
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiple heteroatom-doped few-layer carbons for the electrochemical oxygen reduction reaction†
Baobing
Huang
a,
Yuchuan
Liu
a,
Xing
Huang
*bc and
Zailai
Xie
 *a
*a
aState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: zlxie@fzu.edu.cn
bDepartment of Inorganic Chemistry, Fritz Haber Institute of Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: xinghuang@fhi-berlin.mpg.de; xinghuang0214@mail.ipc.ac.cn
cDepartment of Heterogeneous Reactions, Max Planck Institute for Chemical Energy Conversion, 45470 Mülheim an der Ruhr, Germany
First published on 4th October 2018
Abstract
Heteroatom-doped two-dimensional (2D) carbon materials have been recognized as promising metal-free catalysts for the oxygen reduction reaction (ORR) due to their impressive surface activity. Such materials are often achieved by conventional chemical vapour deposition (CVD) growth followed by post-treatment of heteroatom-doping or obtained by template-guided polymerization and carbonization of organic substances. In this contribution, we report a new strategy that allows a direct conversion of three-dimensional (3D) aggregates of guanine into naturally N/O-doped ultrathin few-layer carbon nanosheets without the use of any template or guiding agent. Moreover, by pre-ionizing guanine with H2SO4/H3PO4, a group of multiple heteroatom-doped (N/O/S, N/O/P, and N/O/S/P) 2D carbons can also be readily realized. Considering the low cost of precursors and the simplicity of the synthetic procedure, large-scale production of such heteroatom-doped 2D carbons is potentially available. The best catalytic performance is obtained from the N/O/S/P-doped carbons which show a half-wave potential (E1/2) of 0.84 V vs. RHE and a diffusion-limited current density (jL) of 5.40 mA cm−2 in 0.1 M KOH, better than those of most graphene-based catalysts and even the commercial Pt/C catalyst. Systematic studies indicate that the observed superior ORR performance arises from a combined effect of the multiple heteroatom-doping, high surface area and abundant structural defects of the electrocatalyst.
Introduction
Heteroatom-doped carbon materials have triggered tremendous research interest due to their potential applications as high performance electrodes in energy-related electrochemical processes.1–7 Since the discovery of the high ORR activity of nitrogen-doped carbon nanotubes (NCNTs) by Dai and co-workers, various versions of heteroatom-doped carbon materials have been explored as very promising substitutes for precious-metal catalysts for the ORR.8–16 It has been proposed that this superior activity originates from the doping-induced modifications of the charge and/or spin distribution of the sp2 lattice, which favours the oxygen adsorption and subsequent breaking of the O–O bond.17 Among others, heteroatom-doped carbon materials with a two-dimensional (2D) morphology have emerged as front runners in an array of electrochemical processes because 2D carbon nanosheets can afford a potentially huge surface area to expose more active sites and possess excellent electrical conductivity, both beneficial to fast interfacial charge transfer and ion diffusion in electrocatalytic reactions.18–20 Therefore, current research has been focused on searching for efficient synthetic techniques that allow the fabrication of doped 2D carbon catalysts with sufficiently high electro-catalytic activity.21–23To date, the chemical vapour deposition (CVD) method has been one of the most favoured methodologies for bottom-up synthesis of 2D carbons.24,25 Nevertheless, the CVD approach needs a substrate with a limited surface area for growth and generally suffers from low yield and complicated post-treatments.26,27 Particularly, a catalytic CVD growth results in impurities due to the use of metal supports, which is undesirable in many heterogeneous catalytic reactions.28,29 Another promising way to obtain 2D carbons or graphene is the pyrolysis or carbonization of organic substances with the assistance of suitable templates. For example, it has been shown that pyrolysis of mixtures of glucose with carbon nitride (C3N4) or molten-salts (metal chlorides) acting as templates can lead to generation of 2D porous carbon at elevated temperature.18,21,30–33 Likewise, the carbonization of predesigned 2D covalent organic frameworks (COFs) in the presence of a structure-guiding agent has also been demonstrated to be able to yield nanosized 2D graphitic carbon nanosheets.34 Although the above strategies provide promising opportunities for fabricating heteroatom-doped carbon nanosheets, inevitable disadvantages, such as the necessary use of a template or structure-guiding agent, make the synthesis a sophisticated and time consuming process and thus difficult to be scaled up.18,21,30–35 In view of these, development of an innovative strategy that allows a facile, low-cost and scalable fabrication of heteroatom-doped 2D carbons is still highly sought after.
Herein, we report a template-free and scalable approach towards synthesis of a new family of multiple heteroatom-doped (N/O, N/S/O, N/P/O, and N/S/P/O) 2D carbons as metal-free catalysts for the high performance ORR by direct pyrolysis of biomolecule guanine aggregates and their salts obtained by ionizing with H2SO4, H3PO4 and their mixture. Guanine is known as a planar molecule with each molecule having five hydrogen bonds in the plane of the molecule (Scheme 1). This molecule has a strong tendency to form molecular aggregates held together by in-plane hydrogen bonds and out-of-plane stacking interactions between the aromatic rings.36–40 The current study, for the first time, demonstrates the utilization of guanine as the carbon precursor that unprecedentedly facilitates the formation of ultrathin and large-scale 2D carbon nanosheets with naturally high-level nitrogen doping. Moreover, the combination of guanine with a dopant-containing acidic solution can give rise to formation of dopant-containing ionic compounds which, after pyrolysis, are transformed into multiple-heteroatom-doped (N/S/O, N/P/O, and N/S/P/O) 2D carbons. Finally, the electrochemical properties of the customized catalyst series are investigated. The best performance is obtained from the N/S/P/O-doped carbon nanosheets, which show a remarkable ORR activity under universal pH conditions, better than that of most previously reported carbon-based catalysts and the commercial Pt/C catalyst. Our strategy may open a new horizon towards synthesis of a kind of high performance carbon-based electrocatalyst as promising candidates to replace precious metal catalysts for application in fuel cells or metal–air batteries.
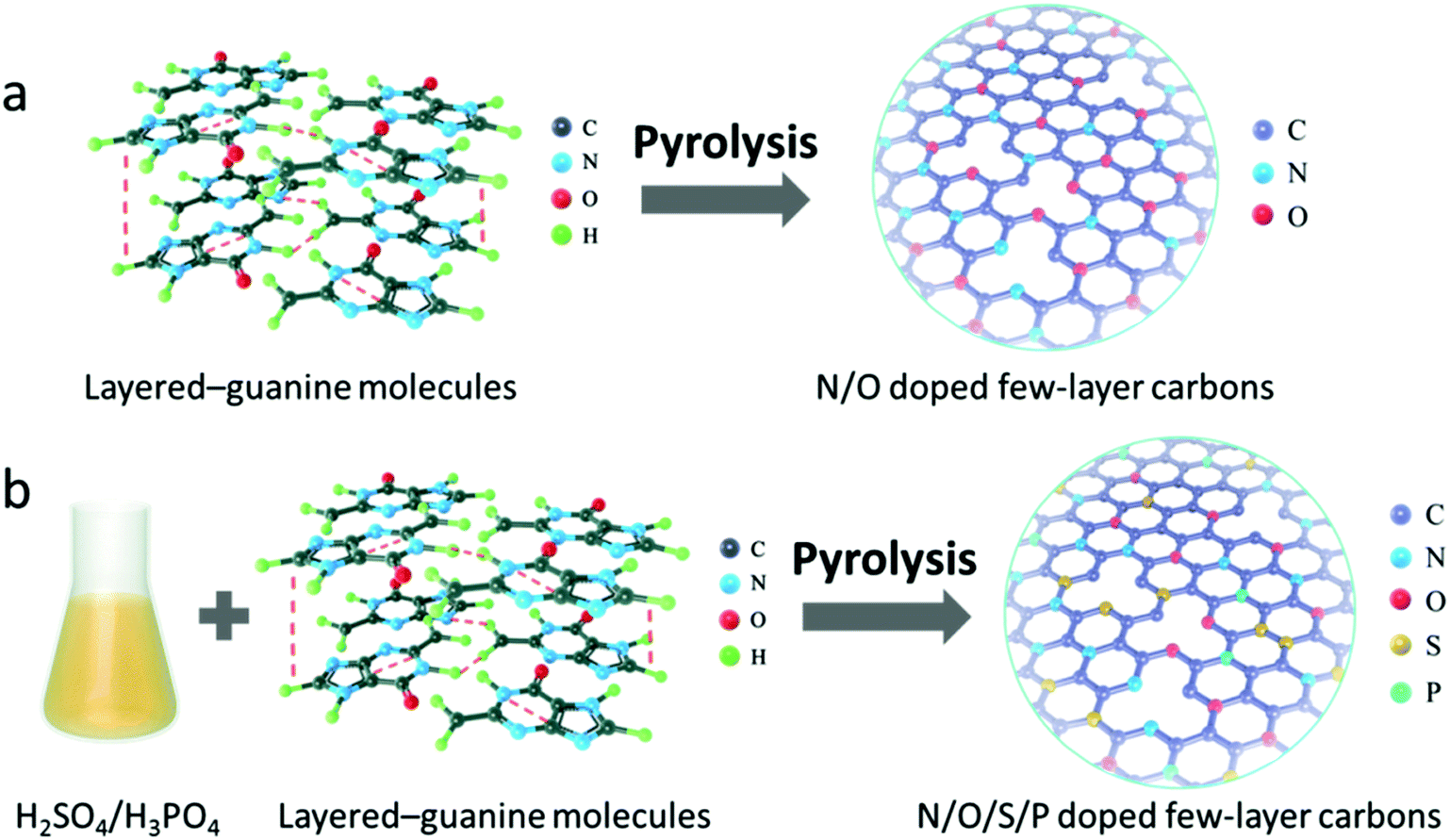 | ||
| Scheme 1 Schematic illustration of the fabrication process of (a) N/O bi-doped and (b) N/O/S/P tetra-doped few-layer carbon nanosheets. | ||
Experimental section
Materials synthesis
In our synthesis, the biomolecule guanine was used as the source for fabrication of 2D carbon materials. It was purchased from Adamas (99+%) and used without further purification. The 2D N/O-doped carbons were prepared by direct pyrolysis of aggregates of guanine at 400, 550, 700, 800, 900, and 1000 °C in a N2 atmosphere for 1 h at a heating rate of 5 °C min−1. For the synthesis of modified 2D carbons, 6 g guanine was dispersed in 60 mL deionized water containing 1.08 mL H2SO4 (98%) and was stirred for 24 hours at room temperature. The solution was then evaporated completely at 75 °C to obtain a solid which was denoted as GS. When replacing 1.08 mL H2SO4 (98%) with 0.7 mL H3PO4 (85%), the as-prepared compound was named GP. When a mixed acid of 1.08 mL H2SO4 (98%) and 0.7 mL H3PO4 (85%) was used, the as-prepared compound was named GSP. The corresponding 2D carbons were produced by pyrolysis of GS/GP/GSP at 1000 °C for 1 h in a N2 atmosphere at a heating rate of 5 K min−1. To remove the possible impurities (e.g. phosphorus compound), the P-doped 2D carbons were soaked in 1 M HCl for 10 hours. All of them were further subjected to graphitization at 1000 °C for another 1 h in a N2 atmosphere to stabilize the structure. The final products were denoted as G-1000, GS-1000, GP-1000 and GSP-1000, respectively.Physical characterization
X-ray photoelectron spectroscopy (XPS) was conducted on a Thermo Scientific ESCALAB 250 (USA) with a monochromatized Al Kα line source (200 W). Scanning electron microscopy (SEM) images were acquired on an S-4800 (Hitachi, Japan). Nitrogen adsorption–desorption isotherms were measured with a Micromeritics ASAP 2020 plus/2060, and the specific surface area was calculated via the Brunauer–Emmett–Teller (BET) method. Atomic force microscopy (AFM) images were obtained with a Dimension Icon (Bruker Nano Inc.) in ScanAsyst mode. X-ray diffraction (XRD) patterns were recorded in reflection mode (CuKα radiation) on a Bruker D8 diffractometer between 5 and 80°. Raman spectra were recorded by using a Thermo Scientific DXR Raman microscope with a 532 nm laser.Transmission electron microscopy (TEM) images, high-resolution TEM (HRTEM) images, and high angle annular dark field scanning TEM (HAADF-STEM) images were recorded on an aberration-corrected JEOL ARM-200CF transmission electron microscope operated at 200 kV. The elemental mapping measurements were carried out using a high angle silicon drift EDX detector (with a solid angle up to 0.98 steradians and a detection area of 100 mm2) equipped with the JEOL TEM. An in situ TEM heating experiment was performed inside the column of a Cs-corrected Titan 80–300 transmission electron microscope operated at 200 kV by using an MEMS-based heating holder (DENSsolutions). The heating chip is composed of a metallic spiral coil which is embedded inside the SiNx thin membrane (see Fig. S5a†). The ceramic SiNx membrane contains several elongated holes along the spiral coil across which the sample is suspended for low background imaging.
Electrochemical measurements
The electrochemical activities of the as-synthesized materials towards the ORR were obtained with an IviumStat multichannel electrochemical analyzer (Netherlands) in a three-electrode cell at room temperature, in which a Ag/AgCl (saturated KCl) electrode, Pt foil (1 cm2) and a rotating disk electrode (RDE) were selected as the reference electrode, the counter electrode and the working electrode, respectively. For the rotating disk electrode (RDE) experiment, a glassy carbon electrode (GCE, d = 4 mm) was pre-polished and rinsed thoroughly. For working electrode preparation, a 5 mg carbon sample was dispersed in a mixture of 0.25 mL deionized water, 0.75 mL ethanol and 0.1 mL Nafion (5 wt%), and then sonicated for 1 h to get a homogeneous catalyst ink. 8.3 μL of the as-prepared inks was drop cast onto the surface of the GCE and dried in air for electrochemical tests. The catalytic measurements were conducted in O2-saturated 0.1 M KOH, 0.1 M HClO4 and 0.1 M PBS solutions, respectively. The loading amount of catalysts was about 0.3 mg cm−2. Commercial 20 wt% Pt/C with a loading of 0.1 mg cm−2 was prepared for comparison. All of the measured potentials were converted to reversible hydrogen electrode potentials using E (RHE) (V) = E (Ag/AgCl) + 0.198 + 0.059 pH. The calculation of the electron transfer number is based on the Koutecky–Levich equations: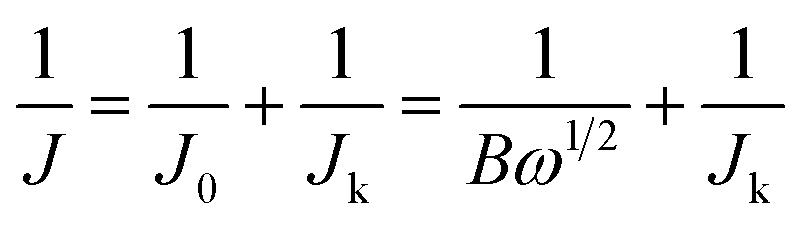
| B = 0.62nFC0(D0)2/3(ν)−1/6 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−3 mol L−1), D0 is the diffusion coefficient of O2 (1.9 × 10−5 cm2 s−1), and ν is the kinematic viscosity of the electrolyte (0.01 cm2 s−1).
485C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−3 mol L−1), D0 is the diffusion coefficient of O2 (1.9 × 10−5 cm2 s−1), and ν is the kinematic viscosity of the electrolyte (0.01 cm2 s−1).
With regard to the rotating ring-disk electrode (RRDE) experiment, the ring-disk electrode composed of a glassy carbon disk (d = 4 mm) and a Pt ring (5 mm inner diameter and 7 mm outer diameter) served as the working electrode with the same catalyst loading as that used in the RDE measurements. The ring potential was kept at 1.5 V (vs. RHE). The number of electrons transferred was evaluated from the following equations:

wherein Ir and Id are the ring and disk currents, respectively, and N is the ring collection efficiency.

Results and discussion
The fabrication process of 2D carbon nanosheets is illustrated in Scheme 1a. It involves only one-step heating of guanine in a N2 atmosphere at a heating rate of 5 °C min−1 up to a desired temperature followed by subsequent annealing for a given time (see the ESI† for more details; the final sample is denoted as G-1000). The synthesis of multiple heteroatom-doped carbon nanosheets is conducted under otherwise similar conditions except for pre-treatment of guanine with H2SO4, H3PO4 and their mixture (Scheme 1b). It is worth mentioning that all these syntheses can be scaled up considering the low cost of precursors and the simplicity of the method.To the best of our knowledge, direct transformation from guanine to ultrathin 2D carbon nanosheets has not been reported before. In order to learn more about this interesting transformation, temperature-dependent pyrolytic treatments are additionally conducted. The SEM images of the samples obtained at different temperatures are displayed in Fig. 1a–g and S1–S4.† One can see that the particles are stable up to 400 °C without showing detectable changes in the morphology (Fig. 1b). These modifications are observed to set in when the temperature is increased to 550 °C, at which most of the particles present a concave shape on their surfaces. In addition, a growth in particle size and a decreased amount of small particles are identified, suggesting the incorporation of small particles into big ones. The clear formation of sheet-like structures with further increased size is observed after pyrolysis at 700 °C. Yet, there still some particles do not form, but in their way to the 2D sheets. The complete transformation is found at 800 °C, and the nanosheets become more crumpled when compared to those at 700 °C. Further temperature increase to 1000 °C finally results in the formation of ultrathin, highly wrinkled and crumpled nanosheets, as indicated in Fig. 1g. The AFM characterization reveals the ultrathin nature of the sheets, namely, a thickness of about 1 nm.
 | ||
| Fig. 1 (a–g) SEM images of guanine-derived materials obtained at different temperatures: (a) RT, (b) 400 °C, (c) 550 °C, (d) 700 °C, (e) 800 °C, (f) 900 °C, and (g) 1000 °C. (h) AFM image of G-1000. (i) Schematic illustration of morphological evolution from 3D guanine precursor particles to 2D few-layer carbon nanosheets. (j and k) HAADF-STEM image and superimposed elemental map of C, N, and O; (l) HRTEM image and SAED pattern of G-1000. | ||
Although the above temperature-dependent ex situ experiments have provided valuable information about the morphological evolution, the dynamics details are still missing due to the lack of continued observation from an identical place. In order to gain more insights into the transformation process, an in situ heating experiment inside the column of a conventional TEM using a MEMS-based heating holder is additionally performed (Fig. S5†). The sample is programmably heated up to 1000 °C following the temperature profile shown in Fig. S5a.† Real-time imaging clearly reveals that the initial aggregated precursor particles are transformed into large size sheet-like structures at elevated temperature. This transformation starts at around 100 °C for small particles with a size of about 200–300 nm and then extends to larger ones (micrometer sizes) at around 450 °C (Movie 1†). Dynamic variation of the image contrast is observed over the particles because of the thickness change. When the temperature is over 700 °C, the morphological change becomes minor (Fig. S5f–i†). Still, the image contrast is reducing, an indication of a further decreased thickness. The final product shows a highly crumpled 2D sheet-like morphology with a lateral size larger than that of individual precursor particles, which is in good agreement with the results of ex situ experiments. To sum up, the combination of both ex situ and in situ experiments reveals rich dynamics involved in the morphological transformation, including the coalescence of precursor particles, and thinning and crumpling of nanosheets, which eventually result in the formation of ultrathin 2D carbon nanosheets. The schematic view of the formation process of 2D carbons is illustrated in Fig. 1i. It is worth mentioning that the morphological transformation during the in situ experiment takes place at relatively lower temperature compared with that in ex situ experiments, which is probably related to the high vacuum conditions in the chamber of the TEM (ex situ experiments were conducted in N2).
The structural and chemical analyses of the sample (G-1000) are performed. The high-resolution TEM (HRTEM) image shows that the sample exists in a highly defective graphitic structure with a thickness of about 3–4 atomic layers (Fig. 1l). This defective structure can also be reflected by the selective-area electron diffraction (SAED) pattern that shows diffused ring patterns (see the inset of Fig. 1l). The elemental analysis of the as-obtained carbon nanosheets reveals the presence of C, N and O, and they are distributed homogeneously over the entire nanosheets (Fig. 1j and k).
In the following, we demonstrate that such 2D carbons can be further functionalized with multiple dopants by mixing guanine with inorganic acids as precursors (H2SO4, H3PO4 or their mixture), prior to the pyrolysis. The contents of heteroatoms can be readily altered by changing the ratio of the acid and natural base. Depending on the acids used, the obtained samples are denoted as GS-1000 (H2SO4), GP-1000 (H3PO4) and GSP-1000 (H2SO4 and H3PO4). Akin to the G-1000 (Fig. 2a), all functionalized samples present a highly crumpled 2D morphology (Fig. 2b–d). AFM characterization reveals that the average thickness of carbon nanosheets is ca. 1 nm, that is, a thickness of three carbon layers (Fig. 2h). Fig. 2e shows a low magnification TEM image of the GSP-1000 and the corresponding SAED pattern. The transparency of the nanosheets to an electron beam indicates the thin thickness of the sample, which, according to the TEM image shown in Fig. 2f, is about 1–1.3 nm, therefore in line with the AFM results. The SAED pattern mainly shows three reflections corresponding to (002), (101) and (110) planes of the graphitic structure. Minor deviations of d-spacings are identified, which are likely due to the defective structure and the incorporation of doping elements. The presence of broad and blurred rings suggests the defective and polycrystalline nature of the nanosheets, in agreement with the HRTEM image (Fig. 2f) which displays disordered lattice fringes. In addition, the enlarged HRTEM image (Fig. 2g) clearly reveals the presence of large numbers of pores in the sheets. As evidenced by the elemental analysis, the GSP-1000 shows uniform dispersion of C, N, S, P and O over the whole nanosheets (Fig. 2i–n), indicating the successful incorporation of dopants into the carbon framework. For structural and compositional analysis of GS-1000, please see the ESI for details (Fig. S6†).
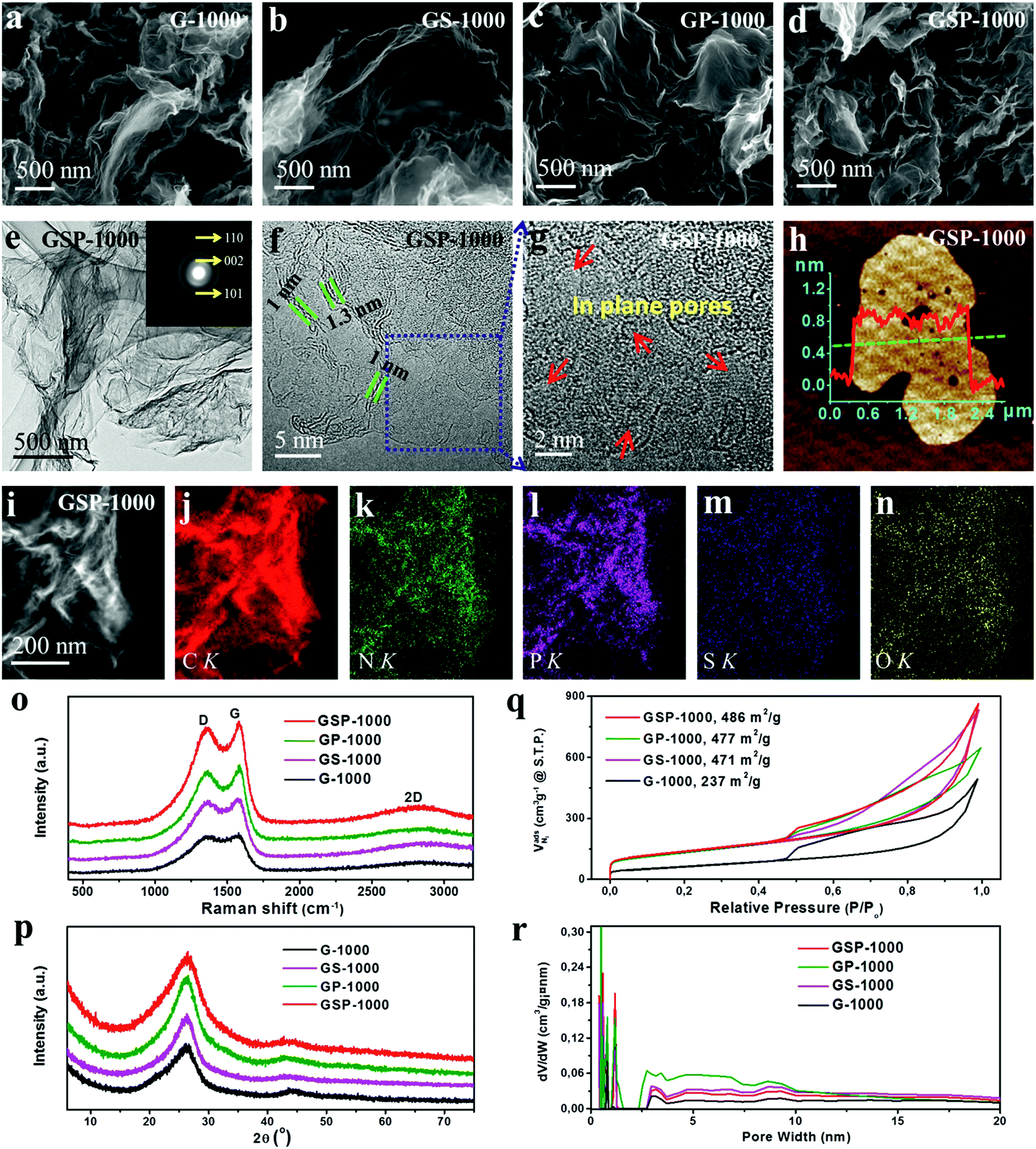 | ||
| Fig. 2 (a–d) SEM images of G-1000, GS-1000, GP-1000 and GSP-1000, showing a 2D sheet-like morphology. (e–g) TEM and HRTEM images of GSP-1000; inset of (e) shows the SAED pattern. (h) AFM image and height profile of GSP-1000. (i–n) HAADF-STEM image of GSP-1000 and the corresponding EDX elemental maps of C, N, P, S, and O. (o) Raman spectra, (p) XRD patterns, (q) nitrogen adsorption/desorption isotherms and (r) pore size distributions (PSDs) of the sample series. | ||
The sample series was also examined by X-ray diffraction (XRD) and Raman spectroscopy, respectively. XRD patterns in Fig. 2p show a strong non-symmetric (002) reflection at ca. 26.0°, together with a relatively weak (100) peak at 43.6° for all samples, suggesting some disordered but mainly sp2 hybridized graphitic structures. The defective graphitic structure can also be reflected by the Raman spectra which show a strong D band located at 1360 cm−1, characteristic of defect induced mode (Fig. 2o). They additionally show a broad 2D band at around 2700 cm−1, which is attributed to the formation of thin carbon films. Therefore, the XRD and Raman results coincide well with those of the SEM and TEM characterization, and they all indicate the ultrathin and defective nature of the as-prepared 2D carbon nanosheets.
X-ray photo-electron spectroscopy (XPS) was performed to analyse the chemical nature of dopants. As shown in Fig. 3a, besides the remarkable presence of C 1s, N 1s and O 1s characteristic peaks in all materials, signals from the S 2p and P 2p can also be visualized in the corresponding materials, demonstrating the successful incorporation of heteroatoms into the graphene skeletons. The elemental composition and weight ratio determined by XPS are shown in Table 1. It is revealed further by high-resolution XPS that the N 1s (Fig. 3b) can be deconvoluted into four characteristic peaks at 398.3 eV, 399.6 eV, 401.0 eV and 402.9 eV, corresponding to pyridinic-N (N1), pyrrolic-N (N2), graphitic-N (N3) and oxidized-N (N4) species, respectively.41 The ratios of four N species in different samples are shown in Fig. 3c. According to previous studies, N1 and N3 species are considered as active configurations that give rise to the enhancement of ORR activity.41,42 The XPS analysis indicates that the G-1000 has the highest amount of N1 and N3 (3.06%), whereas the GP-1000 shows only about 1.54%. In contrast, the GS-1000 and GSP-1000 show amounts in between. Fig. 3d shows the deconvolution of S 2p spectra of GS-1000 and GSP-1000, in which the peaks at 163.9, 165.1 and 168.2 eV can be assigned to –C–S–C– (S 2P3/2), –C–S–C– (S 2P1/2) and –C–SOx–C, respectively.43 It is noted that the S content in GS-1000 is almost two times that of GSP-1000. The spectra of P 2p (Fig. 3e) contains a dominating peak at 131.0 eV and an extra weak peak at 133.7 eV, corresponding to the P–C bond and P–O bond.3 The doping level of P is in close proximity for both GP-1000 and GSP-1000, that is, 1.23% and 1.10%, respectively. The fitting of O 1s spectra (Fig. 3f) also indicates two peaks, i.e., oxygen double bonded to carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 531.8 eV and oxygen single bonded to carbon (C–O) at 533.4 eV.44 The oxygen content among the catalyst series follows an order of GP-1000 > GSP-1000 (GS-1000) > G-1000 (see Table 1 for exact values). It is also interesting to find that the total amount of dopants is indeed very close among samples, which account for about 10% of the total weight. This result, on the other hand, indicates that, for GP-1000, GS-1000 and GSP-1000, the additional doping with S and/or P does not increase the total doping amount but merely changes the relative amount of N and O.
O) at 531.8 eV and oxygen single bonded to carbon (C–O) at 533.4 eV.44 The oxygen content among the catalyst series follows an order of GP-1000 > GSP-1000 (GS-1000) > G-1000 (see Table 1 for exact values). It is also interesting to find that the total amount of dopants is indeed very close among samples, which account for about 10% of the total weight. This result, on the other hand, indicates that, for GP-1000, GS-1000 and GSP-1000, the additional doping with S and/or P does not increase the total doping amount but merely changes the relative amount of N and O.
 | ||
| Fig. 3 (a) XPS survey spectra. (b) High resolution N 1s XPS spectra. (c) The content percentages of different nitrogen species. (d) High resolution S 2p XPS spectra. (e) High resolution P 2p XPS spectra. (f) High resolution O 1s XPS spectra. | ||
| Samples | Porosity data | Elemental compositiond | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S BET [m2 g−1] | V total [cm3 g−1] | V meso [cm3 g−1] | V micro [cm3 g−1] | C | N | O | S | P | N1 + N3 | |
| a From total N2 uptake at P/P0 = 0.95. b From total N2 uptake at P/P0 = 0.95. c From the DFT method. d Determined by XPS, at%. | ||||||||||
| G-1000 | 237 | 0.57 | 0.47 | 0.01 | 93.89 | 4.48 | 1.63 | 0 | 0 | 3.06 |
| GS-1000 | 471 | 0.95 | 0.80 | 0.05 | 92.90 | 3.49 | 3.00 | 0.61 | 0 | 2.78 |
| GP-1000 | 477 | 0.92 | 0.75 | 0.07 | 92.30 | 2.24 | 4.23 | 0 | 1.23 | 1.54 |
| GSP-1000 | 486 | 0.93 | 0.74 | 0.05 | 92.81 | 2.72 | 3.01 | 0.36 | 1.10 | 2.27 |
It is further revealed by the analysis of nitrogen sorption that the functionalization of guanine with different acids leads to not only the successful doping of the carbon lattice with desired heteroatoms, but also the change in the texture of carbons, giving rise to greatly improved specific surface areas, as shown in Table 1. The study of the nitrogen sorption behaviour indicates that all catalysts exhibit similar isotherm shapes and striking hysteresis loops (see Fig. 2q). The G-1000 shows the lowest surface area due to the lack of sufficient micropores. By contrast, the catalysts GS-1000, GP-1000 and GSP-1000 show well-developed micro- and mesopores. The surface areas of these samples are over 470 m2 g−1, which is almost twice as high as that of G-1000. The improved porosity in multiple heteroatom-doped 2D carbons is related to the porogen effect by acid decomposition. In the case of GS-1000, we assume that the releasing of SO2 or SO3 as a result of decomposition of the anion H2SO4 during the carbonization facilitates the development of microporous–mesoporous structures. Likewise, in the formation of GP-1000, the use of phosphoric acid produces H4P2O7, H5P3O10 and polymeric (HPO3)n intermediates by self-dehydration at high temperature, which can induce the formation of microporous textures.45 Since the formation of GSP-1000 involves the use of both H3PO4 and H2SO4, the pore-formation mechanism might combine the above two effects. It is also noted that all the isotherms show an increased N2 uptake at P/P0 > 0.9, indicating the existence of macropores. This is also confirmed by the pore size distributions (PSDs) shown in Fig. 2r.
In order to demonstrate the potential use of the synthesized heteroatom-doped carbon nanosheets in electrode-related applications, the electrocatalytic performance towards the ORR is systematically investigated. The cyclic voltammograms (CVs) recorded in O2- and N2-saturated 0.1 M KOH solutions at a scan rate of 50 mV s−1 are shown in Fig. 4a and S7.† The presence of a striking oxygen reduction cathodic peak at ca. 0.75 V (vs. RHE) and its absence in the N2-saturated alkaline solution suggest the promising ORR activities of the as-synthesized carbon nanosheets. Rotating disk electrode (RDE) technology is further performed to study the ORR kinetics of the different catalysts (Fig. 4b). It is revealed that GSP-1000 and GS-1000 have similar onset potentials (0.99 V vs. RHE and 0.98 V vs. RHE, respectively) and half-wave potentials (0.84 V vs. RHE and 0.82 V vs. RHE), close to those of the commercial 20 wt% Pt/C catalyst (Eonset = 1.03 V vs. RHE and E1/2 = 0.85 V vs. RHE). Intriguingly, when comparing the diffusion-limited current densities, GSP-1000 and GP-1000 (5.4 mA cm−2) are superior to the Pt/C catalyst (5.1 mA cm−2), and they outperform most of the reported graphene-based electrocatalysts (see Table S2†). The positive response of the diffusion-limited current density to the rotating speed (Fig. 4c and S8†) indicates that the ORR is diffusion-controlled. The average electron transfer numbers calculated from the linear Koutecky–Levich (K–L) plots are up to 4 for all the catalysts, forcefully demonstrating that the favourable reduction pathway of oxygen directly to hydroxyl is highly achieved. In particular, the collected rotating ring-disk electrode (RRDE) data in Fig. 4d and e show that the electron transfer numbers of GSP-1000 at different potentials are over 3.8 with yields of H2O2 below 10% throughout the whole potential range, confirming the excellent catalytic selectivity of GSP-1000. The stability and anti-methanol ability of the sample series are also examined. Taking the GSP-1000 as an example, the current retention rate becomes 90.7% of the initial activity after a more than 4 h test (results of a longer stability test are shown in Fig. S9†), whereas it becomes 76.2% for the Pt/C catalyst (Fig. 4f). Besides, unlike the Pt/C catalyst showing significantly decreased current density when methanol is added, GSP-1000 is almost immune to methanol, presenting excellent durability (inset of Fig. 4f).
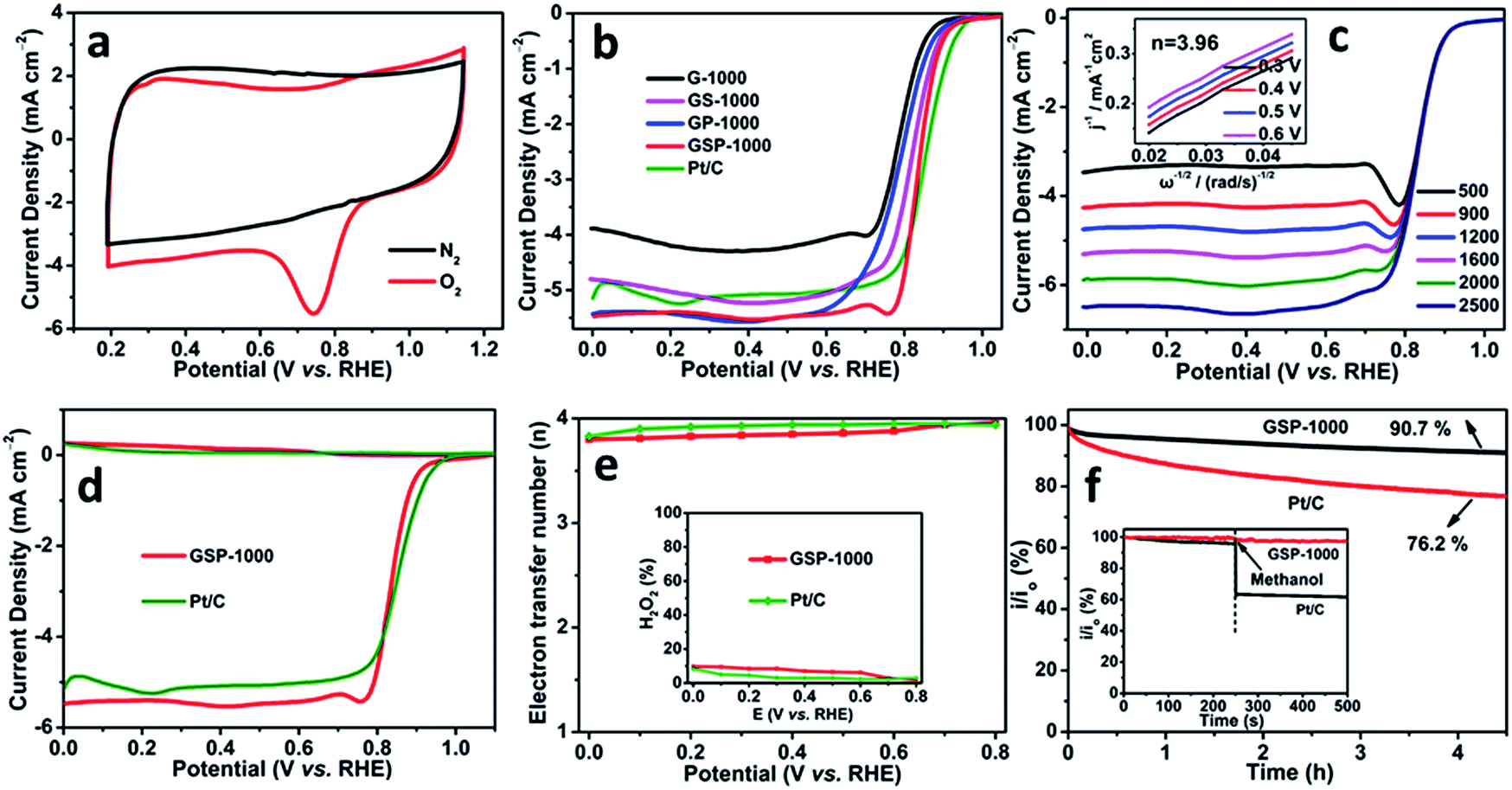 | ||
| Fig. 4 (a) Cyclic voltammograms (CVs) of GSP-1000 at 50 mV s−1. (b) Comparisons of the RDE polarization curves in O2-saturated 0.1 M KOH at 1600 rpm with 10 mV s−1. (c) RDE polarization curves of GSP-1000 at different rotation speeds. (d) RRDE curves at 1600 rpm. (e) The corresponding electron transfer numbers; inset: peroxide percentages. (f) i–t Response at 0.8 V (vs. RHE) at 1600 rpm; inset shows the methanol crossover effect test upon addition of 1 M methanol. | ||
Development of highly efficient electrocatalysts feasible under multiple-pH conditions is of both scientific and technical importance for future practical applications. In this regard, we additionally measured the ORR performances of the as-prepared 2D nanocarbons in acidic and neutral electrolytes (Fig. S10–S15†). Our electrochemical tests clearly reveal that all of the samples exhibit obvious oxygen reduction characteristic peaks (Fig. S10 and S11†) in both O2-saturated 0.1 M HClO4 and 0.1 M PBS (phosphate buffer solution, pH = 7.0). It is particularly shown that the GSP-1000 has superior ORR activity and durability compared to other catalysts in the series and also the commercial Pt/C (Fig. S12†). The detailed discussion of the catalytic performances in acidic and neutral electrolytes is included in the ESI.† We are therefore convinced to state that the multiple-heteroatom-doped 2D carbon nanosheets are promising candidates as highly efficient pH-universal electrocatalysts towards the ORR.
In the following, we discuss and address the possible reasons for the excellent ORR activity of multiple heteroatom-doped 2D carbon nanosheets. As revealed above, the ORR activities of GP-1000, GS-1000 and GSP-1000 all got improved at different degrees compared to that of G-1000 following the order of G-1000 < GP-1000 < GS-1000 < GSP-1000. Based on the above investigations, it is reasonable to propose that both heteroatom-doping and doping-induced improved porosity are responsible for the remarkably enhanced activity.46–48 From the viewpoint of porosity, the low activity of G-1000 can be understood by the lower specific surface due to the absence of a microporous–mesoporous structure, resulting in the insufficient exposure of active sites and unsatisfactory ion transport. In contrast, GS-1000, GP-1000 and GSP-1000 possess well-developed micro- and mesoporous structures with a significantly increased surface area, thus having more active sites exposed. Improvement of ion diffusion is also expected due to the improved porosity. Therefore, the combination of these two aspects gives rise to the enhanced activity of GS-1000, GP-1000 and GSP-1000. Considering the fact that GS-1000, GP-1000 and GSP-1000 have similar BET surface areas and pore size distributions, their activity difference thus should be mostly related to the synergistic effect of different kinds of heteroatoms. Generally speaking, dopants work in a way that they introduce the altered charge or spin distribution of the sp2 carbon plane, which facilitates the oxygen adsorption and/or the O–O bond breaking, leading to an improved ORR activity.17 Among various dopants, N has been demonstrated to be the most promising dopant for improving the activity of carbon-based materials. In particular, pyridinic N with a lone electron pair and graphitic N are considered as active configurations.49,50 The GSP-1000, which shows the best activity, however does not possess the highest amount of pyridinic and graphitic N (see Table 1). This result suggests that the incorporation of the configuration of active N species (N1 + N3) is more important than the gross content.17,42,50 On the other hand, dopants such as O, S, and P have been established to be beneficial for boosting the ORR activity. In particular, simultaneous incorporation of several kinds of heteroatoms into carbon materials has been demonstrated as an effective way to promote the activity.9,51 In our study, the observation of the highest activity from N/O/S/P tetra-doped 2D carbons (GSP-1000) evidences again the beneficial effect of multiple-heteroatom-doping on improving the ORR activity. It is reasonable to believe that the co-existence of several kinds of dopants can lead to generation of more active sites due to increased charge/spin densities. Moreover, the synergistic coupling effect between different dopants can enhance the activity of single active sites, thus leading to further improved catalytic performance towards the ORR.52–54 It is also important to mention that the structural defects, either existing intrinsically or introduced during the doping process, can also induce the electron deformation and contribute to the ORR activity.14,15,55–58 According to the XRD, Raman and TEM characterization, the as-prepared few-layer carbon nanosheets are rich in structural defects. In particular, the HRTEM images (Fig. 1l and 2f) show the presence of highly defective edges that may serve as active sites for achieving a good ORR activity. Overall, we believe that the combined effect of multiple heteroatom-doping, high surface area and abundant structural defects provides the N/O/S/P-doped 2D carbons an extraordinary ORR activity under pH-universal conditions, making them appealing candidates as metal-free electrocatalysts for practical applications.
Conclusions
In summary, we demonstrate that the pyrolysis of biomolecule guanine can lead to direct formation of naturally N/O-doped ultrathin 2D carbon nanostructures. We further show that the combination of guanine with H2SO4/H3PO4 enables the fabrication of multiple heteroatom-doped 2D carbons with a significantly increased surface area and abundant heteroatom sites. The current study could be a breakthrough for a simple, cheap and sustainable synthesis of heteroatom-doped 2D carbon materials. Systematic characterization using multiple techniques is conducted to study the morphology (including morphological transformation), structure, composition, and chemical nature of the dopants, and the specific surface area, porosity, and finally the catalytic performances of the catalyst series. The N/O/S/P-doped carbon nanosheets are recognized as preferable candidates as high performance metal-free ORR catalysts due to their high catalytic activity with exceptional half-wave potentials and diffusion-limited current densities under universal pH conditions (e.g. 0.84 V vs. RHE and 5.40 mA cm−2 in 0.1 M KOH, 0.67 V vs. RHE and 5.48 mA cm−2 in 0.1 M HClO4, and 0.70 V vs. RHE and 4.83 mA cm−2 in 0.1 M PBS). Compared with the commercial Pt/C catalyst, the N/O/S/P-doped carbon nanosheets also show higher durability and better anti-methanol performance. Based on our investigations, we propose that the excellent catalytic performances are a result of synergetic effects combining the heteroatom-doping, large surface area and defective structure of the N/O/S/P-doped 2D carbons. The present work may open a new avenue for rational design and large-scale synthesis of heteroatom-doped 2D carbons as efficient metal-free electrocatalysts for application in fuel cells and metal–air batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC grant number 21571035), the Award Program for Fujian Minjiang Scholar Professorship, the open Project Foundation of Key Laboratory of Physical Chemistry of Solid Surfaces of Xiamen University (2015–18), and the open Project Foundation of Key Laboratory of Structural Chemistry of FJIRSM. Dr Xing Huang would like to thank the financial support from Fritz Haber Institute of Max Planck Society and Max Planck Institute for Chemical Energy Conversion. Open Access funding provided by the Max Planck Society.Notes and references
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- G.-L. Tian, Q. Zhang, B. Zhang, Y.-G. Jin, J.-Q. Huang, D. S. Su and F. Wei, Adv. Funct. Mater., 2014, 24, 5956–5961 CrossRef CAS.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- B. Zhang, H.-H. Wang, H. Su, L.-B. Lv, T.-J. Zhao, J.-M. Ge, X. Wei, K.-X. Wang, X.-H. Li and J.-S. Chen, Nano Res., 2016, 9, 2606–2615 CrossRef CAS.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 2230–2234 CrossRef CAS PubMed.
- Y. Liu, B. Huang, X. Lin and Z. Xie, J. Mater. Chem. A, 2017, 5, 13009–13018 RSC.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed.
- P. Chen, T. Y. Xiao, Y. H. Qian, S. S. Li and S. H. Yu, Adv. Mater., 2013, 25, 3192–3196 CrossRef CAS PubMed.
- K. Elumeeva, N. Fechler, T. P. Fellinger and M. Antonietti, Mater. Horiz., 2014, 1, 588–594 RSC.
- C. Han, J. Wang, Y. Gong, X. Xu, H. Li and Y. Wang, J. Mater. Chem. A, 2014, 2, 605–609 RSC.
- S. Zhang, M. S. Miran, A. Ikoma, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2014, 136, 1690–1693 CrossRef CAS PubMed.
- H. Zhao, C. Sun, Z. Jin, D.-W. Wang, X. Yan, Z. Chen, G. Zhu and X. Yao, J. Mater. Chem. A, 2015, 3, 11736–11739 RSC.
- C. Tang, H. F. Wang, X. Chen, B. Q. Li, T. Z. Hou, B. Zhang, Q. Zhang, M. M. Titirici and F. Wei, Adv. Mater., 2016, 28, 6845–6851 CrossRef CAS PubMed.
- B. Huang, L. Peng, F. Yang, Y. Liu and Z. Xie, J. Energy Chem., 2017, 26, 712–718 CrossRef.
- C. Tang and Q. Zhang, Adv. Mater., 2017, 29, 1604103 CrossRef PubMed.
- X. Liu and M. Antonietti, Adv. Mater., 2013, 25, 6284–6290 CrossRef CAS PubMed.
- J. Zhang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 13296–13300 CrossRef CAS PubMed.
- H. Tao, Y. Gao, N. Talreja, F. Guo, J. Texter, C. Yan and Z. Sun, J. Mater. Chem. A, 2017, 5, 7257–7284 RSC.
- X. H. Li, S. Kurasch, U. Kaiser and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 1–5 CrossRef.
- J. Xu, J. Zhu, X. Yang, S. Cao, J. Yu, M. Shalom and M. Antonietti, Adv. Mater., 2016, 28, 6727–6733 CrossRef CAS PubMed.
- J. Zhang, H. Zhou, X. Liu, J. Zhang, T. Peng, J. Yang, Y. Huang and S. Mu, J. Mater. Chem. A, 2016, 4, 15870–15879 RSC.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- A. K. Singh, M. W. Iqbal, V. K. Singh, M. Z. Iqbal, J. H. Lee, S. H. Chun, K. Shin and J. Eom, J. Mater. Chem., 2012, 22, 15168–15174 RSC.
- A. Reina, X. T. Jia, J. Ho, D. Nezich, H. B. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 30–35 CrossRef CAS PubMed.
- G. V. Bianco, M. Losurdo, M. M. Giangregorio, P. Capezzuto and G. Bruno, Phys. Chem. Chem. Phys., 2014, 16, 3632–3639 RSC.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- G. L. Tian, M. Q. Zhao, D. Yu, X. Y. Kong, J. Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 2251–2259 CrossRef CAS PubMed.
- X. Liu, C. Giordano and M. Antonietti, Small, 2014, 10, 193–200 CrossRef CAS PubMed.
- Z. Ling, Z. Wang, M. Zhang, C. Yu, G. Wang, Y. Dong, S. Liu, Y. Wang and J. Qiu, Adv. Funct. Mater., 2016, 26, 111–119 CrossRef CAS.
- M. Antonietti and M. Oschatz, Adv. Mater., 2018, 1706836 CrossRef PubMed.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. Waterhouse, Y. Zhao, C. Zhou, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080–5086 CrossRef CAS PubMed.
- Q. Xu, Y. Tang, X. Zhang, Y. Oshima, Q. Chen and D. Jiang, Adv. Mater., 2018, 1706330 CrossRef PubMed.
- X. Li, Y. Fang, S. Zhao, J. Wu, F. Li, M. Tian, X. Long, J. Jin and J. Ma, J. Mater. Chem. A, 2016, 4, 13133–13141 RSC.
- A. Levy-Lior, B. Pokroy, B. Levavi-Sivan, L. Leiserowitz, S. Weiner and L. Addadi, Cryst. Growth Des., 2008, 8, 507–511 CrossRef CAS.
- Y. Oaki, S. Kaneko and H. Imai, J. Mater. Chem., 2012, 22, 22686–22691 RSC.
- A. Hirsch, D. Gur, I. Polishchuk, D. Levy, B. Pokroy, A. J. Cruz-Cabeza, L. Addadi, L. Kronik and L. Leiserowitz, Chem. Mater., 2015, 27, 8289–8297 CrossRef CAS.
- D. Gur, B. A. Palmer, S. Weiner and L. Addadi, Adv. Funct. Mater., 2017, 27, 1603514 CrossRef.
- A. Hirsch, B. A. Palmer, N. Elad, D. Gur, S. Weiner, L. Addadi, L. Kronik and L. Leiserowitz, Angew. Chem., Int. Ed., 2017, 56, 1–6 CrossRef PubMed.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 RSC.
- L. Ye, G. Chai and Z. Wen, Adv. Funct. Mater., 2017, 27, 1606190 CrossRef.
- K. Qu, Y. Zheng, S. Dai and S. Z. Qiao, Nano Energy, 2016, 19, 373–381 CrossRef CAS.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef.
- M. Kılıç, E. Apaydın-Varol and A. E. Pütün, Appl. Surf. Sci., 2012, 261, 247–254 CrossRef.
- D.-W. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576–591 RSC.
- Z. Zhang, G. M. Veith, G. M. Brown, P. F. Fulvio, P. C. Hillesheim, S. Dai and S. H. Overbury, Chem. Commun., 2014, 50, 1469–1471 RSC.
- M. Qiao, C. Tang, G. He, K. Qiu, R. Binions, I. P. Parkin, Q. Zhang, Z. Guo and M. M. Titirici, J Mater. Chem. A, 2016, 4, 12658–12666 RSC.
- D. H. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- M. Wu, K. Wang, M. Yi, Y. Tong, Y. Wang and S. Song, ACS Catal., 2017, 7, 6082–6088 CrossRef CAS.
- C. Tang, M. M. Titirici and Q. Zhang, J. Energy Chem., 2017, 26, 1077–1093 CrossRef.
- D. C. Higgins, M. A. Hoque, F. Hassan, J.-Y. Choi, B. Kim and Z. Chen, ACS Catal., 2014, 4, 2734–2740 CrossRef CAS.
- R. Li, Z. Wei and X. Gou, ACS Catal., 2015, 5, 4133–4142 CrossRef CAS.
- S. S. Shinde, C. H. Lee, A. Sami, D. H. Kim, S. U. Lee and J. H. Lee, ACS Nano, 2017, 11, 347–357 CrossRef CAS PubMed.
- Y. Jiang, L. Yang, T. Sun, J. Zhao, Z. Lyu, O. Zhuo, X. Wang, Q. Wu, J. Ma and Z. Hu, ACS Catal., 2015, 5, 6707–6712 CrossRef CAS.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed.
- L. Tao, Q. Wang, S. Dou, Z. Ma, J. Huo, S. Wang and L. Dai, Chem. Commun., 2016, 52, 2764–2767 RSC.
- Z. Liu, Z. Zhao, Y. Wang, S. Dou, D. Yan, D. Liu, Z. Xia and S. Wang, Adv. Mater., 2017, 29, 1606207 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta06743k |
| This journal is © The Royal Society of Chemistry 2018 |