
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of the FeCp(CO)2+ moiety on the dynamics of the metalloid [Ge9(Si(SiMe3)3)3]− cluster in thf: synthesis and characterization by time-resolved absorption spectroscopy†‡
Nadine C.
Michenfelder
a,
Christian
Gienger
b,
Andreas
Schnepf
 *b and
Andreas-Neil
Unterreiner
*a
*b and
Andreas-Neil
Unterreiner
*a
aInstitut für Physikalische Chemie, Karlsruher Institut für Technologie (KIT), Kaiserstr. 12, 76131 Karlsruhe, Germany. E-mail: andreas.unterreiner@kit.edu; Tel: +49 721 608 47807
bInstitut für Anorganische Chemie, Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: andreas.schnepf@uni-tuebingen.de; Fax: +49 (7071) 28-2436; Tel: +497071 29 76635
First published on 18th July 2019
Abstract
A neutral tetrasubstituted Ge9 cluster with a covalently bound transition metal substituent was synthesized successfully via a salt metathesis reaction. Photoexcitation of [Ge9(Si(SiMe3)3)3FeCp(CO)2] induces excited state dynamics of the compound that was analysed by extended broadband fs absorption spectroscopy in the UV-Vis-NIR region. After UV or Vis excitation, an electron is detached from the [Ge9(Si(SiMe3)3)3]−-entity and localizes within few hundred fs. Recombination of this cluster-electron-pair occurs in about 7–9 ps. Finally, a third component can be attributed to complete ground state recovery within roughly 150 ps. This is much shorter compared to a longer-lived component within Li[Ge9(Si(SiMe3)3)3], whose transient absorption exceeds the ns timescale after UV excitation. This observation emphasizes a strong influence of the Fe moiety.
Introduction
Metalloid germanium-clusters1 of the general formula GenRm (n > m; R = organic or metal–organic substituents like Dipp2-C6H3 (Dipp = 2,4-iPr2-C6H3), Si(SiMe3)3 or N(SiMe3)2) are ideal model compounds for germanium nanoparticles, exhibiting novel physical or chemical properties.2–5 Among these clusters in recent years a fruitful chemistry has been mainly observed for [Ge9(Hyp)3]− (Hyp = Si(SiMe3)3) and its derivatives.6 This highly stable Ge9 compound was first synthesized by us under inert conditions via the disproportionation reaction of a Ge(I) halide.7 However, a more convenient synthetic route starting from the parent Zintl anion Ge94− and HypCl was established later on by Sevov et al.8Via this route also differently substituted metalloid germanium clusters with nine germanium atoms could be synthesized where lately also phosphine substituents are bound to the Ge9 core opening the door for further coordination chemistry.9 Also the disubstituted cluster [Ge9(Hyp)2]2− (ref. 10) is obtained via this synthetic route by varying the experimental conditions. The latter opened the door to “mixed”-substituted clusters like [Ge9(Hyp)2(HypPh)]− (HypPh = Si(SiMe3)2(SiPh3),9 [Ge9(HypPh)2(Hyp)]−,11 [Ge9(Hyp)2(PtBu2Cu-NHCDipp)]−,12 [Ge9(Hyp)2(PtBu2)]−,13 [Ge9(Hyp)2(SiPh2-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)]− (ref. 14) and connected Ge9(Hyp)2 units in [(Hyp)2Ge9-Me2Si-C6H4-SiMe2-Ge9(Hyp)2K]−.15 The so far obtained clusters might be further connected to dimers via transition metal atoms like in [AuGe18(Hyp)6]− (ref. 16) or might be oxidatively coupled to give the neutral metalloid germanium cluster Ge18(Hyp)6.17 Additionally, the introduction of a fourth substituent can lead to neutral clusters of the form Ge9(Hyp)3R (R = Et,18 (CH2)3(CHCH2),19 (CH2)3–C6H5,20 SnPh3,21 and ZnCp*
CH2)]− (ref. 14) and connected Ge9(Hyp)2 units in [(Hyp)2Ge9-Me2Si-C6H4-SiMe2-Ge9(Hyp)2K]−.15 The so far obtained clusters might be further connected to dimers via transition metal atoms like in [AuGe18(Hyp)6]− (ref. 16) or might be oxidatively coupled to give the neutral metalloid germanium cluster Ge18(Hyp)6.17 Additionally, the introduction of a fourth substituent can lead to neutral clusters of the form Ge9(Hyp)3R (R = Et,18 (CH2)3(CHCH2),19 (CH2)3–C6H5,20 SnPh3,21 and ZnCp*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22) and also a pentafunctionlized cluster Ge9(Hyp)3Et(PdPPh3) is known.23 The addition of a transition metal substituent can also lead to cluster enlargement to give a distorted bicapped square antiprismatic [MGe9] core.24–26 The bonding of a transition metal atom to a Ge9(Hyp)3 unit strongly alters the electronic situation of the cluster and thus the color. Lately it became obvious that the number of transition metal atoms bound to a Ge9(Hyp)3 unit has a greater influence on the optical properties than the kind of the transition metal atom. Thus a compound exhibiting a M-Ge9(Hyp)3-M unit shows a strong absorption band around 450 nm while a compound exhibiting a M-Ge9(Hyp)3 unit shows an absorption band around 400 nm.27 To further check the influence of a transition metal fragment on the electronic situation of a [Ge9(Hyp)3]− unit we wondered if it is possible to introduce a fourth cationic substituent to obtain a neutral [Ge9(Hyp)3R] compound where R is a transition metal based substituent. In the following we present first results in this respect. The metalloid cluster [Ge9(Hyp)3]− with a cation like Li+ has a strong absorption band in the UV region. As known from earlier publications in the gas phase, photodetachment of an electron from a Ge9− entity occurs at 3.37 eV.28 Hence, excitation in the liquid phase at wavelengths near 400 nm or shorter may lead to an abstraction of an electron from the cluster core governing ultrafast dynamics. The localization of this electron and the following processes can be elegantly investigated via transient absorption spectroscopy in the UV-Vis and NIR spectral range. After photoexcitation into the UV band the transient response on a fs to ps timescale reveals cluster dynamics but also longer-lived states such as solvated electrons or contact pairs, which have absorption spectra in the near-infrared (NIR) spectral range.28 For example, the solvated electron in thf peaks around 2120 nm at room temperature with its high-energy tail extended to the Vis regime,29 while contact pairs30 have been observed in the NIR region, e.g. around 1180 nm for [Li, e−].31 The newly synthesized cluster compound was investigated by broadband fs-transient absorption spectroscopy in the visible and NIR spectral region to explore the influence of a transition metal substituent on the excited state dynamics of such a cluster using [Ge9(Hyp)3]− as a reference system.
22) and also a pentafunctionlized cluster Ge9(Hyp)3Et(PdPPh3) is known.23 The addition of a transition metal substituent can also lead to cluster enlargement to give a distorted bicapped square antiprismatic [MGe9] core.24–26 The bonding of a transition metal atom to a Ge9(Hyp)3 unit strongly alters the electronic situation of the cluster and thus the color. Lately it became obvious that the number of transition metal atoms bound to a Ge9(Hyp)3 unit has a greater influence on the optical properties than the kind of the transition metal atom. Thus a compound exhibiting a M-Ge9(Hyp)3-M unit shows a strong absorption band around 450 nm while a compound exhibiting a M-Ge9(Hyp)3 unit shows an absorption band around 400 nm.27 To further check the influence of a transition metal fragment on the electronic situation of a [Ge9(Hyp)3]− unit we wondered if it is possible to introduce a fourth cationic substituent to obtain a neutral [Ge9(Hyp)3R] compound where R is a transition metal based substituent. In the following we present first results in this respect. The metalloid cluster [Ge9(Hyp)3]− with a cation like Li+ has a strong absorption band in the UV region. As known from earlier publications in the gas phase, photodetachment of an electron from a Ge9− entity occurs at 3.37 eV.28 Hence, excitation in the liquid phase at wavelengths near 400 nm or shorter may lead to an abstraction of an electron from the cluster core governing ultrafast dynamics. The localization of this electron and the following processes can be elegantly investigated via transient absorption spectroscopy in the UV-Vis and NIR spectral range. After photoexcitation into the UV band the transient response on a fs to ps timescale reveals cluster dynamics but also longer-lived states such as solvated electrons or contact pairs, which have absorption spectra in the near-infrared (NIR) spectral range.28 For example, the solvated electron in thf peaks around 2120 nm at room temperature with its high-energy tail extended to the Vis regime,29 while contact pairs30 have been observed in the NIR region, e.g. around 1180 nm for [Li, e−].31 The newly synthesized cluster compound was investigated by broadband fs-transient absorption spectroscopy in the visible and NIR spectral region to explore the influence of a transition metal substituent on the excited state dynamics of such a cluster using [Ge9(Hyp)3]− as a reference system.
Results and discussion
The reaction of a thf solution of [Ge9(Hyp)3]− with Br-FeCp(CO)2 leads to an instant color change of the reaction solution and after work-up we were able to obtain the anticipated products [Ge9(Hyp)3FeCp(CO)2] 1 in the form of dark-red single crystals in 27% yield.
1 crystallizes in the triclinic crystal system in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and from the molecular structure (Fig. 1) it is obvious that the FeCp(CO)2 substituent is bound to the Ge9 core via a Ge–Fe single bond of 240.3 pm, similar to the single bond found in Ge12[FeCp(CO)2]8[FeCp(CO)]2 (240.7–249.4 pm).32 The Ge9 core in 1 is best described as a distorted monocapped square antiprismatic arrangement of nine germanium atoms, where the capping germanium atom (Ge8) is bound to a Hyp substituent via a Ge–Si single bond of 236.9 pm. Within the capped square of the antiprism the Ge–Ge distances are much longer (285–309 pm) than the Ge–Ge distances within the non-capped square (249–257 pm). The arrangement is thus similar to the one found in the isoelectronic compound [Ge9(Hyp)3-Cr(CO)5]−224 and Ge9(Hyp)3-R (R = Et 3,18 (CH2)3(CHCH2) 4,19 (CH2)3–C6H5520). However, in the case of 2 the transition metal germanium bond is a coordinative bond and in the case of 1 the transition metal fragment is bound covalently via a 2c2e bond to the Ge9(Hyp)3 unit. Within the IR spectrum two strong absorptions are found in the carbonyl region which are comparable to those found for the trigonal prismatic cluster Ge6[FeCp(CO)2]633 where the FeCp(CO)2-units are also bound via 2e2c bonds to the germanium atoms.
and from the molecular structure (Fig. 1) it is obvious that the FeCp(CO)2 substituent is bound to the Ge9 core via a Ge–Fe single bond of 240.3 pm, similar to the single bond found in Ge12[FeCp(CO)2]8[FeCp(CO)]2 (240.7–249.4 pm).32 The Ge9 core in 1 is best described as a distorted monocapped square antiprismatic arrangement of nine germanium atoms, where the capping germanium atom (Ge8) is bound to a Hyp substituent via a Ge–Si single bond of 236.9 pm. Within the capped square of the antiprism the Ge–Ge distances are much longer (285–309 pm) than the Ge–Ge distances within the non-capped square (249–257 pm). The arrangement is thus similar to the one found in the isoelectronic compound [Ge9(Hyp)3-Cr(CO)5]−224 and Ge9(Hyp)3-R (R = Et 3,18 (CH2)3(CHCH2) 4,19 (CH2)3–C6H5520). However, in the case of 2 the transition metal germanium bond is a coordinative bond and in the case of 1 the transition metal fragment is bound covalently via a 2c2e bond to the Ge9(Hyp)3 unit. Within the IR spectrum two strong absorptions are found in the carbonyl region which are comparable to those found for the trigonal prismatic cluster Ge6[FeCp(CO)2]633 where the FeCp(CO)2-units are also bound via 2e2c bonds to the germanium atoms.
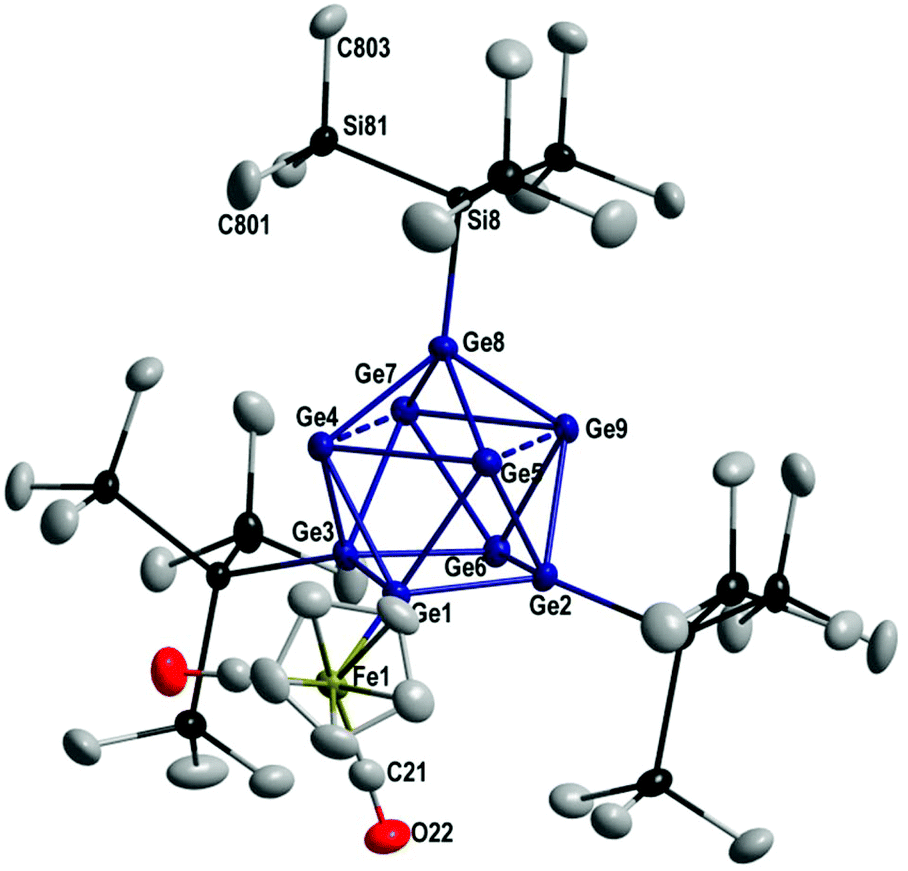 | ||
| Fig. 1 Molecular structure of [Ge9(Hyp)3FeCp(CO)2] 1; hydrogen atoms are omitted for clarity and all atoms are shown with thermal ellipsoids at 50% probability. Selected bond lengths [pm] and angles [°]: Ge1–Ge2: 249.35(4); Ge1–Ge3: 250.03(4); Ge2–Ge6: 256.81(4); Ge3–Ge6: 256.53(4); Ge4–Ge5: 284.65(4); Ge7–Ge9: 281.06(4); Ge4–Ge7: 309.73(5); Ge5–Ge9: 298.97(5); Ge4–Ge8: 250.72(4) Ge5–Ge8: 250.81; Ge7–Ge8: 252.04(4); Ge8–Ge9: 252.20(4); Ge1–Ge4: 261.72(4); Ge1–Ge5: 265.26(4); Ge2–Ge9: 252.93(4); Ge6–Ge7: 263.83(4); Ge1–Fe1: 240.36(5); Ge8–Si8: 236.92(8); Fe1–C21: 175.9(3); C21–O22: 114.7(4); Si8–Si81: 235.01(11); Si81–C801: 187.0(4); Si81–C803: 187.8(3); Ge1–Ge2–Ge6: 93.987(13); Ge2–Ge6–Ge3: 84.333(13); Ge4–Ge5–Ge9: 90.670(13); Ge4–Ge7–Ge8: 51.777(11); Ge5–Ge2–Ge9: 70.439(12); Ge4–Ge3–Ge7: 72.815(12); Ge7–Ge8–Ge9: 67.751(12); Ge7–Ge8–Ge5: 111.524(14). | ||
The addition of a fourth substituent leads to a different environment of the Hyp substituents and thus more than one signal in proton NMR should be observed for 1. At room temperature only one signal is detected, showing that 1 is dynamic in solution. However, at low temperature the signal splits, leading to two signals with an intensity ratio of 2:1 showing that the dynamics can be frozen at low temperature (ESI Fig. S9‡). A similar behavior was observed for Ge9(Hyp)3-R 3–5, where the dynamics is already frozen between 0 °C and −15 °C. In the case of 1 the compound is dynamic until −45 °C, showing that here a more flexible system is present with respect to 3–5, giving the opportunity to further enlighten the mechanism of the dynamics in solution, which is the scope of ongoing research in this direction.
Steady-state spectroscopy in solution
A UV-Vis-NIR absorption spectrum of 1 is shown in Fig. 2. The UV-Vis part is dominated by a band, which extends from the UV to the visible range with shoulders at 340 and 560 nm while in the NIR region no noticeable absorption can be found. It is similar to the spectrum of the metalloid cluster [Ge9Hyp3]−,20,28 which shows a comparable UV band – with one shoulder at 260 nm – that extends to visible but without significant absorption beyond 450 nm (see Fig. S1‡). When compared with [FeCp(CO)2I]34 or [FeCp(CO)2]2,35 the additional bands of 1 in the visible range can be readily attributed to electronic transitions of the (FeCp(CO)2)+-unit at 340 nm and an MLCT transition from Fe to the CO-ligands at 560 nm in line with ref. 36. UV-Vis absorption studies on other Ge9− clusters20 reveal UV absorption similar to the one found here which is evidence for the assignment to the Ge9− entity. On the other hand, transitions in the Vis range were assigned to originate from the Ge92−-cluster in ref. 20, which is not present in our case. Therefore, we assign the band peaking around 560 nm (Fig. 2) to the Fe entity of 1.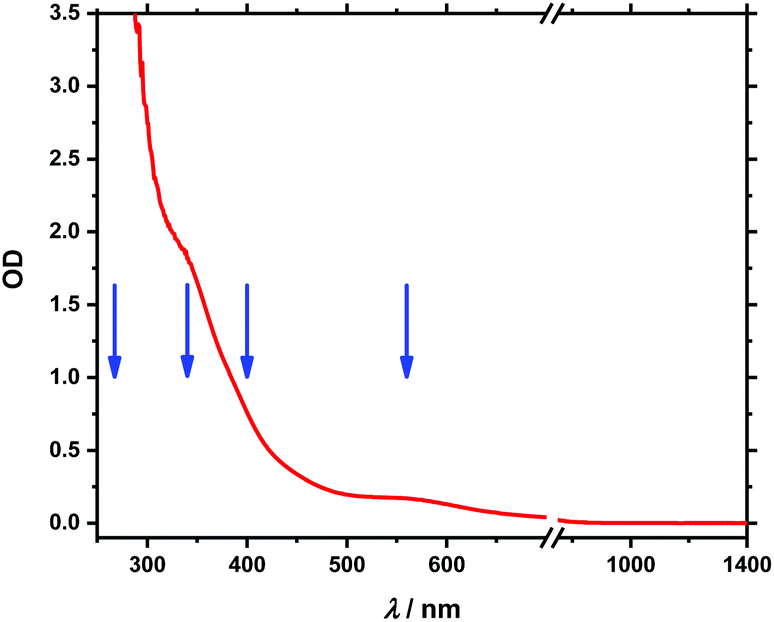 | ||
| Fig. 2 Stationary absorption spectrum of Fe(CO)2CpGe9Hyp3 between 250 and 1400 nm. Blue arrows mark excitation wavelengths at 258/267, 340, 388/400 and 560 nm, respectively. | ||
Transient absorption spectroscopy in solution
Transient absorption (TA) spectra after excitation at 267 nm (for TA in the visible region) and at 258 nm (for TA in the NIR region), respectively, are shown in Fig. 3. In the visible part of the spectrum, a positive transient response throughout the measured spectral range can be seen. It fully recovers within 1 ns. Likewise, the NIR spectral region between 900 and 1400 nm is governed by transient absorption, which seems to degrade faster by at least one order of magnitude than that in the UV-Vis region. Similarly, excitation at 400/388 nm leads to a positive transient response as well (see Fig. 4). This broad absorption both in the visible and NIR regions decays on timescales comparable to the spectral signature after UV excitation. There is, however, one noticeable difference. After 100 ps delay time, there is a small band appearing between 500 and 600 nm (see also decay associated spectra in Fig. S6 and S7‡). In the analysis of both transient spectra (Fig. 3 and 4) care should be taken with the interpretation at 410 nm and below. In this part, ground state bleach – resulting in a negative response – superimposes the positive transient response leading to an apparent maximum at 410 nm whereby the real maximum is probably further shifted towards the UV region, i.e. lower than the accessible probing wavelength of 350 nm.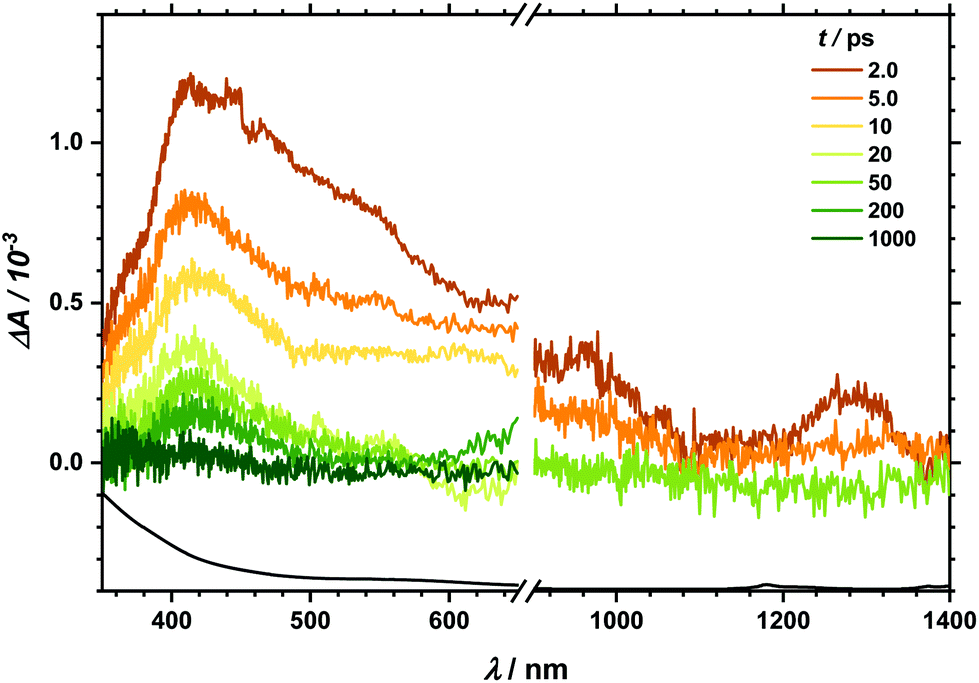 | ||
| Fig. 3 TA spectra at given delay times after excitation with 267 (258) nm in the visible and NIR spectral regions. For better clarity in NIR only three significant spectra are given. In the bottom, the absorption spectrum in arbitrary units is given for comparison. For further description see text. | ||
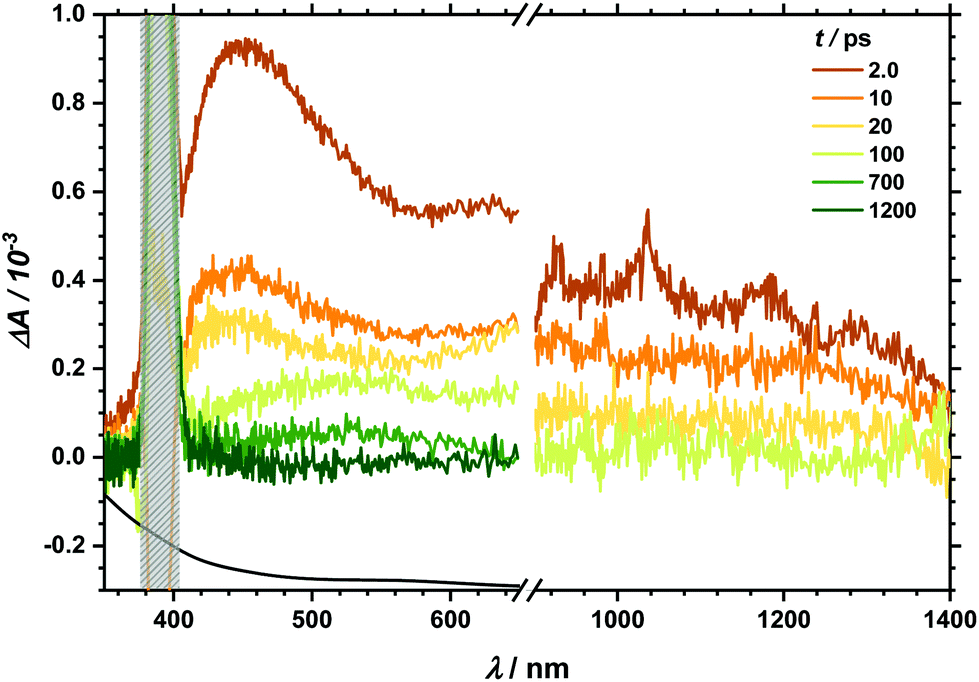 | ||
| Fig. 4 TA spectra after 400/388 nm excitation at given delay times. Grey box marks the artefact from the pump pulse at 400 nm. | ||
Excitation with 340 nm – though much more noisy due to weaker pump pulse intensity – results in a spectrum similar to the one after 267 nm excitation (see Fig. S2‡) while 560 nm excitation (see Fig. S3‡) reveals a TA spectrum comparable to 400 nm excitation if one identifies the onset between 450 and 500 nm after 100 ps delay time as the additional small band was observed between 500 and 600 nm after 400 nm excitation (Fig. 4).
The quantitative analysis concentrated first on single transients of the transient spectrum that show highest amplitudes, e.g. at a probe wavelength of 415 nm (Fig. S4‡). Additionally, Fig. S5‡ represents results at a probe wavelength of 1000 nm after 267 and 400 nm excitations. Please note that no TA spectra were recorded for 340 and 560 nm in the NIR region. Next, all transients could be fitted to multi-exponential fit functions (typically with three time constants) as summarized in Table 1 in the ESI.‡ The shortest time constant shows the strongest amplitude and is equal to or below 0.5 ps whereas the second time constant ranges from 7 to 9 ps. Finally, a third time constant is determined to be 150 ± 50 ps.
Similar dynamics has been found in previous experiments on [Ge9(Hyp)3Li],28 (see also the absorption in Fig. S1 and the transients of Fig. S5‡). Therefore, we also refer to the fact that the electron detachment energy of the reference Ge9-cluster has been determined to be 3.37 eV (368 nm) in the gas phase, i.e. the electron is completely removed from the cluster.28 Moreover and in line with calculations37 one has to consider both dynamics of the germanium cluster core and the ligand shell. As a consequence, we attribute the first two time constants to localization of the electron with subsequent vibrational relaxation in the electronically excited state and geminate recombination. Consequently, the first two processes are attributed to dynamics of the Ge9-entity in thf solution. We note that the second time constant is a factor 3 to 4 higher compared to the reference compound, [Ge9(Hyp)3Li]28 indicating an influence of the FeCp(CO)2 moiety.
As mentioned above, a transient absorption of a solvated electron in thf or cation-electron contact pairs has been found at 870 nm and beyond with lifetimes exceeding 1 ns.29–31 Hence, inspection of the NIR spectral range is essential. In contrast to the reference system, the LiGe9-compound,28 no long lived signature (>1 ns) is found neither in the UV-Vis nor in the NIR spectral range making the formation of solvated electrons or contact pairs in the present case rather unlikely. For the same reason, CO release which is typical of [FeCp(CO)2X] and [FeCp(CO)2]234,38 preventing complete ground state recovery can be excluded too. Instead, the observation of a complete recovery of the transient response with a time constant of ∼150 ps at all wavelengths inspected suggests another efficient process. To find its origin, one should remember that the covalent attachment of the FeCp(CO)2 substituent is the only difference compared to the reference system [Ge9Hyp3]−. In collaboration with the Powell group,39 earlier and comparable time-resolved studies from our group on Fe-clusters and -complexes – so called Fe10Ln10 nano-toruses – revealed three time constants in the same order of magnitude. Similar to the present case, the first one was assigned to vibrational relaxation and formation of trap states. Geminate recombination from the conduction band back into the valence band could be observed within few ps. The third time constant – on a 100 ps timescale – was attributed to the lifetime of trap states. This third process resembles very much our case, where the trap is an Fe-moiety although with a different formal oxidation state (+II in the present study vs. +III in ref. 39). Thus, for 1 the trapping would lead to a reduction of the iron to form a charge-transfer complex. Fe is also known in an oxidation state of +I from e.g. [FeCp(CO)2]2, which is an easily formed and stable product from the [FeCp(CO)2X]-complex (X = halogen) with electron excess in electrochemical experiments.40 In such a scenario, the relatively long lifetime (τ3 ∼ 150 ps) of this charge-transfer-complex can be explained by the low electron withdrawing effect of the Ge90-entity, i.e. an electron is transiently detached from [Ge9(Hyp)3]− and localized on the Fe-moiety. Generally, trap states lie under the band edge in the energy gap;39 only low pump energy is needed to excite into these levels. Hence, the longer-lived band between 500 and 600 nm after 400 and 560 nm excitations can be assigned to the formation of this charge-transfer-complex. Finally, UV excitation results in a decay associated spectrum (DAS) where the amplitude of the second and third time constants look rather similar (Fig. S6‡). This is in contrast to the Vis-excited DAS (Fig. S7‡) where the corresponding amplitudes are different. Considering that the energy depth of such trap states is quite low, typically in the order of 0.4 eV for band gaps above 3.5 eV,39 one may speculate whether this is a consequence of different channel population – 400 and 560 nm excitation leading to charge-transfer states and 258/340 nm excitation favoring local cluster core dynamics. It will therefore remain to future detailed studies to deal with this issue. In all cases, however, the detachment energy to obtain longer-lived states is not high enough, even after 4.7–4.8 eV excitation, which is in contrast to the reference [Ge9(Hyp)3]− with no FeCp(CO)2 attached.
Conclusions
[Ge9(Hyp)3]− with an additional transition metal substituent FeCp(CO)2 was synthesized successfully via a salt metathesis reaction, leading to a structural change of the germanium atoms from a tricapped trigonal prismatic arrangement within [Ge9(Hyp)3]− to a monocapped square antiprismatic arrangement in [Ge9(Hyp)3FeCp(CO)2] 1.The stationary UV-Vis-NIR spectrum shows two bands in addition to UV absorption of the Ge9-entity at 340 and 560 nm which can be attributed to excited state absorption of the FeCp(CO)2-substituent and an MLCT transition in the visible part of the spectrum. fs transient absorption spectroscopy revealed on the fs to tens of ps timescale a similar transient response like the unsubstituted [Ge9(Hyp)3]− which can be attributed to localization of an electron within the cluster assembly and a recombination process. However, in sharp contrast to the unsubstituted compound, no long lasting transient response exceeding 1 ns could be observed. From this finding we conclude that electron detachment into the solvent is hindered by the FeCp(CO)2+ moiety as this is the only considerable change within these clusters. Our future efforts will concentrate on electron transfer onto this moiety. This will have to be accompanied by additional fs studies of reference systems such as [FeCp(CO)2]2 and [FeCp(CO)2X] under comparable conditions. Also, the question arises whether one can identify ligand situations which do not inhibit but promote electron detachment. Such studies would open a new way to contribute to elucidating mechanistic steps of the long-lasting issue of how electron detachment and localization in polar liquids can be quantitatively understood.41–45
Experimental
General considerations
All reactions were performed under a nitrogen atmosphere by using Schlenk techniques. Toluene and thf were dried with sodium and pentane was dried with CaH2. All organic solvents were freshly distilled under nitrogen prior to use. Bruker DRX-250, ASX-300, AV-400 and AVII-500 spectrometers were used to obtain 1H, 13C, and 29Si NMR spectra. 1H, 13C, and 29Si chemical shifts δ are given in ppm and are referenced to Me4Si. NMR spectra were recorded at room temperature. KGe9(Hyp)38 and BrFeCp(CO)246 were synthesized via literature procedures.
Synthesis of [Ge9(Hyp)3FeCp(CO)2] 1
In a round bottom flask with a magnetic stirrer BrFeCp(CO)2 (69 mg, 269 μmol) and KGe9(Hyp)3 (300 mg, 190 μmol) are dissolved in thf at room temperature and stirred for 4 h. The reaction mixture turns from red to brown and is dried in vacuo. The black residue is extracted with pentane, leading to a dark red pentane extract. Storing the pentane extract at −30 °C gives [Ge9(Hyp)3FeCp(CO)2] 1 in the form of orange-black needles (80 mg, 27%).NMR (thf-d8): 1H (300 MHz): 0.35 ppm (s, 81 H, SiMe3), 5.17 ppm (s, 5 H, C5H5), 13C (62.9 MHz, dept-135): 3.19 ppm (SiMe3), 85.56 ppm (C5H5), 29Si (49.7 MHz, inept-nd): −8.23 ppm (decet, Si(SiMe3)3, 2JSi–H = 6.6 Hz), −103.15 ppm (s, Si(SiMe3)3).
Steady-state spectroscopy in solution
Absorption spectra were obtained with a UV/Vis/NIR spectrometer Cary 500 (Varian) in thf as a solvent in a wavelength range between 200 and 2000 nm. Spectra were measured at room temperature in cuvettes made of fused silica (Hellma) with a 1 mm optical path length.Transient absorption spectroscopy in solution
To obtain time resolved spectra in the UV-Vis wavelength range, an experimental setup described elsewhere47 was used. Briefly, the Astrella laser system (Coherent) generates 38 fs pulses with 800 nm central wavelength at a repetition rate of 1 kHz. One small part of the 7 mJ laser output was used to generate a white light continuum between 350 and 720 nm in a movable 2 mm CaF2 crystal. After passing the sample, the white-light is refracted by a fused silica prism, so as to be recorded by a CCD Camera (Series 2000, Si Photodetector, Entwicklungsbüro Stresing). Another part of the laser output is used to pump a non-collinear optic parametric amplifier to obtain pump wavelengths in the Vis region (here 560 nm) and after second harmonic generation in a BBO crystal in the UV spectral part (here 340 nm). Pump wavelengths at 400 and 267 nm were generated by second and third harmonic generation of the fundamental laser wavelength. The spot size in the sample was about 200 μm, which was more than twice the white-light spot size. Excitation energies were for all wavelengths 400 nJ per pulse, except for 340 nm, where it was only 200 nJ per pulse. Delay of the pump pulse was managed by a computer controlled translation stage (maximum delay ∼1.2 ns, Thorlabs), whereby every second pulse was blocked with an optical chopper (Thorlabs), resulting in spectra with and without excitation. Differentiation results in ΔA spectra with a time resolution better than 100 fs. Data were collected with an in-house written Labview program.For recording transient absorption (TA) spectra in the NIR spectral range a CPA 2210 (Clark-MXR) with output pulse energies of 1.3 mJ at a central wavelength of 775 nm, a repetition rate of 1 kHz and a pulse length of 200 fs was used. A 5 μJ part of the laser output, used as a probe pulse, was led through a computer controlled translation stage (maximum delay range 1.4 ns, Physical Instruments PI) to manage the time delay between pump and probe pulses. Afterwards, the 775 nm laser pulse was focused with a 30 mm lens into a 4 mm YAG crystal (nortus optronic) to generate a white-light continuum between 900 and 1600 nm, which was collected by using a CCD Camera (Series 2000, InGaAs Photodetector, Entwicklungsbüro Stresing) after refraction in a SF10 prism. Data were processed by using the same program like in the other systems adapted to the needs for NIR detection. Pump pulses were generated by second and third harmonic generation of the fundamental laser wavelength of 775 nm. Excitation energies lie in the range of 1 μJ per pulse and spot sizes in the sample around 500 μm which, again, were roughly two times the white light spot size. Long pulses and a large group velocity difference resulted in a time resolution of roughly 200 fs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Deutsche Forschungsgemeinschaft through project UN108/6-1 and SCHN738/9-1. We thank Dr Claudio Schrenk for helpful discussion and support during measuring and solving the crystal structures.Notes and references
- A. Schnepf, Clusters – Contemporary Insight in Structure and Bonding, Springer International Publishing, 2017 Search PubMed.
- A. Schnepf and H. Schnöckel, Angew. Chem., 2002, 114, 3682–3704 CrossRef.
- E. G. Mednikov and L. F. Dahl, Philos. Trans. R. Soc., A, 2010, 368, 1301–1332 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, T. Chen and J. Xie, Coord. Chem. Rev., 2016, 329, 1–15 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- O. Kysliak and A. Schnepf, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2017 Search PubMed.
- A. Schnepf, Angew. Chem., 2003, 115, 2728–2729 CrossRef.
- F. Li and S. C. Sevov, Inorg. Chem., 2012, 51, 2706–2708 CrossRef CAS PubMed.
- F. S. Geitner, W. Klein and T. F. Fässler, Angew. Chem., Int. Ed., 2018, 57, 14509–14513 CrossRef CAS PubMed.
- O. Kysliak and A. Schnepf, Dalton Trans., 2016, 45, 2404–2408 RSC.
- O. Kysliak, T. Kunz and A. Schnepf, Eur. J. Inorg. Chem., 2017, 2017, 805–810 CrossRef CAS.
- F. S. Geitner, J. V. Dums and T. F. Fässler, J. Am. Chem. Soc., 2017, 139, 11933–11940 CrossRef CAS PubMed.
- F. S. Geitner, C. Wallach and T. F. Fässler, Chem. – Eur. J., 2018, 24, 4103–4110 CrossRef CAS PubMed.
- K. Mayer, L. J. Schiegerl, T. Kratky, S. Günther and T. F. Fässler, Chem. Commun., 2017, 53, 11798–11801 RSC.
- O. Kysliak, C. Schrenk and A. Schnepf, Inorg. Chem., 2017, 56, 9693–9697 CrossRef CAS PubMed.
- C. Schenk and A. Schnepf, Angew. Chem., Int. Ed., 2007, 46, 5314–5316 CrossRef CAS PubMed.
- O. Kysliak, C. Schrenk and A. Schnepf, Angew. Chem., Int. Ed., 2016, 55, 3216–3219 CrossRef CAS PubMed.
- F. Li and S. C. Sevov, J. Am. Chem. Soc., 2014, 136, 12056–12063 CrossRef CAS PubMed.
- S. Frischhut and T. F. Fässler, Dalton Trans., 2018, 47, 3223–3226 RSC.
- S. Frischhut, J. G. Machado de Carvalho, A. J. Karttunen and T. F. Fässler, Z. Anorg. Allg. Chem., 2018, 644, 1337–1343 CrossRef CAS.
- F. Li, A. Munoz-Castro and S. C. Sevov, Angew. Chem., 2012, 124, 8709–8712 CrossRef.
- K. Mayer, L. J. Schiegerl and T. F. Fässler, Chem. – Eur. J., 2016, 22, 18794–18800 CrossRef CAS PubMed.
- F. Li, A. Muñoz-Castro and S. C. Sevov, Angew. Chem., Int. Ed., 2016, 55, 8630–8633 CrossRef CAS PubMed.
- C. Schenk and A. Schnepf, Chem. Commun., 2009, 3208–3210 RSC.
- F. Henke, C. Schenk and A. Schnepf, Dalton Trans., 2011, 40, 6704–6710 RSC.
- S. Frischhut, F. Kaiser, W. Klein, M. Drees, F. E. Kühn and T. F. Fässler, Organometallics, 2018, 37, 4560–4567 CrossRef CAS.
- O. Kysliak, D. D. Nguyen, A. Z. Clayborne and A. Schnepf, Inorg. Chem., 2018, 57, 12603–12609 CrossRef CAS PubMed.
- M. Klinger, C. Schenk, F. Henke, A. Clayborne, A. Schnepf and A. N. Unterreiner, Chem. Commun., 2015, 51, 12278–12281 RSC.
- F. Y. Jou and L. M. Dorfman, J. Chem. Phys., 1973, 58, 4715–4723 CrossRef CAS.
- M. C. Cavanagh, R. E. Larsen and B. J. Schwartz, J. Phys. Chem. A, 2007, 111, 5144–5157 CrossRef CAS PubMed.
- B. Bockrath and L. M. Dorfman, J. Phys. Chem., 1975, 79, 1509–1512 CrossRef CAS.
- C. Schenk, F. Henke and A. Schnepf, Angew. Chem., Int. Ed., 2013, 52, 1834–1838 CrossRef CAS PubMed.
- L. Preißing, C. Schrenk and A. Schnepf, Dalton Trans., 2019, 48, 3831–3834 RSC.
- C. E. Borja, V. Jakúbek and A. J. Lees, Inorg. Chem., 1998, 37, 2281–2284 CrossRef CAS PubMed.
- B. E. Bursten, M. L. Drummond and J. Li, Faraday Discuss., 2003, 124, 1–24 RSC.
- A. Pilon, P. Gírio, G. Nogueira, F. Avecilla, H. Adams, J. Lorenzo, M. H. Garcia and A. Valente, J. Organomet. Chem., 2017, 852, 34–42 CrossRef CAS.
- P. Andre Clayborne and H. Hakkinen, Phys. Chem. Chem. Phys., 2012, 14, 9311–9316 RSC.
- C. J. Arnold, T.-Q. Ye, R. N. Perutz, R. E. Hester and J. N. Moore, Chem. Phys. Lett., 1996, 248, 464–469 CrossRef CAS.
- A. Baniodeh, Y. Liang, C. E. Anson, N. Magnani, A. K. Powell, A.-N. Unterreiner, S. Seyfferle, M. Slota, M. Dressel, L. Bogani and K. Goß, Adv. Funct. Mater., 2014, 24, 6280–6290 CrossRef CAS.
- B. Rong, W. Zhong, E. Gu, L. Long, L. Song and X. Liu, Electrochim. Acta, 2018, 283, 27–35 CrossRef CAS.
- R. M. Young and D. M. Neumark, Chem. Rev., 2012, 112, 5553–5577 CrossRef CAS PubMed.
- D. Luckhaus, Y.-i. Yamamoto, T. Suzuki and R. Signorell, Sci. Adv., 2017, 3, e1603224 CrossRef PubMed.
- L. Mones, G. Pohl and L. Turi, Phys. Chem. Chem. Phys., 2018, 20, 28741–28750 RSC.
- J. Stähler, J.-C. Deinert, D. Wegkamp, S. Hagen and M. Wolf, J. Am. Chem. Soc., 2015, 137, 3520–3524 CrossRef PubMed.
- P. Vöhringer, Annu. Rev. Phys. Chem., 2015, 66, 97–118 CrossRef PubMed.
- J.-P. Barras, S. G. Davies, M. R. Metzler, A. J. Edwards, V. M. Humphreys and K. Prout, J. Organomet. Chem., 1993, 461, 157–165 CrossRef CAS.
- C. Schweigert, O. Babii, S. Afonin, T. Schober, J. Leier, N. C. Michenfelder, I. V. Komarov, A. S. Ulrich and A. N. Unterreiner, ChemPhotoChem, 2019, 3, 403–410 CAS.
Footnotes |
| † Dedicated to the occasion of Prof. Annie Powell's 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1917230. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C9DT02091H |
| This journal is © The Royal Society of Chemistry 2019 |