and water (ε = 80.0) to compare with the vacuum (ε = 1.0) phase. ΔEsolvation is the relative energy difference of GO_TC (or RGO_TC) systems in a vacuum and these solvents:| ΔEsolvation = Ead(solvent) − Ead(vacuum) |
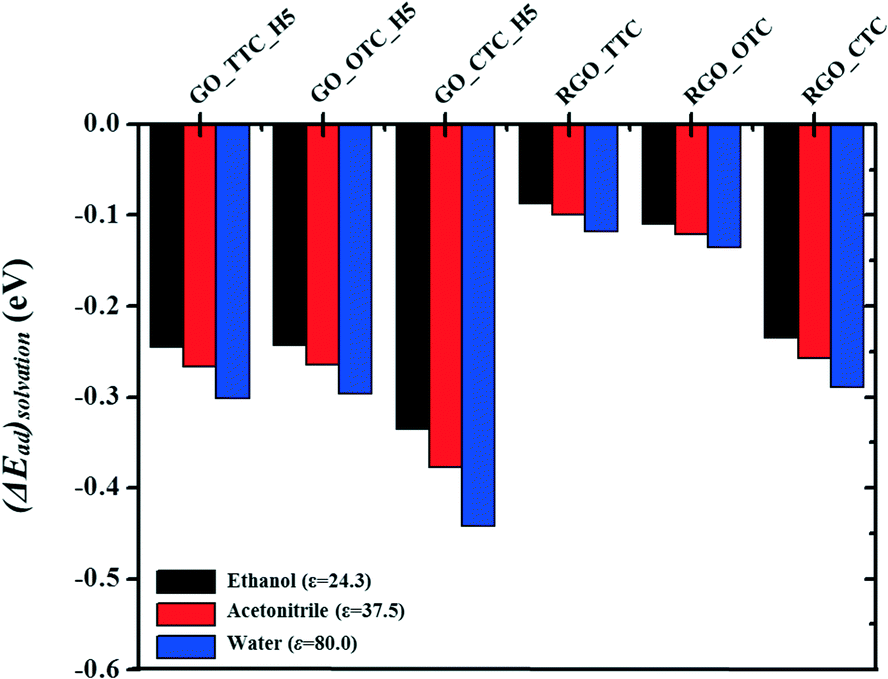 | ||
| Fig. 8 The solution effect (ΔEsolvation) in different solvents for GO_TC and RGO_TC systems. | ||
3.5. Molecular dynamics simulations
Fig. 9a–c show the summary statistics for the minimum distance between the different TCs and GO over time. From the graph we can see that the final equilibrium distance for the adsorption of TCs toward GO is around 0.3 nm. However, a small number of exceptions exist in the TTC and OTC systems exhibiting an equilibrium range of 0.8–1.2 nm. If we look through the snapshots of the MD trajectory in Fig. S8,† these exceptions are on account of the intermolecular accumulation which stunted the effective adsorption. Hence, the dynamic interaction between the different TCs may have an influence on the adsorption. Besides, significant differences were found in the adsorption behavior of the TC molecules. The CTC molecules seem to be more competitive than the other TCs and reach the stable equilibrium within 20 ns. By contrast, due to the fast adsorption of CTC and the intermolecular interactions, there were slight fluctuations in the distances for the TTC and OTC systems.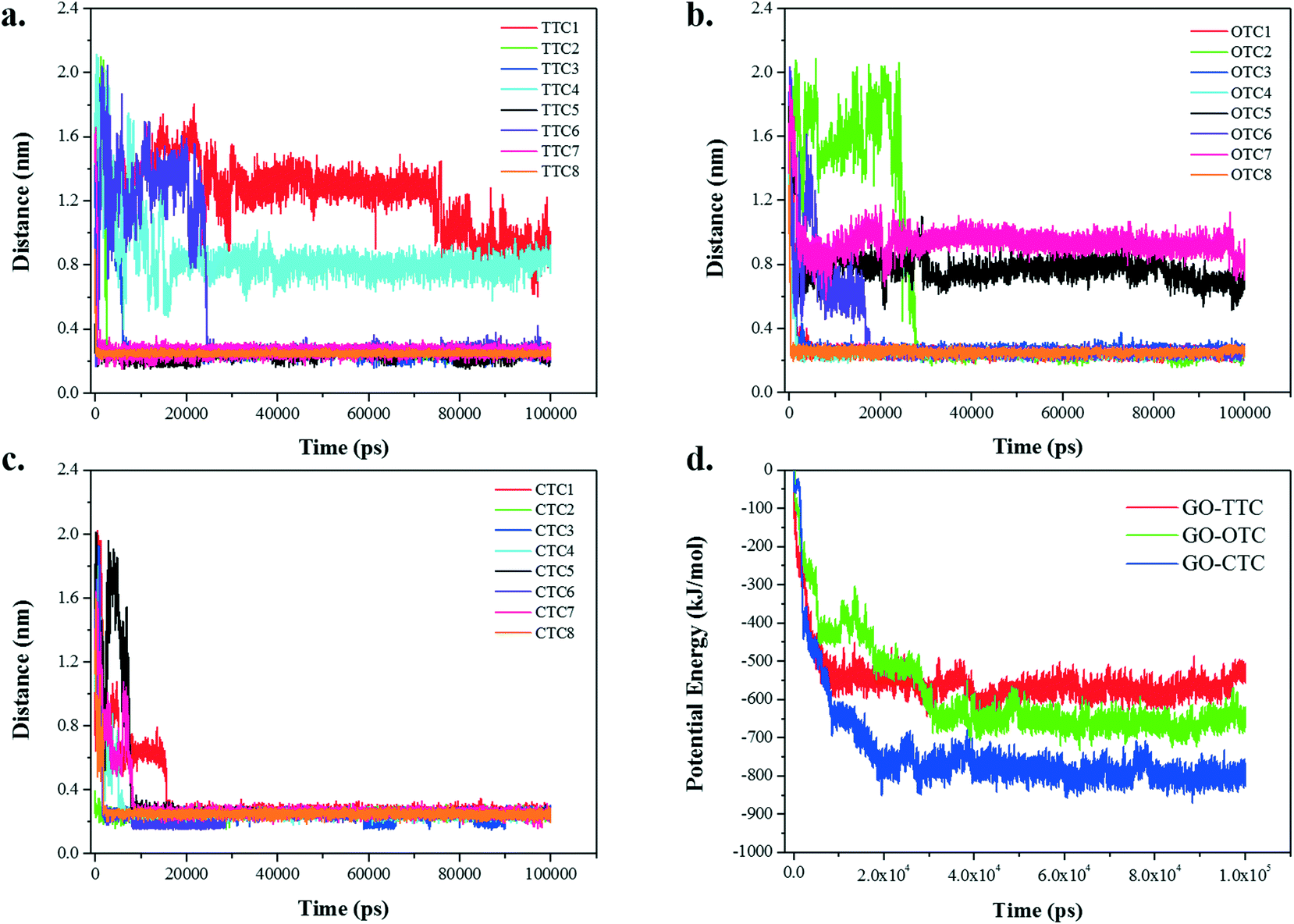 | ||
| Fig. 9 (a–c) The minimum distance between TC molecules and GO; (d) the potential energy between GO and TCs. | ||
The radial distribution functions (RDFs) and running coordination numbers are shown in Fig. 10. The first peak (about 0.5 nm) in Fig. 10a corresponds to the first adsorption layer which is the main adsorption layer. Accordingly, from Fig. 10b, in the simulation of the GO_CTC system with 8 molecules of CTC, 7 molecules form the first layer and the last remaining CTC made the second layer. Comparatively, in the first adsorption layer, the running coordination numbers of TTC and OTC are 5 and 4, respectively. These data also correspond to the slight fluctuations of TTC and OTC molecules in Fig. 9. Meanwhile, it can be found that GO shows more obvious adsorption ability for CTC molecules.
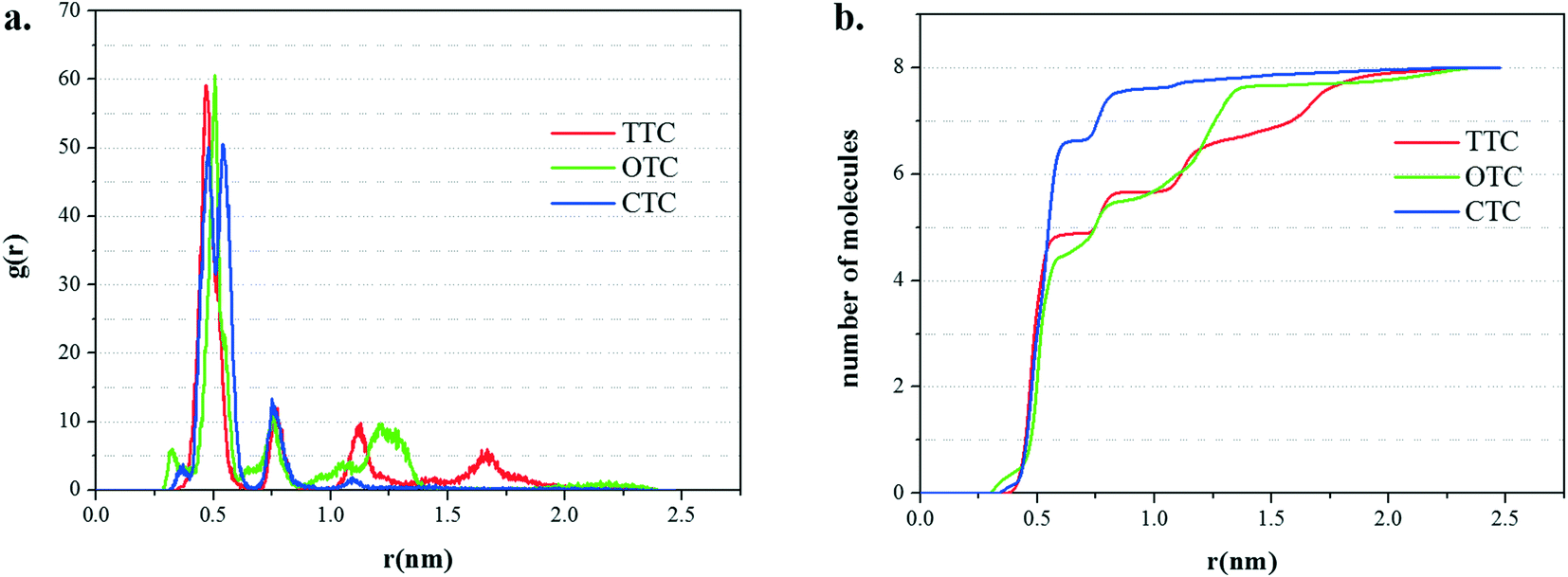 | ||
| Fig. 10 (a) The radial distribution function (RDF) of the three TC molecules on the GO surface; (b) the running coordination numbers of the three TCs to the surface of GO. | ||
Fig. 9d shows the potential energies between GO and TC molecules over time together with the specific values of potential energy summarized in Table 1. As the simulation progresses, the interaction energy decreases rapidly in the initial 30 ns and then remains in equilibrium. The numerical values of the van der Waals (vdW) interaction in the GO_TC systems accounted for more than 95% of the potential energies, which was much higher than the Coulomb interaction, indicating that the vdW interaction was the dominant force between GO and TC molecules.
| GO_TTC | GO_OTC | GO_CTC | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Energy time | Coulomb interaction (kJ mol−1) | vdW interaction (kJ mol−1) | Potential energy (kJ mol−1) | Coulomb interaction (kJ mol−1) | vdW interaction (kJ mol−1) | Potential energy (kJ mol−1) | Coulomb interaction (kJ mol−1) | vdW interaction (kJ mol−1) | Potential energy (kJ mol−1) |
| 0 ns | 0.01 | −12.41 | −12.40 | 0 | 0 | 0 | 2.31 | −6.41 | −4.10 |
| 20 ns | −15.58 | −520.38 | −535.97 | −1.94 | −457.56 | −459.50 | −19.98 | −760.40 | −780.38 |
| 40 ns | −8.85 | −589.21 | −598.06 | −3.50 | −604.69 | −608.19 | −4.86 | −770.20 | −775.07 |
| 60 ns | −10.97 | −540.54 | −551.51 | −11.786 | −660.61 | −672.39 | −20.47 | −755.76 | −776.23 |
| 80 ns | −21.32 | −583.83 | −605.15 | −9.54 | −646.47 | −656.01 | 0.621 | −794.88 | −794.26 |
| 100 ns | −9.43 | −515.74 | −525.17 | −12.10 | −626.06 | −638.16 | −1.54 | −766.34 | −767.88 |
MD simulations show that GO has superior adsorption performance towards the three TC molecules, especially excellent for CTC. Almost all TCs show good adsorption stability during the 100 ns simulations, and the adsorption is mainly concentrated in the first adsorption layer. The adsorption of the GO_TC systems are through the Coulomb interaction and vdW interaction, and the vdW interaction is the dominant force.
4. Conclusion
In summary, in this article, the adsorption behavior and interaction mechanisms of TC molecules on RGO and GO were investigated by both the DFT method and MD simulations. Firstly, the DFT results showed that the adsorption performance of GO towards three TC molecules was stronger than that of RGO, and the adsorption energies followed the order CTC > TTC > OTC and TTC > CTC > OTC for the GO and RGO systems, respectively. Static calculations also confirmed that the preferential adsorption site of TCs is located on H5 when interacting with GO. Meanwhile, the adsorption mechanism was examined and the π–π EDA interaction and hydrogen bonds were found to play significant roles in the GO_TC systems. In the meantime, we also propose that C–H⋯π interaction may contribute to the adsorption mechanism, although it is much weaker than other chemical interactions. Regarding the effects of the pH value and solvent on the adsorption process, the computed results showed that low pH values were suitable for the adsorption of TCs on GO (or RGO) due to the reinforced π–π EDA interaction, hydrogen bonds and cation–π interaction. And low polarity solvents were verified to be more beneficial to the adsorption process. Finally, MD simulations showed that a competitive adsorption process existed in TCs and the CTC molecules displayed the most competitive ability towards the GO material. Besides, the dominant force in the dynamic adsorption process was the van der Waals interactions. This work can offer new insights into the knowledge gaps in the microscopic binding configuration, molecular-scale mechanism and competitive adsorption behavior during the adsorption process of TCs onto graphene-based materials. Our results are helpful in making the experimental design goal-oriented and may provide a theoretical basis for the removal of tetracycline antibiotics in the environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21777039), the National Key Research and Development Program of China (2017YFA0207002), the Fundamental Research Funds for the Central Universities (2017YQ001) and the MOE Key Laboratory of Resources and Environmental Systems Optimization (NCEPU). The partial computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at [SNIC CENTER].References
- A. K. Sarmah, M. T. Meyer and A. B. A. Boxall, A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment, Chemosphere, 2006, 65, 725–759 CrossRef CAS.
- L. Ji, F. Liu, Z. Xu, S. Zheng and D. Zhu, Adsorption of pharmaceutical antibiotics on template-synthesized ordered micro- and mesoporous carbons, Environ. Sci. Technol., 2010, 44, 3116–3122 CrossRef CAS PubMed.
- K. Kummerer, Antibiotics in the aquatic environment--a review--part II, Chemosphere, 2009, 75, 435–441 CrossRef PubMed.
- R. E. Alcock, A. Sweetman and K. C. Jones, Assessment of organic contaminant fate in waste water treatment plants I: Selected compounds and physicochemical properties, Chemosphere, 1999, 38, 2247–2262 CrossRef CAS.
- T. Heberer, Occurrence, fate, and removal of pharmaceutical residues in the aquatic environment: a review of recent research data, Toxicol. Lett., 2002, 131, 5–17 CrossRef CAS.
- M. Förster, V. Laabs, M. Lamshöft, J. Groeneweg, S. Zühlke, M. Spiteller, M. Krauss, M. Kaupenjohann and W. Amelung, Sequestration of manure-applied sulfadiazine residues in soils, Environ. Sci. Technol., 2009, 15, 1824 CrossRef PubMed.
- S. Manzetti and R. Ghisi, The environmental release and fate of antibiotics, Mar. Pollut. Bull., 2014, 79, 7–15 CrossRef CAS.
- M. Boonsaner and D. W. Hawker, Transfer of oxytetracycline from swine manure to three different aquatic plants: implications for human exposure, Chemosphere, 2015, 122, 176–182 CrossRef CAS PubMed.
- P. A. Blackwell, P. Kay, R. Ashauer and A. B. Boxall, Effects of agricultural conditions on the leaching behaviour of veterinary antibiotics in soils, Chemosphere, 2009, 75, 13–19 CrossRef CAS.
- E. E. Ghadim, F. Manouchehri, G. Soleimani, H. Hosseini, S. Kimiagar and S. Nafisi, Adsorption properties of tetracycline onto graphene oxide: equilibrium, kinetic and thermodynamic studies, PLoS One, 2013, 8, e79254 CrossRef PubMed.
- L. Wollenberger, B. Halling-Sørensen and K. O. Kusk, Acute and chronic toxicity of veterinary antibiotics to Daphnia magna, Chemosphere, 2000, 40, 723–730 CrossRef CAS.
- H. Y. Kim, S. Yu, T. Y. Jeong and S. D. Kim, Relationship between trans-generational effects of tetracycline on Daphnia magna at the physiological and whole organism level, Environ. Pollut., 2014, 191, 111–118 CrossRef CAS PubMed.
- H. Wang, N. Wang, B. Wang, Q. Zhao, H. Fang, C. Fu, C. Tang, F. Jiang, Y. Zhou, Y. Chen and Q. Jiang, Antibiotics in Drinking Water in Shanghai and Their Contribution to Antibiotic Exposure of School Children, Environ. Sci. Technol., 2016, 50, 2692–2699 CrossRef CAS PubMed.
- S. Aydin, B. Ince and O. Ince, Development of antibiotic resistance genes in microbial communities during long-term operation of anaerobic reactors in the treatment of pharmaceutical wastewater, Water Res., 2015, 83, 337–344 CrossRef CAS PubMed.
- J. Giammarco, V. N. Mochalin, J. Haeckel and Y. Gogotsi, The adsorption of tetracycline and vancomycin onto nanodiamond with controlled release, J. Colloid Interface Sci., 2016, 468, 253–261 CrossRef CAS PubMed.
- Y. Chen, C. Hu, J. Qu and M. Yang, Photodegradation of tetracycline and formation of reactive oxygen species in aqueous tetracycline solution under simulated sunlight irradiation, J. Photochem. Photobiol., A, 2008, 197, 81–87 CrossRef CAS.
- S. Kim, P. Eichhorn, J. N. Jensen, A. S. Weber and D. S. Aga, Removal of Antibiotics in Wastewater: Effect of Hydraulic and Solid Retention Times on the Fate of Tetracycline in the Activated Sludge Process, Environ. Sci. Technol., 2005, 39, 5816–5823 CrossRef CAS PubMed.
- J. Pang, Y. Chao, H. Chang, H. Li, J. Xiong, Q. Zhang, G. Chen, J. Qian, W. Zhu and H. Li, Silver Nanoparticle-Decorated Boron Nitride with Tunable Electronic Properties for Enhancement of Adsorption Performance, ACS Sustainable Chem. Eng., 2018, 6, 4948–4957 CrossRef CAS.
- Y. Gao, Y. Li, L. Zhang, H. Huang, J. Hu, S. M. Shah and X. Su, Adsorption and removal of tetracycline antibiotics from aqueous solution by graphene oxide, J. Colloid Interface Sci., 2012, 368, 540–546 CrossRef CAS PubMed.
- M. F. Li, Y. G. Liu, G. M. Zeng, S. B. Liu, X. J. Hu, D. Shu, L. H. Jiang, X. F. Tan, X. X. Cai and Z. L. Yan, Tetracycline absorbed onto nitrilotriacetic acid-functionalized magnetic graphene oxide: Influencing factors and uptake mechanism, J. Colloid Interface Sci., 2017, 485, 269–279 CrossRef CAS PubMed.
- X. Li, Y. Liu, C. Zhang, T. Wen, L. Zhuang, X. Wang, G. Song, D. Chen, Y. Ai, T. Hayat and X. Wang, Porous Fe2O3 microcubes derived from metal organic frameworks for efficient elimination of organic pollutants and heavy metal ions, Chem. Eng. J., 2018, 336, 241–252 CrossRef CAS.
- L. Wang, H. Song, L. Y. Yuan, Z. J. Li, Y. J. Zhang, J. K. Gibson, L. R. Zheng, Z. F. Chai and W. Q. Shi, Efficient U(VI) Reduction and Sequestration by Ti2CTx MXene, Environ. Sci. Technol., 2018, 52, 10748–10756 CrossRef CAS.
- Z. W. Huang, Z. J. Li, Q. Y. Wu, L. R. Zheng, L. M. Zhou, Z. F. Chai, X. L. Wang and W. Q. Shi, Simultaneous elimination of cationic uranium(VI) and anionic rhenium(VII) by graphene oxide–poly(ethyleneimine) macrostructures: a batch, XPS, EXAFS, and DFT combined study, Environ. Sci.: Nano, 2018, 5, 2077–2087 RSC.
- L. Wang, H. Song, L. Y. Yuan, Z. J. Li, P. Zhang, J. K. Gibson, L. R. Zheng, H. Q. Wang, Z. F. Chai and W. Q. Shi, Effective Removal of Anionic Re(VII) by Surface-Modified Ti2CTx MXene Nanocomposites: Implications for Tc(VII) Sequestration, Environ. Sci. Technol., 2019, 53, 3739–3747 CrossRef CAS PubMed.
- L. Y. Yuan, M. Tan, J. H. Lan, X. Z. Cao, X. L. Wang, Z. F. Chai, J. K. Gibson and W. Q. Shi, Defect engineering in metal–organic frameworks: a new strategy to develop applicable actinide sorbents, Chem. Commun., 2018, 54, 370–373 RSC.
- L. Mei, F. Z. Li, J. H. Lan, C. Z. Wang, C. Xu, H. Deng, Q. Y. Wu, K. Q. Hu, L. Wang, Z. F. Chai, J. Chen, J. K. Gibson and W. Q. Shi, Anion-adaptive crystalline cationic material for 99TcO4− trapping, Nat. Commun., 2019, 10, 1532 CrossRef PubMed.
- J. Yu, L. Yuan, S. Wang, J. Lan, L. Zheng, C. Xu, J. Chen, L. Wang, Z. Huang and W. J. C. C. Tao, Phosphonate-Decorated Covalent Organic Frameworks for Actinide Extraction: A Breakthrough Under Highly Acidic Conditions, 2019, pp. 286–293 Search PubMed.
- X. Wang, Y. Liu, H. Pang, S. Yu, Y. Ai, X. Ma, G. Song, T. Hayat, A. Alsaedi and X. Wang, Effect of graphene oxide surface modification on the elimination of Co(II) from aqueous solutions, Chem. Eng. J., 2018, 344, 380–390 CrossRef CAS.
- Y. Ai, Y. Liu, W. Lan, J. Jin, J. Xing, Y. Zou, C. Zhao and X. Wang, The effect of pH on the U(VI) sorption on graphene oxide (GO): A theoretical study, Chem. Eng. J., 2018, 343, 460–466 CrossRef CAS.
- G. Zhao, T. Wen, C. Chen and X. Wang, Synthesis of graphene-based nanomaterials and their application in energy-related and environmental-related areas, RSC Adv., 2012, 2, 9286–9303 RSC.
- G. Zhao, T. Wen, X. Yang, S. Yang, J. Liao, J. Hu, D. Shao and X. Wang, Preconcentration of U(VI) ions on few-layered graphene oxide nanosheets from aqueous solutions, Dalton Trans., 2012, 41, 6182–6188 RSC.
- S. Chen, J. Hong, H. Yang and J. Yang, Adsorption of uranium (VI) from aqueous solution using a novel graphene oxide-activated carbon felt composite, J. Environ. Radioact., 2013, 126, 253–258 CrossRef CAS PubMed.
- S. Yadav, N. Goel, V. Kumar, K. Tikoo and S. Singhal, Removal of fluoroquinolone from aqueous solution using graphene oxide: experimental and computational elucidation, Environ. Sci. Pollut. Res., 2018, 25, 2942–2957 CrossRef CAS PubMed.
- X. Zhang, J. Shen, N. Zhuo, Z. Tian, P. Xu, Z. Yang and W. Yang, Interactions between Antibiotics and Graphene-Based Materials in Water: A Comparative Experimental and Theoretical Investigation, ACS Appl. Mater. Interfaces, 2016, 8, 24273–24280 CrossRef CAS PubMed.
- W. Song, T. Yang, X. Wang, Y. Sun, Y. Ai, G. Sheng, T. Hayat and X. Wang, Experimental and theoretical evidence for competitive interactions of tetracycline and sulfamethazine with reduced graphene oxides, Environ. Sci.: Nano, 2016, 3, 1318–1326 RSC.
- L. He, F. Liu, M. Zhao, Z. Qi, X. Sun, M. Z. Afzal, X. Sun, Y. LI, J. Hao and S. Wang, Electronic-property dependent interactions between tetracycline and graphene nanomaterials in aqueous solution, J. Environ. Sci., 2018, 66, 286–294 CrossRef PubMed.
- B. Peng, L. Chen, C. Que, K. Yang, F. Deng, X. Deng, G. Shi, G. Xu and M. Wu, Adsorption of Antibiotics on Graphene and Biochar in Aqueous Solutions Induced by pi-pi Interactions, Sci. Rep., 2016, 6, 31920 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmiiller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- X. Wu, M. C. Vargas, S. Nayak, V. Lotrich and G. Scoles, Towards extending the applicability of density functional theory to weakly bound systems, J. Chem. Phys., 2001, 115, 8748 CrossRef CAS.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- M. Pekka and L. Nilsson, Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef.
- L. J. William, S. M. David and T.-R. Julian, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef.
- N. Sukumar and J. E. Pask, Classical and enriched finite element formulations for Bloch-periodic boundary conditions, Int. J. Numer. Method Biomed. Eng., 2009, 77, 1121–1138 CrossRef.
- G. J. Martyna, D. J. Tobias and M. L. Klein, Constant pressure molecular dynamics algorithms, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, GROMACS: fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A Well Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: the RESP Model, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- P. Sjoberg and P. Politzer, Use of the electrostatic potential at the molecular surface to interpret and predict nucleophilic processes, J. Phys. Chem. A, 1990, 94, 3959–3961 CrossRef CAS.
- L. He, F. F. Liu, M. Zhao, Z. Qi, X. Sun, M. Z. Afzal, X. Sun, Y. Li, J. Hao and S. Wang, Electronic-property dependent interactions between tetracycline and graphene nanomaterials in aqueous solution, J. Environ. Sci., 2018, 66, 286–294 CrossRef PubMed.
- A. Amat, S. Fantacci, F. D. Angelis, B. Carlotti and F. Elisei, DFT/TDDFT investigation of the stepwise deprotonation in tetracycline: pKa assignment and UV–vis spectroscopy, Theor. Chem. Acc., 2012, 131, 1218 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Partially optimized structures, density of states, and ESP values of RGO_TC and GO_TC systems, together with the GO model construction and some snapshots in MD simulations. See DOI: 10.1039/c9en00866g |
| This journal is © The Royal Society of Chemistry 2019 |