
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransforming PPh3 into bidentate phosphine ligands at Ru–Zn heterobimetallic complexes†‡
Niall
O'Leary
,
Fedor M.
Miloserdov
 *,
Mary F.
Mahon
* and
Michael K.
Whittlesey
*
*,
Mary F.
Mahon
* and
Michael K.
Whittlesey
*
Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: f.miloserdov@bath.ac.uk; m.f.mahon@bath.ac.uk; m.k.whittlesey@bath.ac.uk
First published on 26th August 2019
Abstract
The reaction of [Ru(PPh3)3Cl2] with excess ZnMe2 led to P–C/C–H bond activation and P–C/C–C bond formation to generate a chelating diphenylphosphinobenzene ligand as well as a cyclometallated (diphenylphosphino)biphenyl group in the final product of the reaction, [Ru(dppbz)(PPh2(biphenyl)′)(ZnMe)] (1; dppbz = 1,2-bis(diphenylphosphino)benzene); PPh2(biphenyl)′ = cyclometallated PPh2(biphenyl). The mechanism of reaction was studied and C–C coupling to give a bidentate 2,2′-bis(diphenylphosphino)biphenyl (BIPHEP) ligand was suggested to be one of the key steps of the process. This was confirmed by the reaction of [Ru(BIPHEP)(PPh3)HCl] with ZnMe2, which also gave 1. An analogous set of steps took place upon addition of ZnMe2 to [Ru(rac-BINAP)(PPh3)HCl] (rac-BINAP = racemic(2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) to give [Ru(dppbz)(PPh2(binaphthyl)′)ZnMe] (3). H2 and the C–H bond of PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH added across the Ru–Zn bond of 1, and also reversed the phosphine cyclometallation, to give [Ru(dppbz)(Ph2P(biphenyl))(H)2(H)(ZnMe)] (4) and [Ru(dppbz)(Ph2P(biphenyl))(CCPh)2(H)(ZnMe)] (5) respectively.
CH added across the Ru–Zn bond of 1, and also reversed the phosphine cyclometallation, to give [Ru(dppbz)(Ph2P(biphenyl))(H)2(H)(ZnMe)] (4) and [Ru(dppbz)(Ph2P(biphenyl))(CCPh)2(H)(ZnMe)] (5) respectively.
Introduction
Alkane elimination resulting from the treatment of a transition metal hydride (TM-H) complex with a main group hydrocarbyl (MG-R) reagent represents an established, but still under-utilised synthetic route to TM-MG heterobimetallic complexes.1 We have recently employed this approach to good effect with ruthenium N-heterocyclic carbene (NHC) and/or phosphine hydride precursors and MG(alkyl)n (n = 2, 3) compounds to generate a number of new Ru–MG (MG = Zn, In, Ga) complexes (Scheme 1).2–4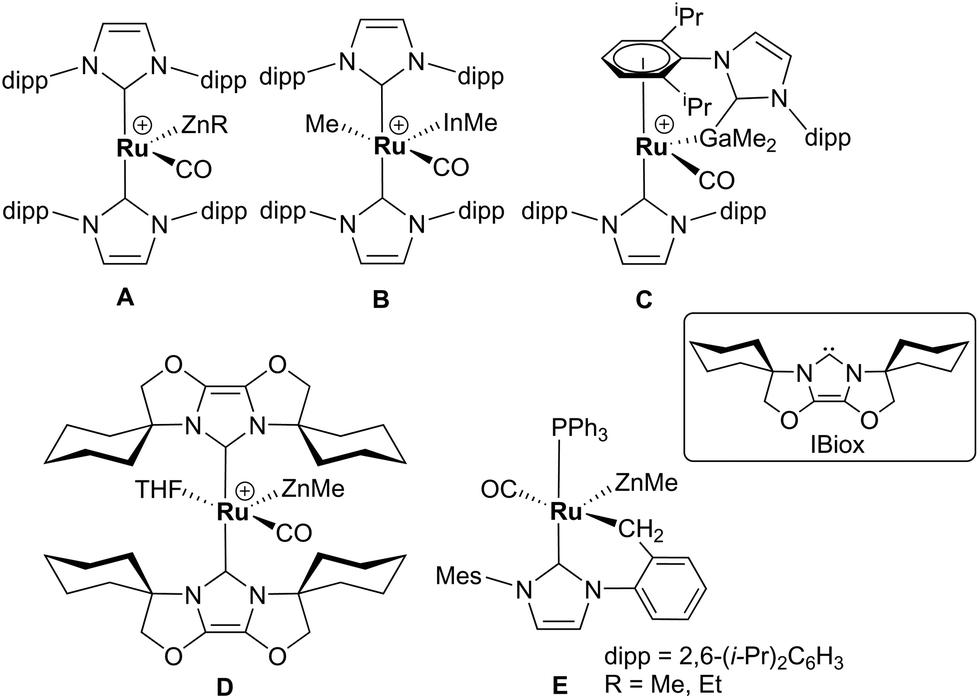 | ||
| Scheme 1 Examples of our recently reported Ru–MG heterobimetallic complexes. The [BArF4] anion in each of the charged species has been omitted for clarity. | ||
With the cationic hydride precursors [Ru(NHC)2(CO)H][BArF4] (NHC = IPr, IBiox (see Scheme 1 for structures); [BArF4] = [B(3,5-(F3C)2C6H3)4]), elimination of a single equivalent of C2H6 or CH4 took place upon addition of ZnR2 (R = Et, Me) to give [Ru(NHC)2(CO)ZnR][BArF4] (A, D).2,3 These complexes contain unsupported Ru–Zn bonds, which in the case of the IPr compounds, react readily with a range of E–H (E = H, B, Si) bonds. Addition of the group 13 trialkyls MMe3 (M = Ga, In) to [Ru(IPr)2(CO)H]+ resulted in chemistry that was different not only to that of ZnR2, but also to each other. With InMe3, methane loss was followed by methyl migration from In to Ru to generate [Ru(IPr)2(CO)(InMe)Me][BArF4] (B). With GaMe3, a more complex set of reactions occurred that led ultimately to migration of an IPr ligand from Ru to Ga to yield C.4
The addition of excess ZnMe2 to the neutral mixed NHC/phosphine precursor [Ru(IMes)(PPh3)(CO)HCl] induced loss of two equivalents of CH4, as well as ZnMeCl, to give the cyclometallated carbene complex [Ru(IMes)′(PPh3)(CO)(ZnMe)] E ((IMes)′ = cyclometallated IMes).3 We have also established that the elimination of more than one equivalent of alkane is a feature of the reactions of [Ru(PPh3)3HCl] with ZnMe2 (as well as LiCH2SiMe3 and MgMe2), the second equivalent of alkane now arising from Ru–H intermediates generated upon cyclometallation of a PPh3 ligand.5 Given the wealth of Ru–PPh3 precursors, we turned our attention to a non-hydride containing example to probe whether PPh3 metalation still took place. Herein, we show that the reaction of [Ru(PPh3)3Cl2] with ZnMe2 results in a remarkable series of C–H and P–C bond activation, as well as P–C and C–C bond formation, steps to generate [Ru(dppbz)(PPh2(biphenyl)′)(ZnMe)] (1; dppbz = 1,2-bis(diphenylphosphino)benzene; PPh2(biphenyl)′ = cyclometallated PPh2(biphenyl)). The order of these transformations on the pathway to formation of the chelating diphenylphosphinobenzene and cyclometallated (diphenylphosphino)biphenyl ligands has been probed by 1H and 31P NMR spectroscopy. Initial studies of the reactivity of 1 show that both H2 and the C–H bond of an alkyne can add across the Ru–Zn bond, with reversal of Ph2P(biphenyl) metalation also taking place.
Results and discussion
Characterisation of [Ru(dppbz)(PPh2(biphenyl)′)ZnMe] (1) and trapping with CO
Treatment of [Ru(PPh3)3Cl2] with ZnMe2 (5 equiv.)6 in THF led to the evolution of a single red product over 48 h, which was characterised as [Ru(dppbz)(PPh2(biphenyl)′)ZnMe] (1, Scheme 2) arising from the remarkable transformation of two of the PPh3 ligands into a bidentate bis(diphenylphosphino)benzene ligand, and conversion of the third PPh3 into a (diphenylphosphino)biphenyl ligand, which undergoes cyclometallation at the ruthenium centre.7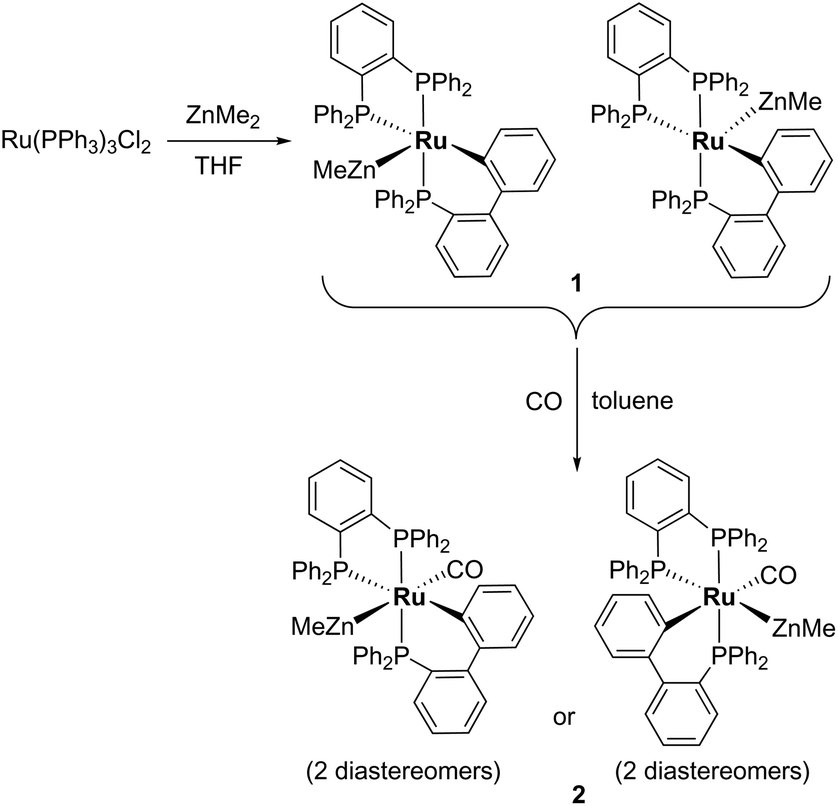 | ||
| Scheme 2 Formation of [Ru(dppbz)(PPh2(biphenyl)′)ZnMe] (1) and trapping by CO. | ||
Fig. 1 shows the X-ray structure of 1. The geometry can be described as a distorted square pyramid based on maximum deviations of +0.264 Å and −0.297 Å for P1 and C2, respectively, either side of the mean-plane containing atoms Ru1, P1, P2, P3 and C2. The apical position in the structure is occupied by the ZnMe ligand, which is trans to a vacant site.8 The Ru–Zn distance of 2.3713(3) Å is shorter than that in A (R = Me; 2.4069(7) Å), D (2.3997(8) Å) or E (2.3819(4) Å).3
 | ||
| Fig. 1 Molecular structure of 1. Ellipsoids are represented at 30% probability. Hydrogen atoms and guest solvent have been omitted for clarity. | ||
Dissolution of 1 in C6D6 gave a 31P{1H} NMR spectrum (ESI‡) comprising of two sets of three resonances in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture, which were assigned as diastereomers (Scheme 2) by comparison to the results from the reaction of [Ru(BINAP)(PPh3)HCl] with ZnMe2 described below. The major isomer of 1 exhibited a doublet of doublets (2JPP = 257, 3 Hz) resonance at δ 65, assigned to one terminus of the dppbz ligand. The large 2JPP splitting of 257 Hz arises from coupling to the trans-cyclometallated Ph2P(biphenyl)′ ligand, which itself appeared as a doublet of doublets (2JPP = 257, 18 Hz) at δ 51.9 The small couplings of 18 and 3 Hz showed that both phosphorus nuclei were cis to the second terminus of the dppbz ligand, which appeared at δ 79. The minor diastereomer exhibited analogous signals at δ 78, 62 and 69 respectively, with similar sized coupling constants. Interconversion of the two diastereomers was shown by 31P{1H} EXSY measurements (ESI‡).
:1 mixture, which were assigned as diastereomers (Scheme 2) by comparison to the results from the reaction of [Ru(BINAP)(PPh3)HCl] with ZnMe2 described below. The major isomer of 1 exhibited a doublet of doublets (2JPP = 257, 3 Hz) resonance at δ 65, assigned to one terminus of the dppbz ligand. The large 2JPP splitting of 257 Hz arises from coupling to the trans-cyclometallated Ph2P(biphenyl)′ ligand, which itself appeared as a doublet of doublets (2JPP = 257, 18 Hz) at δ 51.9 The small couplings of 18 and 3 Hz showed that both phosphorus nuclei were cis to the second terminus of the dppbz ligand, which appeared at δ 79. The minor diastereomer exhibited analogous signals at δ 78, 62 and 69 respectively, with similar sized coupling constants. Interconversion of the two diastereomers was shown by 31P{1H} EXSY measurements (ESI‡).
Trapping of 1 by CO gave the coordinatively saturated carbonyl complex, [Ru(dppbz)(PPh2(biphenyl)′)(CO)ZnMe] (2, Scheme 2) in a rapid reaction that was accompanied by colour change from red to pale yellow. Complex 2 was characterised by a combination of NMR and IR data, but proved elusive to crystallographic characterisation. As in the case of 1, 2 was present in solution as what we believe is a mixture of two diastereomers, in a ratio of ca. 1:1.3. The minor isomer appeared in the room temperature 31P{1H} NMR spectrum as two sharp, overlapping doublets at δ 67, with couplings of 243 and 18 Hz, and a sharp doublet of doublets at δ 47 (2JPP = 243, 18 Hz), whereas the major isomer displayed three very broad signals. These resolved upon cooling to 223 K into a doublet of doublets at δ 75 (2JPP = 241, 12 Hz), a broad triplet at δ 59 and a second doublet of doublets at δ 46 (2JPP = 241, 18 Hz). The solution IR spectrum of 2 (in C6D5CD3) showed a carbonyl stretch for each of the two isomers at 1934 cm−1 and 1911 cm−1 (ESI‡).
When 1 was reacted with 13CO, each of the 31P resonances of 2-13CO exhibited an additional doublet with a 2JPC splitting of 7–11 Hz (ESI‡). A geometry in which all three phosphorus nuclei are cis to the carbonyl ligand is implied by the magnitude of these couplings, but the absence of an X-ray structure precluded us from being able to elucidate whether the ZnMe was trans to CO or a phosphine (Scheme 2). In the 13C{1H} NMR spectrum of 2-13CO, the carbonyl resonance of the minor diastereomer appeared as a sharp quartet (2JCP = 8 Hz) at δ 204 at room temperature (this broadened upon cooling), whereas that of the major diastereomer only ever appeared as a broad resonance at δ 209, even down at 211 K.
Mechanism of formation of 1
31P NMR monitoring of the reaction of [Ru(PPh3)3Cl2]·PPh3 and ZnMe2 showed that the formation of 1 proceeded through two sets of intermediates (Fig. 2). One set formed very early in the reaction and was identified at low temperature by NMR spectroscopy. The second set appeared later in the reaction and proved amenable to room temperature spectroscopic scrutiny.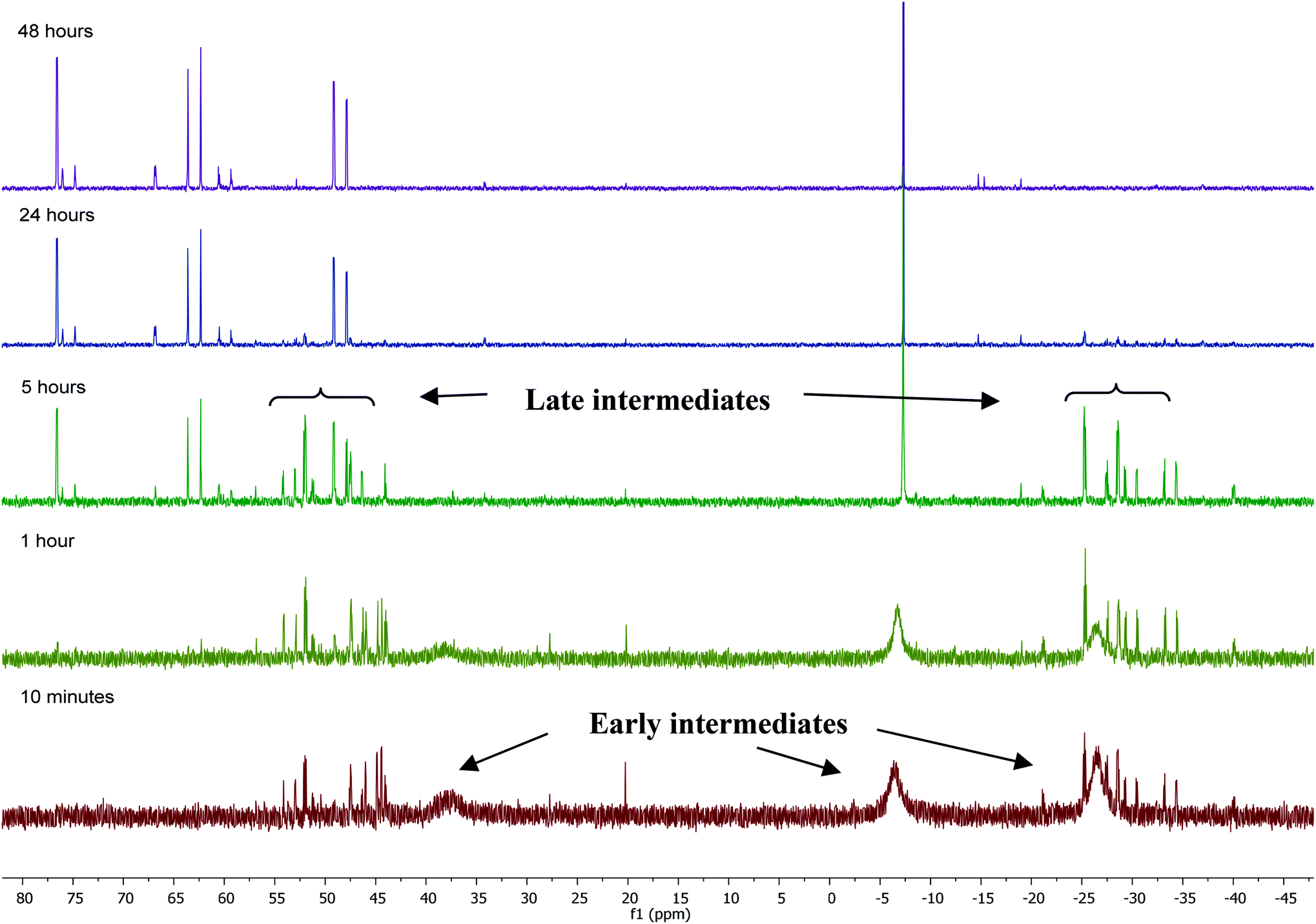 | ||
| Fig. 2 31P{1H} NMR spectra (202 MHz, THF-d8, 298 K) showing the progress of the reaction between [Ru(PPh3)3Cl2]·PPh3 and ZnMe2 with time. | ||
Characterisation of these two sets of intermediates (see below) led us to the mechanistic hypothesis depicted in Scheme 3. The initial addition of ZnMe2 to [Ru(PPh3)3Cl2]·PPh3 gives the ‘early' intermediates I–III, which all feature two ortho-metallated PPh3 ligands (step i). Further reaction with ZnMe2, followed by C–C coupling,10 generates the late intermediates IV–VI (step ii) which contain 2,2′-bis(diphenylphosphino)biphenyl (BIPHEP) ligands. A formal P–C/Ru–C metathesis reaction11 involving BIPHEP and PPh2(C6H4) ligands in step iii12 ultimately yields 1.13 Support for the formation of BIPHEP containing species on the mechanistic pathway was provided by the reaction of ZnMe2 with an in situ generated sample of [Ru(BIPHEP)(PPh3)HCl] (Scheme 3, step iv) which produced NMR signals for IV–VI, as well as the final product 1 (ESI‡).
 | ||
| Scheme 3 Proposed mechanism for the formation of 1. | ||
Characterisation of intermediates I–VI
A low temperature (246 K) reaction of ZnMe2 and [Ru(PPh3)3Cl2]·PPh3 generated intermediates I–III, which appeared in the form of three very broad 31P NMR signals at δ 51, −27 and −39. As shown in Fig. 3, these resolved into five low and four higher frequency resonances at 210 K. Intermediates I–III were assigned as bis-cyclometallated species (Scheme 4) based on the number of resonances, their chemical shifts (the low frequency signals are diagnostic of four-membered ring cyclometallated species)9,14 and 31P COSY measurements (ESI‡). Geometries were based upon the magnitudes of 2JPP. The pale yellow colour of the reaction mixture (cf. the red colour of 1) supports I–III being coordinatively saturated, although the exact nature of the two-electron donor X in species I remains unknown; ZnMe2, ZnMeCl or THF are the most likely candidates.15 The intermediates I–III were present in a ratio of ca. 3.5:5.5:1 and were shown to be in exchange by 31P{1H} EXSY measurements.
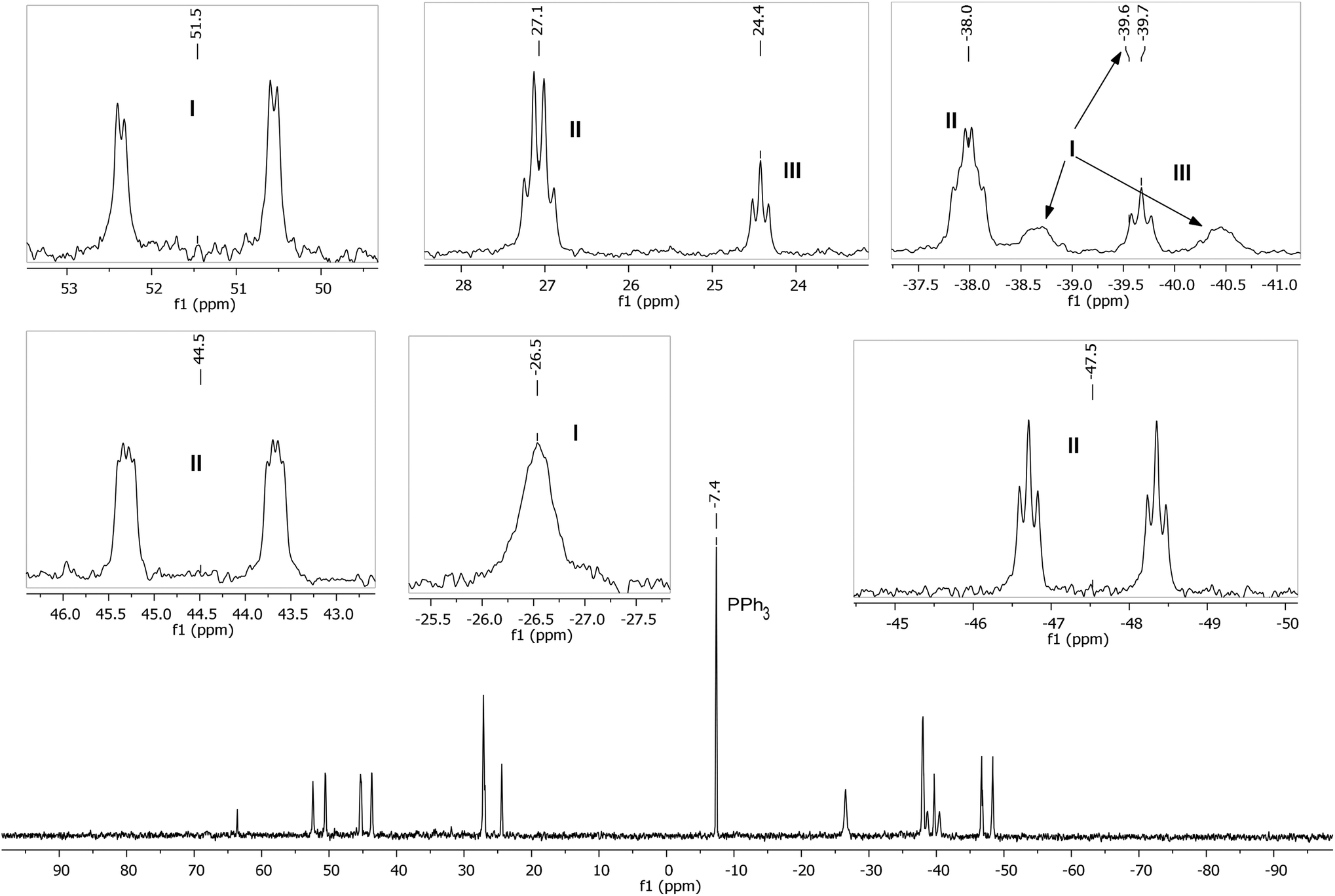 | ||
| Fig. 3 Low temperature (210 K) 31P{1H} NMR spectrum (162 MHz, THF-d8) showing intermediates I–III, with assignments and multiplicities indicated in the insets. | ||
 | ||
| Scheme 4 Proposed structure for intermediates I–III. | ||
The formation of a second set of intermediates IV–VI was established in a separate experiment in which a 1:5 molar ratio of [Ru(PPh3)3Cl2]·PPh3 and ZnMe2 were combined at room temperature. Tellingly, IV and V each showed just a single cyclometallated 31P{1H} NMR resonance, together with two other higher frequency signals (ESI‡). We propose that IV and V must also contain ZnMe ligands in order to remain as Ru(II). Structures consistent with these data are shown in Scheme 5, although NMR spectra do not allow them to be differentiated. The identity of VI was more straightforward given the presence of one high and two low frequency 31P{1H} resonances, and a doublet of triplets hydride resonance (δ −8.05 ppm, 2JHP = 46.8 Hz, 2JHP = 13.4 Hz; ESI‡). VI is related to IV and V by an additional cyclometallation reaction. The proposed structures of the three intermediates were based upon a comparison of chemical shifts and coupling constants to triphenylphosphine derivatives formed in the reaction of [Ru(PPh3)3HCl] with ZnMe2.5
 | ||
| Scheme 5 Proposed structures for intermediates IV–VI. | ||
Synthesis and characterisation of [Ru(dppbz)(PPh2(binaphthyl)′)ZnMe] (3)
Given the reactivity of [Ru(BIPHEP)(PPh3)HCl] (Scheme 3), ZnMe2 was added to the racemic BINAP derivative [Ru(rac-BINAP)(PPh3)HCl] (rac-BINAP = racemic(2,2′-bis(diphenylphosphino)-1,1′-binaphthyl).16 An instantaneous colour change from red-orange to deep green was observed,17 which reverted back to red upon heating at 70 °C (24 h). 31P{1H} NMR spectroscopy (ESI‡) showed the presence of two new species in a 1:1 ratio, both of which exhibited three high frequency doublet of doublet resonances (δ 77, 69, 63; δ 79, 68, 53). The signals were assigned to two diastereomers (vide infra for crystallographic confirmation) of [Ru(dppbz)(PPh2(binaphthyl)′)ZnMe] (3, Scheme 6). In contrast to 1, the diastereomers did not exchange by 31P EXSY (ESI‡).
 | ||
| Scheme 6 Formation of [Ru(dppbz)(PPh2(binaphthyl)′)ZnMe] (3). | ||
Compound 3 exhibited a broadly analogous structure to 1 (Fig. 4), in terms of the co-ordination geometry about the central ruthenium,18,19 however, the square based pyramid was substantially less distorted than that in 1, as evidenced by maximum deviations of +0.17 Å and −0.087 Å for Ru1 and P3, respectively, either side of the mean-plane containing the ruthenium centre, the three P atoms and the cyclometallated carbon. The Ru–Zn distance was even further reduced in 3 compared to 1 (to 2.2867(6) Å), although the two Ru–Cmetallated bond lengths were similar in both cases (1: 2.111(2) Å; 3: 2.120(2) Å). Both of these values are noticeably longer than the equivalent metric of 2.051(4) Å reported for a cycloruthenated Cy2P(binaphthyl)′ ligand.18 The crystal structure of 3 was heavily disordered such that the asymmetric unit contains an overlay of two diastereomers (Fig. 4). The two components (53:47 occupancy ratios) are related by an approximate (non-crystallographic) mirror which bisects the P3–Ru1–P3A angle. As the disorder only impacts, in a minor way, on the asymmetric ligand that contains atom P1, the disordered fractions are not mirror images of each other.
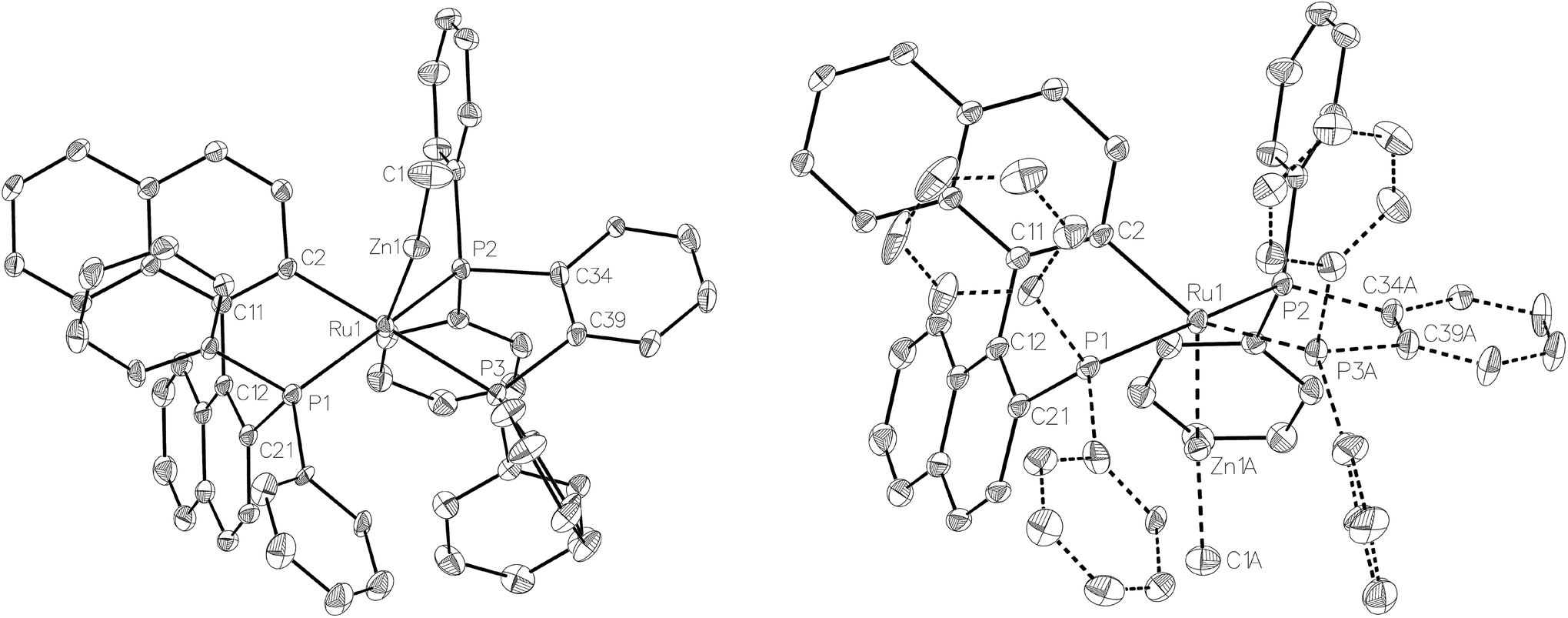 | ||
| Fig. 4 Molecular structure of the two, mutually disordered, diastereomers present in the structure of 3, from approximately similar viewpoints. Ellipsoids are represented at 30% probability. Hydrogen atoms and guest solvent have been omitted for clarity. | ||
Reactivity of 1 with H2 and PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CH
CH
The presence of both unsaturated Ru and Zn centres afforded an opportunity to compare the reactivity of 1 with that previously reported for A and E (summarised in Scheme 7).2,3,20
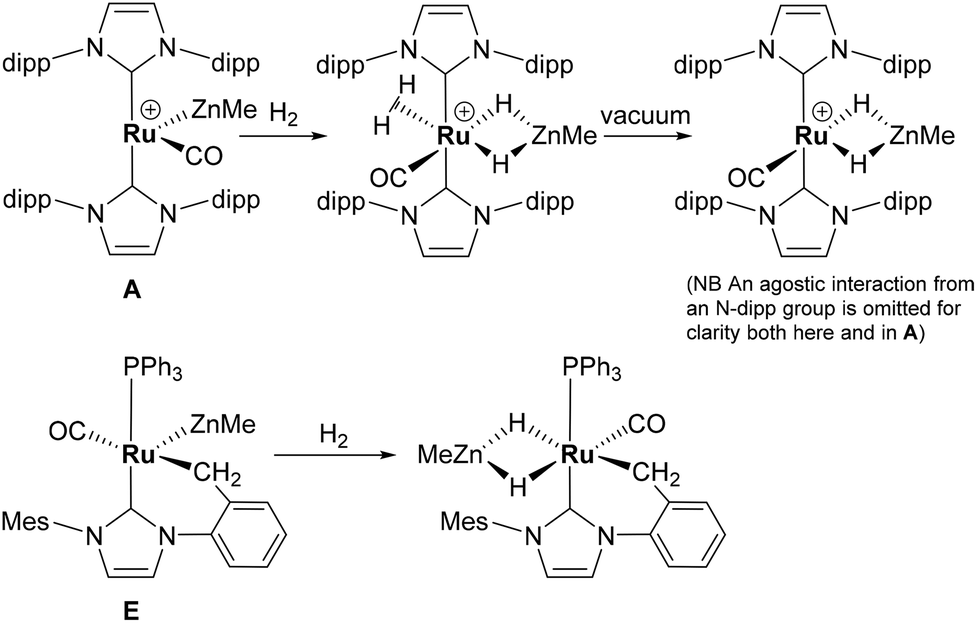 | ||
| Scheme 7 Summary of the reactions of [Ru(IPr)2(CO)ZnMe]+ and [Ru(IMes)′(PPh3)(CO)(ZnMe)] with H2. | ||
The addition of 1 atm H2 to a toluene-d8 solution of 1 brought about an instantaneous change from the red colour of the coordinatively unsaturated starting material to colourless, again indicative of coordinatively saturated Ru. Three new broad, low frequency hydride resonances were present in the 1H NMR spectrum of the reaction mixture. Cooling to 246 K partially resolved these signals and showed they were present in a 1:1:1 ratio (ESI‡). Together with the 31P{1H} resonances (δ 86 (dd, 2JPP = 242, 24 Hz), 81 (t, 2JPP = 25 Hz), 58 (dd, 2JPP = 242, 26 Hz); ESI‡), these data are consistent with the formation of mer-[Ru(dppbz)(Ph2P(biphenyl))(H)2(H)(ZnMe)] (4), arising through a reaction with two equivalents of H2. One equivalent added across the Ru–Zn bond to afford a Ru(H)2Zn moiety, while the second added across the Ru–C bond to reverse the cyclometallation of the Ph2P(biphenyl)′ ligand, also generating a terminal Ru–H ligand in the process (Scheme 8).
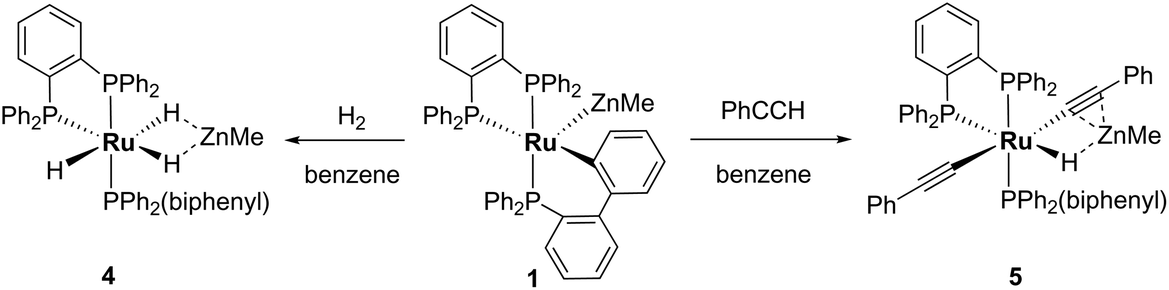 | ||
| Scheme 8 Reactivity of 1 with H2 and PhCCH. | ||
Complex 1 also reacted with two molecules of PhCCH (Scheme 8) to form the structurally characterised mer-isomer of [Ru(dppbz)(Ph2P(biphenyl))(CCPh)2(H)(ZnMe)] (5). Compound 5 results from C–H activation of one equivalent of alkyne across the Ru–Zn bond,21 together with C–H activation of a second equivalent, leading to reversal of biphenyl group cyclometallation and generation of a terminal Ru-acetylide ligand. The X-ray structure (Fig. 5) revealed several features worthy of comment. The Zn atom was asymmetrically bound to the μ–η2:η1-CCPh ligand, with Zn(1)–C(2) and Zn(1)–C(3) distances of 2.1123(17) Å and 2.4415(18) Å, respectively. These differences mirror what is seen in the few other examples of compounds in which there is an apparent side-on interaction involving zinc and a CC bond.22,23 Similarly, the interaction with zinc led to a slightly less linear Ru–CC unit (167.83°) than in the terminal acetylide ligand (175.11°). The two CC distances were similar within 3σ. The presence of two different acetylide groups was also apparent from the presence of two CC vibrations (2082, 2012 cm−1)22 in the IR spectrum.
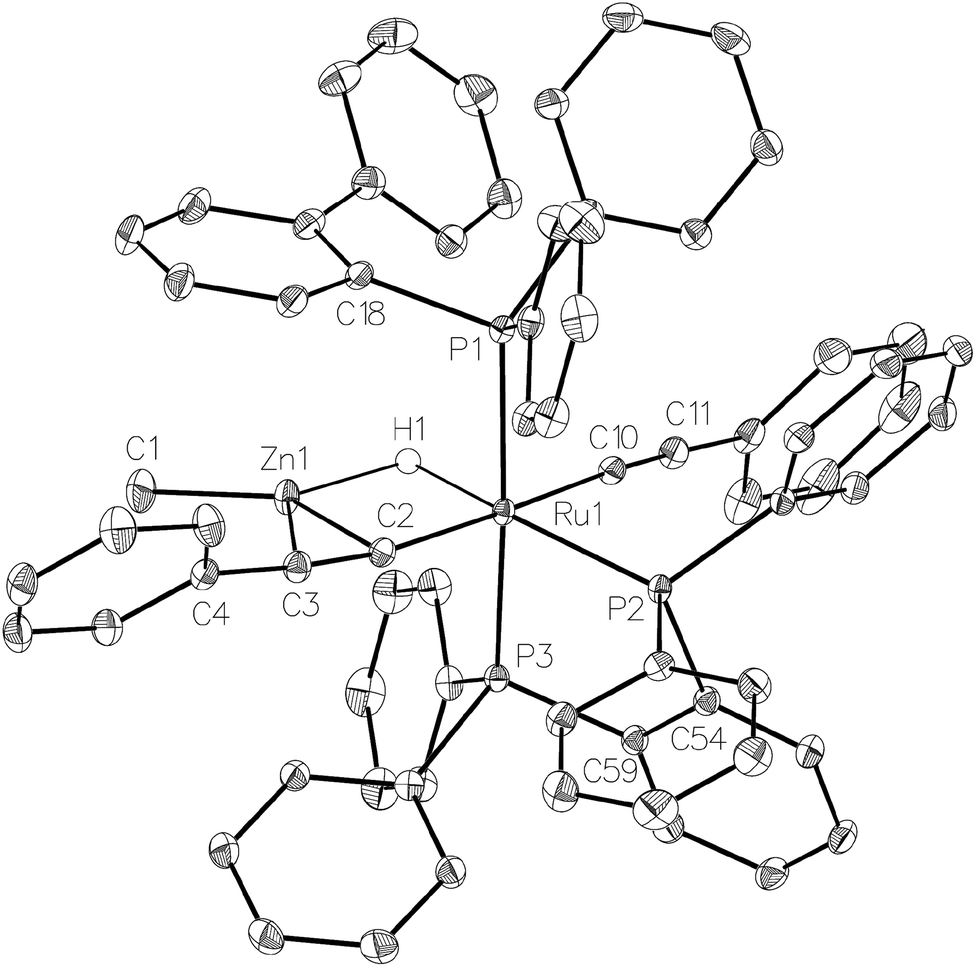 | ||
| Fig. 5 Molecular structure of 5. Ellipsoids are represented at 30% probability. Guest solvent and hydrogen atoms, with the exception of H1, have been omitted for clarity. | ||
Both 4 and 5 displayed only limited stability, slowly degrading over time in solution (5 also slowly degraded in the solid-state) at room temperature.
Conclusions
In contrast to the ‘simple’ Ru–Zn products A, D and E (Scheme 1) which are formed upon addition of ZnMe2 to [Ru(NHC)2(CO)H]+ (NHC = IPr, IBiox) and [Ru(NHC)(PPh3)(CO)HCl] (NHC = IMes), [Ru(PPh3)3Cl2] reacts through a series of C–H activation, C–C coupling, P–C activation and P–C bond forming steps to give [Ru(dppbz)(PPh2(biphenyl)′)(ZnMe)] (1). Although the individual reactions involved in the overall transformation of three PPh3 ligands into Ph2PC6H4PPh2 and Ph2P(biphenyl) groups have been observed separately,11 to the best of our knowledge, there are no examples in which they have been observed in concert with one another. The role of ZnMe2 is central to the observation of the chemistry, as it provides a means to bring about the elimination of both CH4 and ZnMeCl, allowing access to highly reactive, low-valent ruthenium fragments capable of performing the array of bond activation/formation steps observed. Additional studies of novel Ru–Zn heterobimetallic complexes that highlight this even further will be reported in due course.Experimental
All manipulations were carried out using standard Schlenk, high vacuum and glovebox techniques. Solvents were purified using an MBraun SPS solvent system (hexane, Et2O) or under a nitrogen atmosphere from sodium benzophenone ketyl (benzene, THF). C6D6, toluene-d8 and THF-d8 were vacuum transferred from potassium. CD2Cl2 was dried over CaH2. NMR spectra were recorded on Bruker Avance 400 and 500 NMR spectrometers at 298 K (unless stated otherwise) and referenced as follows; 1H: chemical shifts of residual protio solvent resonances (C6D6δ 7.16, THF-d8δ 3.58, CD2Cl2δ 5.32, C6D5CD3δ 2.08); 13C{1H}: solvent signal for C6D5CD3 (δ 20.4); 31P{1H}: externally to 85% H3PO4 (δ 0.0). IR spectra were recorded in C6D5CD3 solution or in KBr discs on a Nicolet Nexus spectrometer. Elemental analyses were performed by Elemental Microanalysis Ltd, Okehampton, Devon, UK. Literature methods were used to prepare [Ru(PPh3)3Cl2]·PPh3,6a,24 [Ru(PPh3)3Cl2],24 and [Ru(PPh3)3HCl]·toluene.25 Racemic [Ru(rac-BINAP)(PPh3)HCl]·0.35BINAP was prepared by an analogous process to that described for [Ru(R-BINAP)(PPh3)HCl].16[Ru(dppbz)(PPh2(biphenyl)′)ZnMe] (1)
ZnMe2 (1.25 mL of 2.0 M toluene solution, 2.5 mmol) was added to a THF (25 mL) suspension of [Ru(PPh3)3Cl2]·PPh3 (610 mg, 0.5 mmol) and the mixture stirred for 42 h. After removal of the solvent and residual ZnMe2 under vacuum, the red residue was redissolved in THF (10 mL), filtered and the filtrate layered with hexane (14 mL). A red crystalline solid was collected, washed with Et2O (6 mL) and dried under vacuum. Yield: 230 mg (48%). Employing [Ru(PPh3)3Cl2] as the precursor gave 236 mg of the title product as a red solid (49% yield). 1 exists in solution as an equilibrium mixture of 2 diastereomers in a 5:1 ratio (referred to as maj and min below). 1H NMR: δH (500 MHz, C6D6) 7.71–7.62 (m, 1Hmaj, 1Hmin, Ar), 7.62–7.53 (m, 5Hmaj, 5Hmin, Ar), 7.51–7.19 (m, 9Hmaj, 9Hmin, Ar), 7.12–6.71 (m, 24Hmaj, 26Hmin, Ar), 6.68 (t, J = 7.5 Hz, 2Hmaj, Ar), 6.62 (t, J = 7.3 Hz, 1Hmaj, Ar), 6.34 (t, J = 7.2 Hz, 1Hmin, Ar), 0.08 (s, 3Hmin, ZnCH3), −1.00 (s, 3Hmaj, ZnCH3). 31P{1H} NMR: δP (202 MHz, C6D6) 79.1 (dd, 2JPP = 18, 3 Hz, dppbzmaj), 77.6 (dd, 2JPP = 250, 7 Hz, dppbzmin), 68.9 (dd, 2JPP = 18, 7 Hz, dppbzmin), 65.3 (dd, 2JPP = 257, 3 Hz, dppbzmaj), 62.0 (dd, 2JPP = 250, 18 Hz, PPh2(biphenyl)′min), 50.6 (dd, 2JPP = 257, 18 Hz, PPh2(biphenyl)′maj). Anal. Calcd for C55H45P3ZnRu: C, 68.43, H, 4.70%; Found: C, 67.89, H, 4.84%.
[Ru(dppbz)(PPh2(biphenyl)′)(CO)ZnMe] (2)
A J. Young's resealable NMR tube was charged with 1 (10 mg, 0.01 mmol) in C6D5CD3 (0.5 mL), and placed under 1 atm CO (or 13CO). Vigorous shaking brought about a colour change from deep red to pale yellow and complete dissolution of starting material was achieved within a few minutes. The solution was analysed by 1H and 31P{1H} NMR spectroscopy, which showed that 2 was present as a mixture of 2 diastereomers in a ratio of ca. 1.3:1 (referred to as maj and min below). Selected 1H NMR: δH (400 MHz, C6D5CD3, 223 K) −0.52 (s, ZnCH3maj), −1.54 (s, ZnCH3min). 31P{1H} NMR: δP (162 MHz, C6D5CD3, 223 K) 75.0 (dd, 2JPP = 241, 12 Hz, dppbzmaj), 68.1 (d, 2JPP = 18 Hz, dppbzmin), 68.0 (d, 2JPP = 241 Hz, dppbzmin), 58.7 (t, 2JPP = 15 Hz, dppbzmaj), 48.6 (dd, 2JPP = 241, 18 Hz, PPh2(biphenyl)′min), 46.3 (dd, 2JPP = 241, 18 Hz, PPh2(biphenyl)′maj). 2-13CO: Selected 13C{1H} NMR: δC (101 MHz, C6D5CD3, 223 K) 208.7 (m, RuCOmaj), 203.9 (q, 2JPC = 8 Hz, RuCOmin). 31P{1H} NMR: δP (162 MHz, C6D5CD3, 223 K) 75.0 (dt, 2JPP = 242, 11 Hz, 2JPC = 11 Hz, dppbzmaj), 68.1 (dd, 2JPP = 18 Hz, 2JPC = 7 Hz, dppbzmin), 68.0 (dd, 2JPP = 241 Hz, 2JPC = 7 Hz, dppbzmin), 58.7 (m, dppbzmaj), 48.6 (ddd, 2JPP = 241, 18 Hz, 2JPC = 8 Hz, PPh2(biphenyl)′min), 46.3 (ddd, 2JPP = 242, 18 Hz, 2JPC = 11 Hz, PPh2(biphenyl)′maj). IR (C6D5CD3, cm−1): 1934 (νCO), 1911 (νCO).
Detection and characterisation of intermediates I–III
ZnMe2 (42 μL of 1.2 M toluene solution, 0.05 mmol) in THF-d8 (0.2 mL) was vacuum transferred into a J. Young's resealable NMR tube containing a THF-d8 (0.3 mL) solution of [Ru(PPh3)3Cl2]·PPh3 (12 mg, 0.01 mmol). The solution was maintained at <200 K prior to insertion into a pre-cooled (210 K) NMR spectrometer. After warming to 246 K for 15 min to initiate reaction, the temperature was returned to 210 K to prevent further reaction and 1H, 31P{1H}, 31P COSY and 31P EXSY measurements of intermediates I–III recorded. The reaction mixture was then warmed to room temperature. After 8 h, 31P{1H} NMR spectroscopy revealed the formation of intermediates IV–VI (see below), as well as the final product 1. 31P{1H} NMR of I: δP (162 MHz, THF-d8, 210 K) 51.5 (dd, 2JPP = 292, 14 Hz), −26.5 (br s), −39.6 (br d, 2JPP = 290 Hz). 31P{1H} NMR of II: δP (162 MHz, THF-d8, 210 K) 44.5 (ddd, 2JPP = 265, 18, 9 Hz), 27.1 (q, 2JPP = 19 Hz), −38.0 (td, 2JPP = 19, 10 Hz), −47.5 (dt, 2JPP = 266, 19 Hz). 31P{1H} of III: δP (162 MHz, THF-d8, 210 K) 24.4 (t, 2JPP = 16 Hz), −39.7 (t, 2JPP = 16 Hz).Detection and characterisation of intermediates of IV–VI
A J. Young's resealable NMR tube was charged with [Ru(PPh3)3Cl2]·PPh3 (12 mg, 0.01 mmol) in THF-d8 (0.5 mL) and ZnMe2 (42 μL of 1.2 M toluene solution, 0.05 mmol) was added. After standing for 3.5 h at room temperature, 1H, 31P{1H}, 31P HMQC and 31P COSY measurements were used to characterise the three intermediates IV–VI. 31P{1H} NMR of IV: δP (202 MHz, THF-d8) 55.4 (dd, 2JPP = 240, 15 Hz), δ 49.4 (dd, 2JPP = 20, 15 Hz), −31.9 (dd, 2JPP = 240, 20 Hz). 31P{1H}NMR of V: δP (202 MHz, THF-d8) 48.8 (dd, 2JPP = 234, 18 Hz), 45.9 (dd, 2JPP = 22, 18 Hz), −28.0 (dd, 2JPP = 234, 22 Hz). Selected 1H NMR of VI: δH (500 MHz, THF-d8) −8.05 (dt, 2JHP = 46.8, 13.4 Hz, 1H, RuH). 31P{1H} NMR of VI: δP (202 MHz, THF-d8) δ 53.9 (dd, 2JPP = 24, 21 Hz), −23.4 (dd, 2JPP = 21, 19 Hz), −26.7 (dd, 2JPP = 24, 19 Hz).Reaction of [Ru(BIPHEP)(PPh3)HCl] and ZnMe2
[Ru(BIPHEP)(PPh3)HCl] was prepared in situ upon heating (40 °C, 24 h) [Ru(PPh3)3HCl]·toluene (10 mg, 0.01 mmol) and BIPHEP (5 mg, 0.01 mmol) in CD2Cl2 (0.6 mL). The resulting red-orange solution comprised ca. 80% [Ru(BIPHEP)(PPh3)HCl], together with [Ru(PPh3)3HCl] (ca. 12%) and a third species attributed to [Ru(BIPHEP)2HCl] (ca. 8%). After removal of solvent, the red residue was redissolved in THF-d8 (0.5 mL) and ZnMe2 (42 μL of 1.2 M toluene solution, 0.05 mmol) added. A 31P{1H} NMR spectrum recorded 6 h later showed the presence of intermediates IV–VI, along with 1. Selected 1H NMR of [Ru(BIPHEP)(PPh3)HCl]: δH (500 MHz, CD2Cl2) −22.78 (dt, 2JHP = 37, 23 Hz, 1H, RuH). 31P{1H} NMR of [Ru(BIPHEP)(PPh3)HCl]: δP (202 MHz, CD2Cl2) 87.0 (dd, 2JPP = 38, 21 Hz), 41.2 (dd, 2JPP = 305, 38 Hz), 35.5 (dd, 2JPP = 305, 21 Hz). Selected 1H NMR of [Ru(BIPHEP)2HCl]: δH (500 MHz, CD2Cl2) −16.47 (tt, 2JHP = 24, 15 Hz, 1H, RuH). 31P{1H} NMR of [Ru(BIPHEP)2HCl]: δP (202 MHz, CD2Cl2) 35.8 (t, 2JPP = 35 Hz), 22.2 (t, 2JPP = 35 Hz).[Ru(dppbz)(PPh2(binaphthyl)′)ZnMe] (3)
ZnMe2 (0.50 mL of 1.2 M toluene solution, 0.6 mmol) was added to a THF (6 mL) suspension of racemic [Ru(rac-BINAP)(PPh3)HCl]·0.35BINAP (120 mg, 0.10 mmol), leading to an immediate colour change from red-orange to deep green. The solution was then heated for 24 h at 70 °C. After removal of the solvent, the red residue was redissolved in THF (1.5 mL), filtered through a pad of Celite® and the filtrate treated with hexane (3 mL). After standing for 24 h, free BINAP was separated by filtration through Celite® and a further 1 mL hexane added to the filtrate to give, over 24 h, a red crystalline solid, which was isolated and dried under vacuum. Yield: 58 mg, ca. 90% pure (49% yield). A sample of the product (20 mg) was recrystallised from THF/hexane to give 15 mg of pure material. 3 exists as a mixture of two diastereomers (referred to as A and B below) in a ratio of ca. 1:1 ratio. These were assigned by comparison with the NMR signals of minor and major diastereomers of 1. 1H NMR: δH (500 MHz, C6D6) 7.96 (d, J = 8.5 Hz, 1H, Ar), 7.82 (t, J = 8.5 Hz, 2H, Ar), 7.73–7.34 (m, 25H, Ar), 7.33–7.18 (m, 5H, Ar), 7.15–6.82 (m, 39H, Ar), 6.81–6.68 (m, 10H, Ar), 6.67–6.49 (m, 8H, Ar), 6.23 (t, J = 7.1 Hz, 2H, Ar), 0.15 (s, 3HA, ZnCH3), −1.40 (s, 3HB, ZnCH3). 31P{1H} NMR: δP (202 MHz, C6D6) 78.5 (dd, 2JPP = 18, 3 Hz, dppbzB), 77.1 (dd, 2JPP = 248, 11 Hz, dppbzA), 68.6 (dd, 2JPP = 17, 11 Hz, dppbzA), 68.2 (dd, 2JPP = 255, 3 Hz, dppbzB), 63.4 (dd, 2JPP = 248, 17 Hz, PPh2(binaphthyl)′A), 52.8 (dd, 2JPP = 255, 18 Hz, PPh2(binaphthyl)′B). Anal. Calcd for C63H49P3ZnRu: C, 71.02; H, 4.64%; Found: C, 71.25; H, 5.05%.
[Ru(dppbz)(Ph2P(biphenyl))(H)2(H)(ZnMe)] (4)
A J. Young's resealable NMR tube was charged with 1 (10 mg, 0.01 mmol) in C6D5CD3 (0.5 mL) and placed under 1 atm H2. Vigorous shaking brought about a colour change from deep red to pale yellow. Complete dissolution of starting material took place in <10 min. The resulting fine suspension was filtered through a pad of Celite® and rapidly (<30 min) analysed by 1H and 31P{1H} NMR spectroscopy, which showed clean formation of 4. Slow decomposition of the product took place in solution at room temperature, precluding isolation. 1H NMR: δH (400 MHz, C6D5CD3, 246 K) 8.54 (t, J = 9.0 Hz, 2H, Ar), 8.15 (dd, J = 12.5, 8.2 Hz, 1H, Ar), 7.99 (t, 8.5 Hz, 2H, Ar), 7.57–7.37 (m, 6H, Ar), 7.36–6.92 (m, 16H, Ar; overlapped with residual solvent signals in C6D5CD3), 6.91–6.80 (m, 6H, Ar), 6.79–6.65 (m, 8H, Ar), 6.59 (t, J = 7.2 Hz, 2H, Ar), −0.89 (s, 3H, ZnCH3), −7.08 (quint, 2JHP = 8.9 Hz, 1H, RuH), −8.37 (dt, 2JHP = 52.1 Hz, 19.4 Hz, 1H, RuH), −8.62 (m, 1H, RuH). 31P{1H} NMR: δP (162 MHz, C6D5CD3, 246 K) 85.5 (dd, 2JPP = 242, 24 Hz), 81.2 (t, 2JPP = 25 Hz), 57.9 (dd, 2JPP = 242, 26 Hz).[Ru(dppbz)(Ph2P(biphenyl))(CCPh)2(H)(ZnMe)] (5)
A suspension of 1 (97 mg, 0.1 mmol) in C6H6 (2 mL) was treated with PhCCH (33 μL, 0.3 mmol) and the mixture stirred vigorously for 3 h. The resulting yellow fine suspension was filtered through a pad of Celite®, diluted with hexane (7.5 mL) and crystallized at −35 °C. Yellow crystalline solid was separated, washed with hexane (1.5 mL × 2) and dried under vacuum. Yield: 106 mg (90%). The product undergoes slow decomposition both in solution and in the solid-state at room temperature. 1H NMR: δH (500 MHz, C6D6) 8.76 (dd, J = 13.8, 8.0 Hz, 1H, Ar), 8.62 (t, J = 9.1 Hz, 2H, Ar), 8.20 (br t, J = 8.3 Hz, 2H, Ar), 7.66 (t, J = 7.3 Hz, 1H, Ar), 7.57 (t, J = 7.6 Hz, 1H, Ar), 7.44–7.29 (m, 6H, Ar), 7.22 (d, J = 7.4 Hz, 2H, Ar), 7.19–6.60 (m, 38H, Ar), −0.47 (s, 3H, ZnCH3), −6.91 (dt, 2JHP = 59.6, 16.2 Hz, 1H, RuH). 31P{1H} NMR: δP (202 MHz, C6D6) 73.8 (dd, 2JPP = 275, 20 Hz), 55.8 (dd, 2JPP = 24, 20 Hz), 37.9 (dd, 2JPP = 275, 24 Hz). IR (KBr, cm−1): 2082 (νCC), 2012 (νCC). Anal. Calcd for C71H57P3ZnRu: C, 72.91; H, 4.91%; Found: C, 73.03; H, 4.89%.
X-ray crystallography
Data for 1 were collected on an Agilent Xcalibur diffractometer (using Mo-Kα radiation) while those for 3 and 5 were obtained using an Agilent SuperNova instrument and a Cu-Kα source. All experiments were conducted at 150 K. Using Olex2,26 all structures were solved with the olex2·solve27 structure solution program and subsequently refined using the SHELXL program.28Points of note include the fact that the asymmetric unit in 1 contains a portion of solvent in addition to one molecule of the complex. While the former was identifiable as a hexane moiety, the disorder was so extensive that modelling would have resulted in excessive parameterization. Hence, the solvent was treated using the solvent mask in Olex2. However, the formula as presented, herein, accounts for the presence of a 1:1 ratio of the title compound to hexane in the crystal.
The asymmetric unit in 3 resolved beautifully, once the rampant disorder was addressed. In particular, Zn1, P3, the methyl group based on C1 and the phenyl groups based on C34, C40, C41, C45 and C58 were all treated for 53:47 disorder. Some distance and ADP restraints were included in the model (on merit), in disordered regions – to assist convergence. There was also a modicum of disordered solvent in the asymmetric unit, which did not lend itself to being readily modelled. As above, this was treated with the solvent mask algorithm available in Olex2, and an allowance for one molecule of THF per unit cell has been made in the formula as presented – to account for the pre-squeeze electron density evident in the difference Fourier electron density map. It merits note that strenuous checks were performed regarding the diffraction pattern symmetry in this structure, given the level of disorder present. Integration of the data in the triclinic, reduced cell [a = 10.8687(3), b = 12.1871(3), c = 20.5157(5) Å, α = 79.451(2), β = 81.396(2), γ = 80.252(2)°] afforded two molecules in the asymmetric unit (as expected) in P1 plus some solvent. Both of the complex molecules exhibited similar disorder to that presented for the Pbca solution/refinement reported herein. Ultimately, the exercise of testing in space-group P1 served as reassurance that the evident disorder is real. H1 in the structure of 5 was located and refined freely. Three guest molecules of benzene were also found in the asymmetric unit for this crystal.
Crystallographic data for all compounds have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications CCDC 1937663–1937665 for 1, 3 and 5, respectively.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the EU (Marie Curie Individual Fellowship to FMM (792674 H2020-MSCA-IF 2017)) for financial support.References
- For reviews, see: (a) Molecular Metal-Metal Bonds: Compounds, Synthesis, Properties, ed. S. T. Liddle, Wiley-VCH, Weinheim, 2015 Search PubMed; (b) A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef CAS PubMed. For specific examples, see: (c) F. N. Tebbe, J. Am. Chem. Soc., 1973, 95, 5412–5414 CrossRef CAS; (d) J. N. St. Denis, W. Butler, M. D. Glick and J. P. Oliver, J. Organomet. Chem., 1977, 129, 1–16 CrossRef; (e) P. H. M. Budzelaar, A. A. H. Vanderzeijden, J. Boersma, G. J. M. Vanderkerk, A. L. Spek and A. J. M. Duisenberg, Organometallics, 1984, 3, 159–163 CrossRef CAS; (f) J. W. Bruno, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1984, 106, 444–445 CrossRef CAS; (g) D. L. Thorn and R. L. Harlow, J. Am. Chem. Soc., 1989, 111, 2575–2580 CrossRef CAS; (h) M. D. Fryzuk, D. H. McConville and S. J. Rettig, Organometallics, 1990, 9, 1359–1360 CrossRef CAS; (i) M. D. Fryzuk, D. H. McConville and S. J. Rettig, Organometallics, 1993, 12, 2152–2161 CrossRef CAS; (j) J. T. Golden, T. H. Peterson, P. L. Holland, R. G. Bergman and R. A. Andersen, J. Am. Chem. Soc., 1998, 120, 223–224 CrossRef CAS; (k) R. A. Fischer, D. Weiss, M. Winter, I. Müller, H. D. Kaesz, N. Frölich and G. Frenking, J. Organomet. Chem., 2004, 689, 4611–4623 CrossRef CAS; (l) M. Ohashi, K. Matsubara and H. Suzuki, Organometallics, 2007, 26, 2330–2339 CrossRef CAS; (m) C. J. Durango-García, J. O. C. Jiménez-Halla, M. López-Cardoso, V. Montiel-Palma, M. A. Muñoz-Hernández and G. Merino, Dalton Trans., 2010, 39, 10588–10589 RSC.
- I. M. Riddlestone, N. A. Rajabi, J. P. Lowe, M. F. Mahon, S. A. Macgregor and M. K. Whittlesey, J. Am. Chem. Soc., 2016, 138, 11081–11084 CrossRef CAS PubMed.
- M. Espinal-Viguri, V. Varela-Izquierdo, F. M. Miloserdov, I. M. Riddlestone, M. F. Mahon and M. K. Whittlesey, Dalton Trans., 2019, 48, 4176–4189 RSC.
- I. M. Riddlestone, N. A. Rajabi, S. A. Macgregor, M. F. Mahon and M. K. Whittlesey, Chem. – Eur. J., 2018, 24, 1732–1738 CrossRef CAS PubMed.
- F. M. Miloserdov, N. A. Rajabi, J. P. Lowe, M. F. Mahon, S. A. Macgregor and M. K. Whittlesey, unpublished results.
- (a) Both forms of [Ru(PPh3)3Cl2] (i.e. with and without co-crystallized PPh3) could be utilized for the reaction and gave the same yield of 1. H. Samouei, F. M. Miloserdov, E. C. Escudero-Adan and V. V. Grushin, Organometallics, 2014, 33, 7279–7283 CrossRef CAS; (b) The overall reaction requires 3 equivalents of ZnMe2; 2 equivalents to abstract chloride in the form of ZnMeCl, and the third to provide the ZnMe ligand in the product. An excess (5 equivalents) of ZnMe2 was typically employed to drive the formation of 1 to completion.
- The formation of 1 contrasts markedly with the outcome of the reaction of [Ru(PPh3)3Cl2] and AlMe3 in arene solvents which yields [(η6-arene)Ru(PPh3)2Me][AlCl2Me2] salts as the products. X. G. Fang, J. G. Watkin, B. L. Scott, K. D. John and G. J. Kubas, Organometallics, 2002, 21, 2336–2339 CrossRef CAS.
- C. J. Pell, W. C. Shih, S. Gatard and O. V. Ozerov, Chem. Commun., 2017, 53, 6456–6459 RSC.
- (a) P. E. Garrou, Inorg. Chem., 1975, 14, 1435–1439 CrossRef CAS; (b) P. E. Garrou, Chem. Rev., 1981, 81, 229–266 CrossRef CAS.
- P. A. Shaw, G. J. Clarkson and J. P. Rourke, Chem. Sci., 2017, 8, 5547–5558 RSC.
- Y. H. Lee and B. Morandi, Coord. Chem. Rev., 2019, 386, 96–118 CrossRef CAS.
- (a) L. M. Alcazar-Roman, J. F. Hartwig, A. L. Rheingold, L. M. Liable-Sands and I. A. Guzei, J. Am. Chem. Soc., 2000, 122, 4618–4630 CrossRef CAS; (b) S. Z. Ge, R. A. Green and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 3300–3300 CrossRef CAS PubMed.
- Such a process can be mediated by transition metal phosphido complexes. P. E. Suess, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 4746–4760 CrossRef PubMed.
- F. Mohr, S. H. Priver, S. K. Bhargava and M. A. Bennett, Coord. Chem. Rev., 2006, 250, 1851–1888 CrossRef CAS.
- We envisage ZnMe2 or ZnMeCl coordinating in Ru⋯Zn complexes in which Me and/or Cl groups bridge the two metal centres (ref. 5).
- K. Abdur-Rashid, A. J. Lough and R. H. Morris, Organometallics, 2001, 20, 1047–1049 CrossRef CAS.
- Studies to elucidate the structure of the initially formed green complex are ongoing and will be the subject of a subsequent publication.
- N. Feiken, P. S. Pregosin, G. Trabesinger and M. Scalone, Organometallics, 1997, 16, 537–543 CrossRef CAS.
- For other examples of P–C activation in Ru(BINAP) complexes, see: (a) T. J. Geldbach, P. S. Pregosin, A. Albinati and F. Rominger, Organometallics, 2001, 20, 1932–1938 CrossRef CAS; (b) T. J. Geldbach and P. S. Pregosin, Eur. J. Inorg. Chem., 2002, 1907–1918 CrossRef CAS; (c) T. J. Geldbach, P. S. Pregosin and A. Albinati, Organometallics, 2003, 22, 1443–1451 CrossRef CAS; (d) T. J. Geldbach, F. Breher, V. Gramlich, P. G. A. Kumar and P. S. Pregosin, Inorg. Chem., 2004, 43, 1920–1928 CrossRef CAS PubMed.
- IBiox complex D decomposes upon addition of H2 (ref. 3).
- For recent examples of alkyne C–H addition across Pt–Au/Ag bonds, see: (a) J. Campos, J. Am. Chem. Soc., 2017, 139, 2944–2947 CrossRef CAS PubMed; (b) N. Hidalgo, C. Maya and J. Campos, Chem. Commun., 2019, 55, 8812–8815 RSC.
- H. Lang, N. Mansilla and G. Rheinwald, Organometallics, 2001, 20, 1592–1596 CrossRef CAS.
- E. E. Wilson, A. G. Oliver, R. P. Hughes and B. L. Ashfeld, Organometallics, 2011, 30, 5214–5221 CrossRef CAS.
- P. S. Hallman, T. A. Stephenson and G. Wilkinson, Inorg. Synth., 1970, 12, 237–238 CAS.
- R. A. Schunn, E. R. Wonchoba and G. Wilkinson, Inorg. Synth., 1972, 13, 131–134 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef; G. M. Sheldrick, SHELXL-97, a computer program for crystal structure refinement, University of Göttingen, 1997 Search PubMed.
Footnotes |
| † MKW dedicates this paper to Professor Robin Perutz FRS on the occasion of his 70th birthday. Thank you Robin for the enthusiasm, guidance, encouragement and friendship over the last 30 years. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1937663–1937665. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03106e |
| This journal is © The Royal Society of Chemistry 2019 |