
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu2+ selective chelators relieve copper-induced oxidative stress in vivo†
Ananya
Rakshit
 a,
Kaustav
Khatua
a,
Vinit
Shanbhag
b,
Peter
Comba
c and
Ankona
Datta
*a
a,
Kaustav
Khatua
a,
Vinit
Shanbhag
b,
Peter
Comba
c and
Ankona
Datta
*a
aDepartment of Chemical Sciences, Tata Institute of Fundamental Research, 1 Homi Bhabha Road, Colaba, Mumbai-400005, India. E-mail: ankona@tifr.res.in
bDepartment of Biochemistry, Christopher S. Bond Life Science Center, University of Missouri, Columbia, USA
cUniversität Heidelberg, Anorganisch-Chemisches Institut, Interdisciplinary Center for Scientific Computing, INF 270, D-69120 Heidelberg, Germany
First published on 2nd October 2018
Abstract
Copper ions are essential for biological function yet are severely detrimental when present in excess. At the molecular level, copper ions catalyze the production of hydroxyl radicals that can irreversibly alter essential bio-molecules. Hence, selective copper chelators that can remove excess copper ions and alleviate oxidative stress will help assuage copper-overload diseases. However, most currently available chelators are non-specific leading to multiple undesirable side-effects. The challenge is to build chelators that can bind to copper ions with high affinity but leave the levels of essential metal ions unaltered. Here we report the design and development of redox-state selective Cu ion chelators that have 108 times higher conditional stability constants toward Cu2+ compared to both Cu+ and other biologically relevant metal ions. This unique selectivity allows the specific removal of Cu2+ ions that would be available only under pathophysiological metal overload and oxidative stress conditions and provides access to effective removal of the aberrant redox-cycling Cu ion pool without affecting the essential non-redox cycling Cu+ labile pool. We have shown that the chelators provide distinct protection against copper-induced oxidative stress in vitro and in live cells via selective Cu2+ ion chelation. Notably, the chelators afford significant reduction in Cu-induced oxidative damage in Atp7a−/− Menkes disease model cells that have endogenously high levels of Cu ions. Finally, in vivo testing of our chelators in a live zebrafish larval model demonstrate their protective properties against copper-induced oxidative stress.
1. Introduction
Redox-active metal ions are critical for biological function. Copper (Cu), iron (Fe), and manganese (Mn) ions act as co-factors for essential enzymes. Recent reports show that labile or weakly bound pools of redox active metal ions also participate in neuronal signaling and removal of reactive oxygen species.1–4 While absolutely essential for biological activity both in protein-bound and labile forms, excess redox-active metal ions have been associated with severe neuro-degenerative disorders.5–8 A significant route by which excess redox-active metal ions affect cellular function is via the Fenton reaction.9–13 Cu+/Cu2+ and Fe2+/Fe3+ catalyze the production of reactive hydroxyl radicals which cause permanent modification of cellular lipids, nucleic acids, and proteins leading to severe oxidative damage (Fig. 1A and B).9,10,12,14–16 Previous studies have shown that metal ion chelators can alleviate oxidative stress associated symptoms of metal-induced disorders.17–23 However, a major shortcoming of chelation therapy is the non-specific removal of other biologically essential metal ions and metal co-factors from metallo-enzymes leading to multiple side-effects.12,24–28 Therefore, cell-permeable metal selective chelators that can specifically remove excess metal ions responsible for oxidative stress would be extremely valuable motifs for chelation therapy (Fig. 1C).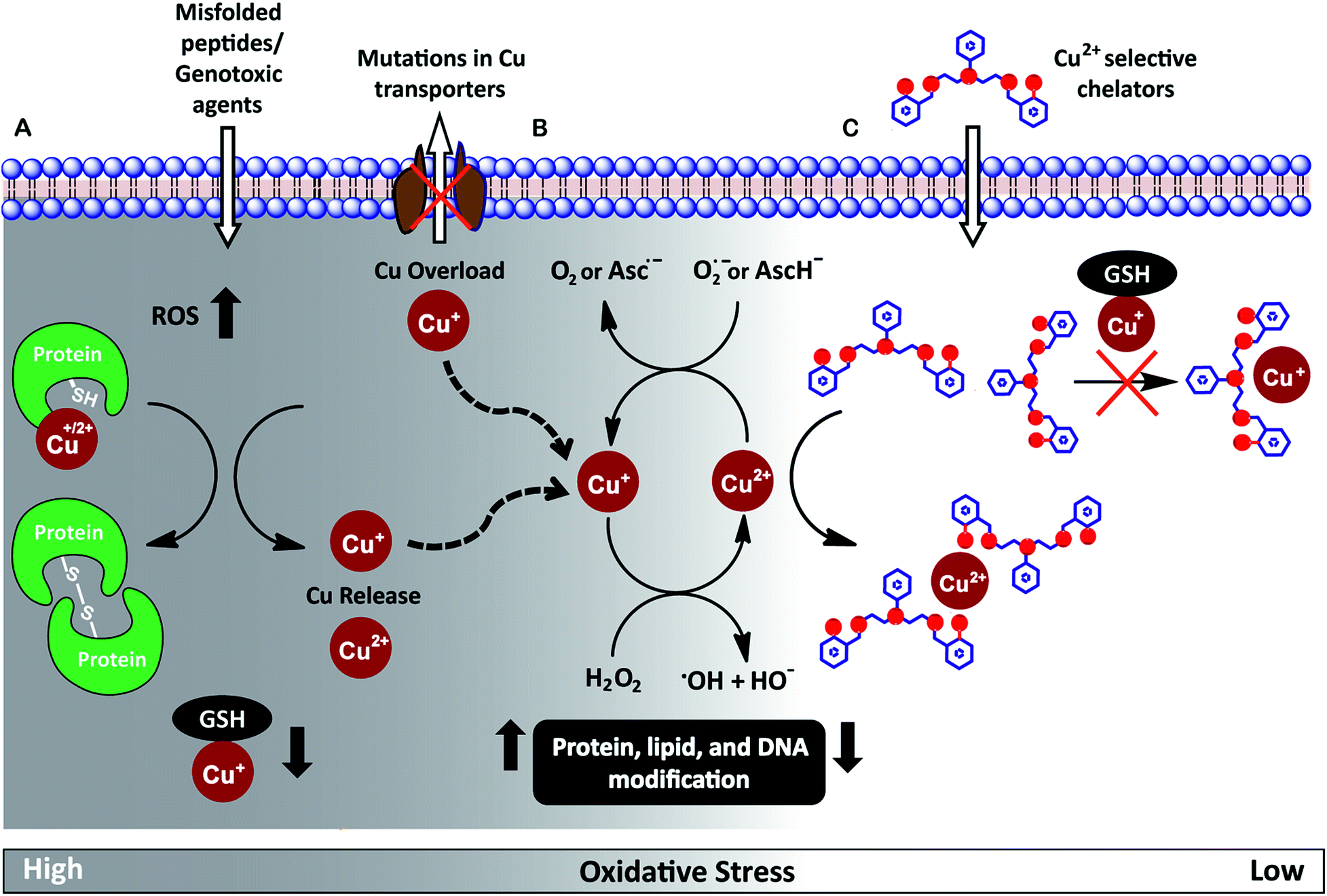 | ||
| Fig. 1 (A) Scheme highlighting mechanisms that can increase intracellular Cu ion levels. Exposure to external agents and mis-folded peptides can cause increased levels of reactive oxygen species (ROS) leading to oxidative stress. Increased oxidative stress leads to protein oxidation releasing Cu ions from proteins and also reduces levels of glutathione (GSH) bound Cu+. Mutations in Cu ion transporters lead to elevated intracellular Cu ion levels. (B) Cu+/Cu2+ catalyzed Fenton reaction produces reactive hydroxyl radicals, HO˙. Cu2+ is reduced by cellular reductants like superoxide (O2˙−) and hydroascorbate (AscH−) to complete the catalytic cycle. (C) Proposed cell-permeable Cu2+ chelators that alleviate oxidative stress via selective Cu2+ chelation. | ||
We have worked on the development of selective cell-permeable chelators for redox-active metal ions with a focus on Cu ion chelation. The motivation for our work derives from the fact that while Cu+/Cu2+ ions are absolutely essential for biological activity, abnormal Cu homeostasis is implicated in severe neurological and metabolic disorders like Alzheimer's disease,7,29–32 amyotrophic lateral sclerosis,33–37 cancer,38,39 Menkes disease,5,40,41 and Wilson's disease.8,42–44
Intra-cellular Cu ions are tightly regulated such that ‘free’ Cu+–aqua species are almost non-existent.45–49 Labile Cu ions are mostly bound to available biological ligands like glutathione and exist in the Cu+ oxidation state under the normal reducing physiological conditions.50,51 These ‘natural’ ligands tune the reduction potential of Cu such that the physiologically available labile pool does not participate in Fenton chemistry and the cell is able to efficiently regulate Cu ions to minimize oxidative damage.12,13,52 However, the Cu balance gets compromised during disease conditions via multiple mechanisms leading to Cu ion overload (Fig. 1A).5,13,35,36,41,43,52 Under Cu dysregulation conditions, excess Cu ions can bind to other biologically available ligands and misfolded proteins forming copper complexes with nM to pM affinities.15,36,53 Importantly, several of these newly formed species have redox potentials that now allow Cu-assisted catalysis of the Fenton reaction.15,29,36,54
Since the aberrant labile Cu ion pool is also bound to available ligands, strong chelators are required for effective chelation. However, such strong chelators would also affect the benign non-redox cycling labile Cu+ ion pool, like Cu+–glutathione,12,55 further exacerbating the pathophysiological condition. A novel approach to overcome this challenge is to develop chelators that target the Cu2+ labile pool. This pool is mostly non-existent inside cells during physiological conditions,55 but would be generated as a result of redox cycling during Fenton reaction catalysed by the aberrant Cu ion pool (Fig. 1B and C). Such a chelator would also be expected to protect against Cu2+ liberated during protein mis-metallation under oxidative stress conditions.13,52 Therefore, we set forth to develop cell-permeable chelators that would be able to specifically bind Cu2+ over Cu+. Such chelators would not only serve as scaffolds for the development of therapeutics but would also act as strategic chemical tools for elucidating Cu ion homeostasis.
Achieving Cu2+ selective chelation against this backdrop would also necessitate the removal of aberrant labile Cu2+ without affecting the labile pools of other biological metal ions. The concentration of the labile pools of metal ions like Ca2+, Mn2+, Zn2+ and Fe2+ range from mM to nM levels.46,56 Since the ionic radii of divalent transition metal ions fall within 0.7–0.8 Å and all of these metal ions can bind to N and O donor atom containing ligands, designing a selective chelator with a Cu2+ binding affinity that is several orders of magnitude higher compared to other metal ions is a significant coordination chemistry challenge.
We have designed a modular synthetic strategy and developed mixed N- and O-donor atom containing Cu2+ selective chelators (Scheme 1). The binding constants of the novel chelators toward Cu2+ are 108 times higher than that for other biologically relevant metal ions including Cu+. Notably, the binding affinities of the chelators for other divalent metal ions are below the concentrations of their respective labile pools46,56 and the chelators do not remove protein bound Cu2+. The chelators are cell-permeable and do not affect cell-viability under applied concentrations. These novel molecules suppress Cu2+ triggered oxidative stress both in live cells and in vivo. Importantly, the chelators were also able to suppress Cu-induced oxidative stress in Menkes disease model Atp7a−/− cells57 which have endogenously high levels of Cu ions. Hence, our ligands stand out not only in affording exquisite selectivity for Cu2+ but also for their applicability to biological systems which we have proven all the way from in cell disease models to a live multicellular vertebrate system.
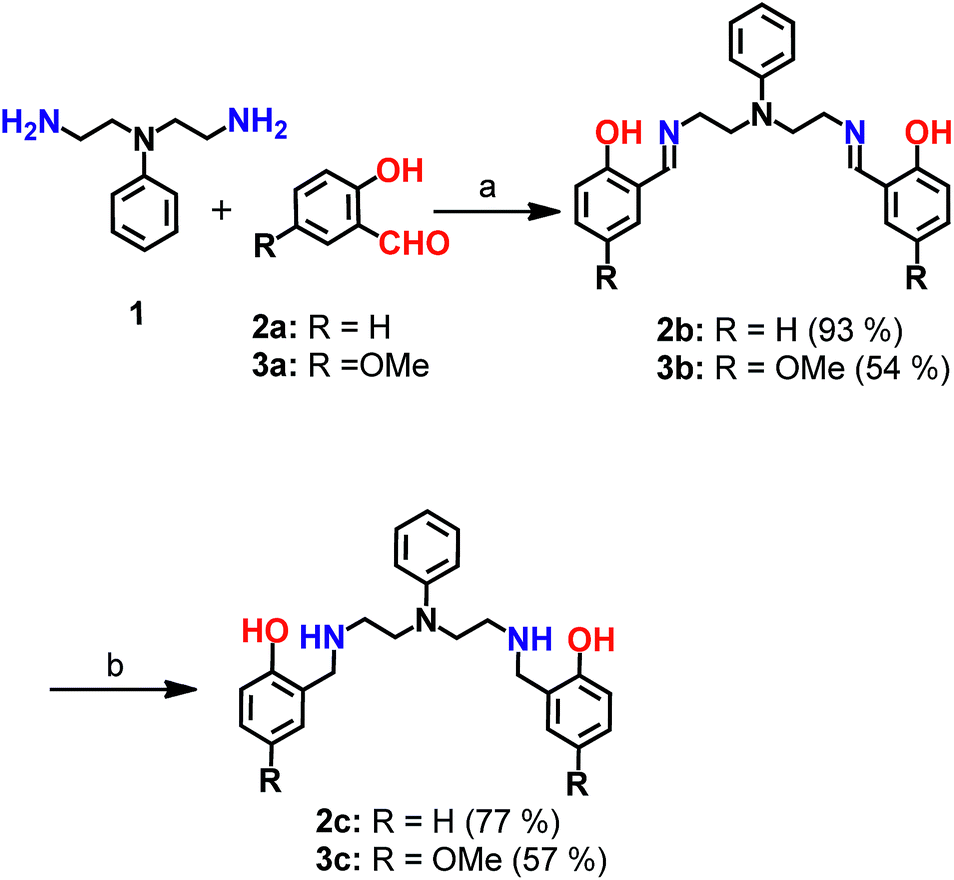 | ||
| Scheme 1 Synthesis of chelators 2c and 3c. aReagents and conditions: (a) sodium acetate buffer, pH 4.5 and methanol, 100 °C; (b) sodium borohydride in dry methanol, 0 °C. | ||
2. Results and discussion
Chelator synthesis and stability in physiological buffer
Mixed N- and O-donor atom containing ligands were synthesized via a modular strategy as shown in Scheme 1. The basic design criterion was to keep the number of synthetic steps minimal. Further, S-donor atoms were avoided to introduce selectivity for Cu2+ over Cu+. The precursors 2b58 and 3b were synthesized in a single step by a Schiff base condensation reaction between the amine 1 and either aldehyde 2a or 3a (Scheme 1). Compounds 2b and 3b were obtained in 93% and 54% yields, respectively. An advantage of this strategy is that the substituent on the aromatic aldehyde could be varied to generate ligands with potentially different metal ion binding affinities. Compounds 2b and 3b were reduced in the presence of sodium borohydride. The reduced novel ligands 2c and 3c were obtained in 77% and 57% yields respectively starting from 2b and 3b (Scheme 1, Fig. S1 and S2†).Since the chelators would be finally applied in living cells and biological systems, stability in aqueous media was an important criterion.59 We, therefore, assessed the stability of the molecules over time, using LC/ESI-MS. Both compounds 2c and 3c were stable in aqueous buffer as well as in cell-culture medium for over 12 h as shown in Fig. S3.†
Ligands 2c and 3c bind Cu2+ ions
The molecules 2c and 3c were taken forward for studying Cu2+ chelation properties. Absorption titrations for both ligands were performed in the presence of Cu2+ ions in aqueous buffer. The absorption spectrum of compound 2c showed peaks at 247 nm, 277 nm, and a shoulder at 301 nm (Fig. S4†). For compound 3c, peaks were observed at 247 nm and 295 nm (Fig. S4†).A distinct new band was observed for both compounds 2c and 3c in the presence of Cu2+ at 420 nm (ε, 736 M−1 cm−1) and 450 nm (ε, 832 M−1 cm−1), respectively (Fig. 2a and b, Table S2†). Upon titrating Cu2+ to both ligands, the new bands at 420 nm and 450 nm showed increase in intensity with a sharp transition and saturation at 2 equivalents of ligand relative to Cu2+ ions (Fig. 2a and b, inset). Other spectral changes were also observed upon Cu2+ addition to both ligands. For example, an increase in the intensity of the 275 nm peak was observed for ligand 2c. Further, the 247 nm peak shifted to 237 nm with concomitant intensity increase (Fig. 2a). Compound 3c afforded an increase in intensity for both the 247 nm and 295 nm peaks (Fig. 2b). A visible color change was observed from colorless to greenish upon Cu2+ addition to both ligands (Fig. S5†). Closer inspection of the absorption spectrum revealed new bands at 623 nm (ε, 173 M−1 cm−1) and 626 nm (ε, 275 M−1 cm−1), respectively for 2c and 3c, which explain the observed color change (Fig. 2c and inset, Table S2†).
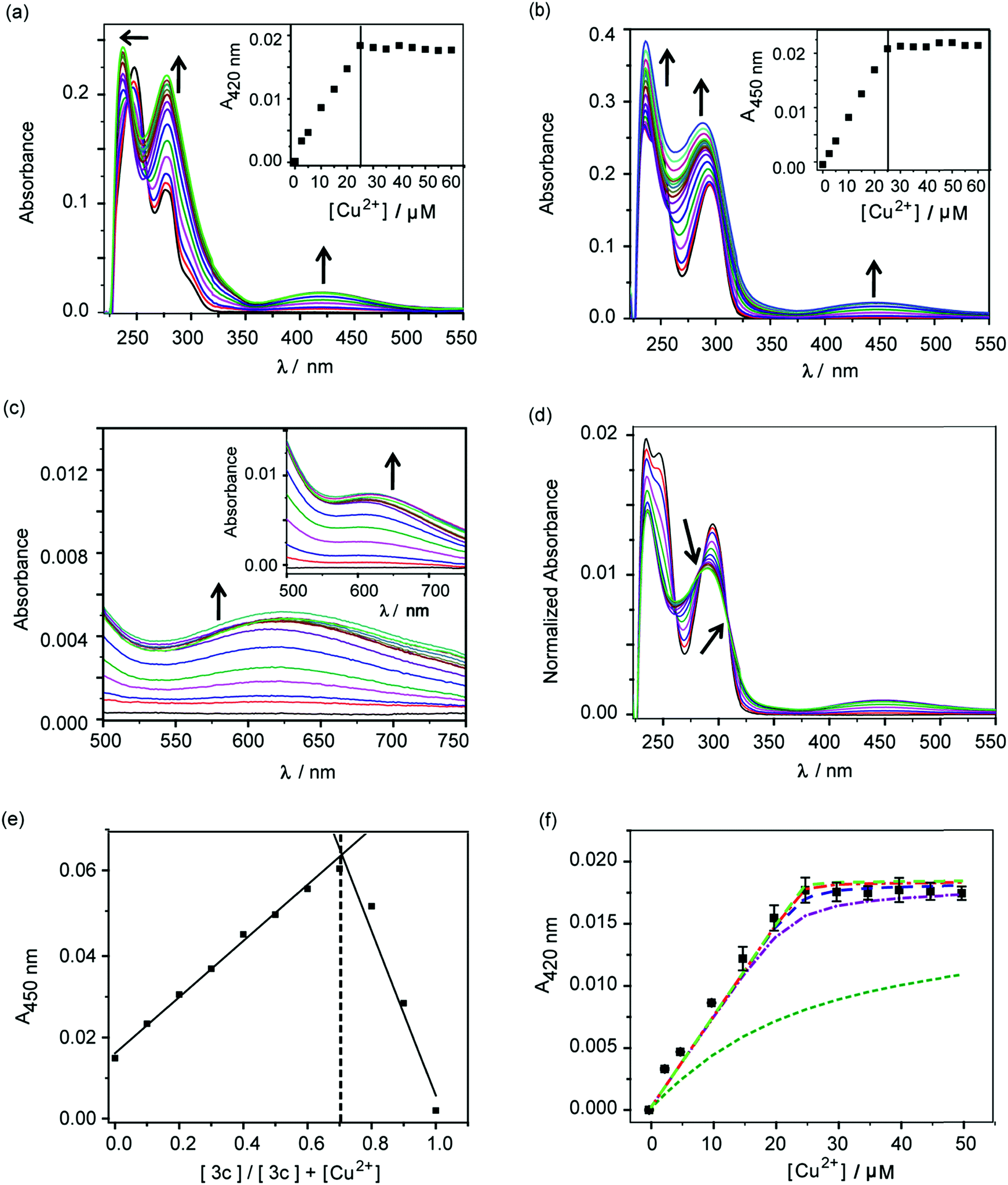 | ||
Fig. 2 Absorbance response of ligands 2c and 3c with Cu2+. (a) Ultraviolet (UV)-visible titration of 2c (50 μM) with CuCl2 (0–60 μM) in buffer. Black arrows indicate spectral changes upon addition of Cu2+. The inset shows the plot of absorbance at 420 nm versus concentration of Cu2+. (b) UV-visible titration of 3c (50 μM) with CuCl2 (0–60 μM) in buffer. Black arrows indicate spectral changes upon addition of Cu2+. The inset shows the plot of absorbance at 450 nm versus concentration of Cu2+. (c) Visible-near infra-red (NIR) absorbance spectra for titration of 2c with Cu2+. The inset shows visible-NIR absorbance spectra for titration of 3c with Cu2+. (d) Area normalized absorbance spectra representing titration of CuCl2 to 3c. Black arrows indicate isosbestic points. (e) Job's plot depicting absorbance versus mole fraction for the Cu2+–3c system. (f) Absorption binding plot for titration of Cu2+ to 2c (50 μM, black squares) with simulated fits (fitted to equation: conditional stability constant β = [ML2]/[M][L]2), using log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β as 9 (green dashed line), 11 (purple dashed line), 12 (blue dashed line), 13 (red dashed line), 14 (light green dashed line). Buffer used 20 mM HEPES (100 mM NaCl, pH 7.0). β as 9 (green dashed line), 11 (purple dashed line), 12 (blue dashed line), 13 (red dashed line), 14 (light green dashed line). Buffer used 20 mM HEPES (100 mM NaCl, pH 7.0). | ||
Area normalized absorption spectra for Cu2+ titration into both ligands showed isosbestic points indicating the presence of distinct Cu2+ complexes (Fig. 2d and S6†). The stoichiometry of the complexes was confirmed to be 1:2 Cu2+:ligand by the method of continuous variation from Job's plots (Fig. 2e and S7†). The formation of 1:2 Cu2+:ligand complexes was further confirmed by ESI/MS analysis (Fig. S8 and S9†). The sharp transitions in absorption titrations (Fig. 2a and b, inset) and Job's plots for 2c and 3c with Cu2+ (Fig. 2e and S7†) indicated the formation of Cu2+ complexes with high binding affinity toward the metal ion. We, therefore, proceeded to determine the stability constants of the ligands with Cu2+. Since, the absorption titrations for both 2c and 3c indicated sharp transitions we determined the lower limits for the Cu2+ conditional stability constants (β) by simulating binding curves using a 1:2 metal:ligand binding model (see ESI†). As shown in Fig. 2f, the data for the titration of Cu2+ to 2c fitted to simulated curves generated by considering logβ values ≥12. Similarly, the logβ value for Cu2+ binding to 3c was determined to be ≥13 (Fig. S10†).
We also performed electron paramagnetic resonance (EPR) spectroscopy of the Cu2+–2c complex in order to obtain insight into its coordination geometry (Fig. S21†). The spin Hamiltonian parameters obtained by simulating the experimental spectrum of the Cu2+–2c complex indicate that the Cu2+ ion has a tetragonal geometry (pseudo-square planar; g‖ = 2.2220; g⊥ = 2.0502). The value of g‖/Azz is 120.6 cm. This value is used to assess the degree of geometric distortion and values between 105–135 cm are typical of Cu2+ complexes with tetragonal (square planar) geometry.60 Further, the EPR data indicate that neither un-complexed Cu2+ ions nor other Cu2+ complexes like di-nuclear Cu2+ species are present.
Computed structures of the Cu2+ complexes
The UV-visible-NIR titration experiments indicate that 2c and 3c coordinate to Cu2+ in 1:2 Cu2+:ligand stoichiometry. The isosbestic points suggest that the 1:2 complexes have a higher stability than the corresponding 1:1 complexes (Fig. 2d and S6†). The UV-visible-NIR spectra of the two complexes indicate that phenolate donors are coordinated to Cu2+, when compared to other ligand systems exhibiting Cu2+–phenolate coordination.61,62 The electronic transitions at 420 nm (ε, 736 M−1 cm−1) and 450 nm (ε, 832 M−1 cm−1), respectively, can be assigned to phenolate-based charge-transfer (CT) transitions, while those with significantly lower intensity at 623 nm (ε, 173 M−1 cm−1), and 626 nm (ε, 275 M−1 cm−1) respectively, are due to d–d transitions. The EPR data indicate the formation of a tetragonal (pseudo square-planar) complex. Taken together the UV-visible-NIR titration and the EPR data indicate the formation of a 1:2 Cu2+:ligand complex with a pseudo-square-planar geometry, where two N-and phenolate O-donor atoms each are coordinated to the central metal ion.
In order to obtain further information about the coordination geometries and explore reasons for the formation of 1:2 Cu2+:ligand complexes, we used computational methods to predict the corresponding structures. The two ligands 2c and 3c are potentially penta-dentate and an important question to answer is: why do the ligands only use either two or three donor atoms each to coordinate to Cu2+? Since ligand-imposed steric strain seems to be a major reason, force field calculations are an ideal approach to get possible answers to this question.
In order to obtain strain energies for comparison between different structures, we have optimized possible 1:1 complexes with 2c as an O,N bidentate (12), an O,N,N (23,33) or N,N,N tridentate (43), an O,N,N,O (54) or an O,N,N,N tetradentate (64) and a pentadentate ligand (75; not all possible isomers were considered, for details see Fig. S34†). We have used the well-established MOMEC program and force field for the calculations.63–67 Additional parameters involving Cu2+–Ophenolate bonding were obtained from known structures61,62 (see ESI, Table S5†). All Cu2+ centers were computed as Jahn–Teller elongated hexa-coordinate geometries with water molecules completing the coordination spheres, and the conformational space was evaluated deterministically. Details of the computational analysis have been included in the ESI.†
The structure with 2c coordinated as an O,N bidentate ligand (12) afforded the lowest strain energy. The only other structures that might compete in terms of steric strain were those with 2c as a tridentate or tetradentate ligand involving amino-salicylate donor groups (23 and 54), and these structures were 15 and 11 kJ mol−1 higher in energy, respectively than the 12 structure (Fig. S34 and Table S6†). However, these structures involve relatively flexible 8-membered chelate rings and therefore are entropically disfavored. Due to severe van der Waals repulsion by the phenyl substituent of the tertiary amine donor, all other isomers are destabilized by an additional 15–20 kJ mol−1. The optimized structures of the lowest energy 1:1 and 1:2 Cu2+:2c complexes are presented in Fig. 3 and ESI Video.† Hence, we infer that these ligands coordinate to Cu2+ in a bidentate mode with high 1:2 metal:ligand Cu2+ binding constants. Although we cannot completely rule out the possibility of the formation of minor ternary complexes with available ligands from the media, we note that the structure of the 1:2 Cu2+:2c complex is in agreement with the UV-visible-NIR spectra, the spectrophotometric titration, the ESI-MS spectra, and the EPR spectrum.
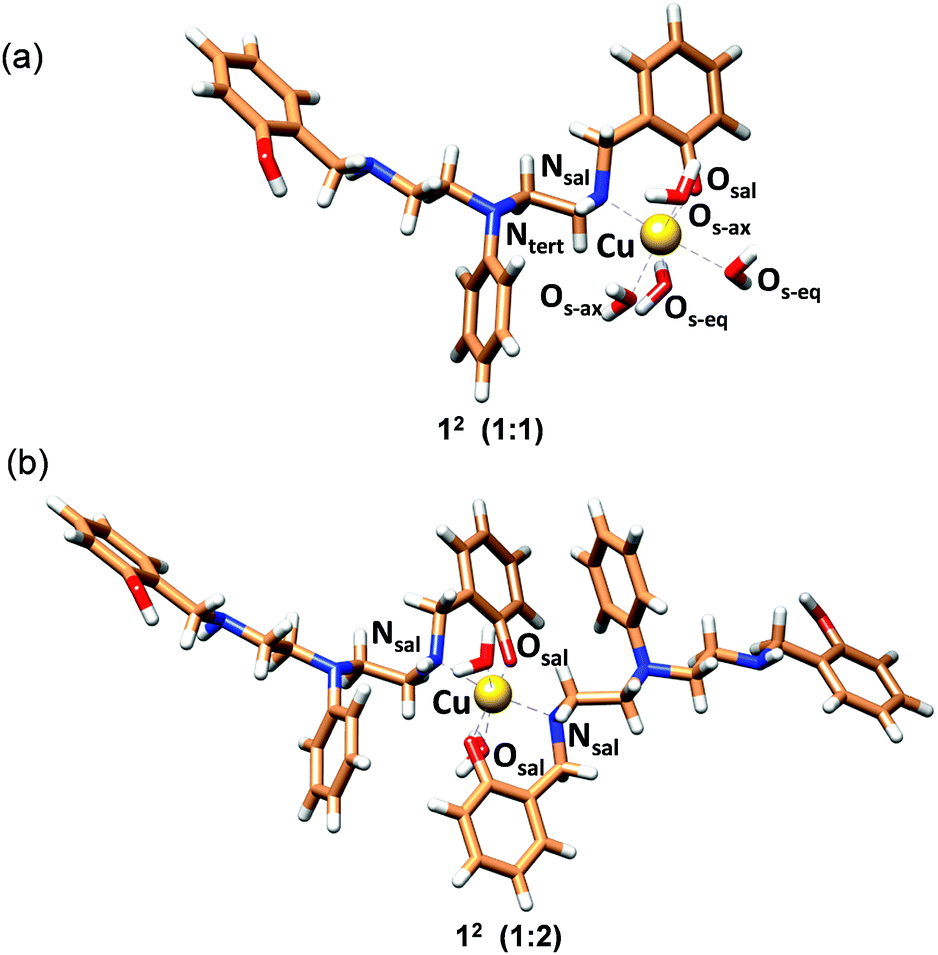 | ||
| Fig. 3 MOMEC optimized structures for (a) [Cu2+(2c)(OH2)4] (12; 1:1) and (b) [Cu2+(2c)2(OH2)2] (12; 1:2) complexes. Ligand 2c is in the phenolate form. In the structures, carbon, nitrogen, oxygen and hydrogen atoms are represented in light brown, blue, red and white colors, respectively. Copper ions are represented in gold. | ||
Competition experiment of 2c and 3c with a high affinity Cu2+ chelator and protein-bound Cu2+
We next performed a control experiment in which ethylene-diamine tetra-acetic acid (EDTA) was titrated into a solution containing 1:2 Cu2+:ligand 3c. The absorption band at 450 nm decreased with increasing concentrations of EDTA and the band completely attenuated at ∼0.75 equivalent of EDTA (Fig. S11†). The Cu2+–EDTA (1:1) complex has a logβ of 15.8.68 This control experiment indicated that the ligands would not be able to chelate out Cu2+ from essential enzymes that have Cu2+ binding affinities in the aM–fM regime.50 To further confirm that the chelators will not affect enzyme bound copper, we titrated the chelators to Cu2+–azurin complex which has a Cu2+ dissociation constant of 25 fM.69 Cu2+–azurin complex has a characteristic absorption peak at 628 nm due to cysteine–Cu2+ charge-transfer.70 The absorption of the peak remained unaffected even upon addition of upto 12 equivalents of the chelators 2c and 3c (Fig. S12†). Therefore, the chelators 2c and 3c will be able to remove excess labile copper without affecting enzyme bound copper.
Selectivity of the chelators toward Cu2+
Encouraged by the Cu2+ binding data we evaluated the conditional stability constants of 2c and 3c toward other biologically relevant metal ions that can bind to ligands with mixed N- and O-donor atoms. Absorption titrations for both ligands were performed in the presence of Cu+, Ca2+, Fe3+, Fe2+, Ni2+, Zn2+, Mn2+ and Co2+ metal ions (Fig. S13–S20†). The titration curves indicated weak binding and the data could be fitted to a 1:1 binding model. The conditional stability constants ranged between logβ values of 2.53 to 4.48 (Table 1) indicating that the stability constants of the ligands toward Cu2+ were at least 108 times higher than the stability constants toward Cu+, Ca2+, Zn2+, and other transition metal ions. The binding data indicated that 2c and 3c were indeed highly selective Cu2+ chelators (Table 1).
β) for the chelators with different metal ions
| Compound | Cu2+ | Cu+ | Mn2+ | Co2+ | Zn2+ | Ca2+ | Fe2+ | Fe3+ | Ni2+ |
|---|---|---|---|---|---|---|---|---|---|
| a No binding was detected. Conditional stability constant, β1n = [MLn]/[M][L]n; n = 2 for Cu2+ and n = 1 for other metal ions. All the absorbance measurements were carried out in 20 mM HEPES buffer (100 mM NaCl, pH 7.0). | |||||||||
| 2c | ≥12 | 4.19 | n.ba | 3.97 | 3.93 | n.ba | 3.63 | 3.39 | 2.53 |
| 3c | ≥13 | 4.48 | 4.20 | n.ba | 3.90 | n.ba | 4.05 | 3.89 | 3.10 |
None of the existing commercial Cu2+ chelators that are used for either therapy of Cu-overload diseases or for mechanistic studies in elucidating Cu regulation afford such distinct selectivity toward Cu2+ (Table S3†).27,71 In fact, when we compared the reported binding constants of these commercial ligands for Cu2+versus other metal ions we found that most of them bind more strongly to Fe3+ and also have very high affinities toward Zn2+ and Ni2+.27,71 We also compared our chelators to the few other previously reported reduced salen chelators23,68 (Table S4†). Unlike our chelators the metal binding constants of these reduced salen chelators for most biologically relevant metal ions had not been reported. We found that our ligands stood out in their selectivity toward Cu2+ relative to the majority of this group as well.
Since the spectroscopic characterization and computed structures of the Cu2+–2c complex indicate that only half of the ligand comprising a phenolate O atom and the secondary N atom coordinate to the metal ion we investigated if a minimal control binding unit, 2-((benzylamino)methyl)phenol, would be sufficient to explain the observed selectivity. Absorption titration data of Cu2+ to the control molecule (performed under same experimental conditions as 2c and 3c, Fig. S22†) indicated that the ligand can bind to Cu2+ ions in 1:2 Cu2+:ligand stoichiometry with a logβ value of 7.0. However, interestingly, the ligand did not afford a change in its absorption spectrum in the presence of other essential metal ions under identical experimental conditions, suggesting that the stability constants of the ligand toward other metal ions were very low (Fig. S23†). This data indicates that the control molecule, although a weaker Cu2+ binder (logβ 7.0) compared to 2c and 3c (logβ ≥ 12), retains its selectivity for Cu2+ binding. This result suggests that the other half of the molecules 2c and 3c would be important in pre-orienting the binding unit for Cu2+ coordination thereby increasing the binding affinity. Further, the computational studies show that the steric strain provided by the central N-phenyl moiety drives the binding toward a bidentate mode and disfavors the use of other donor atoms in the ligand. This is a key feature that leads to Cu2+ selectivity since the resultant pseudo square-planar (tetragonal) geometry adopted by the Cu2+ complex is a highly preferred ligand orientation for d9 Cu2+ systems when compared to other metal ions. We next proceeded to test the ability of our chelators to alleviate Cu-induced oxidative stress.
Chelators reduce Cu ion catalyzed hydroxyl radical production in vitro
First, an in vitro deoxyribose-based assay72 was performed to test whether the chelators could suppress Cu ion catalyzed formation of hydroxyl radicals. In this assay, deoxyribose reacts with in situ generated hydroxyl radicals to produce malondialdehyde. Malondialdehyde reacts with thiobarbituric acid to produce a pink chromophore with absorbance maxima at 532 nm (Fig. S24†). If a ligand is able to chelate Cu2+ from the reaction mixture then the production of the chromophore would be reduced. Importantly, Cu2+ chelation would reduce the production of hydroxyl radicals provided the metal complex does not promote further Fenton reaction. The absorbance of the chromophore at 532 nm is therefore used as a readout for Cu ion catalyzed production of hydroxyl radicals.Both ligands 2c and 3c suppressed the production of hydroxyl radicals as seen from the reduction of the absorbance at 532 nm (Fig. 4). Desferrioxamine mesylate (DFO) was used as a positive control since DFO is a known chelator for redox active metal ions like iron and copper. A related ligand desferrioxamine B which does not have the mesylate groups has a stability constant logβ value of 14.1 for a 1:1 Cu2+:ligand complex.71 The plots for absorbance at 532 nm versus chelator concentration for ligands 2c and 3c followed a similar trend as that of DFO with greater than 90% reduction in absorbance at concentrations above 50 μM (Fig. 4). On the other hand, salicyaldehyde, a weaker chelator (logβ for Cu2+ 7.4)71 only partially reduced the formation of hydroxyl radicals (Fig. 4). The deoxyribose assay clearly indicated that the ligands 2c and 3c would be able to chelate Cu2+ and suppress Cu ion induced hydroxyl radical formation. Importantly, unlike chelators such as DFO which have extremely high binding affinities for most metal ions,71,73 our chelators are highly selective for Cu2+. The in vitro assay also indicated that the Cu2+ complexes of 2c and 3c would not promote further Fenton chemistry. This is an important criterion for Cu chelation targeted toward assuaging Cu-induced oxidative stress since there are multiple examples of chelators that can bind to Cu2+ and promote the production of hydroxyl radicals.52,74–77 Hence, our chelators can be potentially useful for treating Cu ion overload associated disorders.
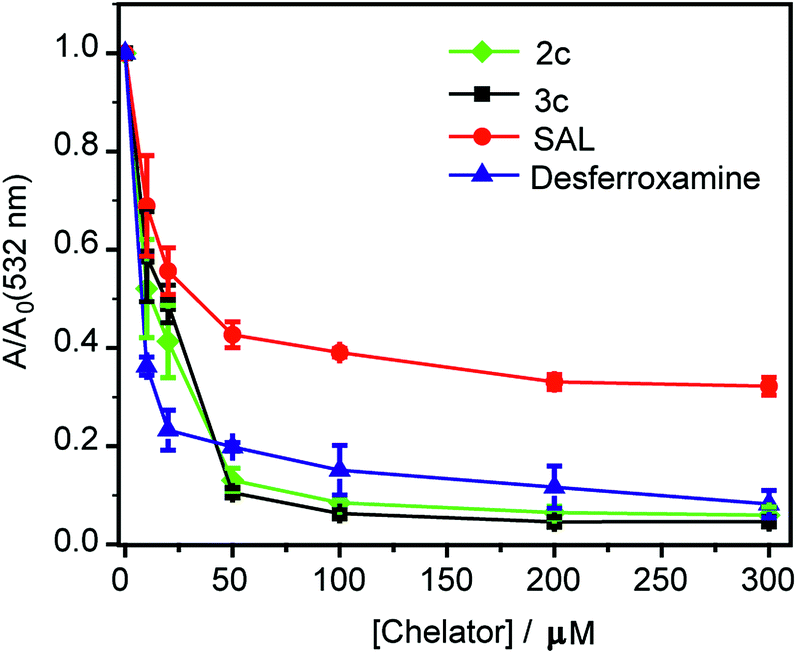 | ||
| Fig. 4 Effect of chelators 2c and 3c on deoxyribose degradation via Cu ion mediated hydroxyl radical formation. Salicylaldehyde (SAL) and desferroxamine mesylate (DFO) were used as controls. Ratio of values of absorbance in the presence of chelator (A) versus absorbance in the absence of chelator (Ao) <1 signify protection against hydroxyl radical mediated deoxyribose degradation. Conditions: CuSO4 (10 μM), 2-deoxyribose (15 mM), H2O2 (100 μM), ascorbic acid (2 mM), at pH 7.4 in phosphate buffer. | ||
Chelators remove Cu ions from Cu2+ exposed living cells
As a first step toward evaluating the chelators in living systems, we wanted to test whether the chelators could remove Cu ions from Cu2+ overloaded cells. Phen Green FL diacetate is a cell permeable fluorescent dye that is quenched in the presence of Cu ions, and has been used effectively to image intracellular Cu ions in Cu2+ overloaded biological systems.78–80 Human embryonic kidney cells (HEK293T) were used for the experiments. Cells were incubated with Phen Green FL and imaged in a confocal microscope. In comparison to control untreated cells the Cu2+-treated cells showed a significant decrease in fluorescence intensity (Fig. 5a and c, ***p ≤ 0.001). When the Cu2+-treated cells were incubated with the chelators 2c and 3c for 30 min the Phen Green fluorescence intensities afforded significant recovery and were similar to the Cu2+ untreated controls (Fig. 5b and c). The recovery of Phen Green fluorescence upon chelator treatment in Cu2+ overloaded cells along with the distinct Cu2+ selectivity of our chelators indicate that these chelators will be able to efficiently remove Cu2+ ions from cells under oxidative stress conditions.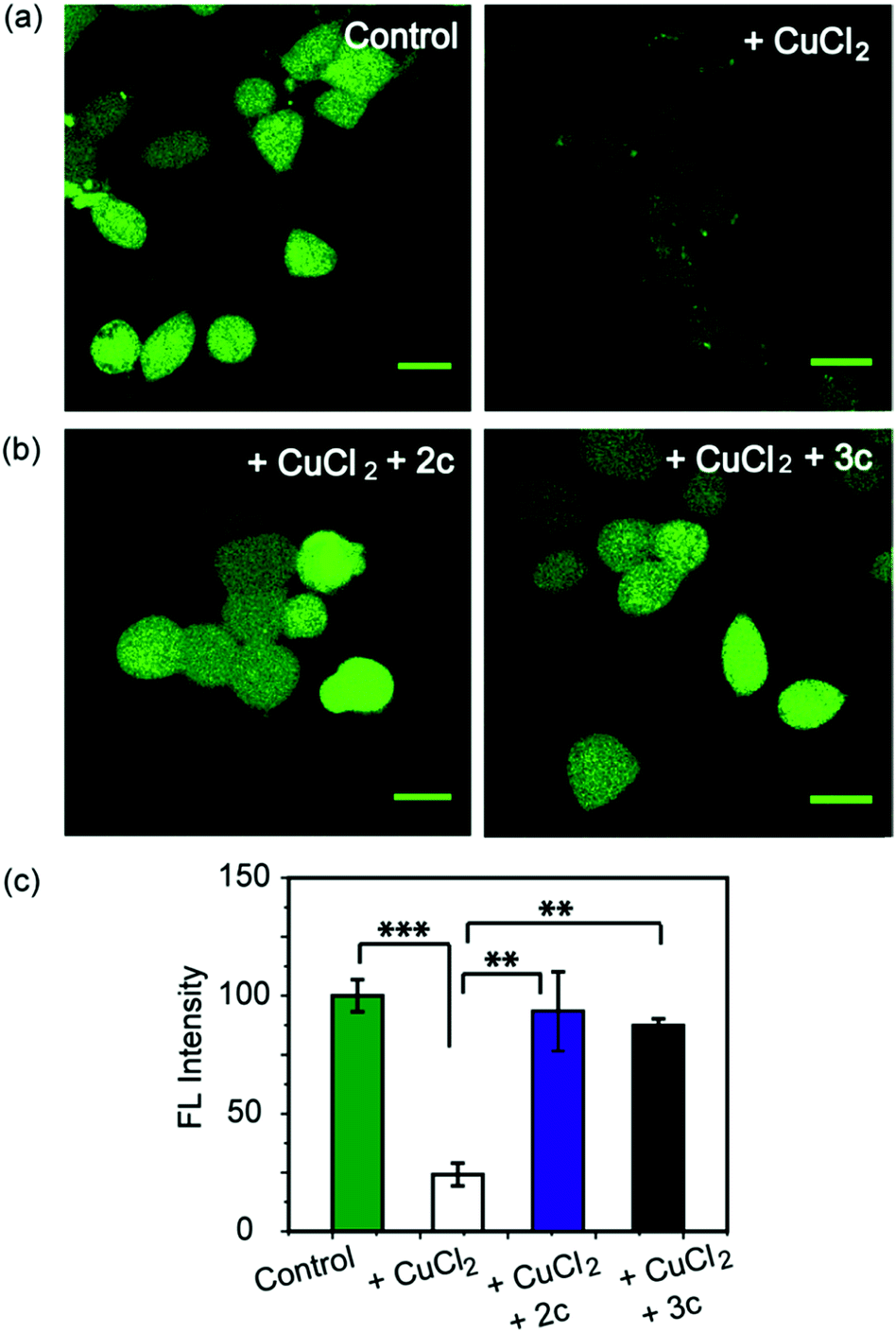 | ||
| Fig. 5 Confocal fluorescence images of live HEK293T cells to show the effect of Cu2+ chelators using a Cu ion responsive dye Phen Green FL. All cells were incubated with Phen Green FL dye (5 μM) initially for 30 min before any further treatments. (a) Left to right: control cells; cells treated with CuCl2 (20 μM) for 30 min. (b) Left to right: cells treated with CuCl2 (20 μM) for 30 min followed by 2c (80 μM) for 30 min; CuCl2 (20 μM) for 30 min followed by 3c (80 μM) for 30 min. Cells were washed three times with PBS buffer after each addition and then imaged (λex/em: 488/498−600 nm). Fluorescence intensity analyses of confocal images were carried out by using ImageJ software. (c) Bar plots represent the intensity analyses results. Intensity data were normalized to intensity of control untreated cells. Error bars denote SEM, n = 3. Statistical analyses were performed using an unpaired, two-tailed Student's t-test (**p ≤ 0.01, ***p ≤ 0.001). Images were acquired using a 40× objective; scale bar: 20 μm. | ||
Chelators suppress Cu induced oxidative stress in living cells
Our in vitro data had shown that the chelators could attenuate Cu induced production of hydroxyl radicals which is a major pathway by which Cu ions cause oxidative damage. We have also validated that our chelators would be able to remove Cu2+ ions from live cells. The next key step was to test the ability of the chelators to alleviate Cu2+ induced oxidative stress in living cells. HEK293T cells were incubated with Cu2+ and the level of reactive oxygen species (ROS) in the cells was monitored using CellROX® Deep Red81 which emits in the red (λex: 633 nm, λem: 641–700 nm) upon oxidation with ROS (Fig. 6 and S25†). Fig. 6 depicts confocal images of live HEK293T cells. The basal level of ROS in cells was determined by using control cells that had not been treated with Cu2+. Addition of the chelators to cells did not cause any increase in the emission intensity of CellROX indicating that the chelators alone did not induce any oxidative stress (Fig. 6). Cu2+ treated cells induced a significant increase in ROS over control untreated cells (****p ≤ 0.0001, Fig. 6). Both CuCl2 (Fig. 6) and CuSO4 (Fig. S25†) showed similar CellROX responses indicating that the oxidative stress was due to Cu2+, rather than the anion. Finally, when the Cu2+ treated cells were incubated with the chelators a significant decrease in the CellROX emission intensity was observed indicating that both ligands 2c and 3c were able to rescue the cells from Cu2+ induced oxidative stress conditions (Fig. 6 and S25†). Moreover, pre-treating the cells with the chelators prior to Cu2+ incubation also protected the cells from Cu-induced oxidative stress (Fig. S26†).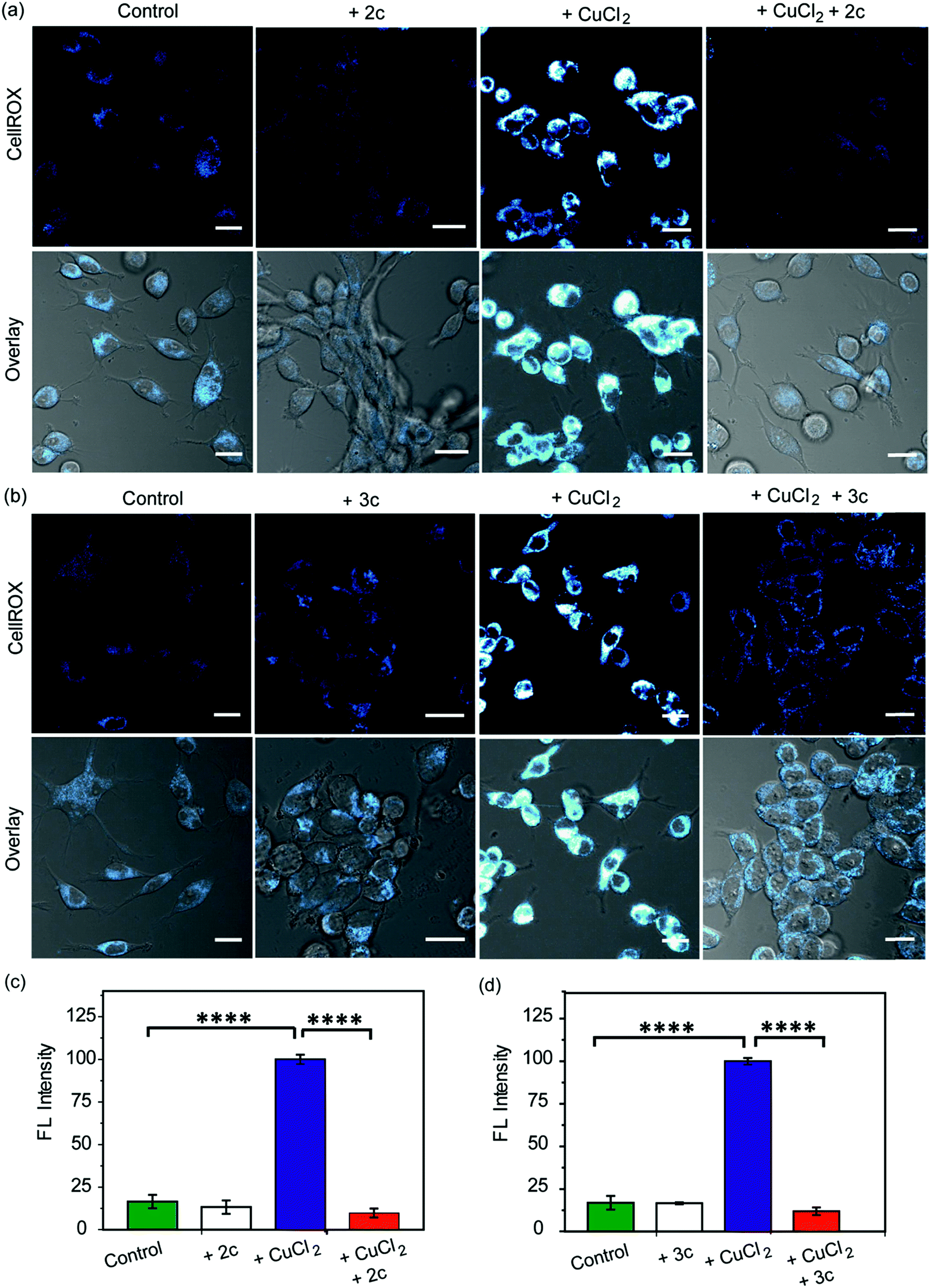 | ||
| Fig. 6 Confocal fluorescence images of live HEK293T cells in presence of chelators and a ROS sensitive dye. (a) Left to right: control cells; cells treated with chelator 2c (40 μM) for 30 min; with CuCl2 (20 μM) for 30 min; with CuCl2 (20 μM) for 30 min followed by 2c (40 μM) for 30 min. (b) Left to right: control cells; cells treated with 3c (40 μM) for 30 min; CuCl2 (20 μM) for 30 min; CuCl2 (20 μM) for 30 min followed by 3c (40 μM) for 30 min. Cells were stained with CellROX (5 μM) for 30 min at the final step before imaging. (λex/em: 633/641–700 nm). Cells were washed three times with PBS buffer after each addition and then imaged. Lower panels of (a) and (b) shows bright field images of cells overlaid with confocal images. Fluorescence intensity analyses of confocal images were carried out by using ImageJ software. Bar plots represent the intensity analyses results for (c) chelator 2c and (d) chelator 3c. Intensity data were normalized to intensity of CuCl2 treated cells. Error bars denote SEM, n = 3. Statistical analyses were performed using an unpaired, two-tailed Student's t-test (****p ≤ 0.0001). Images were acquired using a 40× objective; scale bar: 20 μm. | ||
Fe2+ ions also catalyse Fenton reaction. However, our chelators being Cu2+ specific should not alleviate Fe-induced oxidative stress. A control experiment in the presence of Phen Green which also responds to Fe2+ indicated that the chelators could not remove Fe ions from cells treated with an Fe2+ salt (Fig. S27†). Further, CellROX fluorescence also increased in cells incubated with Fe2+ indicating increased oxidative stress. However, the chelators could not reduce CellROX fluorescence (Fig. S28†) confirming the selectivity of our chelators toward Cu2+ in live cells.
As a positive control, D-penicillamine (D-Pen)27,82 a cell-permeable non-selective Cu2+ chelator (Table S3†) also suppressed Cu2+ induced oxidative stress to a similar extent as chelators 2c and 3c (Fig. 6a–d and S29a, b†). However, our chelators are highly selective toward Cu2+ and hence have a significant advantage over other non-specific Cu2+ chelators. In order to further confirm that the chelators 2c and 3c indeed reduce Cu2+-induced oxidative stress via Cu2+ chelation and not via the removal of ROS through reactions with the phenolic moieties in the chelators,83,84 we performed an additional control experiment by monitoring the effect of the chelators on lipopolysaccharide induced oxidative stress. Lipopolysaccharides generate ROS via the activation of toll-like receptors which in turn activate NADPH oxidase leading to oxidative stress.85,86 Both chelators 2c and 3c had no effect on LPS induced oxidative stress in live cells demonstrating that the chelators function via Cu2+ chelation in alleviating Cu-induced oxidative stress (Fig. S29c and d†).
Further, we simultaneously imaged Cu ions and ROS in live cells to firstly show that regions of high ROS in Cu-overloaded cells colocalize with Cu ions; and secondly to show that the chelators can access these regions, chelate out Cu2+, and efficiently reduce the ROS burden. Phen Green FL and CellROX Deep Red were used to image Cu ions and ROS respectively. Cu2+ untreated cells showed high Phen Green intensity and low CellROX intensity indicating low Cu2+ and low ROS levels, respectively (Fig. 7A, B and C, a panels). Cells treated with Cu2+ showed low Phen Green intensity due to quenching of the Phen Green fluorescence by Cu ions throughout the cells (Fig. 7A, B and C, b panels). CellROX intensity was high in the cytoplasm indicating oxidative stress conditions (Fig. 7A, B and C, b panels). Upon treatment with the chelators, the Phen Green intensity recovered with a simultaneous decrease in the CellROX intensity from the cytoplasm indicating that the chelators can distinctly reduce Cu2+ induced oxidative stress in live cells via Cu2+ chelation (Fig. 7A, B and C, c and d panels).
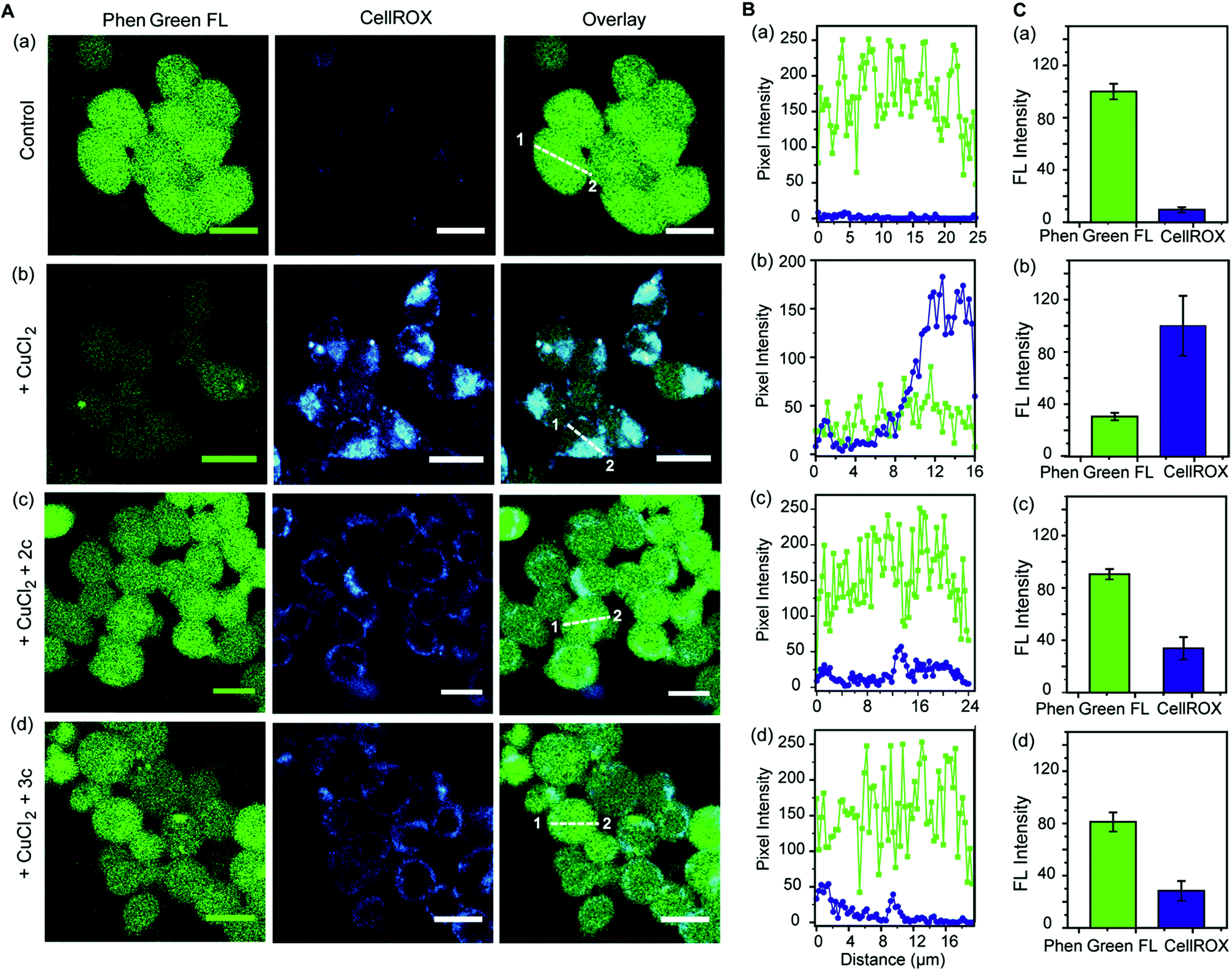 | ||
| Fig. 7 Simultaneous imaging of copper levels and oxidative stress in live HEK293T cells in the presence and absence of chelators 2c and 3c. Phen Green FL was used to image Cu ions and CellROX was used to image ROS. (A) Top to bottom (a–d): confocal images of control untreated cells; cells treated with CuCl2 (20 μM) for 30 min; cells treated with CuCl2 (20 μM) for 30 min followed by 2c (80 μM) for 30 min; cells treated with CuCl2 (20 μM) for 30 min followed by 3c (80 μM) for 30 min. All cells were incubated with Phen Green FL (5 μM) for 30 min prior to any other treatment (λex/em: 488/498–600 nm) and incubated with CellROX (5 μM) for 30 min at the final step before imaging (λex/em: 633/641–700 nm). Cells were washed three times with PBS buffer after each addition and then imaged. Number of washings was identical for all experiments. Left to right: HEK293T cells showing Phen Green FL emission, CellROX emission, and overlaid confocal images. (B) The white lines in the overlaid images were used to calculate the fluorescence intensity of Phen Green FL (green) and CellROX (blue) in the line scan along the direction of 1 to 2. (C) Bar plots represent the intensity analyses results for panel (A, a–d). Intensity data were normalized to intensity of control untreated or CuCl2 treated cells. Error bars denote SEM, n = 3. Images were acquired using a 40× objective; scale bar: 20 μm. | ||
Finally, we checked whether the chelators would affect cell-viability. More than 93% cells were viable up to 4 h of incubation with 100 μM of the chelators (Fig. S30†). Hence, the chelators would have minimal toxicity when applied up to micro molar levels (Fig. S30†). All of the confocal imaging experiments, intensity analyses of the images, and cell-viability data taken together highlight the efficacy of the novel chelators toward alleviating Cu ion induced oxidative stress conditions and indicate the potential of the chelators toward therapeutic applications. We, therefore, took the chelators a next step forward and tested their applicability in a cellular disease model representing endogenous Cu ion overload.
Effect of chelators on oxidative stress in an Atp7a−/− cell model of Menkes disease
In order to ascertain the efficacy of our Cu2+ selective chelators in a disease model of copper overload, we tested the ligands 2c and 3c in mouse embryonic fibroblast cells lacking the Atp7a gene, which is mutated in Menkes disease (Fig. S31† and 8).57 ATP7A is a P-type ATPase required for copper export. Loss of ATP7A function causes copper hyper-accumulation,41,87 and previous ICP-MS studies have shown that the Atp7a−/− mouse embryonic fibroblast cells accumulate 6-fold more copper in the cytoplasm compared to isogenic wildtype cells.57 Higher Cu levels would induce oxidative stress provided the excess Cu ions are bound to cellular ligands that promote redox cycling leading to Cu ion catalyzed ROS production. Therefore, we decided to check the level of ROS in the Atp7a−/− mouse embryonic fibroblast cells. The Atp7a−/− cells showed significantly higher oxidative stress compared to the wildtype cells (Fig. 8a–d left panels, and Fig. 8e and f). This data implied that the increased endogenous level of Cu ions in the Atp7a−/− cells were capable of promoting oxidative stress. Importantly, both 2c and 3c were able to reduce the levels of oxidative stress in the Atp7a−/− cells (Fig. 8a–d right panels, Fig. 8e and f). This data demonstrates the efficacy of 2c and 3c against endogenous Cu ion overload induced oxidative stress.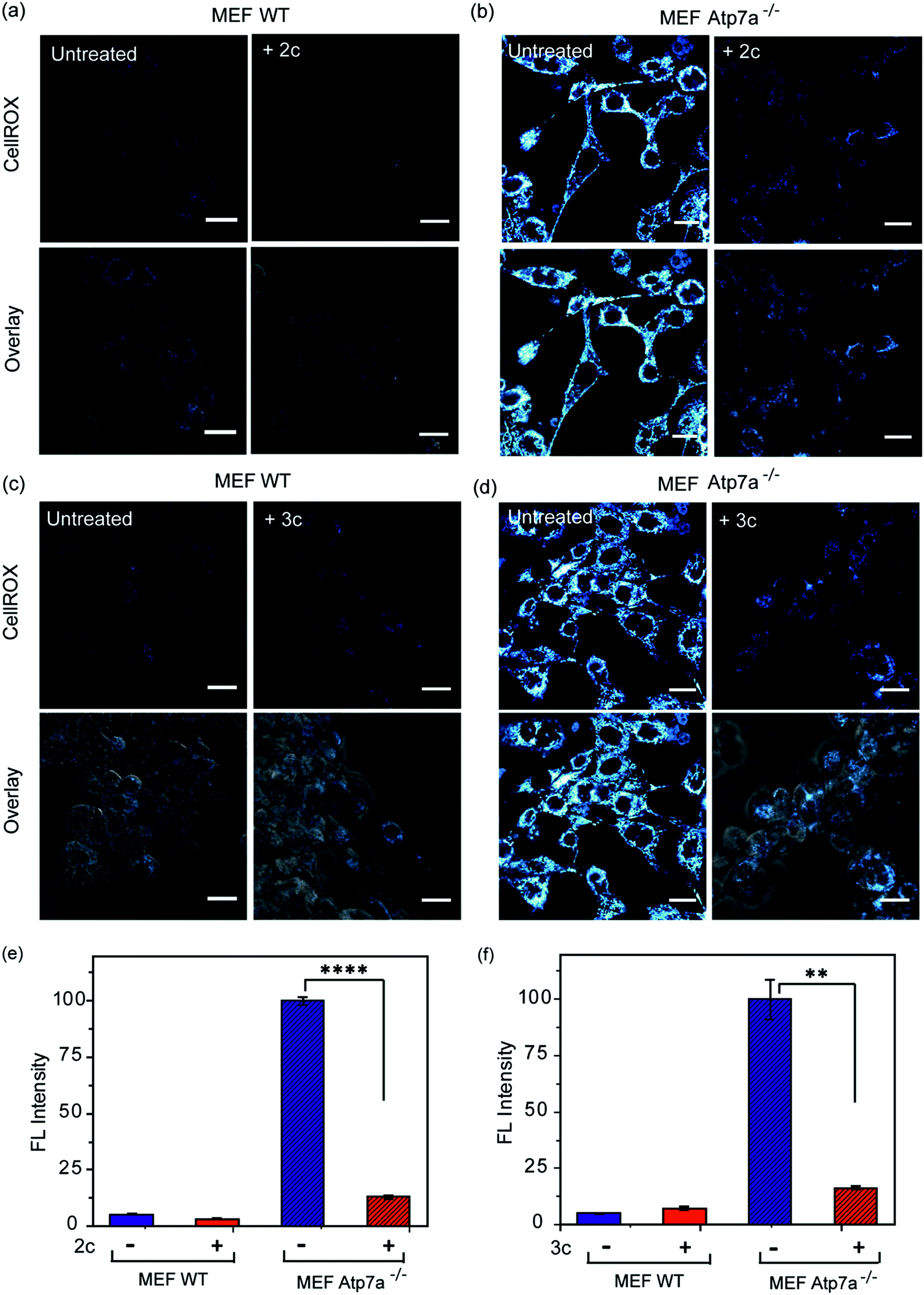 | ||
| Fig. 8 Confocal fluorescence imaging in mouse embryonic fibroblast wildtype (MEF WT) and mouse embryonic fibroblast Atp7a knockout (MEF Atp7a−/−) cells in presence of chelators and a ROS sensitive dye. (a) MEF WT cells: left to right, untreated cells; cells treated with chelator 2c (40 μM) for 60 min. (b) MEF Atp7a−/− cells: left to right, untreated cells; cells treated with chelator 2c (40 μM) for 60 min. (c) MEF WT cells: left to right, untreated cells; cells treated with chelator 3c (40 μM) for 60 min. (d) MEF Atp7a−/− cells: left to right, untreated cells; cells treated with chelator 3c (40 μM) for 60 min. Cells were stained with CellROX (5 μM) for 30 min at the final step before imaging (λex/em: 633/641–700 nm). Cells were washed three times with PBS buffer after each addition and then imaged. Lower panels of (a)–(d) shows bright field images of cells overlaid with confocal images. Fluorescence intensity analyses of confocal images were carried by using ImageJ software. Bar plots represent the intensity analyses of fluorescence images for chelators (e) 2c and (f) 3c. Fluorescence intensities were normalized to intensity of MEF Atp7a−/− cells. Error bars denote SEM, n = 3. Statistical analyses were performed using an unpaired, two-tailed Student's t-test (**p ≤ 0.01, ****p ≤ 0.0001). Images were acquired using a 40× objective; scale bar: 20 μm. | ||
The fact that our chelators reduce oxidative stress via Cu2+ ion chelation has been established both directly (Fig. 7A–C) and through control experiments in the presence of LPS (Fig. S29c and d†). These results when combined with the data on the Atp7a−/− cells, indicate that Cu ion overload is indeed associated with increased oxidative stress catalyzed by Cu species that can perform redox cycling to generate ROS. Our chelators catch the Cu ions in the +2 oxidation state and generate a chelated species that blocks the redox cycling to alleviate Cu-induced oxidative stress. Hence, we decided to check the efficacy of the chelators in a multicellular, vertebrate model system.
Chelators alleviate oxidative stress in vivo in a zebrafish larval model
To evaluate the efficacy of 2c and 3c compounds in vivo, we investigated their ability to modulate pathways of redox signaling using a zebrafish model. Zebrafish larvae have emerged as a robust vertebrate model system for toxicological studies aimed at understanding the effects and mechanisms underlying oxidative stress.88–90 Pathways related to redox signaling and lipid metabolism in zebrafish are highly analogous to those in humans.91,92 According to literature reports zebrafish have an enhanced oxidative stress response.93–95 One of the major factors responsible for the increased oxidative stress response is the presence of comparatively high levels of unsaturated fatty acids that help fish maintain the fluidity of cell membranes at lower temperatures.92 The optical transparency of zebrafish larvae make them suitable for optical imaging with ROS sensitive optical probes like CellROX.81 We, therefore, tested the efficacy of the chelators toward alleviating Cu2+ induced oxidative stress in live zebrafish larvae.3.5 days post fertilization (dpf) wildtype zebrafish larvae were exposed to Cu2+ by incubating the larvae in a medium containing copper salts. The larvae treated with copper salts exhibited increased oxidative stress as observed by increased intensity of CellROX. Highest intensities were observed in the tail muscles, gut, lateral fins, and around the eyes of the larvae (Fig. 9a). The swimming motion of zebrafish larvae are well-established where the larvae perform C-start motions during which the tail fin is bent into a C-shape.96,97 This movement helps the fish propel itself forward and dictates the speed of swimming.96,97 The C-start movement is an energy-demanding process for the muscles in the tail fin implying high mitochondrial activity in this region. Further, the lateral fins help the fish maintain balance during swimming which is also associated with increased muscle activity in these regions.98 The gut undergoes spontaneous peristaltic movements starting from 2 dpf. These peristaltic movements are driven by the gut muscles which are controlled by the autonomous nervous system.99 Finally, the zebrafish larvae actively scan their lateral field of view as they swim to visualize their environment which requires extra-ocular muscles that help in eye movement.100 In summary, we noted that the anatomical regions that showed increased oxidative stress in the zebrafish larvae upon Cu exposure were regions of high muscle activity and hence would be associated with greater mitochondrial density.81,101 Since, hydrogen peroxide production increases with increased mitochondrial activity,102,103 these regions would be more susceptible to metal induced oxidative stress via the Fenton pathway in accordance with our observation (although other regions might also accumulate metal ions).
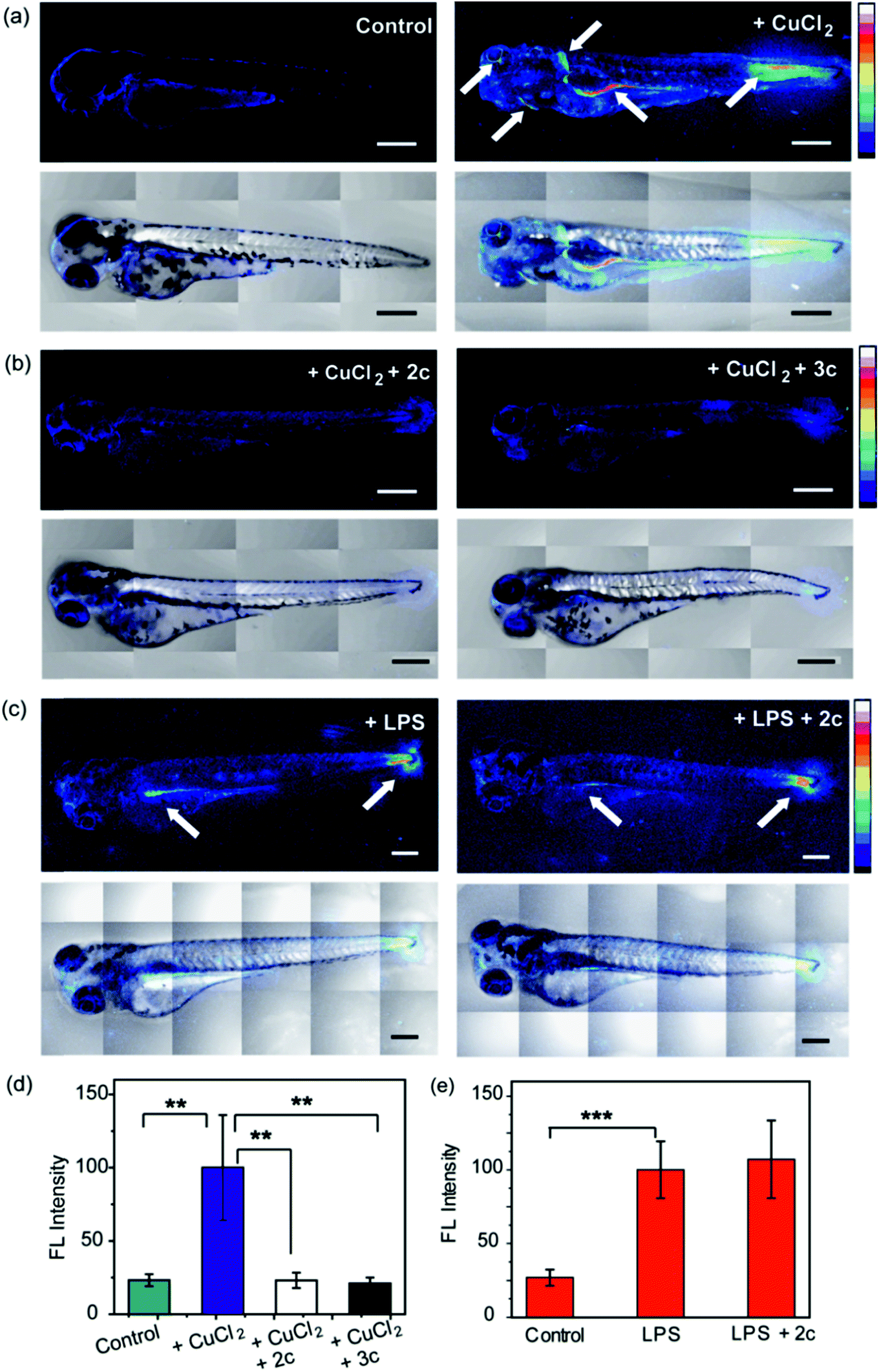 | ||
| Fig. 9 Confocal fluorescence images depicting the effect of chelators on live 3.5 dpf zebrafish larvae treated with copper salts. (a–c) Upper panels depict Z stacked confocal fluorescence images and lower panels depict bright field images overlaid with Z stacked confocal fluorescence images. (a) Control Cu-untreated larvae (left) and larvae treated with CuCl2 (30 μM) for 30 min (right). (b) Larvae treated with CuCl2 (30 μM) for 30 min followed by 2c (60 μM) for 30 min (left) and larvae treated with CuCl2 (30 μM) for 30 min followed by 3c (60 μM) for 30 min (right). (c) Larvae treated with LPS (10 μg mL−1) for 30 min (left) and larvae treated with LPS (10 μg mL−1) for 30 min followed by 2c (60 μM) for 30 min (right). White arrows indicate regions exhibiting high oxidative stress. Larvae were stained with CellROX (10 μM) for 30 min in the final step before imaging. (d) Bar plots representing average fluorescence intensities obtained from intensity analysis of confocal images of the zebrafish larvae experiments for which representative images are shown in panels (a and b). Intensity data were normalized to intensity of CuCl2 treated larvae. (e) Quantification of fluorescence intensities of confocal images of zebrafish larvae for which representative images are shown in panel (c). Intensity data were normalized to LPS treated larvae. For both panels (d and e) intensity analyses were carried out on Z-stacked confocal images of zebrafish larvae. ImageJ software (NIH, USA) was used for image analysis. For fluorescence quantification, a region of interest (ROI) was created, and fluorescence intensity was measured. The reported fluorescence intensity was determined by averaging the measured intensity of at least fifteen different ROIs from experiments on at least six different zebrafish larvae. Error bars denote SEM, n = 6. Statistical analyses were performed using an unpaired, two-tailed Student's t-test (**p ≤ 0.01, ***p ≤ 0.001). 20× objective was used for imaging; scale bar: 200 μm. | ||
We were pleased to note that the level of oxidative stress in the Cu-treated larvae significantly decreased in the presence of the chelators to levels similar to untreated larvae (Fig. 9b and d). The 2c and 3c chelators alone did not induce any oxidative stress in the larvae (Fig. S32†), and their ability to suppress copper-stimulated ROS was similar to D-Pen (Fig. S33†). Control experiments with LPS treated larvae showed that 2c and 3c could not rescue LPS induced oxidative stress (Fig. 9c and e). Therefore ligands 2c and 3c suppress Cu ion induced oxidative stress via Cu2+ chelation in a multicellular, vertebrate organism indicating the in vivo applicability of the chelators.
3. Concluding remarks
The regulation of redox active metal ions involves the intricate interplay of multiple proteins and biological ligands and exhibits many shades of subtle homeostatic balance.56,104 While the concept of chelation therapy has a long history, the appreciation of the balance between the labile and the protein bound pool and the regulatory factors that maintain this balance is very recent.105 Hence, most chelation therapy based approaches for treating metal-overload disorders are based on chelators that are non-specific.27 These non-specific chelators are also frequently used for mechanistic studies in unravelling metal ion regulation, leading to equivocal information on metal ion homeostasis.104,105 The development of selective chelators for a specific oxidation state of a redox-active metal ion remains a significant challenge, but is extremely worthwhile given the wealth of mechanistic information that can be obtained by the judicious use of such chelators and the tremendous potential in minimizing the side-effects associated with chelation therapy.By analyzing the literature on Cu-induced oxidative stress and Cu ion speciation in cells,56,105 we realized that chelating out Cu2+ ions which would mostly be generated under oxidative stress conditions inside cells would provide access to selective Cu ion chelation without affecting the essential intracellular metal pool. An important criterion was that the chelated Cu2+ should not further promote Fenton chemistry. Using our redox-state selective chelation strategy, we have therefore developed novel chelators that exhibit unique Cu2+ selectivity and can alleviate metal overload induced oxidative stress both in vitro and in live cells. Importantly, we were able to show that the chelators reduce oxidative stress via Cu2+ chelation rather than by direct ROS scavenging, and were able to suppress ROS production in a disease model of cellular Cu overload. Finally, the chelators were able to reduce Cu-induced oxidative stress in zebrafish larvae indicating their ability to function in vivo.
The high selectivity of our chelators for Cu2+ over Cu+ and other biologically relevant metal ions makes these scaffolds valuable chemical tools for exploring pathways of Cu ion regulation. Moreover, these highly specific Cu2+ chelators can be applied as chemical tools to modulate Cu homeostatic pathways and Cu induced oxidative stress response in neurodegenerative conditions like Alzheimer's disease and in Cu-overload disorders like Wilson's disease. Finally, the molecular design of the Cu2+ binders, synthesized via a simple modular strategy, can serve as scaffolds for design and development of molecules aimed at chelation therapy for Cu-overload induced diseases.
Animal ethics statement
Zebrafish were bred and maintained and experiments were performed in accordance with guidelines and protocols approved by the institutional animal ethics committee of the Tata Institute of Fundamental Research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. D. acknowledges financial support from TIFR and Department of Atomic Energy, India. The authors acknowledge Prof. Michael J. Petris for generously sharing MEF Atp7a−/− cells; Dr Marion Kerscher for her help with the illustration of MOMEC optimized structures and Dr Sreelaja Nair for helpful discussions on zebrafish larvae results. The authors acknowledge Cell culture and Imaging facility, Department of Chemical Sciences, TIFR, India; National NMR facility, TIFR, India; Electron spin resonance laboratory, Department of Chemical Sciences, TIFR; Biophotonics Laboratory, Department of Chemical Sciences, TIFR; and Mass Laboratory, Chemistry Department, Indian Institute of Technology, Bombay, India.Notes and references
- C. J. Chang, Nat. Chem. Biol., 2015, 11, 744–747 CrossRef CAS PubMed.
- M. J. Daly, E. K. Gaidamakova, V. Y. Matrosova, A. Vasilenko, M. Zhai, A. Venkateswaran, M. Hess, M. V. Omelchenko, H. M. Kostandarithes, K. S. Makarova, L. P. Wackett, J. K. Fredrickson and D. Ghosal, Science, 2004, 306, 1025–1028 CrossRef CAS PubMed.
- K. Barnese, E. B. Gralla, J. S. Valentine and D. E. Cabelli, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6892–6897 CrossRef CAS PubMed.
- Y. Hatori, Y. Yan, K. Schmidt, E. Furukawa, N. M. Hasan, N. Yang, C.-N. Liu, S. Sockanathan and S. Lutsenko, Nat. Commun., 2016, 7, 10640 CrossRef CAS PubMed.
- M. L. Schlief, T. West, A. M. Craig, D. M. Holtzman and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14919–14924 CrossRef CAS PubMed.
- A. I. Bush, C. L. Masters and R. E. Tanzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11193–11194 CrossRef CAS PubMed.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS PubMed.
- S. Lutsenko, Curr. Opin. Chem. Biol., 2010, 14, 211–217 CrossRef CAS PubMed.
- I. Fridovich, Science, 1978, 201, 875–880 CrossRef CAS PubMed.
- Q. Liu, U. Berchner-Pfannschmidt, U. Möller, M. Brecht, C. Wotzlaw, H. Acker, K. Jungermann and T. Kietzmann, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4302–4307 CrossRef CAS PubMed.
- B. Halliwell and J. M. Gutteridge, Biochem. J., 1984, 219, 1–14 CrossRef CAS PubMed.
- S. J. Stohs and D. Bagchi, Free Radical Biol. Med., 1995, 18, 321–336 CrossRef CAS PubMed.
- K. Jomova and M. Valko, Toxicology, 2011, 283, 65–87 CrossRef CAS PubMed.
- L. Macomber and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8344–8349 CrossRef CAS PubMed.
- M. F. Poyton, A. M. Sendecki, X. Cong and P. S. Cremer, J. Am. Chem. Soc., 2016, 138, 1584–1590 CrossRef CAS PubMed.
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef CAS PubMed.
- L. K. Charkoudian, D. M. Pham and K. J. Franz, J. Am. Chem. Soc., 2006, 128, 12424–12425 CrossRef CAS PubMed.
- T. Storr, M. Merkel, G. X. Song-Zhao, L. E. Scott, D. E. Green, M. L. Bowen, K. H. Thompson, B. O. Patrick, H. J. Schugar and C. Orvig, J. Am. Chem. Soc., 2007, 129, 7453–7463 CrossRef CAS PubMed.
- Y. Wei and M. Guo, Angew. Chem., Int. Ed., 2007, 46, 4722–4725 CrossRef CAS PubMed.
- M. R. Jones, E. Mathieu, C. Dyrager, S. Faissner, Z. Vaillancourt, K. J. Korshavn, M. H. Lim, A. Ramamoorthy, V. Wee Yong, S. Tsutsui, P. K. Stys and T. Storr, Chem. Sci., 2017, 8, 5636–5643 RSC.
- K. J. Franz, Curr. Opin. Chem. Biol., 2013, 17, 143–149 CrossRef CAS PubMed.
- M. Eckshtain-Levi, R. Lavi, D. S. Yufit, B. Daniel, O. Green, O. Fleker, M. Richman, S. Rahimipour, A. Gruzman and L. Benisvy, Chem. Commun., 2016, 52, 2350–2353 RSC.
- A. Robert, Y. Liu, M. Nguyen and B. Meunier, Acc. Chem. Res., 2015, 48, 1332–1339 CrossRef CAS PubMed.
- A. Roberts Eve and L. Schilsky Michael, Hepatology, 2008, 47, 2089–2111 CrossRef CAS PubMed.
- V. Prachayasittikul, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Drug Des., Dev. Ther., 2013, 7, 1157–1178 CrossRef PubMed.
- P. Leanderson and C. Tagesson, Carcinogenesis, 1996, 17, 545–550 CrossRef CAS PubMed.
- X. Ding, H. Xie and Y. J. Kang, J. Nutr. Biochem., 2011, 22, 301–310 CrossRef CAS PubMed.
- A. J. Baxter and E. P. Krenzelok, Clin. Toxicol., 2008, 46, 1083–1084 CrossRef PubMed.
- C. Cheignon, P. Faller, D. Testemale, C. Hureau and F. Collin, Metallomics, 2016, 8, 1081–1089 RSC.
- M. A. Greenough, A. Ramirez Munoz, A. I. Bush and C. M. Opazo, Metallomics, 2016, 8, 831–839 RSC.
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52, 12193–12206 CrossRef CAS PubMed.
- D. Pramanik, C. Ghosh and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 15545–15552 CrossRef CAS PubMed.
- J. P. Taylor, R. H. Brown Jr and D. W. Cleveland, Nature, 2016, 539, 197–206 CrossRef PubMed.
- S. Sangwan, A. Zhao, K. L. Adams, C. K. Jayson, M. R. Sawaya, E. L. Guenther, A. C. Pan, J. Ngo, D. M. Moore, A. B. Soriaga, T. D. Do, L. Goldschmidt, R. Nelson, M. T. Bowers, C. M. Koehler, D. E. Shaw, B. G. Novitch and D. S. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8770–8775 CrossRef CAS PubMed.
- J. S. Valentine and P. J. Hart, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3617–3622 CrossRef CAS PubMed.
- N. Fujimaki, F. Kitamura and H. Takeuchi, Biochemistry, 2013, 52, 5184–5194 CrossRef CAS PubMed.
- P. M. Roos, O. Vesterberg, T. Syversen, T. P. Flaten and M. Nordberg, Biol. Trace Elem. Res., 2013, 151, 159–170 CrossRef CAS PubMed.
- S. Ishida, P. Andreux, C. Poitry-Yamate, J. Auwerx and D. Hanahan, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19507–19512 CrossRef CAS PubMed.
- A. Gupte and R. J. Mumper, Cancer Treat. Rev., 2009, 35, 32–46 CrossRef CAS PubMed.
- Y. Wang, S. Zhu, G. A. Weisman, J. D. Gitlin and M. J. Petris, PLoS One, 2012, 7, e43039 CrossRef CAS PubMed.
- A. Bhattacharjee, H. Yang, M. Duffy, E. Robinson, A. Conrad-Antoville, Y.-W. Lu, T. Capps, L. Braiterman, M. Wolfgang, M. P. Murphy, L. Yi, S. G. Kaler, S. Lutsenko and M. Ralle, J. Biol. Chem., 2016, 291, 16644–16658 CrossRef CAS PubMed.
- R. Linz and S. Lutsenko, J. Bioenerg. Biomembr., 2007, 39, 403–407 CrossRef CAS PubMed.
- E. V. Polishchuk, M. Concilli, S. Iacobacci, G. Chesi, N. Pastore, P. Piccolo, S. Paladino, D. Baldantoni, S. C. D. van Ijzendoorn, J. Chan, C. J. Chang, A. Amoresano, F. Pane, P. Pucci, A. Tarallo, G. Parenti, N. Brunetti-Pierri, C. Settembre, A. Ballabio and R. S. Polishchuk, Dev. Cell, 2014, 29, 686–700 CrossRef CAS PubMed.
- M. L. Graper, D. Huster, S. G. Kaler, S. Lutsenko, M. L. Schilsky and D. J. Thiele, Ann. N. Y. Acad. Sci., 2014, 1314, v–vi CrossRef PubMed.
- L. A. Finney and T. V. Halloran, Science, 2003, 300, 931–936 CrossRef CAS PubMed.
- A. W. Foster, R. Pernil, C. J. Patterson, A. J. P. Scott, L.-O. Palsson, R. Pal, I. Cummins, P. T. Chivers, E. Pohl and N. J. Robinson, Nat. Chem. Biol., 2017, 13, 409–414 CrossRef CAS PubMed.
- A. Hong-Hermesdorf, M. Miethke, S. D. Gallaher, J. Kropat, S. C. Dodani, J. Chan, D. Barupala, D. W. Domaille, D. I. Shirasaki, J. A. Loo, P. K. Weber, J. Pett-Ridge, T. L. Stemmler, C. J. Chang and S. S. Merchant, Nat. Chem. Biol., 2014, 10, 1034–1042 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Kozyreva, K. Zovo and P. Palumaa, Nature, 2010, 465, 645–648 CrossRef CAS PubMed.
- A. K. Boal and A. C. Rosenzweig, Chem. Rev., 2009, 109, 4760–4779 CrossRef CAS PubMed.
- J. J. A. Cotruvo, A. T. Aron, K. M. Ramos-Torres and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4400–4414 RSC.
- L. Yang, R. McRae, M. M. Henary, R. Patel, B. Lai, S. Vogt and C. J. Fahrni, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11179–11184 CrossRef CAS PubMed.
- M. C. Linder, Mutat. Res., 2001, 475, 141–152 CrossRef CAS PubMed.
- B. Alies, E. Renaglia, M. Rózga, W. Bal, P. Faller and C. Hureau, Anal. Chem., 2013, 85, 1501–1508 CrossRef CAS PubMed.
- S. Rabizadeh, E. B. Gralla, D. R. Borchelt, R. Gwinn, J. S. Valentine, S. Sisodia, P. Wong, M. Lee, H. Hahn and D. E. Bredesen, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3024–3028 CrossRef CAS.
- P. Delangle and E. Mintz, Dalton Trans., 2012, 41, 6359–6370 RSC.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed.
- S. Jia, K. M. Ramos-Torres, S. Kolemen, C. M. Ackerman and C. J. Chang, ACS Chem. Biol., 2017, 13, 1844–1852 CrossRef PubMed.
- K. Harry and J. D. Spivack, US Pat., No. US2928876, 1960.
- Q. Wang and K. J. Franz, Bioorg. Med. Chem. Lett., 2017, 27, 4165–4170 CrossRef CAS PubMed.
- E. Faggi, R. Gavara, M. Bolte, L. Fajarí, L. Juliá, L. Rodríguez and I. Alfonso, Dalton Trans., 2015, 44, 12700–12710 RSC.
- M. Tarnai, PhD thesis, Heidelberg University, 2006.
- S. Y. Ebrahimipour, I. Sheikhshoaie, J. Castro, W. Haase, M. Mohamadi, S. Foro, M. Sheikhshoaie and S. Esmaeili-Mahani, Inorg. Chim. Acta, 2015, 430, 245–252 CrossRef CAS.
- P. Comba, T. W. Hambley and B. Martin, in Molecular Modeling of Inorganic Compounds, Wiley-VCH Verlag GmbH & Co. KGaA, 3rd edn with a tutorial, based on MOMEC3, 2009, pp. 215–292 Search PubMed.
- P. Comba, M. Ströhle and T. W. Hambley, Helv. Chim. Acta, 1995, 78, 2042–2047 CrossRef CAS.
- P. Comba, B. Martin and A. Sanyal, J. Comput. Chem., 2013, 34, 1598–1608 CrossRef CAS PubMed.
- J. E. Bol, C. Buning, P. Comba, J. Reedijk and M. Ströhle, J. Comput. Chem., 1998, 19, 512–523 CrossRef CAS.
- P. V. Bernhardt and P. Comba, Inorg. Chem., 1992, 31, 2638–2644 CrossRef CAS.
- M. G. D. Leed, N. Wolkow, D. M. Pham, C. L. Daniel, J. L. Dunaief and K. J. Franz, J. Inorg. Biochem., 2011, 105, 1161–1172 CrossRef CAS PubMed.
- J. Marks, I. Pozdnyakova, J. Guidry and P. Wittung-Stafshede, J. Biol. Inorg. Chem., 2004, 9, 281–288 CrossRef CAS PubMed.
- O. Farver and I. Pecht, Coord. Chem. Rev., 2011, 255, 757–773 CrossRef CAS.
- L. G. Sillen and A. E. Martell, Stability Constants of Metal Ion Complexes, Chemical Society, 17th edn, 1964 Search PubMed.
- B. Halliwell, J. M. C. Gutteridge and O. I. Aruoma, Anal. Biochem., 1987, 165, 215–219 CrossRef CAS PubMed.
- E. Farkas, É. A. Enyedy and H. Csóka, Polyhedron, 1999, 18, 2391–2398 CrossRef CAS.
- A. I. B. Romo, D. S. Abreu, T. d. F. Paulo, M. S. P. Carepo, E. H. S. Sousa, L. Lemus, C. Aliaga, A. A. Batista, O. R. Nascimento, H. D. Abruña and I. C. N. Diógenes, Chem.–Eur. J., 2016, 22, 10081–10089 CrossRef CAS PubMed.
- N. Berthet, V. Martel-Frachet, F. Michel, C. Philouze, S. Hamman, X. Ronot and F. Thomas, Dalton Trans., 2013, 42, 8468–8483 RSC.
- Y. Ma, T. Ogino, T. Kawabata, J. Li, K. Eguchi and S. Okada, Free Radical Biol. Med., 1999, 27, 227–233 CrossRef CAS PubMed.
- Y. Ma, L. Cao, T. Kawabata, T. Yoshino, B. B. Yang and S. Okada, Free Radical Biol. Med., 1998, 25, 568–575 CrossRef CAS PubMed.
- J. Zheng, J. R. Lou, X.-X. Zhang, D. M. Benbrook, M. H. Hanigan, S. E. Lind and W.-Q. Ding, Cancer Lett., 2010, 298, 186–194 CrossRef CAS PubMed.
- P. A. Castro, A. Ramirez, F. J. Sepúlveda, C. Peters, H. Fierro, J. Waldron, S. Luza, J. Fuentealba, F. J. Muñoz, G. V. De Ferrari, A. I. Bush, L. G. Aguayo and C. M. Opazo, Front. Aging Neurosci., 2014, 6, 319 Search PubMed.
- P. Chavez-Crooker, N. Garrido and G. A. Ahearn, J. Exp. Biol., 2001, 204, 1433–1444 CAS.
- V. Mugoni, A. Camporeale and M. M. Santoro, J. Visualized Exp., 2014, 89, 51328 Search PubMed.
- R. Squitti, P. M. Rossini, E. Cassetta, F. Moffa, P. Pasqualetti, M. Cortesi, A. Colloca, L. Rossi and A. Finazzi-Agro', Eur. J. Clin. Invest., 2002, 32, 51–59 CrossRef CAS PubMed.
- A. Barzegar and A. A. Moosavi-Movahedi, PLoS One, 2011, 6, 26012 CrossRef PubMed.
- Y. Aboul-Enein Hassan, I. Kruk, A. Kładna, K. Lichszteld and T. Michalska, Biopolymers, 2007, 86, 222–230 CrossRef CAS PubMed.
- G. Yücel, Z. Zhao, I. El-Battrawy, H. Lan, S. Lang, X. Li, F. Buljubasic, W.-H. Zimmermann, L. Cyganek, J. Utikal, U. Ravens, T. Wieland, M. Borggrefe, X.-B. Zhou and I. Akin, Sci. Rep., 2017, 7, 2935 CrossRef PubMed.
- A. Ngkelo, K. Meja, M. Yeadon, I. Adcock and P. A. Kirkham, J. Inflammation, 2012, 9, 1 CrossRef CAS PubMed.
- C. White, J. Lee, T. Kambe, K. Fritsche and M. J. Petris, J. Biol. Chem., 2009, 284, 33949–33956 CrossRef CAS PubMed.
- M. Priyadarshini, L. A. Orosco and P. J. Panula, PLoS One, 2013, 8, 81851 CrossRef PubMed.
- R. Zhang, J. Zhao, G. Han, Z. Liu, C. Liu, C. Zhang, B. Liu, C. Jiang, R. Liu, T. Zhao, M.-Y. Han and Z. Zhang, J. Am. Chem. Soc., 2016, 138, 3769–3778 CrossRef CAS PubMed.
- S. Wu, G. Ji, J. Liu, S. Zhang, Y. Gong and L. Shi, Environ. Toxicol., 2016, 31, 1241–1249 CrossRef CAS PubMed.
- J. L. Anderson, J. D. Carten and S. A. Farber, in Methods. Cell. Biol., ed. H. W. Detrich, M. Westerfield and L. I. Zon, Academic Press, 2016, vol. 133, pp. 165–178 Search PubMed.
- L. Fang and Y. I. Miller, Free Radical Biol. Med., 2012, 53, 1411–1420 CrossRef CAS PubMed.
- M. E. Hahn, A. G. McArthur, S. I. Karchner, D. G. Franks, M. J. Jenny, A. R. Timme-Laragy, J. J. Stegeman, B. R. Woodin, M. J. Cipriano and E. Linney, PLoS One, 2014, 9, 113158 CrossRef PubMed.
- Q. Wang, X. Tan, S. Jiao, F. You and P.-J. Zhang, PLoS One, 2014, 9, 102492 CrossRef PubMed.
- M. Hagedorn, M. McCarthy, V. L. Carter and S. A. Meyers, PLoS One, 2012, 7, 39397 CrossRef PubMed.
- A. Roberts, B. Bill and D. Glanzman, Front. Neural Circuits, 2013, 7, 126 CrossRef PubMed.
- R. C. Eaton, R. K. K. Lee and M. B. Foreman, Prog. Neurobiol., 2001, 63, 467–485 CrossRef CAS PubMed.
- M. H. Green, R. K. Ho and M. E. Hale, J. Exp. Biol., 2011, 214, 3111–3123 CrossRef PubMed.
- H. A. Field, K. A. Kelley, L. Martell, A. M. Goldstein and F. C. Serluca, Neurogastroenterol. Motil., 2009, 21, 304–312 CrossRef CAS PubMed.
- W. Mo, F. Chen, A. Nechiporuk and T. Nicolson, BMC Neurosci., 2010, 11, 110 CrossRef PubMed.
- V. Romanello and M. Sandri, Front. Physiol., 2015, 6, 422 Search PubMed.
- E. Cadenas, Mol. Aspects Med., 2004, 25, 17–26 CrossRef CAS PubMed.
- E. J. Anderson and P. D. Neufer, Am. J. Physiol. Cell Physiol., 2006, 290, C844–C851 CrossRef CAS PubMed.
- C. M. Ackerman and C. J. Chang, J. Biol. Chem., 2018, 13, 4628–4635 CrossRef PubMed.
- C. J. Chang, Acc. Chem. Res., 2017, 50, 535–538 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and tables, details of LC methods for stability and purity analysis of chelators, ESI-MS spectrum of copper complexes, Job's plot, absorbance data and stability constant determination with different metal ions, EPR spectra, control experiments in cells and zebrafish larvae, and 1H NMR, 13C NMR are included. See DOI: 10.1039/c8sc04041a |
| This journal is © The Royal Society of Chemistry 2018 |