
Pd-Catalyzed decarboxylative cross-coupling reactions of epoxides with α,β-unsaturated carboxylic acids†
Xiao-Yu
Lu
 *ab,
Jin-Song
Li
a,
Shi-Qun
Wang
a,
Yu-Jing
Zhu
a,
Yue-Ming
Li
a,
Lu-Yu
Yan
a,
Jia-Mei
Li
a,
Jin-Yu
Wang
a,
Hai-Pin
Zhou
a and
Xiu-Tao
Ge
a
*ab,
Jin-Song
Li
a,
Shi-Qun
Wang
a,
Yu-Jing
Zhu
a,
Yue-Ming
Li
a,
Lu-Yu
Yan
a,
Jia-Mei
Li
a,
Jin-Yu
Wang
a,
Hai-Pin
Zhou
a and
Xiu-Tao
Ge
a
aSchool of Materials and Chemical Engineering, ChuZhou University, Chu Zhou, 239000, China. E-mail: xiaoyulu@mail.ustc.edu.cn
bSchool of Chemistry and Chemical Engineering, AnHui University, He Fei, 230601, China
First published on 16th August 2019
Abstract
A Pd-catalyzed decarboxylative cross-coupling of α,β-unsaturated carboxylic acids with cyclic and acyclic epoxides has been developed. Both β-monosubstituted and β-disubstituted unsaturated carboxylic acids, as well as conjugated diene unsaturated carboxylic acids are suitable reaction substrates. Substituted homoallylic alcohols were obtained in moderate to good yields. The product was obtained as a mixture of diastereomers favoring the anti diastereomer of the cyclic epoxides. This work provides a method for the modification of complex organic molecules containing α,β-unsaturated carboxylic acids.
Transition metal-catalyzed cross-couplings are among the most vital C–C bond-forming reactions in modern organic synthesis.1 Alcohols are valuable compounds that play vital roles in organic synthesis. One synthesis method for alcohols is through ring-opening reactions of epoxides where C–C bonds are constructed.2 In the past few decades, various epoxide ring-opening reactions have been realized,3 including transition-metal-catalyzed Negishi-type4 and Suzuki-type5 ring-opening/cross-coupling reactions. In addition, reductive ring-opening/coupling reactions of epoxides have been reported.6 For example, reductive coupling of alkynes and epoxides does not break the C–C bond to form an alcohol as implied in Scheme 1a but rather forms the same types of homoallylic alcohols made in this publication.6c Furthermore, Heck-type reaction7 and C–H activation reaction8 of epoxides have also been reported. For example, palladium-catalyzed Heck-type reaction of epoxides is shown in Scheme 1b.7a Carboxylic acids are one of the most important classes of organic structures, and are found in many active molecules.9 Recently, decarboxylative cross-coupling has emerged as a powerful tool for the construction of C–C bonds.10 We hypothesized that decarboxylative cross-couplings could be used with epoxide ring-opening reactions to synthesize alcohols. To the best of our knowledge, there have been no reports on decarboxylative cross-coupling of carboxylic acids with epoxides.11
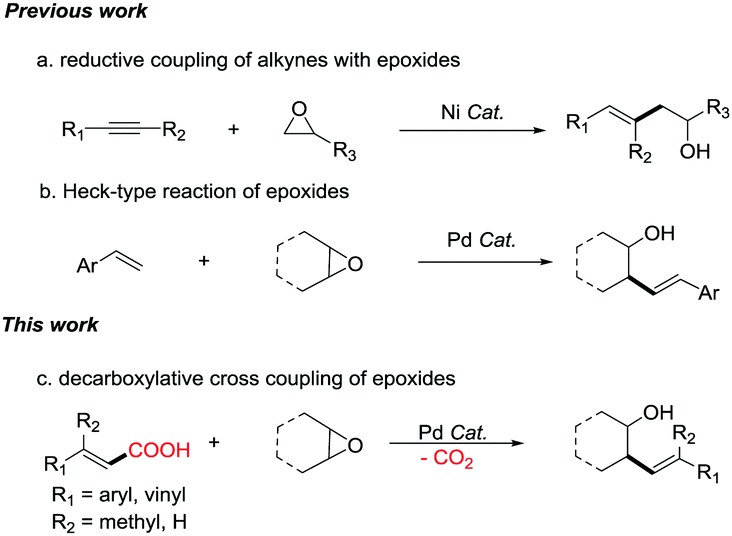 | ||
| Scheme 1 Decarboxylative cross-coupling reactions of epoxides with α,β-unsaturated carboxylic acids. | ||
Herein, we report the first example of Pd-catalyzed decarboxylative cross-coupling reactions of α,β-unsaturated carboxylic acids with epoxides. The product was obtained as a mixture of diastereomers favoring the anti diastereomer of the cyclic epoxides (Scheme 1c). In addition to β-monosubstituted unsaturated carboxylic acids, β-disubstituted unsaturated carboxylic acids and conjugated diene unsaturated carboxylic acids also give products with moderate to good reaction yields. This methodology provides access to a diverse array of synthetically valuable substituted homoallylic alcohols.12 It also provides a method for the modification of complex organic molecules containing α,β-unsaturated carboxylic acids.
We began our study by selecting cinnamic acid (1a) and cyclohexene oxide (2a) as model reaction substrates (Table 1). When using Pd(PPh3)4 as a catalyst, dppf as a ligand, Cy2NMe as a base and PhCF3 as a solvent, the desired product was observed in a low yield (entry 1). First, we assessed the reaction by modifying the ligand and solvent in the presence of Pd(PPh3)4 as the catalyst. Using dioxane instead of PhCF3 as the solvent resulted in almost no increase in reaction yield (entry 2). Switching the ligand to Xantphos decreased the reaction yield (entries 3 and 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Solvent | Yield% 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a (d.r.) :4a (d.r.) |
|
a Reaction conditions: 1a (0.6 mmol), 2a (0.3 mmol), base (1.5 equiv.) in 2 mL solvent at 100 °C for 24 h.
b
1a (0.3 mmol), 2a (0.6 mmol), base (1.5 equiv.) in 2 mL solvent at 100 °C for 24 h.
c
1a (0.3 mmol), 2a (0.6 mmol) in 2 mL solvent (10:1) at 90 °C for 24 h.
d 1.5 mL solvent (5:1), 1a (0.3 mmol), 2a (2.5 equiv.) and NaI/Et3N·HCl at 90 °C for 30 h.
e No NaI. The yield was determined by GC using benzophenone as internal standard. Diox = dioxane.
|
|||||
| 1a | Pd(PPh3)4 | dppf | Cy2NMe | PhCF3 | 28(4.5:1) |
| 2a | Pd(PPh3)4 | dppf | Cy2NMe | Dioxane | 31(3.5:1) |
| 3a | Pd(PPh3)4 | Xantphos | Cy2NMe | PhCF3 | 10(4.2:1) |
| 4a | Pd(PPh3)4 | Xantphos | Cy2NMe | Dioxane | 15(3.2:1) |
| 5b | Pd(PPh3)4 | dppf | Cy2NMe | PhCF3 | 50(4:1) |
| 6b | Pd(PPh3)4 | dppe | Cy2NMe | PhCF3 | 10(4.2:1) |
| 7b | Pd(PPh3)4 | dppp | Cy2NMe | PhCF3 | 2 |
| 8b | Pd(PPh3)4 | PCy3 | Cy2NMe | PhCF3 | Trace |
| 9b | Pd(PPh3)4 | dppf | K3PO4 | PhCF3 | 8(4:1) |
| 10b | Pd(PPh3)4 | dppf | Cs2CO3 | PhCF3 | 5(4:1) |
| 11b | Pd(PPh3)4 | dppf | (iPr)2NEt | PhCF3 | 10(4.1:1) |
| 12c | Pd(PPh3)4 | dppf | Cy2NMe | PhCF3 | 62(5:1) |
| 13c | Pd(PPh3)4 | dppf | Cy2NMe | Toluene | 46(5.5:1) |
| 14c | Pd(PPh3)4 | dppf | Cy2NMe | DMA | 21(4.2:1) |
| 15c | Pd(PPh3)4 | dppf | Cy2NMe | PhCF3/DMA | 28(4.3:1) |
| 16 | Pd(PPh 3 ) 4 | dppf | Cy 2 NMe | PhCF 3 /Diox |
81(3.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
| 17 | — | dppf | Cy2NMe | PhCF3/Diox | 0 |
| 18 | Pd(PPh3)4 | — | Cy2NMe | PhCF3/Diox | (3.1:1) |
| 19e | Pd(PPh3)4 | dppf | Cy2NMe | PhCF3/Diox | Trace |
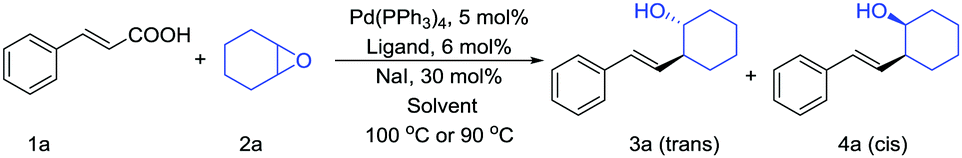
We then adjusted the substrate proportions under the initial reaction conditions (entry 5) and the reaction yield was significantly improved. Using the new substrate proportions, we screened a series of ligands, such as dppe, dppp and PCy3, but they gave reaction yields of less than 10% (entries 6–8). These results indicate that dppf is a relatively good ligand for the targeted reaction. Under the condition of dppf as a ligand, we examined other bases, but saw no improvement in the reaction yield (entries 9–11). Lowering the reaction temperature has certain benefits for improving the reaction yield (entry 12). Finally, we obtained the optimal reaction conditions by using a mixed solvent system of PhCF3 and dioxane (81% GC yield, d.r. = 3.9:1, entry 16). We confirmed the presence of two isomers by NMR (see ESI†). Reactions performed without Pd(PPh3)4 did not result in product formation (entry 17). On removal of dppf, the yield will be significantly reduced (entry 18). A control experiment indicated that the reaction completely failed without the addition of NaI (entry 19). Using other catalysts, such as Pd(OAc)2, significantly reduced the reaction yield (see ESI†).
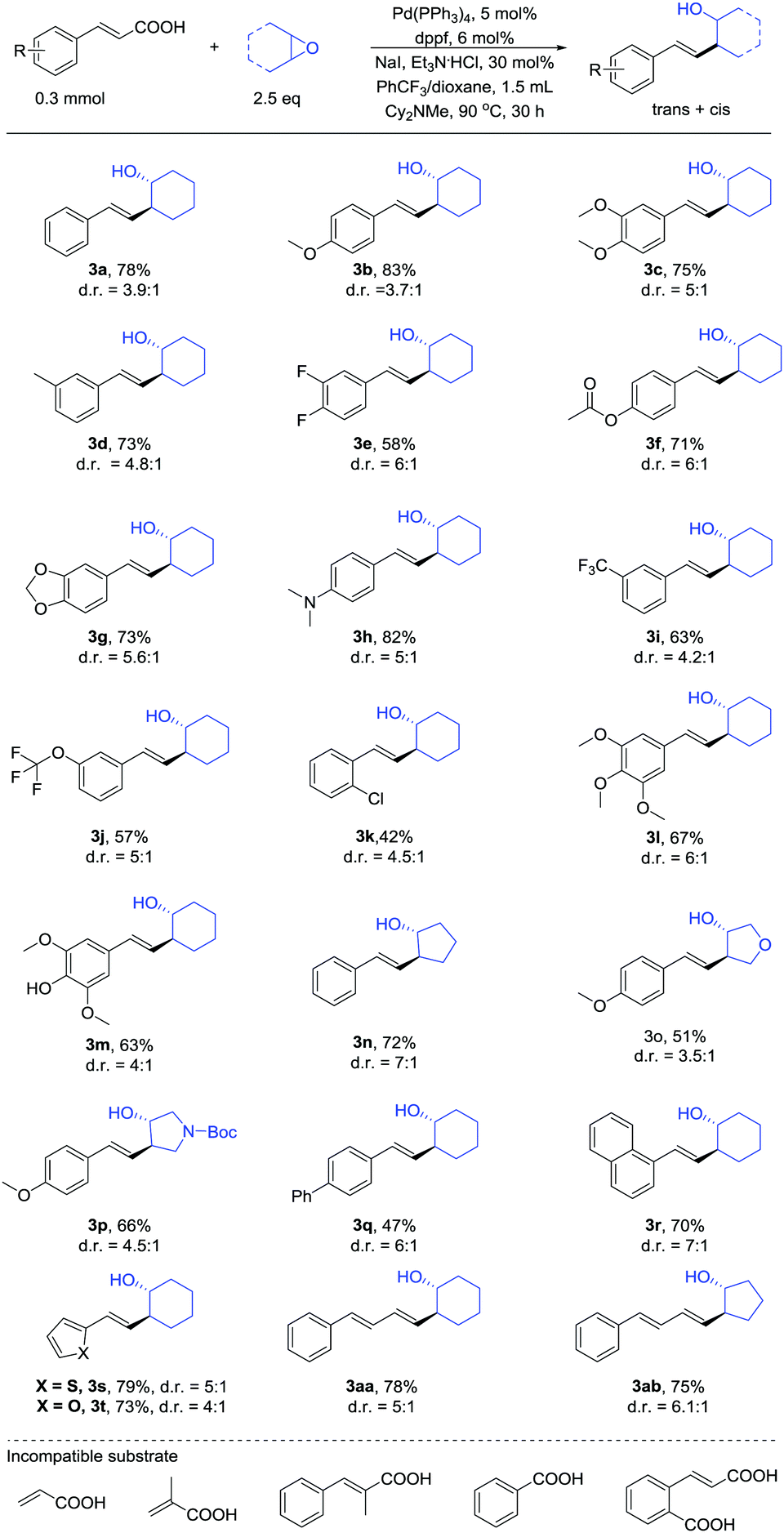
We next examined the substrate scope using the optimized conditions described above (Table 2). Many of the substituted unsaturated carboxylic acids coupled with cyclic epoxides in moderate to good yields and preferential anti-selectivity on the cyclic ring. Both electron-rich and electron-poor unsaturated carboxylic acids afforded the products. The functional groups ether (3b, 3c), halogen (3e), trifluoromethoxy (3j), amino (3h) and trifluoromethyl (3i) were well tolerated and afforded good yields. In particular, the ester (3f), alcohol (3m), ketal (3g) and amide (3p) substitutions were compatible with the reaction conditions. Moreover, carboxylic acids bearing ortho-chlorine (3k), bicyclic naphthyl (3r), thiophene (3s) and furan (3t) groups also participated in the reaction.
|
Conjugated dienes are found in many complex active molecules and pharmaceuticals and play vital roles in organic synthesis and materials science.13 We found that conjugated diene unsaturated carboxylic acids furnished the corresponding products in good yields (3aa and 3ab). In particular, sorbic acid showed satisfactory reactivity towards product formation (Scheme 2), which is notable because it is difficult to synthesize diene substituted homoallylic alcohols by other methods.14
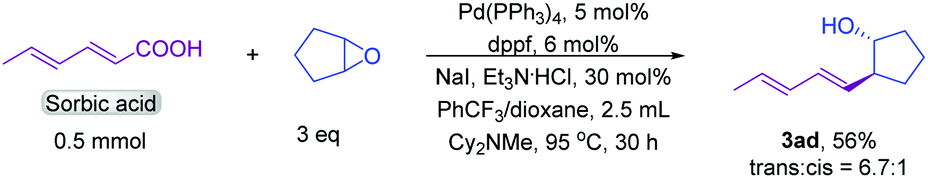 | ||
| Scheme 2 Decarboxylation of sorbic acids. | ||
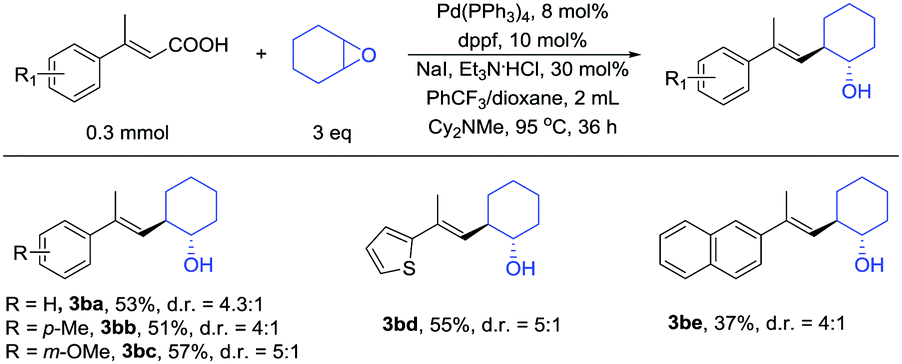
In addition to β-monosubstituted unsaturated carboxylic acids, β-disubstituted unsaturated carboxylic acids also give the products. Many β-disubstituted unsaturated carboxylic acids coupled with epoxides in moderate yields (Table 3). Some substituent groups, such as methyl (3bb) and methoxy (3bc), were tolerated and afforded moderate yields. Thiophene (3bd) and naphthalene (3be) can also be present in the reaction.
|
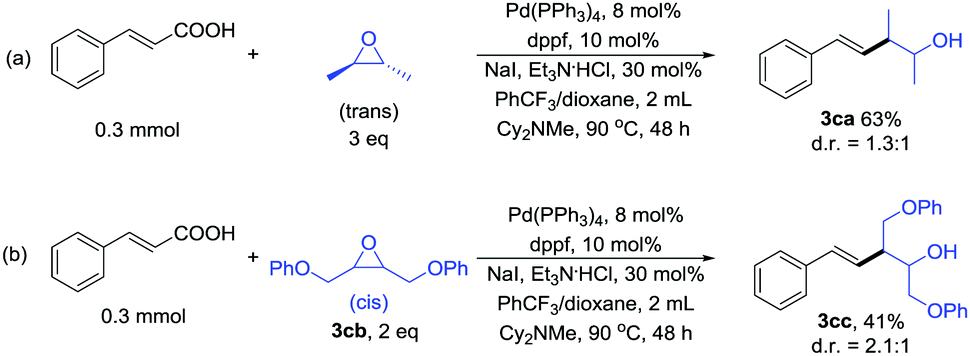
For monosubstituted and 1,1-disubstituted acyclic epoxides the products formed in these reactions are the ester that would result from direct nucleophilic attack of the carboxylic acid on the epoxide (see ESI†). So we tested 1,2-disubstituted acyclic epoxide as the reaction substrate. The trans-2,3-epoxybutane couples with cinnamic acid to form the product in 63% yield (Scheme 3a). The treatment of cis 3cb with cinnamic acid afforded the product in 41% yield (Scheme 3b). These results indicated that the yield of acyclic epoxide was worse than cyclic epoxide. Unfortunately, for styrene epoxides, such as trans-stilbene oxide, we almost got 1,2-diphenylethan-1-one (see ESI†).
 | ||
| Scheme 3 Examples of acyclic epoxides. | ||
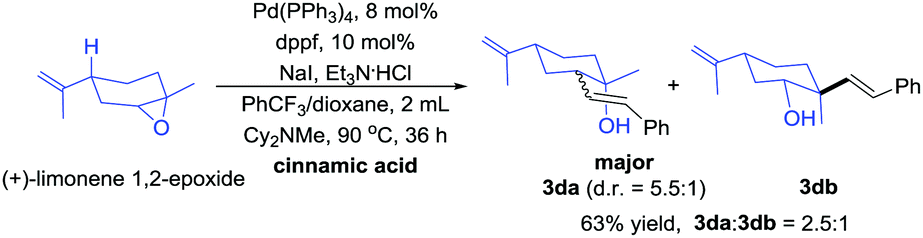
An unsymmetrical epoxy (Scheme 4) substrate was investigated and two regioselective products, tertiary alcohol (3da) and secondary alcohol (3db) were obtained. The regioselectivity at the less-substituted positions was superior (3da:3db = 2.5:1). Therefore, the selectivity of the asymmetric epoxy mainly depends on steric hindrance (the configuration of the isomers was assigned by 1H NMR and H–H COSY spectra).
 | ||
| Scheme 4 Example of asymmetric epoxide. | ||
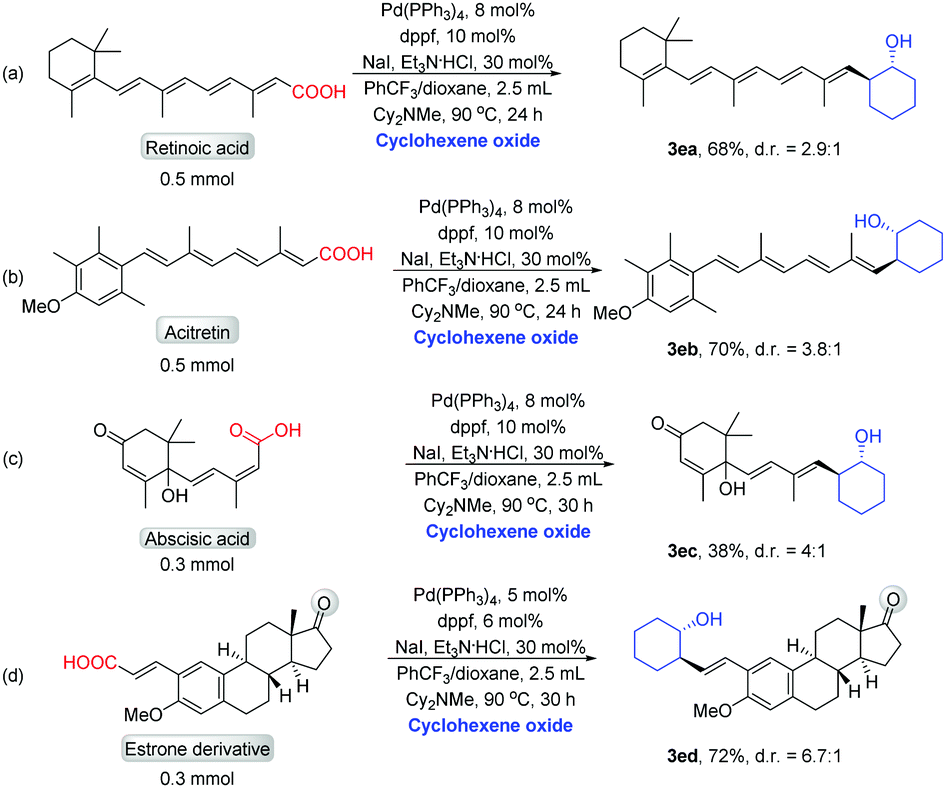
We next demonstrated the decarboxylative cross-coupling reactions of epoxides for late-stage modification of biologically active molecules containing α,β-unsaturated carboxylic acids. The treatment of retinoic acid with cyclohexene oxide afforded the product (3ea) in good yield (Scheme 5a). Modification of acitretin with cyclohexene oxide resulted in the formation of 3eb in high yield (Scheme 5b). The treatment of abscisic acid with cyclohexene oxide obtained the product in 38% yield (Scheme 5c). The treatment of estrone derivative with cyclohexene oxide afforded the product (3ed) in 72% yield (Scheme 5d). These results fully demonstrate the value of this decarboxylative reaction in the modification of complex active molecules.
 | ||
| Scheme 5 The modification of complex molecules. | ||
The mechanism of the reaction was investigated through several experiments. Reactions using 0.5 equiv. of radical trapping agent 2,2,6,6-tetramethylpiperidineoxy (TEMPO) were largely inhibited. Then, we performed a reaction of an in situ formed palladium(0) complex of dppf with iodohydrin. It formed cyclohexanone and cyclohexanol (ESI†). Another reaction of the palladium(0) complex with cyclohexene oxide gave similar results in the presence of 1 equiv. of NaI and Et3N·HCl (ESI†). On removal of NaI, no production of cyclohexanone and cyclohexanol was observed. We observed the product with 16% yield when using iodohydrin instead of the epoxide and in the absence of sodium iodide (see ESI†). These results are similar to previous reports.7a The cyclooctadiene monoxide couples with cinnamic acid to form primarily [3.3.0]-bicyclooctanols (ESI†).6b Moreover, (Z)-cinnamic acid also afforded the same trans-alkene product, and the stereoconvergent reaction was considered to be carried out by a radical pathway (ESI†).15
On the basis of the above results and previous reports,11,16 a possible reaction mechanism is depicted in ESI.† Initially, the β-iodohydrin, which is in situ produced from the epoxide and NaI, reacts with (dppf)palladium(0) species to give a β-hydroxyalkyl radical (I) and an LnPdI complex. Addition of the hydroxyalkyl radical (I) at the α-position of the C—C double bond in cinnamic acid would give the benzylic radical II. Next, the radical II combines with (Ln)PdI to form alkylpalladium(II) species III. Finally, decarboxylation of the Pd(II) species occurs to generate the product while regenerating the Pd(0) species.
In summary, we have developed the first Pd-catalyzed decarboxylative cross-coupling of α,β-unsaturated carboxylic acids with epoxides. The products were obtained as a mixture of diastereomers favoring the anti diastereomer of the cyclic epoxides. Reactions with β-monosubstituted and β-disubstituted unsaturated carboxylic acids proceed with moderate to good yields and conjugated diene unsaturated carboxylic acids showed moderate reactivity. This methodology also provides access to a diverse array of substituted homoallylic alcohols, which are valuable structural fragments in organic synthesis. It also provides a route for the modification of complex organic molecules containing α,β-unsaturated carboxylic acids.
This work was supported by 2017qd11, 16030801108 and KJ2019A0635.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) F. Diederich and P. J. Stang, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 1998 CrossRef; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed; (c) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed; (d) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed; (e) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937 RSC.
- (a) C.-Y. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153 CrossRef CAS PubMed; (b) J. He, J. Ling and P. Chiu, Chem. Rev., 2014, 114, 8037 CrossRef CAS PubMed.
- (a) P. Crotti and M. Pineschi, Aziridines and Epoxides in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 271–313 Search PubMed; (b) V. V. Fokin and P. Wu, Aziridines and Epoxides in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 443–477 Search PubMed; (c) P. A. S. Lowden, Aziridines and Epoxides in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 399–442 Search PubMed; (d) H. Ohno, Aziridines and Epoxides in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 37–71 Search PubMed; (e) B. Olofsson and P. Somfai, Aziridines and Epoxides in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 315–347 Search PubMed.
- (a) C. Y. Huang and A. G. Doyle, J. Am. Chem. Soc., 2012, 134, 9541 CrossRef CAS PubMed; (b) K. L. Jensen, E. A. Standley and T. F. Jamison, J. Am. Chem. Soc., 2014, 136, 11145 CrossRef CAS PubMed; (c) D. K. Nielsen, C. Y. Huang and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 13605 CrossRef CAS PubMed.
- (a) M. L. Duda and F. E. Michael, J. Am. Chem. Soc., 2013, 135, 18347 CrossRef CAS PubMed; (b) X. Y. Lu, C. T. Yang, J. H. Liu, Z. Q. Zhang, X. Lu, X. Lou, B. Xiao and Y. Fu, Chem. Commun., 2015, 51, 2388 RSC; (c) Y. Takeda, Y. Ikeda, A. Kuroda, S. Tanaka and S. Minakata, J. Am. Chem. Soc., 2014, 136, 8544 CrossRef CAS PubMed; (d) E.-A. M. A. Ahmed, X. Lu, T.-J. Gong, Z.-Q. Zhang, B. Xiao and Y. Fu, Chem. Commun., 2017, 53, 909 RSC; (e) A. Ebrahim-Alkhalil, Z.-Q. Zhang, T.-J. Gong, W. Su, X.-Y. Lu, B. Xiao and Y. Fu, Chem. Commun., 2016, 52, 4891 RSC; (f) X.-Y. Lu, J.-S. Li, J.-Y. Wang, S.-Q. Wang, Y.-M. Li, Y.-J. Zhu, R. Zhou and W.-J. Ma, RSC Adv., 2018, 8, 41561 RSC.
- (a) Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2013, 136, 48 CrossRef PubMed; (b) Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2015, 137, 3237 CrossRef CAS PubMed; (c) C. Molinaro and T. F. Jamison, J. Am. Chem. Soc., 2003, 125, 8076 CrossRef CAS PubMed; (d) M. G. Beaver and T. F. Jamison, Org. Lett., 2011, 13, 4140 CrossRef CAS PubMed.
- (a) S. Teng, M. E. Tessensohn, R. D. Webster and J. S. Zhou, ACS Catal., 2018, 8, 7439 CrossRef CAS; (b) Y. Ikeda, H. Yorimitsu, H. Shinokubo and K. Oshima, Adv. Synth. Catal., 2004, 346, 1631 CrossRef CAS.
- G. Cheng, T. J. Li and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 10950 CrossRef CAS PubMed.
- A. Tortajada, F. Julia-Hernandez, M. Borjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed.
- (a) S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2017, 10, 205 CrossRef PubMed; (b) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D. H. Bao, F. L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213 CrossRef CAS PubMed; (c) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322 CrossRef CAS PubMed; (d) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, eaam7355 CrossRef PubMed; (e) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (f) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed.
- (a) S. Han, X. Ren, Q. Wu, A. Liang, J. Li, D. Zou, Y. Wu and Y. Wu, Adv. Synth. Catal., 2018, 360, 2308 CrossRef CAS; (b) R. Kancherla, K. Muralirajan, B. Maity, C. Zhu, P. E. Krach, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3412 CrossRef CAS PubMed; (c) C. Wang, Y. Lei, M. Guo, Q. Shang, H. Liu, Z. Xu and R. Wang, Org. Lett., 2017, 19, 6412 CrossRef CAS PubMed; (d) G. Prina Cerai and B. Morandi, Chem. Commun., 2016, 52, 9769 RSC; (e) K. Xu, Z. Tan, H. Zhang, J. Liu, S. Zhang and Z. Wang, Chem. Commun., 2017, 53, 10719 RSC; (f) J. J. Zhang, J. C. Yang, L. N. Guo and X. H. Duan, Chem. – Eur. J., 2017, 23, 10259 CrossRef CAS PubMed.
- (a) L. F. Tietze, T. Kinzel and C. C. Brazel, Acc. Chem. Res., 2009, 42, 367 CrossRef CAS PubMed; (b) Y. Chen, D. C. Blakemore, P. Pasau and S. V. Ley, Org. Lett., 2018, 20, 6569 CrossRef CAS PubMed; (c) T. Takeda, S. Amarume, I. Sekioka and A. Tsubouchi, Org. Lett., 2015, 17, 1150 CrossRef CAS PubMed; (d) C. Wang and H. Yamamoto, J. Am. Chem. Soc., 2014, 136, 1222 CrossRef CAS PubMed.
- (a) M. A. Bohn, A. Schmidt, G. Hilt, M. Dindaroglu and H. G. Schmalz, Angew. Chem., Int. Ed., 2011, 50, 9689 CrossRef CAS PubMed; (b) L. Liao, R. Jana, K. B. Urkalan and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 5784 CrossRef CAS PubMed; (c) N. J. McAlpine, L. Wang and B. P. Carrow, J. Am. Chem. Soc., 2018, 140, 13634 CrossRef CAS PubMed; (d) E. McNeill and T. Ritter, Acc. Chem. Res., 2015, 48, 2330 CrossRef CAS PubMed; (e) V. T. Nguyen, H. T. Dang, H. H. Pham, V. D. Nguyen, C. Flores-Hansen, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2018, 140, 8434 CrossRef CAS PubMed; (f) A. Tortajada, R. Ninokata and R. Martin, J. Am. Chem. Soc., 2018, 140, 2050 CrossRef CAS PubMed.
- (a) P. A. Wender, D. A. Holt and S. M. Sieburth, J. Am. Chem. Soc., 1983, 105, 3348–3350 CrossRef CAS; (b) J. Montgomery and M. Song, Org. Lett., 2002, 4, 4009 CrossRef CAS PubMed.
- G. Li, T. Wang, F. Fei, Y.-M. Su, Y. Li, Q. Lan and X.-S. Wang, Angew. Chem., Int. Ed., 2016, 55, 3491 CrossRef CAS PubMed.
- (a) J. N. Jaworski, S. D. McCann, I. A. Guzei and S. S. Stahl, Angew. Chem., Int. Ed., 2017, 56, 3605 CrossRef CAS PubMed; (b) J. N. Jaworski, C. V. Kozack, S. J. Tereniak, S. M. M. Knapp, C. R. Landis, J. T. Miller and S. S. Stahl, J. Am. Chem. Soc., 2019, 141, 10462 CrossRef CAS PubMed; (c) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc04795f |
| This journal is © The Royal Society of Chemistry 2019 |