
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsking more from metabolic oligosaccharide engineering
Pierre-André
Gilormini
 a,
Anna R.
Batt
b,
Matthew R.
Pratt
bc and
Christophe
Biot
*a
a,
Anna R.
Batt
b,
Matthew R.
Pratt
bc and
Christophe
Biot
*a
aUniversity of Lille, CNRS UMR 8576, UGSF – Unité de Glycobiologie Structurale et Fonctionnelle, F-59000 Lille, France. E-mail: christophe.biot@univ-lille.fr
bDepartment of Chemistry, University of Southern California, 840 Downey Way, LJS 250 Los Angeles, CA 90089, USA
cDepartment of Biological Sciences, University of Southern California, 840 Downey Way, LJS 250 Los Angeles, CA 90089, USA
First published on 18th September 2018
Abstract
Glycans form one of the four classes of biomolecules, are found in every living system and present a huge structural and functional diversity. As an illustration of this diversity, it has been reported that more than 50% of the human proteome is glycosylated and that 2% of the human genome is dedicated to glycosylation processes. Glycans are involved in many biological processes such as signalization, cell–cell or host pathogen interactions, immunity, etc. However, fundamental processes associated with glycans are not yet fully understood and the development of glycobiology is relatively recent compared to the study of genes or proteins. Approximately 25 years ago, the studies of Bertozzi's and Reutter's groups paved the way for metabolic oligosaccharide engineering (MOE), a strategy which consists in the use of modified sugar analogs which are taken up into the cells, metabolized, incorporated into glycoconjugates, and finally detected in a specific manner. This groundbreaking strategy has been widely used during the last few decades and the concomitant development of new bioorthogonal ligation reactions has allowed many advances in the field. Typically, MOE has been used to either visualize glycans or identify different classes of glycoproteins. The present review aims to highlight recent studies that lie somewhat outside of these more traditional approaches and that are pushing the boundaries of MOE applications.
 Pierre-André Gilormini | Pierre-André Gilormini is currently a temporary teaching and research assistant in the Department of Chemistry at the “Université de Lille”. He obtained his M.S. in Chemistry and Biology in 2014 working on the synthesis of different ManNAc alkyne analogs. He received his PhD in Chemical Biology under the supervision of Christophe Biot in 2017. His work in the Structural and Functional Glycobiology department consists in the development of chemical tools and strategies for the visualization and study of sialic acids, in order to get new insights into their metabolism. |
 Anna R. Batt | Anna Batt is a graduate student in the Pratt lab at the University of Southern California. She received her B.S in Biochemistry and M.S. in Chemistry with a concentration in Biochemistry from San Francisco State University working with Teaster Baird Jr. Her current work in the Pratt lab focuses on the development and characterization of metabolic chemical reporters of glycosylation and the role of O-GlcNAcylation in cellular signaling pathways. |
 Matthew R. Pratt | Matthew Pratt is an Associate Professor of Chemistry and Biological Sciences at the University of Southern California. He obtained his B.S in Biochemistry and Mathematics from the University of Arizona working with Robin Polt on glycosylation methodology. He then moved to UC Berkeley where he received his PhD with Carolyn Bertozzi while applying chemical approaches to understand the roles of complex carbohydrates. Matt then developed chemical methods to control protein function in the laboratory of Tom Muir at the Rockefeller University as an American Cancer Society Postdoctoral Fellow. Matt's lab currently uses both bioorthogonal chemistry and protein semi-synthesis to explore the biology of protein posttranslational modifications, with a particular emphasis on O-GlcNAc modifications. He has received several awards for his work including the Damon Runyon Innovator Award, the American Cancer Society Research Scholar Award, and the David Y. Gin Award from the Carbohydrate Division of the American Chemical Society. |
 Christophe Biot | Christophe Biot is Professor of Organic and Bioorganic Chemistry in the Department of Chemistry at “Université de Lille” where he serves as the Co-director of the master Chemistry and Life Sciences. His earlier work on antimalarial compounds reached human clinical trials. More recently, he applied chemistry to studying biological systems. His scientific interests include glycobiology and bioimaging. He has published more than 100 articles, co-authored 10 book chapters, and given many lectures throughout the country and abroad. |
Introduction
Glycosylation is a major post-translational modification which has been reported in every living system. Glycans present a huge structural and functional diversity and they are involved in many biological events as diverse as immunity, signalization, structuration or storage.1 Structural glycobiology has really emerged with the advent of analytical techniques such as mass spectrometry (MS) and nuclear magnetic resonance (NMR) but the structure–function relationship for glycans remains an open field of research. The last three decades have seen the rapid development of new chemical tools and strategies often designed as Metabolic Oligosaccharide Engineering (MOE). The aim of this review is to highlight some very recent studies which are extending the boundaries of MOE and should open new routes for the future of chemical glycobiology.One can consider that metabolic oligosaccharide engineering (MOE) finds its origins in the early 80's. Very early, several groups made great efforts in order to artificially modulate the cellular glycosylation. Paulson et al. reported such a strategy to study infection mechanisms of myxoviruses and the role of glycans in this process. After enzymatic removal of sialic acids from the erythrocyte membrane, cells were resialylated through the use of different purified sialyltransferases, allowing the restoration of virus infection. Inspired by this pioneering work of Paulson et al.,2 the group of Brossmer was among the first ones to transfer unnatural monosaccharides onto cell-membrane glycans through the action of glycosyltransferases.3,4 The aim at the time was to change the membrane sialylation in order to modulate recognition processes. For instance, Gross and Brossmer showed that desialylated glycoproteins, when resialylated with 9-amino-N-acetyl neuraminic acid, developed resistance toward bacterial sialidases.5
Almost at the same time, a number of monosaccharide analogs such as deoxyglucose or fluoroglucose have been used in order to inhibit glycosylation processes through the cellular biosynthetic machinery.6 While working on mechanisms of inhibition, the group of Reutter found that the elongation of the N-acyl lateral chain of N-acetyl-D-mannosamine (ManNAc) derivatives did not hinder their metabolization.7–9N-Propanoyl-D-mannosamine (ManNProp) was injected into living rats. Then, rat livers were analyzed and N-propanoyl-neuraminic acid (Neu5Prop) could be detected by mass spectroscopy, leading to the conclusion that ManNProp was efficiently used in de novo biosynthesis of sialic acids.9 Unnatural ManNAc analogs were used to modulate the cell sialylation and therefore the host–pathogen interaction. For instance, Keppler et al. showed that the incorporation of N-propanoyl-D-mannosamine, N-butanoyl-D-mannosamine, and N-pentanoyl-D-mannosamine (ManNProp, ManNBut and ManNPent respectively) leads to the formation of the corresponding sialic acids and to the inhibition of polyoma viruses.10 Many efforts have been made in the last two decades in order to investigate the promiscuity of glycan metabolic pathways toward chemical modifications of precursors.11,12 In the same period, the advent of bioorthogonal reactions has allowed for monosaccharides to be used as chemical reporters, enabling the application of monosaccharide analogs in a variety of different contexts.
Since Reutter's pioneering work on monosaccharides, many important biological discoveries have been made over the years using chemical reporters (i.e., functionalized metabolites) as probes for a wide range of biomolecules and bioprocesses, including post-translational modifications, protein synthesis and turn-over, and nucleic-acid analyses.13–15 The vast majority of these approaches involve the installation of reporter tags in a two step process that relies on a range of bioorthogonal reactions,16,17 defined as a unique chemical reaction that can occur in the presence of biomolecules or living systems without interfering with native biochemical processes.18 A range of different protein modifications are amenable to bioorthogonal strategies; however, we will focus here on metabolic oligosaccharide engineering (MOE). The scientific struggle of studying complex biological systems, specifically the intricacies of glycan biosynthesis and metabolism was tremendously alleviated by the development of bioorthogonal chemistry. The first examples exploited the promiscuity of glycan biosynthetic enzymes and used bioorthogonal chemistry to metabolically incorporate unnatural glycan analogs with unique functionalities into cell-surface glycan structures and then subsequently conjugate them to visualization or affinity tags using ketone condensation19 and the Staudinger ligation.20 Since this transformational debut of bioorthogonal chemistry by the Bertozzi lab, an explosion of other newly discovered bioorthogonal chemistries has proven crucial for countless biological discoveries. In general, bioorthogonal chemistries employ either polar reactions between nucleophiles and electrophiles or cycloaddition chemistries.21 Traditionally the most commonly used bioorthogonal chemistries for the installation of different tags into MOE elaborated glycan structures are the Staudinger ligation,20 Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC),22,23 and strain-promoted azide–alkyne cycloaddition (SPAAC) that allows conjugation between azides and cyclooctyne reagents,24,25 and more recently Diels–Alder (DA) cycloaddition chemistries.26,27 Each one of these different chemistries has its own advantages and particular applications including live-cell imaging, protein enrichment, time-resolved cell labeling, etc. (Fig. 1).
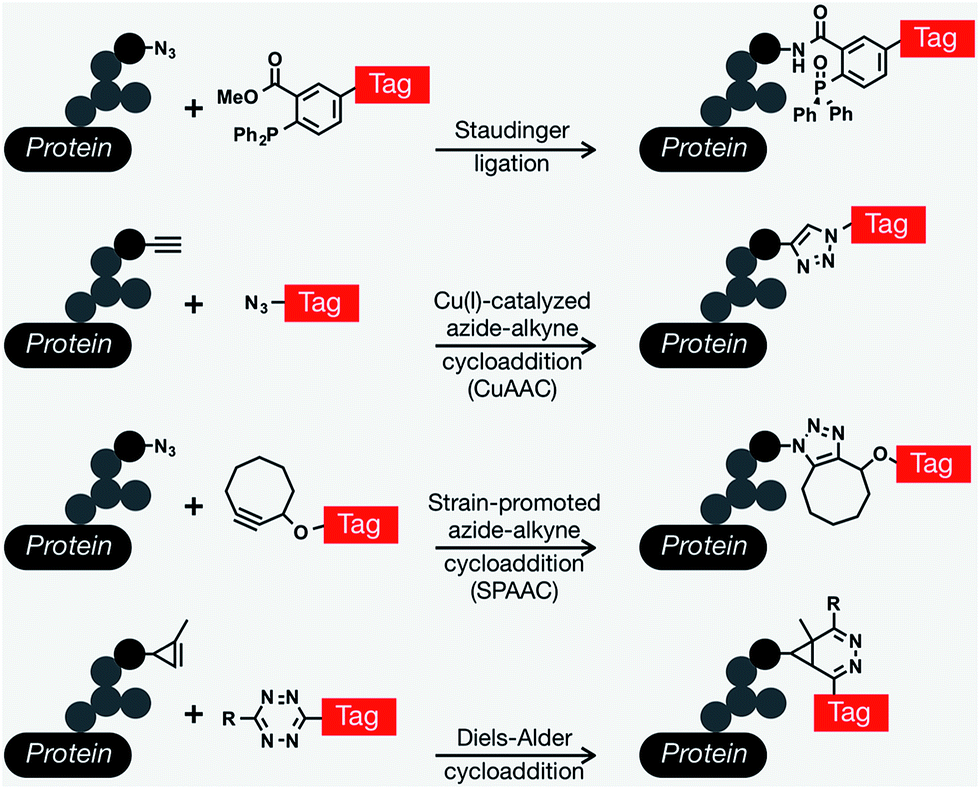 | ||
| Fig. 1 Common bioorthogonal reactions that have been applied to metabolic oligosaccharide engineering. The Staudinger ligation occurs between an azide-modified glycan and a modified triarylphosphine to yield an amide bond. The Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) yields a stable triazole. This versatile reaction can be performed in either orientation, but azido-tags result in a lower background signal. As an alternative to CuAAC, the formation of triazoles can be promoted through ring strain in the strain-promoted azide–alkyne cycloaddition. Finally, tetrazine tags will rapidly undergo an inverse electron-demand Diels–Alder reaction with activated alkynes, like cyclopropenes. | ||
In the past two decades, MOE has been combined with bioorthogonal chemistry to have an unquestionably transformative impact on carbohydrate biology and its role in human health and disease. These unnatural monosaccharides have been shown to be metabolically tolerated and incorporated into glycan pathways, such as biosynthetic and salvage pathways for different monosaccharides that will be ultimately incorporated into cell-surface glycoproteins, glycolipids, and intracellularly modified O-GlcNAc. Remarkably, a range of cellular and in vivo systems, ranging from bacteria28–30 to living animals and plants31,32 are amenable to MOE. As mentioned above, the very first example of a bioorthogonally reactive unnatural sugar for MOE applications was by the Bertozzi laboratory in 1997, when they exploited the fortuitous metabolic tolerance of unnatural N-acyl substituents to incorporate a ketone derivative of ManNAc into the cell surface through the sialic acid biosynthesis pathway.19 The unique ketone functionality on the cell surface allowed for specific conjugation with hydrazide probes gaining a variety of functionalities including fluorescent tagging, biotin enrichment and ligation to new epitopes on the cell surface. Shortly after, additional examples of oligosaccharide chemical reporter functionalities were described for the labelling of sialic acid using uniquely reactive ketones/aldehydes for the display of novel carbohydrate epitopes capable of recognizing and binding protein receptors such as ricin.33 Although the ability to remodel the glycan architecture on the surface of cells using ketone functionalities opened up many new opportunities, the secondary conjugation step produced a relatively high background and had limited use in vivo due to the low pH required for reasonable reaction kinetics and limited selectivity for endogenous aldehyde- and ketone-containing metabolites. To overcome this issue, the Bertozzi group modified the bioorthogonal Staudinger reaction to be compatible with physiological aqueous conditions, achieving what is now known in the chemical biology field as the Staudinger ligation (also referred to as the Staudinger–Bertozzi ligation), by taking advantage of abiotic azides and phosphines.20 This reaction enabled the selective labeling of azide-modified sialic acids on the surface of cells that had been treated with per-O-acetylated N-azidoacetyl-D-mannosamine (Ac4ManNAz).
Shortly after, several non-sialic acid azide-glycan reporters were developed as N-acetylglucosamine (GlcNAc) and N-acetylgalactosamine (GalNAc) analogs such as GlcNAz34,35 and GalNAz,18 respectively. These azide-containing structures have been subsequently expanded to a wide-range of other chemical reporters based on different monosaccharides.36–38 In addition to azide functionalities, a continually growing list of other bioorthogonal handles began to expand what bioorthogonal reactions could be applied to MOE. Alkyne-monosaccharides were of immediate interest as they display a lower background signal when coupled with azide-tags in CuAAC,39 and again, many different reporters have been prepared and incorporated into glycan structures where they could be detected using CuAAC.40,41 Within the last five years there has continued to be an overwhelming amount of research focused on the development of new and unique functionalities that have allowed the use of multiple probes in the same system as well as expanding the conditions that can be used for conjugations. Some of these include the first use of cyclopropenes,42 alkenes,26,43 isonitriles,44 diazo compounds,45 nitrones,46 and norbonenes.47
“Classical” MOE applications such as described in this introduction present some limitations. In this review, we highlight some recent studies addressing these limitations and trying to tackle three main challenges: (i) how can one deal with the complexity of glycan metabolic pathways? Which tools and reporters can be applied to the understanding of complex biological interactions? (ii) How can one target a specific glycan motif? A specific cell or tissue? And how can one efficiently deliver a reporter? (iii) How can one improve the detection of incorporated unnatural monosaccharides?
Finding a way inside the metabolic maze
Glycosylation involves a lot of different and interconnected metabolic routes. Since the work of Leloir,48 it has been known that any monosaccharide needs to be activated under its corresponding donor nucleotide sugar form prior to be transferred onto glycoconjugates by glycosyltransferases. In the last few years, some new strategies have been developed in order to explore this metabolic complexity.Multistaining
With the recent development of bioorthogonal reactions, some groups have successfully combined the simultaneous use of different sugars bearing different chemical moieties in order to perform multi-labelling without unwanted cross-staining. The first example has been reported by Saxon et al. with the combined use of ManNAz and ManNLev, allowing the observation of different incorporation rates between the two reporters.49 More recently, researchers reported the combination of inverse electron demand Diels–Alder reaction (DAinv) and Strain Promoted Azide Alkyne Cycloaddition (SPAAC).26 Niederwieser et al. incubated cells simultaneously with an analog of per-O-acetylated ManNAc bearing a terminal alkene moiety and an azido-analog of N-acetyl glucosamine (GlcNAz). After 3 days of incubation, living cells were reacted with a tetrazine probe and a cyclooctyne probe, allowing orthogonal detection of both incorporated monosaccharides. A similar procedure has been reported with the simultaneous use of cyclopropene (Ac4ManCCP) and azido (Ac4GalNAz) derivatives.50 The two reporters have been incorporated into 4T1 cells which have then been treated with tetrazine and cyclooctyne probes, allowing the detection and localization of the incorporated monosaccharides at the cell membrane. More recently, another dual MOE has been performed to visualize the incorporation of both fucose (Fuc) and 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) into the plant cell wall.51 Alkynyl and azide analogs of fucose (Fuc-Al) and Kdo (Kdo-N3) respectively were synthesized and incorporated into Arabidopsis thaliana seedlings. Two successive CuAAC reactions were then realized. The interest here is the use of a crossed CuAAC (alkyne reporter and azido-probe versus azido-reporter and alkyne probe). Under these particular conditions, no cross-reactions could be observed between the incorporated azido- and alkyne-reporters. Kdo-N3 has also recently been used for multi staining in commensal bacteria. Hudak et al. indeed provided an elegant approach to image at the same time three different macromolecules of the bacteria membrane: the lipopolysaccharide (LPS) using Kdo-N3 and SPAAC reaction, the capsular polysaccharide with a cyclopropene analog of GalNAc and finally the peptidoglycan with a coumarin D-amino acid compound.52 Although this multistaining strategy has been used for the imaging of other biomolecules, there are, to our knowledge, no other examples of multi staining with unnatural monosaccharides. However, this strategy is clearly powerful to observe the simultaneous localization of two different unnatural sugars into a cell or a tissue. One can also use the multi-reporter strategy to get insights into connected metabolic pathways (ManNAc and GlcNAc, or Kdo and GalNAc in the previous examples) allowing new advances in metabolic cross-talk strategies.Metabolic cross-talk
As briefly described in the Introduction, the biosynthesis of monosaccharides into their corresponding donor sugars results in multiple overlapping intermediates that act as branching points for crosstalk between the different metabolic pathways. Although this can lead to complications when trying to study one type of glycan, differences between the metabolic enzymes' ability to tolerate noncanonical structures can be exploited to increase or limit which glycan pathways different MOE probes have access to and consequently their preference for specific glycan types. For example, ManNAc can be made from its biosynthetic precursor, UDP-GlcNAc with the bifunctional enzyme UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) or from GlcNAc through direct conversion by GlcNAc 2-epimerase. Intracellular ManNAc can then enter the sialic acid biosynthetic pathway for the production of CMP-sialic acid. Due to the overlap in these pathways, ManNAc, GlcNAc, and sialic acid analogs can all be used to access the biosynthetic pathway for sialic acid. Several recent examples have shown the use of a sequential bioorthogonal dual strategy where derivatives of both ManNAc as well as Neu5Ac were used to target CMP-Neu5Ac using azides33 as well as alkynes.34,35 Sequential use of both ManNAc and Neu5Ac analogs provided interesting insights into the metabolism rates of sialic acids pathways in different cell lines,53 or helped deciphering the cellular entry mechanisms of sialic acids.54 Besides different uptake routes have been suggested for respectively unprotected ManNAc derivatives and unprotected Neu5Ac derivatives. Likewise there is significant crosstalk between the GlcNAc and GalNAc biosynthetic pathways. More specifically, UDP-GlcNAc and UDP-GalNAc can be reversibly interconverted by the enzyme UDP-glucose/galactose 4-epimerase (GALE). UDP-GlcNAc can be incorporated into the core and branches of N-linked and the branches of mucin-O-linked glycoproteins, heparin sulfate and hyaluronic acid glycans, and intracellular O-GlcNAcylation. In general, unnatural GlcNAc analogs have been shown to have poor labelling of the cell surface36 when compared to intracellular O-GlcNAcylation,22 although some cell surface incorporation is essentially always observed. UDP-GalNAc, in contrast, is typically incorporated into the core of mucin-O-linked glycans and chondroitin sulfate, as well as at low frequency in the branches of cell surface glycoproteins. Notably, the Bertozzi lab was able to exploit the interconversion of UDP-GlcNAc/GalNAc to label both cell surface and O-GlcNAc modified proteins by treatment with GalNAz.37 Remarkably, structural changes to these GlcNAc/GalNAc reporters can bias the amount of crosstalk and therefore the types of glycans that are labeled.27,38 Fucose can be made through the de novo pathway from mannose or used in the fucose salvage pathway to generate GDP-fucose before incorporation into cell surface glycans. Although mannose analogs could theoretically be used to gain access to fucose glycan pathways, it has been shown that 2-azido-2-deoxy-mannose labels the cell surface quite poorly compared to fucose derivatives,21 suggesting that this crosstalk pathway may be more difficult to access. Finally, chemical reporters have also been developed for hexosamine crosstalk between the GlcNAc metabolism and protein acylation, through deacetylation of the 2-N-acetate.39 In this case, a 1-deoxy analog of GlcNAlk was prepared that cannot be converted to a donor sugar and incorporated into glycans. Instead, the 5-pentynoic acid can be enzymatically removed where it has previously been shown to report of protein acetylation.40Addressing the reporter to the right place
One major challenge in Chemical Biology is the conception of selective tools or strategies able to reach specifically the desired target. The target can be a peculiar glycol-motif, a specific cell/tissue or even a given organ. It can be challenging to discriminate between healthy versus pathological conditions. In the last decade, groundbreaking strategies have been developed to tackle these targeting issues.Targeting a specific glyco-motif
In the previous section, we showed how MOE can take advantage of the overlap between the different glycosylation metabolic pathways. This can turn into a major drawback when a specific class or type of glycan is targeted. The use of glycosyltransferases to introduce unnatural monosaccharides onto existing glycans has been pioneered by Paulson and Brossmer and is briefly described in the Introduction. However, it took around 15 years for the first combination between exo-enzymatic labelling and bioorthogonal detection to be reported. In 2003, Khidekel et al. first described the enzymatic incorporation of a ketone-modified galactose residue onto O-GlcNAcylated proteins using an UDP-activated ketone GalNAc analog and galactosyltransferase GalT I. Once introduced, the ketone handle could react with the aminoxy biotin derivative which was subsequently detected with streptavidin–HRP.55 This strategy allowed the discovery of 25 O-GlcNacylated proteins in the brain.56 A few years later, Clark et al. repeated this experiment with activated GalNAz (UDP-GalNAz) and CuAAC reaction. Exo-enzymatic labeling then provided an elegant way to specifically detect N-acetyl-lactosamine on cell-surface glycans with azido-GDP-fucose and a recombinant fucosyltransferase from Helicobacter pylori.57 More recently, GDP-FucAz and H. pylori fucosyltransferase have been used to evidence changes in the expression of N-acetyllactosamine in tissue samples from patients of lung adenocarcinoma. Other commonly found glycan epitopes and motifs have been detected through similar enzymatic strategies such as fucose-α(1–2)galactose (blood groups H1 and H2, Globo H, Fuc, GM1…),58 or Thomsen–Friedenreich antigen (antigen TF)59 and, very recently, Wu et al. imaged several glycan motifs, including heparan sulfate and T/Tn antigens after introduction of activated azido-monosaccharides.60Finally, unnatural activated sialic acids have also been synthesized and the promiscuity of sialyltransferases make them excellent candidates for the development of exo-enzymatic strategies. With the SEEL strategy (Selective Exo Enzymatic Labeling), Mbua et al. opened the way for the transfer of unnatural activated sialic acids immediately followed by bioorthogonal detection.61 Sialyltransferases and azido-CMP-sialic acid have been used to label membrane N-glycans in living cells,61 to remodel antibody's glycans for drug delivery,62 for the display of synthetic ligands on cells,63 or for the assessment and quantification of cell-surface glycoproteins.64 These studies and others have been highlighted in a comprehensive review by Aguilar et al. and the interested reader is invited to refer to this article for more information.65 Whereas recombinant glycosyltransferases combined with unnatural substrates and bioorthogonal ligation are more and more frequently used for the detection of glycans in cells or tissues, only a few applications have been reported for the in vitro studies of glycosyltransferase specificities. As often in the field, Bertozzi's lab opened the way with the probing of galactosyltransferase activity in an ELISA-like assay.66 More recently, sialyltransferases have also been used for in vitro assays.67,68 In our lab, we associated exo-enzymatic labeling with diverse techniques such as LC-MS, radiolabeled monosaccharides, western-blot analysis and fluorescence cytometry and microscopy, in order to develop methodologies for the study of sialyltransferase specificity toward both acceptor and donor substrates.69,70 Such strategies should provide great tools for the characterization of glycosyltransferases by taking advantage of universal donor substrates. Although the extended use of exo-enzymatic labeling combined with bioorthogonal chemistry is relatively recent compared to MOE, it's a growing and very promising tool for the fine study of both glycoconjugates and glycosyltransferases (Fig. 2).
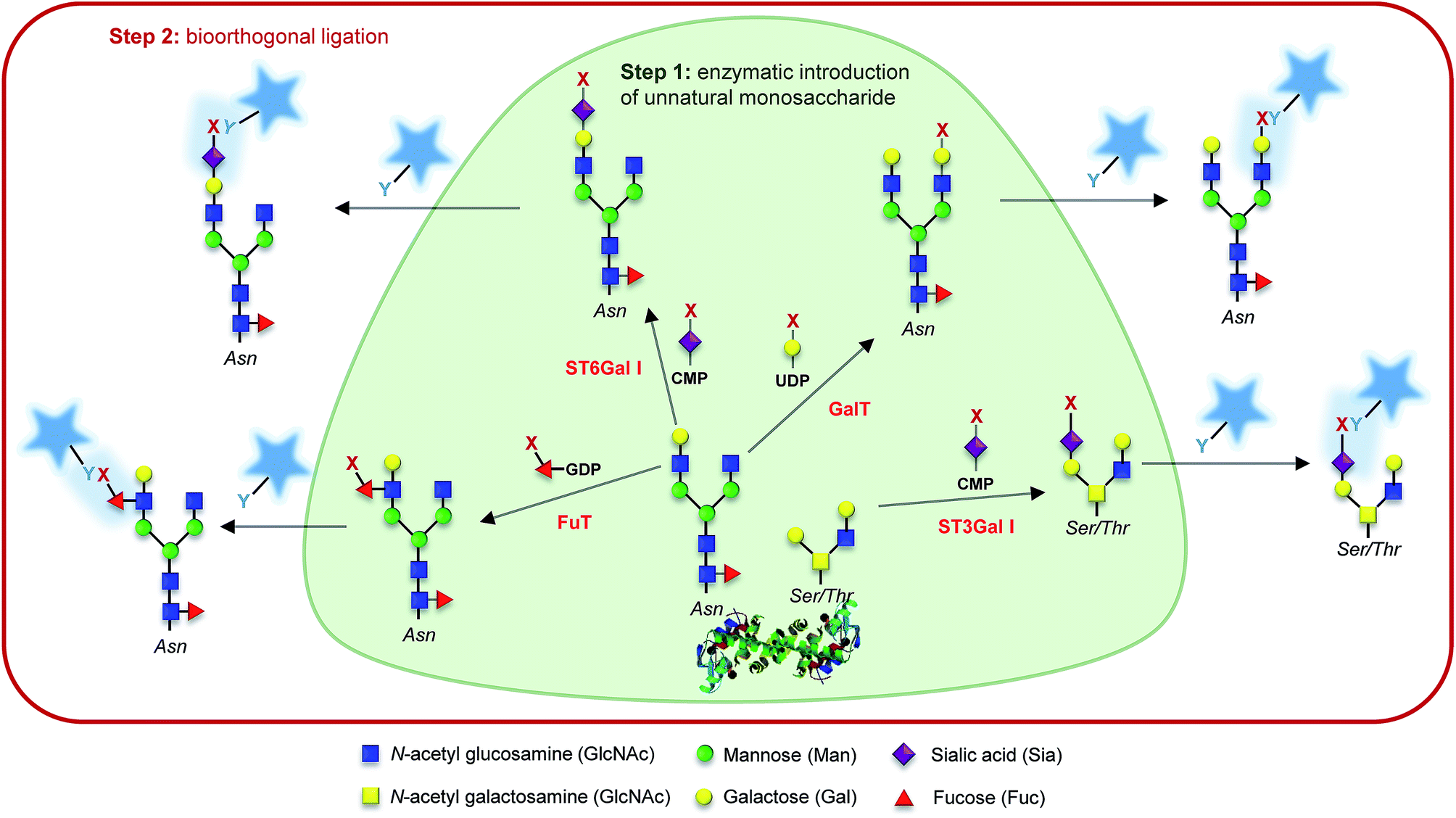 | ||
| Fig. 2 Glyco-motif targeting. Examples of chemo-enzymatic labelling using a given glycosyltransferase (named in red), its associated nucleotide sugar, and the corresponding acceptor glycol-motif. Step 1: an unnatural monosaccharide is transferred from its activated form onto the targeted glycan through the action of a glycosyltransferase. Step 2: the introduced reporter (x) is reacted with a probe bearing a complementary bioorthogonal function (y) allowing its detection. | ||
Targeting a specific cell/tissue/organ
The ability to target a specific cell/tissue or organ is extremely important, especially in the field of therapeutic development. Some groups have therefore focused their efforts on the development of strategies to specifically target MOE-like reporters. For instance, researchers have worked on tools to address unnatural monosaccharides into the brain. Usual sugar-reporters (such as per-O-acetylated ManNAz) do not cross the blood–brain barrier. To this aim, new unnatural monosaccharides have been designed to use specific metabolite transporters through the blood–brain barrier.71 ManNAz analogs have been synthesized by linking at position 1 or 6 either nicotinic acid, valproic acid, or theophylline-7-acetic acid (mimic of caffeine). These metabolites are vectorized to the brain respectively by nicotinate receptors, medium-chain fatty acids transporters, and adenine transporters. The resulting neuroactive reporters could efficiently cross the blood–brain barrier and unspecific esterases allowed the in vivo release of azido-sugars into mice brains (Fig. 3C). Another methodology for specific cell targeting consists in the introduction of a cleavable moiety onto the metabolic reporter which prevents its processing by the enzymatic machinery. The release of this cleavable group under specific conditions then allows the specific incorporation of the reporter into the cells/tissues of interest. Wang et al. proposed such a reporter with the introduction of a photocleavable group onto the anomeric position of Ac4ManNAz.72 In the presence of this labile group, the reporter cannot be incorporated, preventing the introduction of azide function into glycoconjugates. Upon in vivo UV activation, the reductive function is recovered, allowing the incorporation of the modified monosaccharide into a targeted tumor. Another approach is to use specificities of the target as a trigger for the release of the unnatural reporter.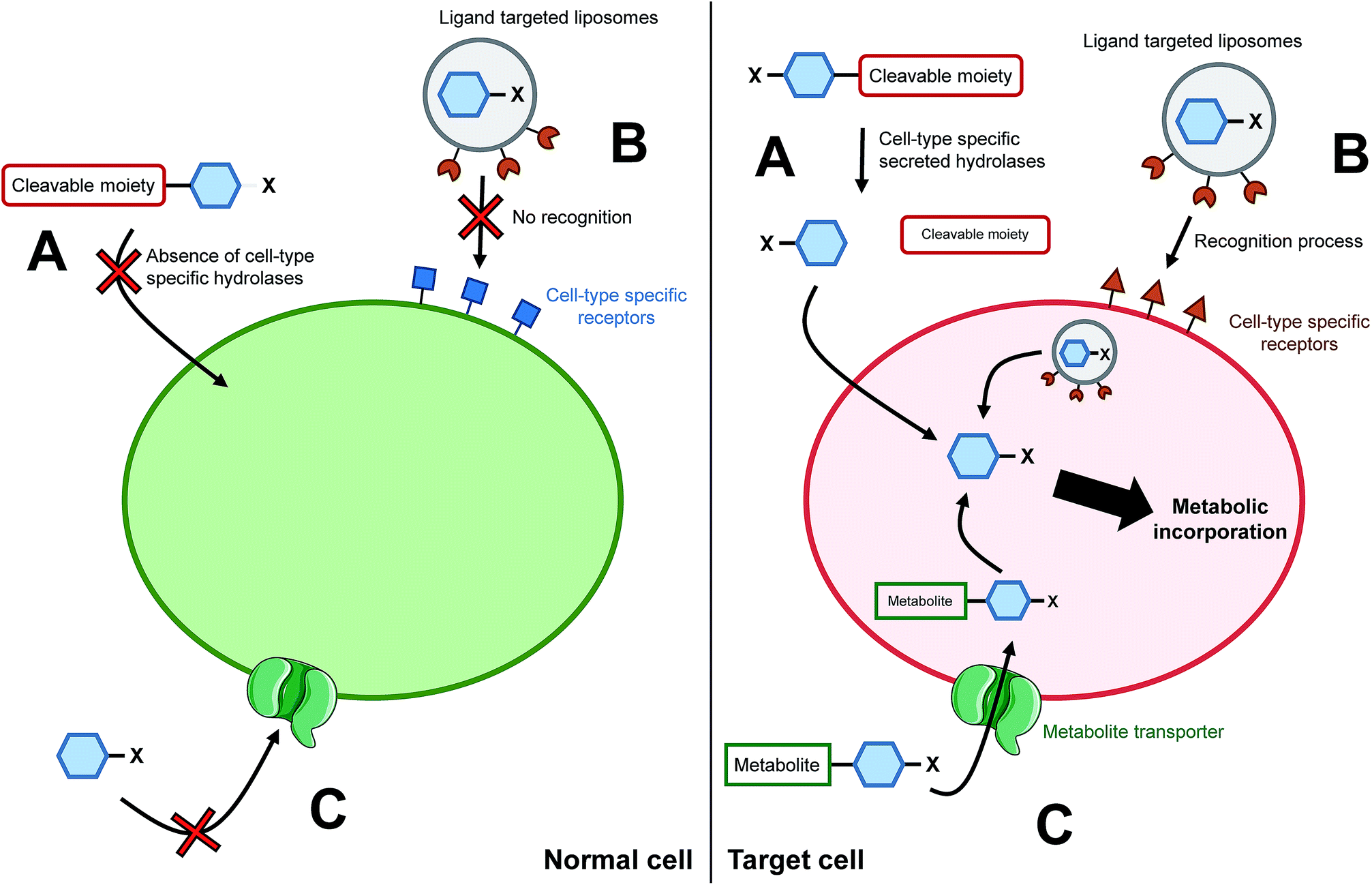 | ||
| Fig. 3 Cell/tissue targeting strategies. (A) The chemically modified monosaccharide (blue hexagon) bears a moiety able to be selectively cleaved off by enzymes secreted only by targeted cells. In normal cells, the chemical reporter does not enter the cell whereas in the target cells, the unnatural monosaccharide does go through the plasma membrane upon cleavage of the moiety. (B) The unnatural monosaccharide is encapsulated into a ligand functionalized liposome. Liposomes' ligands are designed to interact only with receptors specific of the targeted cells, allowing the selective delivery of the monosaccharide into desired cells. (C) Entry of an unnatural monosaccharide functionalized with a metabolite which has a specific carrier. Once into the cytosol, the metabolite is cleaved and the modified sugar can be metabolized. | ||
In 2010, Chang et al. applied such a strategy with a caged metabolic sugar. An azido analog of ManNAc (ManNAz) was functionalized with a peptidic linker, designed to be used as a substrate by a secreted cancer-specific protease. ManNAz could be incorporated only into cancerous cells upon the enzymatic hydrolysis of the peptidic linker (Fig. 3A).73 A similar approach was recently reported with a caspase-3/-7 cleavable peptide conjugated monosaccharide, allowing direct apoptosis specific imaging in living cells.74 Finally, another interesting development for the cell targeting is the encapsulation of the reporter monosaccharide into a delivering system. Xie et al. produced to that aim ligand-targeted liposomes containing a modified monosaccharide.75 ManNAz-containing liposomes were functionalized with folate in order to target cells which expressed folate receptor – folate receptors are overexpressed in many epithelial-derived tumors (Fig. 3B). The authors could observe that the staining was much more significant when the azido-monosaccharide was incorporated into the functionalized liposomes. The strategy has been used for different receptors allowing imaging into organs,76 but also into mice brains in vivo.77 These innovative methodologies provide promising tools to answer one of the major drawbacks when performing chemistry in living systems: to reach the desired target. Specific cell-targeting strategies have great potential for selective imaging and/or eventual clinical applications.
Targeting specific interacting glycans with cross-linking
Cross-linking has been vastly used for the study of interactions between a protein and its ligand. It consists in the formation of a covalent bond between the substrate and the enzyme in order to target and identify a protein. The process reveals to be very powerful, especially for the study of partners which exhibit a weak affinity as it is often the case for glycan–protein binding interactions. The crucial step for cross-linking is the ability to initiate in situ the reaction between the ligand and the protein and photo-triggered reactions are precious tools to reach this specificity.In 1997, Shapiro et al. were able to selectively label tetanus toxin with a synthesized analog of ganglioside GD1b.78 The terminal α2,8 linked sialic acid residue was modified with the introduction of an azido-iodobenzene derivative in place of the glycerol chain of N-acetyl neuraminic acid. After photolysis, the functionalized GD1b was covalently bound to the ganglioside binding domain, which could be subsequently isolated and identified. A few years later, Han et al. proposed the first MOE-combined cross-linking application with the use of a synthetic analog of Neu5Ac bearing a phenyl-azide moiety in position 9 (Fig. 4, 2). This modified sialic acid is indeed metabolized by cell machinery and expressed at the cellular membrane. After UV activation, a cross-linking ligation could be observed between the incorporated reporter and CD22, a sialic acid binding protein.79 In this original study, Han et al. showed that cross-linking is a powerful tool for the in situ study of protein–glycan interactions. However, the size of the phenyl azide moiety hinders the incorporation of the reporter, limiting the application to specific cell lines.
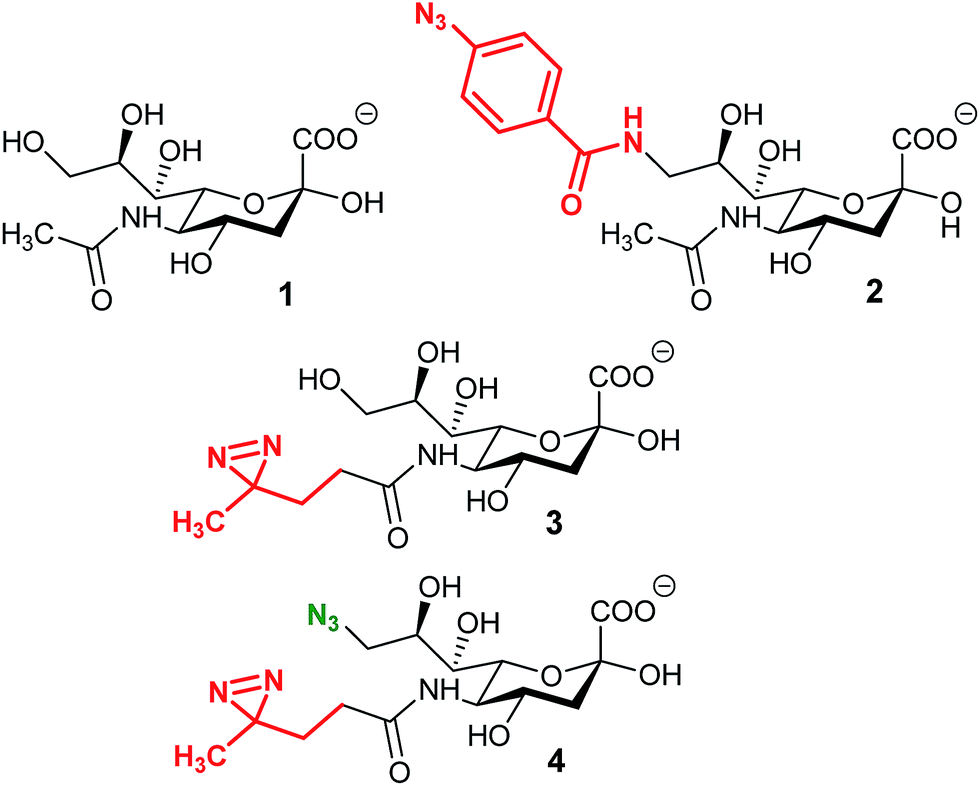 | ||
| Fig. 4 Cross-linking reporters. Structures of reported analogs of sialic acid used for the cross-linking strategy. (1) N-Acetyl neuraminic acid, (2) 9-phenylazido-neuraminic acid, SiaAAz, (3) SiaDAz, and (4) 9-azido-SiaDAz. | ||
An alternative was reported with the synthesis of ManDAz, a diazirine functionalized analog of ManNAc which can be readily metabolized into SiaDaz (Fig. 4, 3).80 The small size and relative stability under physiologic conditions of the diazirine group provides a fine reporter for metabolic incorporation. Once introduced into the glycans, diazirine compounds can be readily excited with UV irradiation (365 nm). Dinitrogen is released resulting in the formation of extremely reactive carbenes which are prone to reaction with electrophilic partners. The proof of concept was obtained with the visualization of interaction between sialylated glycans and the CD22 receptor80 and more recently with cholera toxin subunit B.81 The remaining challenge lies in the protein–glycan complex detection step. Indeed, the strategies described above imply that the nature of the protein partner is known and then can be specifically detected.82 An elegant solution has been proposed with the use of bi-functionalized sialic acid analogs (Fig. 4, 3). First, the diazirine group in position 5 allows the cross-linking reaction with an unknown protein. Then, an azide function introduced onto the position 9 can be used for complex enrichment, detection, and even purification with affinity chromatography.83 Such tools could be very powerful for the identification of unknown proteins involved in transport or recognition processes of glycosylation.
Detecting the reporter
Once installed in the target, using the desired metabolic pathway, the reporter has to be detected. One great advantage of the bioorthogonal reactions previously introduced is their versatility allowing us to specifically conjugate any probe on the reporter. Techniques for glycan imaging using bioorthogonal chemistry have already been reviewed and will not be detailed here.84,85 The recent advances in imaging techniques and especially in optical microscopy have provided great tools for glycoconjugate visualization. Mass spectrometry glycoproteomics also took advantage of MOE for glycoprotein enrichment. This approach has been comprehensively reviewed by Palaniappan and Bertozzi.86 We chose herein to focus on two less widespread strategies: Raman spectroscopy which provides a unique opportunity for direct detection of unnatural monosaccharides, and rolling circle amplification which could provide a way to easily enhance the sensitivity of detection.Direct detection with Raman spectroscopy
Traditional approaches for visualizing glycans rely on a limited number of reagents like fluorescently labeled antibodies and lectins. Bioorthogonal approaches have increased this toolkit, but the direct detection of unnatural sugars would have significant advantages, particularly in living systems. However, without the flexibility of further chemical derivatization, the available probes are limited by the promiscuity of the glycan biosynthetic pathway enzymes. For example, directly detectable metabolic probes with high sensitivity have traditionally included fluorescent dyes; however, these structures are generally too large to be metabolically tolerated as functional groups for metabolic monosaccharide reporters. Recently, Raman microscopy and spectroscopy were reported for the direct detection of unnatural sugars on the cell surface.87 Unlike traditional MOE and fluorescence detection methods, Raman spectroscopy uses Raman scattering to detect specific vibrational signals of molecules of any size, down to a specific bond of a single molecule. Raman signals for natural sugars consist of mostly C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrational stretches which overlap with those from proteins and other biomolecules in the surrounding environment. This high background can be overcome with the use of unnatural monosaccharides with unique Raman signatures outside of the vibrational stretches produced by endogenous biomolecules. Bioorthogonal Raman reporters for glycans are therefore defined as monosaccharides with vibrational signatures in the Raman-silent region of a cell (between 1800 and 2800 cm−1). Classic MOE functional groups such as alkynes and azides have been reported to produce Raman signals within the Raman-silent region of cells making them excellent bioorthogonal Raman reporters.
O vibrational stretches which overlap with those from proteins and other biomolecules in the surrounding environment. This high background can be overcome with the use of unnatural monosaccharides with unique Raman signatures outside of the vibrational stretches produced by endogenous biomolecules. Bioorthogonal Raman reporters for glycans are therefore defined as monosaccharides with vibrational signatures in the Raman-silent region of a cell (between 1800 and 2800 cm−1). Classic MOE functional groups such as alkynes and azides have been reported to produce Raman signals within the Raman-silent region of cells making them excellent bioorthogonal Raman reporters.
The major limitation for expanding the use of Raman detection to glycans is the inherently low signal produced by these Raman probes and therefore low sensitivity. Some of the first successful uses of Raman probes in cells have relied on the high accumulation of the probe within a small area to produce a high local concentration of the Raman signal and subsequently higher sensitivity. Glycans and their monosaccharide building blocks, on the other hand, are dispersed throughout, the entirety of the cell including the surface and cannot be imaged based on a concentrated point. To address this limitation, Raman enhancing techniques have been used to amplify the low signal and visualize glycans on the surface of live cells. For example, surface-enhanced Raman scattering (SERS) for the direct detection of sialylated glycans on live cells.87 Using gold nanoparticles (AuNPs) functionalized with sialic acid-binding 4-mercaptophenylboronic acid (MPBA), they were able to amplify the Raman signals of SiaNAlk and SiaNAz of Ac4ManNAlk and Ac4ManNAz-treated HeLa cells, respectively. They also demonstrated the ability to eliminate the secondary AuNP treatment by culturing cells with the Raman reporters directly on SERS substrates.88 Shortly after, a zone-controllable SERS method tailored cell-surface glycan detection to a specific protein.89 Several other lectin and boronic acid based recognition methods have been combined with SERS for the detection of glycans.90–93 Lectin-based recognition methods have been used in combination with SERS to differentiate and quantify natural glycan types on cell surfaces of both healthy and cancerous cells.92,93 Functionalization of boronic-acid derivatives such as MPBA has recently been shown to detect natural sialic acid structures on the surface of live cells using SERS.90,91 These studies were able to show a correlation of sialic acid expression with cancer cells and Liang et al. have recently optimized this method to monitor real-time cell surface SA expression within a linear detection range using sialidase and the reverse transcriptase inhibitor AZT.91 Due to the fact that natural sialic acid can be detected without the use of azides or other unnatural functionalities, this method can be used as a readout for endogenous levels of modification without perturbing any modification event by using high concentrations of unnatural sugars. In addition to SERS, tip-enhanced Raman scattering (TERS) has been reported for the detection of protein glycosylation as well as the characterization of which specific glycan types were present.94 These amplified signals from combining Raman enhancement techniques with metabolic glycan reporters provided high sensitivity, excellent signal specificity, and compatibility with a broad spectrum of Raman reporter functionalities. The development of bioorthogonal Raman reporters provides an exciting new frontier for MOE direct-detection and additional enhancing techniques are currently expanding its use.94 Currently the application of Raman scattering to glycan MOE is restricted to relatively specialized equipment and techniques; however, the continued development of the technique will undoubtedly increase its application.
Rolling circle amplification strategies
Although being very robust, the MOE strategy may encounter some drawbacks mainly due to the variety of substrates and the labeling efficiency of the target glycan which can lead to a signal too weak for efficient detection/quantification. DNA probes have been recently developed as a potential way to increase the signal in MOE experiments. After incubation with per-O-acetylated ManNAz (and per-O-acetylated GalNAz as negative control) in human breast cancer MCF-7 cells, Chen et al. developed an “hybridization and cleavage” process with DNA barcodes allowing the in situ quantification of epithelial cell adhesion molecule-specific sialic acid on MCF-7.95 A similar strategy was proposed by He et al. who encoded lectins with DNA primers to yield long single-stranded DNA by rolling circle amplification (RCA).96 Analytical information was then obtained by MS. The same RCA strategy was published in the same year but with a CuAAC reaction between the incorporated monosaccharide and a fluorescent probe followed by detection with fluorescence microscopy.97 An elegant strategy named Glyco-seek by Robinson et al. allows the ultrasensitive detection (attomoles of glycoproteins from cell lysates) of O-GlcNAcylation with quantitative polymerase chain reaction (qPCR) after MGE with per-O-acetylated GalNAz and click-chemistry reaction with alkynyl biotin.98 The RCA strategy is an interesting alternative which should allow the detection of small amounts of incorporated unnatural monosaccharides.Conclusions
Metabolic oligosaccharide engineering, simultaneously with the breakthrough in bioorthogonal chemistry, has allowed tremendous progress in glycobiology, notably in terms of visualization, quantification, monitoring and localization of glycans. This review aims to highlight some very recent studies, which all result from the great expansion of MOE in the last two decades. These strategies and methodologies presented and discussed herein share a high contribution from chemical biologists and a strong potential to enhance our understanding of glycosylation: metabolic crosstalk provides tools for great insights into the metabolic pathways and their interconnection. Enzymatic strategies allow at the same time the selection and labelling of in vivo specific glycan-acceptors and the study of the preferences and characteristics of glycosyltransferases. MOE combined with cross-linking opens the way for the comprehension of protein–glycan interactions. Enrichment of glycoconjugates with MOE for mass spectrometry has been and probably will be a powerful tool for glycoproteomics. These different applications, in addition to illustrating the groundbreaking capacities of chemical biology, should contribute to considerable advances in the understanding of glycosylation processes.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- J. C. Paulson, J. E. Sadler and R. L. Hill, J. Biol. Chem., 1979, 254, 2120–2124 CAS.
- R. Brossmer, U. Rose, D. Kasper, T. L. Smith, H. Grasmuk and F. M. Unger, Biochem. Biophys. Res. Commun., 1980, 96, 1282–1289 CrossRef CAS PubMed.
- H.-J. Gross, A. Bünsch, J. C. Paulson and R. Brossmer, Eur. J. Biochem., 1987, 168, 595–602 CrossRef CAS PubMed.
- H. J. Gross and R. Brossmer, Glycoconjugate J., 1988, 5, 411–417 CrossRef CAS.
- A. D. Elbein, Annu. Rev. Biochem., 1987, 56, 497–534 CrossRef CAS PubMed.
- H.-J. Grünholz, E. Harms, M. Opetz and W. Reutter, Carbohydr. Res., 1981, 96, 259–270 CrossRef.
- H. Kayser, C. C. Geilen, C. Paul, R. Zeitler and W. Reutter, FEBS Lett., 1992, 301, 137–140 CrossRef CAS PubMed.
- H. Kayser, R. Zeitler, C. Kannicht, D. Grunow, R. Nuck and W. Reutter, J. Biol. Chem., 1992, 267, 16934–16938 CAS.
- O. T. Keppler, P. Stehling, M. Herrmann, H. Kayser, D. Grunow, W. Reutter and M. Pawlita, J. Biol. Chem., 1995, 270, 1308–1314 CrossRef CAS PubMed.
- J. Du, M. A. Meledeo, Z. Wang, H. S. Khanna, V. D. P. Paruchuri and K. J. Yarema, Glycobiology, 2009, 19, 1382–1401 CrossRef CAS PubMed.
- P. R. Wratil, R. Horstkorte and W. Reutter, Angew. Chem., Int. Ed., 2016, 55, 9482–9512 CrossRef CAS PubMed.
- K. N. Chuh, A. R. Batt and M. R. Pratt, Cell Chem. Biol., 2016, 23, 86–107 CrossRef CAS PubMed.
- K. N. Chuh and M. R. Pratt, Curr. Opin. Chem. Biol., 2015, 24, 27–37 CrossRef CAS PubMed.
- M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475–484 CrossRef CAS PubMed.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14846–14851 CrossRef CAS PubMed.
- L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125–1128 CrossRef CAS PubMed.
- E. Saxon, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC.
- A. Niederwieser, A.-K. Späte, L. D. Nguyen, C. Jüngst, W. Reutter and V. Wittmann, Angew. Chem., Int. Ed., 2013, 52, 4265–4268 CrossRef CAS PubMed.
- H. Wu and N. K. Devaraj, Top. Curr. Chem., 2016, 374, 109 CrossRef PubMed.
- A. Dumont, A. Malleron, M. Awwad, S. Dukan and B. Vauzeilles, Angew. Chem., Int. Ed., 2012, 51, 3143–3146 CrossRef CAS PubMed.
- F. Liu, A. J. Aubry, I. C. Schoenhofen, S. M. Logan and M. E. Tanner, ChemBioChem, 2009, 10, 1317–1320 CrossRef CAS PubMed.
- B. M. Swarts, C. M. Holsclaw, J. C. Jewett, M. Alber, D. M. Fox, M. S. Siegrist, J. A. Leary, R. Kalscheuer and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 16123–16126 CrossRef CAS PubMed.
- S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS PubMed.
- C. T. Anderson, I. S. Wallace and C. R. Somerville, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1329–1334 CrossRef CAS PubMed.
- K. J. Yarema, L. K. Mahal, R. E. Bruehl, E. C. Rodriguez and C. R. Bertozzi, J. Biol. Chem., 1998, 273, 31168–31179 CrossRef CAS PubMed.
- L. K. Mahal, N. W. Charter, K. Angata, M. Fukuda, D. E. Koshland Jr and C. R. Bertozzi, Science, 2001, 294, 380 CrossRef CAS PubMed.
- D. J. Vocadlo, H. C. Hang, E.-J. Kim, J. A. Hanover and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9116–9121 CrossRef CAS PubMed.
- K. W. Dehnert, B. J. Beahm, T. T. Huynh, J. M. Baskin, S. T. Laughlin, W. Wang, P. Wu, S. L. Amacher and C. R. Bertozzi, ACS Chem. Biol., 2011, 6, 547–552 CrossRef CAS PubMed.
- K. N. Chuh, B. W. Zaro, F. Piller, V. Piller and M. R. Pratt, J. Am. Chem. Soc., 2014, 136, 12283–12295 CrossRef CAS PubMed.
- D. L. Shen, T.-W. Liu, W. Zandberg, T. Clark, R. Eskandari, M. G. Alteen, H. Y. Tan, Y. Zhu, S. Cecioni and D. Vocadlo, ACS Chem. Biol., 2017, 12, 206–213 CrossRef CAS PubMed.
- A. E. Speers and B. F. Cravatt, Chem. Biol., 2004, 11, 535–546 CrossRef CAS PubMed.
- T.-L. Hsu, S. R. Hanson, K. Kishikawa, S.-K. Wang, M. Sawa and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2614–2619 CrossRef CAS PubMed.
- B. W. Zaro, Y.-Y. Yang, H. C. Hang and M. R. Pratt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8146–8151 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova, B. Xie, D. N. Kamber and J. A. Prescher, J. Am. Chem. Soc., 2012, 134, 18638–18643 CrossRef CAS PubMed.
- A.-K. Späte, V. F. Schart, S. Schöllkopf, A. Niederwieser and V. Wittmann, Chem.–Eur. J., 2014, 20, 16502–16508 CrossRef PubMed.
- S. Stairs, A. A. Neves, H. Stöckmann, Y. A. Wainman, H. Ireland-Zecchini, K. M. Brindle and F. J. Leeper, ChemBioChem, 2013, 14, 1063–1067 CrossRef CAS PubMed.
- L. Josa-Culleré, Y. A. Wainman, K. M. Brindle and F. J. Leeper, RSC Adv., 2014, 4, 52241–52244 RSC.
- A. R. Sherratt, M. Chigrinova, C. S. McKay, L.-P. B. Beaulieu, Y. Rouleau and J. P. Pezacki, RSC Adv., 2014, 4, 46966–46969 RSC.
- A.-K. Späte, J. E. G. A. Dold, E. Batroff, V. F. Schart, D. E. Wieland, O. R. Baudendistel and V. Wittmann, ChemBioChem, 2016, 17, 1374–1383 CrossRef PubMed.
- L. F. Leloir, Science, 1971, 172, 1299–1303 CrossRef CAS PubMed.
- E. Saxon, S. J. Luchansky, H. C. Hang, C. Yu, S. C. Lee and C. R. Bertozzi, J. Am. Chem. Soc., 2002, 124, 14893–14902 CrossRef CAS PubMed.
- D. M. Patterson, K. A. Jones and J. A. Prescher, Mol. BioSyst., 2014, 10, 1693 RSC.
- M. Dumont, A. Lehner, B. Vauzeilles, J. Malassis, A. Marchant, K. Smyth, B. Linclau, A. Baron, J. Mas Pons, C. T. Anderson, D. Schapman, L. Galas, J.-C. Mollet and P. Lerouge, Plant J., 2016, 85, 437–447 CrossRef CAS PubMed.
- J. E. Hudak, D. Alvarez, A. Skelly, U. H. von Andrian and D. L. Kasper, Nat. Microbiol., 2017, 2, 17099 CrossRef CAS PubMed.
- N. D. Pham, C. S. Fermaintt, A. C. Rodriguez, J. E. McCombs, N. Nischan and J. J. Kohler, Glycoconjugate J., 2015, 32, 515–529 CrossRef CAS PubMed.
- P. A. Gilormini, C. Lion, D. Vicogne, T. Levade, S. Potelle, C. Mariller, Y. Guérardel, C. Biot and F. Foulquier, Chem. Commun., 2016, 52, 2318–2321 RSC.
- N. Khidekel, S. Arndt, N. Lamarre-Vincent, A. Lippert, K. G. Poulin-Kerstien, B. Ramakrishnan, P. K. Qasba and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2003, 125, 16162–16163 CrossRef CAS PubMed.
- N. Khidekel, S. B. Ficarro, E. C. Peters and L. C. Hsieh-Wilson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13132–13137 CrossRef CAS PubMed.
- T. Zheng, H. Jiang, M. Gros, D. Soriano del Amo, S. Sundaram, G. Lauvau, F. Marlow, Y. Liu, P. Stanley and P. Wu, Angew. Chem., Int. Ed., 2011, 50, 4113–4118 CrossRef CAS PubMed.
- J.-L. Chaubard, C. Krishnamurthy, W. Yi, D. F. Smith and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2012, 134, 4489–4492 CrossRef CAS PubMed.
- Q. Li, Z. Li, X. Duan and W. Yi, J. Am. Chem. Soc., 2014, 136, 12536–12539 CrossRef CAS PubMed.
- Z. L. Wu, A. D. Person, M. Anderson, B. Burroughs, T. Tatge, K. Khatri, Y. Zou, L. Wang, T. Geders, J. Zaia and R. Sackstein, Glycobiology, 2018, 28, 69–79 CrossRef PubMed.
- N. E. Mbua, X. Li, H. R. Flanagan-Steet, L. Meng, K. Aoki, K. W. Moremen, M. A. Wolfert, R. Steet and G.-J. Boons, Angew. Chem., Int. Ed., 2013, 52, 13012–13015 CrossRef CAS PubMed.
- X. Li, T. Fang and G.-J. Boons, Angew. Chem., Int. Ed., 2014, 53, 7179–7182 CrossRef CAS PubMed.
- C. J. Capicciotti, C. Zong, M. O. Sheikh, T. Sun, L. Wells and G.-J. Boons, J. Am. Chem. Soc., 2017, 139, 13342–13348 CrossRef CAS PubMed.
- S.-H. Yu, P. Zhao, T. Sun, Z. Gao, K. W. Moremen, G.-J. Boons, L. Wells and R. Steet, J. Biol. Chem., 2016, 291, 3982–3989 CrossRef CAS PubMed.
- A. Lopez Aguilar, J. G. Briard, L. Yang, B. Ovryn, M. S. Macauley and P. Wu, ACS Chem. Biol., 2017, 12, 611–621 CrossRef CAS PubMed.
- H. C. Hang, C. Yu, M. R. Pratt and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 6–7 CrossRef CAS PubMed.
- Z. L. Wu, X. Huang, A. J. Burton and K. A. D. Swift, Carbohydr. Res., 2015, 412, 1–6 CrossRef CAS PubMed.
- Z. L. Wu, X. Huang, A. J. Burton and K. A. D. Swift, Glycobiology, 2016, 26, 329–334 CrossRef CAS PubMed.
- M. Noel, P.-A. Gilormini, V. Cogez, N. Yamakawa, D. Vicogne, C. Lion, C. Biot, Y. Guérardel and A. Harduin-Lepers, ChemBioChem, 2017, 18, 1251–1259 CrossRef CAS PubMed.
- P.-A. Gilormini, C. Lion, M. Noel, M.-A. Krzewinski-Recchi, A. Harduin-Lepers, Y. Guérardel and C. Biot, Glycobiology, 2016, 26, 1151–1156 CAS.
- A. Shajahan, S. Parashar, S. Goswami, S. M. Ahmed, P. Nagarajan and S.-G. Sampathkumar, J. Am. Chem. Soc., 2017, 139, 693–700 CrossRef CAS PubMed.
- H. Wang, R. Wang, K. Cai, H. He, Y. Liu, J. Yen, Z. Wang, M. Xu, Y. Sun, X. Zhou, Q. Yin, L. Tang, I. T. Dobrucki, L. W. Dobrucki, E. J. Chaney, S. A. Boppart, T. M. Fan, S. Lezmi, X. Chen, L. Yin and J. Cheng, Nat. Chem. Biol., 2017, 13, 415–424 CrossRef CAS PubMed.
- P. V. Chang, D. H. Dube, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 9516–9518 CrossRef CAS PubMed.
- M. K. Shim, H. Y. Yoon, S. Lee, M. K. Jo, J. Park, J.-H. Kim, S. Y. Jeong, I. C. Kwon and K. Kim, Sci. Rep., 2017, 7, 16635 CrossRef PubMed.
- R. Xie, S. Hong, L. Feng, J. Rong and X. Chen, J. Am. Chem. Soc., 2012, 134, 9914–9917 CrossRef CAS PubMed.
- R. Xie, L. Dong, R. Huang, S. Hong, R. Lei and X. Chen, Angew. Chem., Int. Ed., 2014, 53, 14082–14086 CrossRef CAS PubMed.
- R. Xie, L. Dong, Y. Du, Y. Zhu, R. Hua, C. Zhang and X. Chen, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5173–5178 CrossRef CAS PubMed.
- R. E. Shapiro, C. D. Specht, B. E. Collins, A. S. Woods, R. J. Cotter and R. L. Schnaar, J. Biol. Chem., 1997, 272, 30380–30386 CrossRef CAS PubMed.
- S. Han, B. E. Collins, P. Bengtson and J. C. Paulson, Nat. Chem. Biol., 2005, 1, 93–97 CrossRef CAS PubMed.
- Y. Tanaka and J. J. Kohler, J. Am. Chem. Soc., 2008, 130, 3278–3279 CrossRef CAS PubMed.
- A. M. Wands, A. Fujita, J. E. McCombs, J. Cervin, B. Dedic, A. C. Rodriguez, N. Nischan, M. R. Bond, M. Mettlen, D. C. Trudgian, A. Lemoff, M. Quiding-Järbrink, B. Gustavsson, C. Steentoft, H. Clausen, H. Mirzaei, S. Teneberg, U. Yrlid and J. J. Kohler, eLife, 2015, 4, e09545 CrossRef PubMed.
- M. R. Bond, H. Zhang, P. D. Vu and J. J. Kohler, Nat. Protoc., 2009, 4, 1044–1063 CrossRef CAS PubMed.
- L. Feng, S. Hong, J. Rong, Q. You, P. Dai, R. Huang, Y. Tan, W. Hong, C. Xie, J. Zhao and X. Chen, J. Am. Chem. Soc., 2013, 135, 9244–9247 CrossRef CAS PubMed.
- A. Godinat, H. Karatas, G. Budin and E. A. Dubikovskaya, in Chemical Ligation, ed. L. D. D'Andrea and A. Romanelli, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2017, pp. 447–483 Search PubMed.
- B. Ovryn, J. Li, S. Hong and P. Wu, Curr. Opin. Chem. Biol., 2017, 39, 39–45 CrossRef CAS PubMed.
- K. K. Palaniappan and C. R. Bertozzi, Chem. Rev., 2016, 116, 14277–14306 CrossRef CAS PubMed.
- L. Lin, X. Tian, S. Hong, P. Dai, Q. You, R. Wang, L. Feng, C. Xie, Z.-Q. Tian and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 7266–7271 CrossRef CAS PubMed.
- M. Xiao, L. Lin, Z. Li, J. Liu, S. Hong, Y. Li, M. Zheng, X. Duan and X. Chen, Chem.–Asian J., 2014, 9, 2040–2044 CrossRef CAS PubMed.
- Y. Chen, L. Ding, W. Song, M. Yang and H. Ju, Chem. Sci., 2016, 7, 569–574 RSC.
- M. Tabatabaei, G. Q. Wallace, F. A. Caetano, E. R. Gillies, S. S. G. Ferguson and F. Lagugné-Labarthet, Chem. Sci., 2016, 7, 575–582 RSC.
- L. Liang, H. Qu, B. Zhang, J. Zhang, R. Deng, Y. Shen, S. Xu, C. Liang and W. Xu, Biosens. Bioelectron., 2017, 94, 148–154 CrossRef CAS PubMed.
- Y. Chen, L. Ding, J. Xu, W. Song, M. Yang, J. Hu and H. Ju, Chem. Sci., 2015, 6, 3769–3774 RSC.
- D. Craig, S. McAughtrie, J. Simpson, C. McCraw, K. Faulds and D. Graham, Anal. Chem., 2014, 86, 4775–4782 CrossRef CAS PubMed.
- D. P. Cowcher, T. Deckert-Gaudig, V. L. Brewster, L. Ashton, V. Deckert and R. Goodacre, Anal. Chem., 2016, 88, 2105–2112 CrossRef CAS PubMed.
- Y. Chen, L. Ding, W. Song, M. Yang and H. Ju, Anal. Chem., 2016, 88, 2923–2928 CrossRef CAS PubMed.
- Z. He, Q. Chen, F. Chen, J. Zhang, H. Li and J.-M. Lin, Chem. Sci., 2016, 7, 5448–5452 RSC.
- X. Zhang, R. Li, Y. Chen, S. Zhang, W. Wang and F. Li, Chem. Sci., 2016, 7, 6182–6189 RSC.
- P. V. Robinson, C. Tsai, A. E. de Groot, J. L. McKechnie and C. R. Bertozzi, J. Am. Chem. Soc., 2016, 138, 10722–10725 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |