DOI:
10.1039/C8SC01943F
(Perspective)
Chem. Sci., 2018,
9, 7027-7042
Adaptive responses of sterically confined intramolecular chalcogen bonds†
Received
29th April 2018
, Accepted 24th July 2018
First published on 25th July 2018
Abstract
The responsive behavior of an entity towards its immediate surrounding is referred to as an adaptive response. The adaptive responses of a noncovalent interaction at the molecular scale are reflected from its structural and functional roles. Intramolecular chalcogen bonding (IChB), an attractive interaction between a heavy chalcogen E (E = Se or Te) centered sigma hole and an ortho-heteroatom Lewis base donor D (D = O or N), plays an adaptive role in defining the structure and reactivity of arylchalcogen compounds. In this perspective, we describe the adaptive roles of a chalcogen centered Lewis acid sigma hole and a proximal Lewis base (O or N) in accommodating built-in steric stress in 2,6-disubstituted arylchalcogen compounds. From our perspective, the IChB components (a sigma hole and the proximal Lewis base) act in synergism to accommodate the overwhelming steric force. The adaptive responses of the IChB components are inferred from the observed molecular structures and reactivity. These include (a) adaptation of a conformation without IChBs, (b) adaptation of a conformation with weak IChBs, (c) twisting the skeletal aryl ring while maintaining IChBs, (d) ionization of the E–X bond (e.g., X = Br) to relieve stress and (e) intramolecular cyclization to relieve steric stress. A comprehensive approach, involving X-ray data analysis, density functional theory (DFT) calculations, reaction pattern analysis and principal component analysis (PCA), has been employed to rationalize the adaptive behaviors of IChBs in arylchalcogen compounds. We believe that the perception of ChB as an adaptive/stimulus responsive interaction would profit the futuristic approaches that would utilise ChB as self-assembly and molecular recognition tools.
| Dr Selvakumar, born in 1980 in Tamil Nadu, India, did his Bachelor of Science (B. Sc) in the year 2001, Vivekananda College, Madurai, India. He did his master's degree at The American College, India in 2004. He carried out his PhD work at IIT Bombay under the mentorship of Prof. Harkesh B. Singh. Then, he moved to Weizmann Institute of Science in the year 2011 to work with Dr David Margulies as a postdoctoral fellow. Currently, he has been working as an INSPIRE Faculty at Central Electrochemical Research Institute, Karaikudi, India, since 2015. His current research interests are chalcogen bonding and biomimetic recognition chemistry. |
| Prof. Harkesh B. Singh, born in 1956 in UP, India, obtained his PhD (1979) from Lucknow University with Prof. T. N. Srivastava. In 1979, he moved to the U.K. and did a second PhD at Aston University, Birmingham, under the supervision of Prof. W. R. McWhinnie (1979–1983). After a brief stay at the Indian Institute of Technology, Delhi, as Pool Scientist with Prof. B. L. Khandelwal, he joined the Indian Institute of Technology, Bombay, in 1984 as a lecturer and rose through the ranks to become a full professor in 1995. He is a Fellow of the National Academy of Sciences (FNASc), Allahabad; Indian Academy of Sciences (FASc), Bangalore; Indian National Academy of Sciences (FNA), New Delhi and the Royal Society of Chemistry (FRSC). He has been a Visiting Scientist at the University of California, Santa Barbara (Prof. Fred Wudl's laboratory), under the BOYS-CAST scheme. His research objectives are centered around design, synthesis, and structural studies of novel organometallic derivatives of sulfur, selenium, and tellurium and their applications. |
1. Introduction
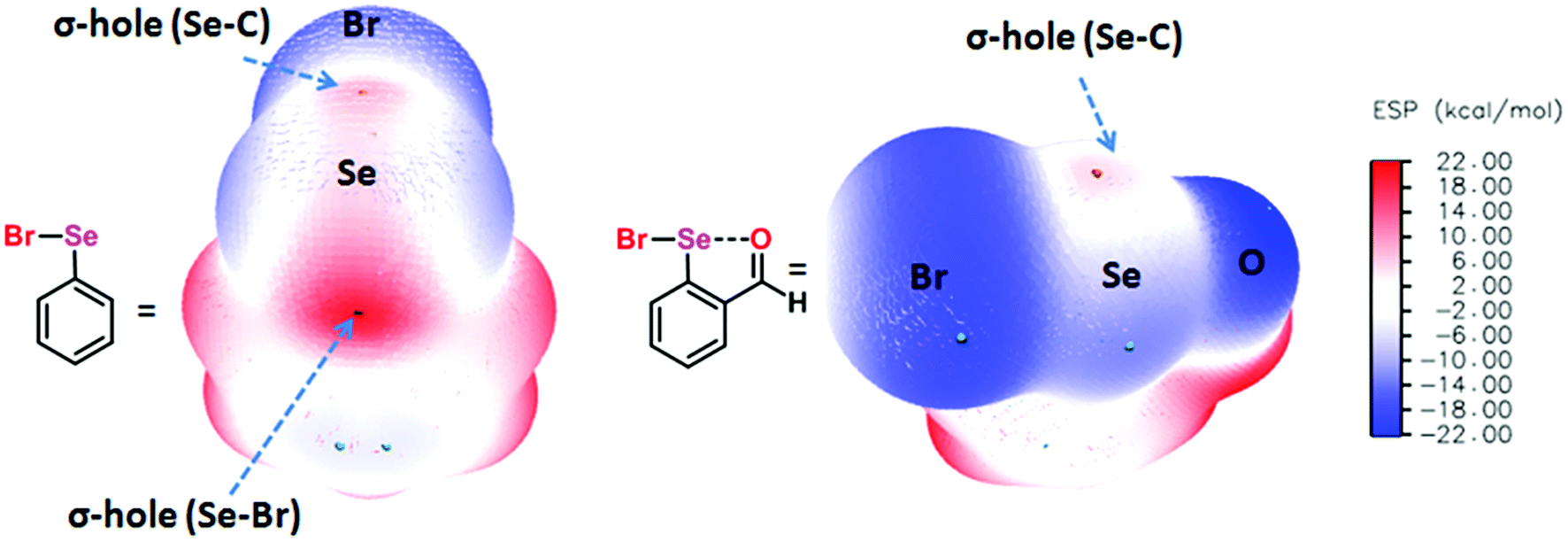
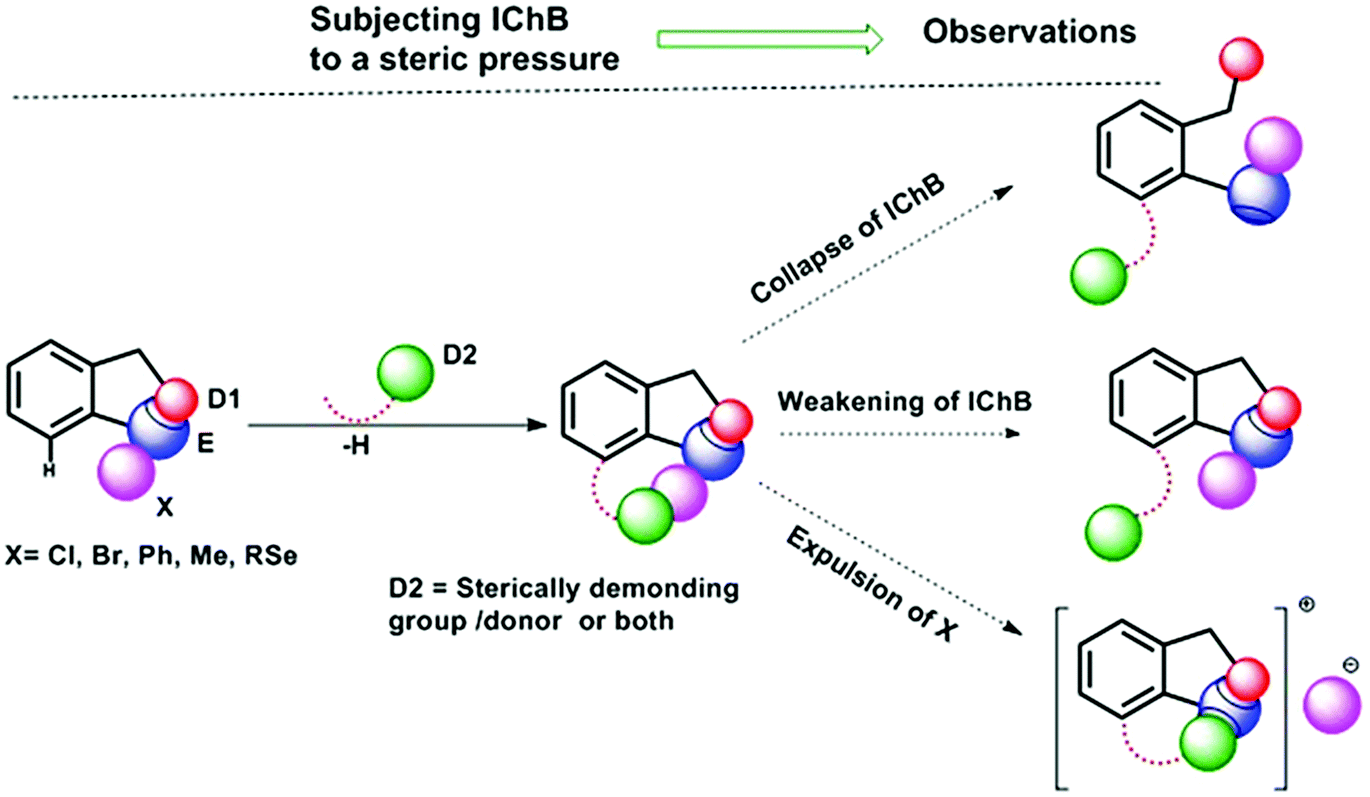
Chalcogen bonding (ChB),1 a sister interaction to halogen bonding,2 is instrumental in modulating the glutathione peroxidase (GPx)-like activity of organochalcogen compounds,3 asymmetric oxyselenenylation reactions,4 Lewis acid catalysis,5 and anion recognition.6 It plays a crucial role in supramolecular self-assemblies of chalcogenadiazoles,7 packing of chalcogen containing molecules in solid states,8 supramolecular vesicles,9 macrocycles,10 and telluroxane clusters,11 and stabilization of radicals in condensed phases.12 In molecules, it acts as a conformational lock to stabilize the systems with distinct intramolecular chalcogen bonding (IChB).13 Its potential to lock molecular conformations plays a crucial role in the stabilization of reactive selenenyl halides,13b selenenate esters,13c radical ions,14 and intermediates formed during cyclic selenium imides.15 This interaction, for many decades, has been popularly described as secondary bonding interaction (SBI) where a low-valent heavy chalcogen (E) interacts with nearby heteroatoms.16 The emergence of halogen bonding as a unique weak interaction2 and also the growing interest in chalcogen–heteroatom interactions1,3–14 have largely influenced the adoption of more specific term ChB to describe the chalcogen–heteroatom interactions. ChB has been described using different bonding models including donor–acceptor17,18 and electrostatic sigma–hole interactions.1,19 The donor–acceptor model invokes charge-transfer from the non-bonding electrons (n) of a proximal donor atom (D) to the anti-bonding orbital (σ*) of the acceptor bond (E–X) (where E = S, Se, and Te and X = Cl, Br, CN, etc.).17,18 This model follows the Rundle–Pimentel description of three center-four electron bonding (3c–4e).20 The poor sensitivity of this interaction towards a range of solvents augments this view that chalcogen bonding has predominant charge-transfer characteristics (n → σ* orbital interaction).21 The Politzer model, which adequately accounts for the structure and reactivity of a diverse class of main group organometallic/metalloid compounds, describes chalcogen bonding as an electrostatic interaction.22 This model describes that heavy main group elements in molecules contain electron deficient sites on their surface just opposite to sigma bonds (E–X) due to their soft polarizable electron clouds. The visual presentation of the anisotropic distribution of electron densities on the main group elements (E) bonded to other elements (X) is the attractive feature of this electrostatic model (Fig. 1), which provides an easy means to comprehend the reactivity of the chalcogen centre towards nucleophiles (H2O, RNH2, RSH, etc.) or towards electrophiles (metal ions and H+). In the electrostatic surface potential map (ESP), as shown in Fig. 1, the red regions represent the electron deficient surface sites, and the blue region indicates the electron-rich surface sites. The electron-deficient site on E at the backside along the extension of a sigma bond is called the sigma hole. For instance, the selenium atom in PhSeBr has two sigma holes on its surface; one at the back side along the extension of the Se–C bond and the other at the back side along the extension of the Se–Br bond (Fig. 1). In this case, the bromine atom is more electronegative than a carbon atom and acts as a strong polarizer of the selenium electron cloud than carbon. As a result, the Se–Br sigma hole serves as a potential Lewis acid site for the attack of external Lewis bases and might contribute to the instability of such molecules towards air and moisture.13b,c,22 We and others have shown in several arylchalcogen compounds that a proximal Lewis base (N or O) present at the 2-position (-ortho) not only locks the molecule in a conformation with intramolecular chalcogen bonding (IChB) but also prevents it from reacting with external nucleophiles.13c,23 For example, the oxygen atom of ortho-CHO in 2-formylphenylselenenyl bromide acts as an intramolecular Lewis base.24 Thereby, it hides the reactive Se–Br sigma hole donor (or the chalcogen bonding donor) from the approach of external nucleophiles. In general, the alignment of atoms involved (e.g. O⋯Se–Br) in IChBs is linear with a bond angle of approximately 180° and the geometry around E is T-shaped.13,18,23,24 Significant deviations from these geometric characteristics apparently reflect either the presence of a very weak IChB or none. Beyond this aspect, IChBs do display fascinating adaptive structural and reactivity modulator roles under sterically demanding conditions.25 As shown in Fig. 2, the elements involved in the D1⋯E–X interaction are present approximately in the same plane of the aryl ring. Upon subjecting it to additional pressure (e.g. incorporating substituent D2, which contains steric/donor components or both) the subjected molecular system naturally follows certain adaptive strategies to accommodate such an unanticipated pressure. As a consequence, both D1 and D2 might turn away from E or only D2 may turn away from E, or X is expelled from E. The primary goal of this article is to analyze the effect of D2 on the D1⋯E–X interaction in 2,6-disubstituted aryl systems using X-ray crystallography data analysis, conformational analysis, reaction pattern analysis, principal component analysis, and DFT calculations. Finally, we show that confining the chalcogen centered sigma hole with steric/donor components or both will pave the way to develop new organoselenium reagents and stimulus-responsive organochalcogen compounds.
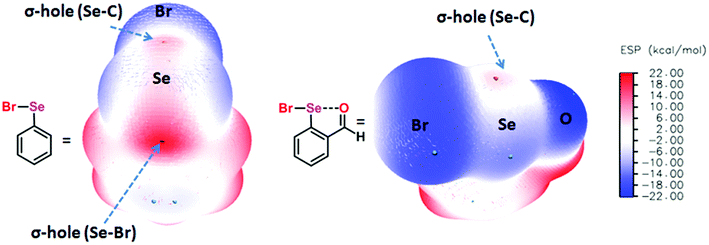 |
| | Fig. 1 The left panel shows the molecular structure of phenylselenenyl bromide and its ESP on the isodensity surface, ρ(r) = 0.001 au, showing two sigma holes on the Se atom. The middle panel shows the molecular structure of 2-formylphenylselenenyl bromide and its ESP on the isodensity surface, ρ(r) = 0.001 au, showing one sigma hole on the Se atom. The right panel shows the color scale of the ESP (e.g. C6H5SeBr; −14. 92 (blue) to 21.46 (red)). | |
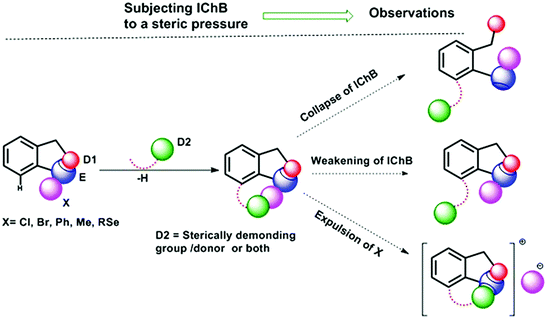 |
| | Fig. 2 The possible outcomes of subjecting IChBs (D1⋯Se–X) to additional steric stress. Replacement of the hydrogen atom present at the 6-position of the aryl ring with a sterically demanding group/donor or both leads to distinct structural and reactivity patterns. | |
2. Adaptation strategies at the molecular scale
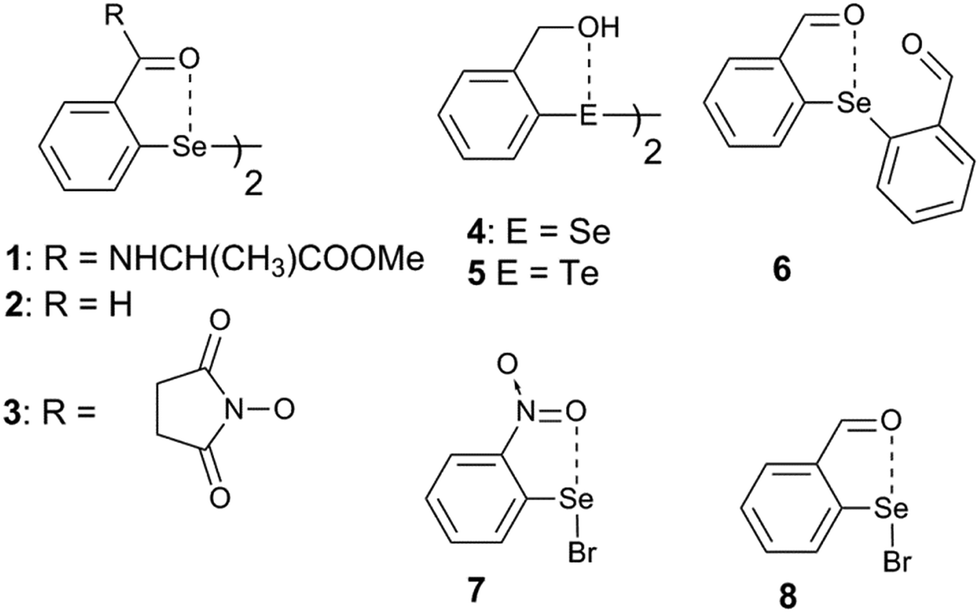
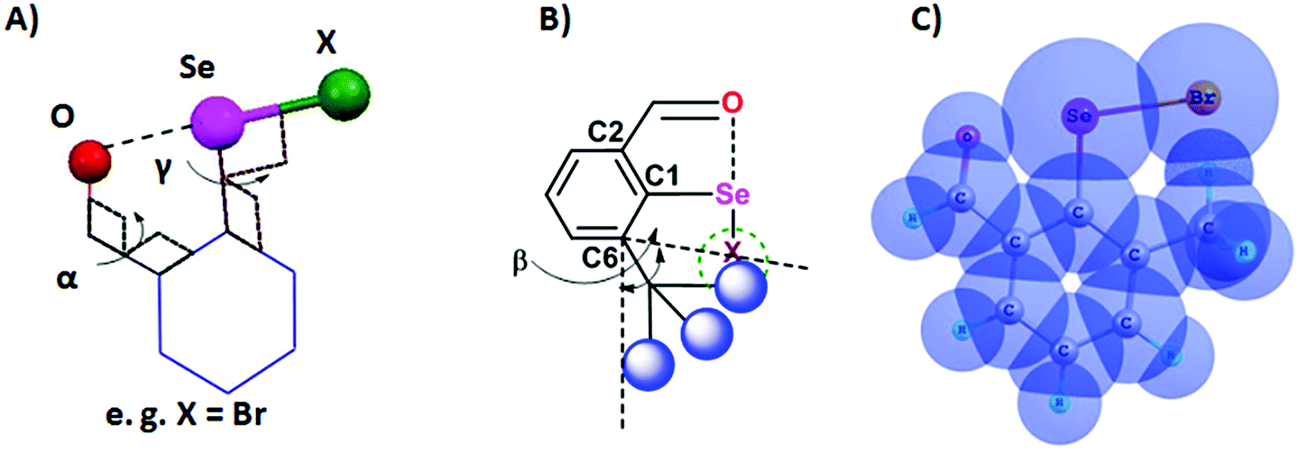
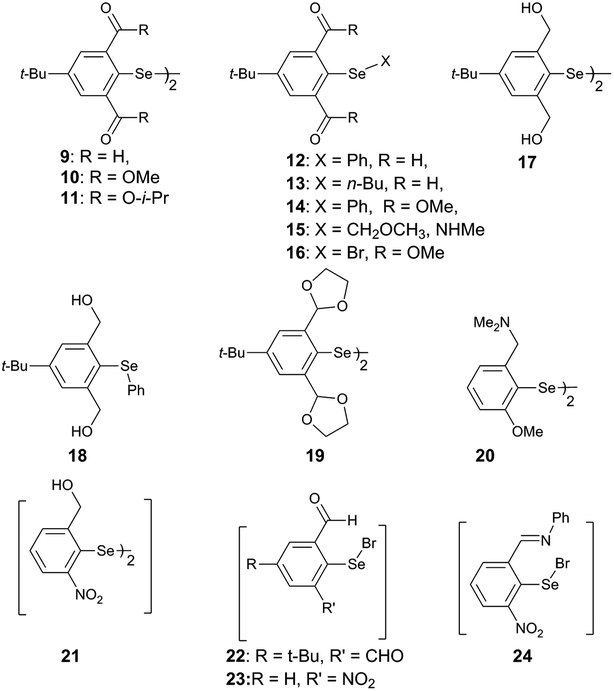
Steric and other weak interactions are the coded information that defines the energetics, structures and reactivity of molecular/supramolecular systems.26 Molecular entities tend to achieve their lowest energy states through the optimization of such interactions via torsion motions. For instance, the energetics of 2-substituted arylchalcogen compounds (1–8, Fig. 3) are largely influenced by two flexible torsion motions across α and γ torsion angles as shown in Fig. 4A. 2-Substituted arylchalcogen compounds (e.g.1–8) invariably achieve molecular structures with IChBs as they afford an additional contribution to the molecular stability. The simplest model system to illustrate the effect of the 6-substituent on IChBs is 2-formyl-6-methylphenylselenenyl bromide (Fig. 4B). In this model, the carbon atom of the methyl group is attached to the 6-carbon of the aryl ring by approximating the distance (C6–CMe) and (C1–C6–CMe) angle as 1.50 Å and 120° respectively. The torsion angle between methyl hydrogen (D2) and C1 carbon is defined as β(C1–C6–C–H). The C6 atom occupies the vertex of the cone formed by the hydrogen atom of the methyl group. The Tolman cone angle27 computed at van der Waals radii of the hydrogen atoms of the methyl group is 105° (Fig. S1, ESI†). If we approximate that the methyl group is an optimal steric function, then, any functional group with a cone angle falling around 105° is most likely to hamper IChB. Despite the space-filling model presenting a perceptible steric clash of the X atom and the 6-functionality (Fig. 4C), predicting its outcome was quite challenging until recently. Thanks to the growing body of X-ray crystallographic structural data and the reactivity pattern displayed by several 2,6-disubstituted systems, such as 9–24 (Fig. 5), our understanding about adaptive behaviors of chalcogen bonding components (the sigma donors and the proximal Lewis base under steric confinement) was enhanced.25,28,29
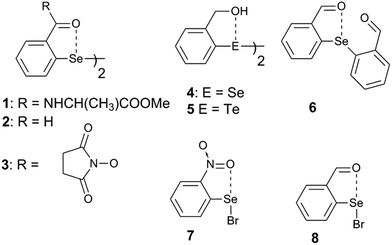 |
| | Fig. 3 2-Substituted arylchalcogen compounds showing the IChB between E and O atoms, where E = Se or Te. | |
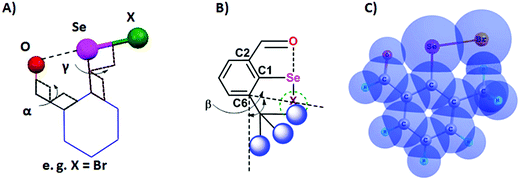 |
| | Fig. 4 (A) Illustration of the O⋯Se–X interaction. (B) Illustration of the cone angle, (C) space-filling model showing the steric collision of methyl hydrogen with bromine (CH)⋯(Br). | |
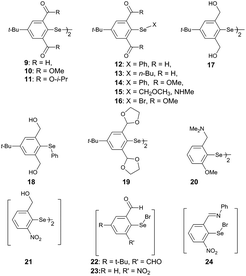 |
| | Fig. 5 2,6-Disubstituted arylchalcogen compounds. | |
The adaptive structural and reactivity patterns observed at the molecular scale include (a) adaptation of a conformation without IChBs, (b) adaptation of a conformation with weak IChBs, (c) twisting the skeletal aryl ring while maintaining IChBs, (d) ionization of the E–X bond and (e) intramolecular cyclization. In the following sections each of these behaviors is supported by experimental evidence.
2.1. Adaptation of a conformation without IChBs
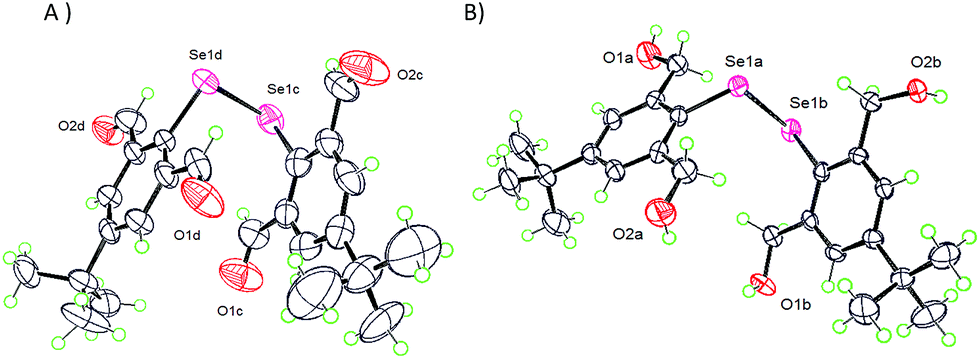
The best way for the IChB components in molecular systems to respond to a sterically overwhelming situation is to retract the O⋯Se–X interaction, such that the X atom moves orthogonally above the aryl plane (γ = 90°) and directs the smallest electrophilic atom (e.g. H atom) closer to the negative electrostatic surface potential around selenium (Fig. S2, ESI†). Five molecular systems 9,2512,2517,281829 and 1925 adhere to this rule. The X-ray crystallographic structure of diselenide 9, having 2,6-diformyl groups, showed that all of the four oxygen atoms have turned away from the selenium centre (Fig. 6A) with α ≈ β ≈ 180°. Similar features have been observed in the structure of diorganyl monochalcogenide 12 with the 2,6-bis(formyl) group.25 Diselenide 17 with the 2,6-dihydroxymethyl group also follows this torsion angle formalism (Fig. 6B).28 Mononoselenide 18 with two 2,6-dihydroxymethyl groups displayed a slightly different orientation of OH groups, where α ≈ 180°, β ≈ 90°, and γ ≈ 90°.29 The differences in –OH orientation in diselenide 17 and monoselenide 18 could be due to the free rotor behavior of the –CH2OH function and the inherent hydrogen bonding properties of the OH groups.
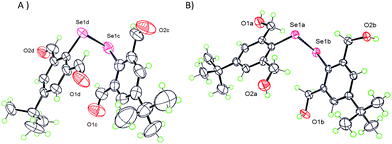 |
| | Fig. 6 X-ray structures showing no O⋯Se–Se interaction in 9 (A) and 17 (B). | |
The more interesting case is 19, the lactal derivative of aldehyde 9, which displayed α ≈ β ≈ 90°, and γ ≈ 90° indicating the projection of oxygen-containing rings orthogonally away from the aryl plane.25 This could be due to the conformational constraints within the lactal ring. All these compounds have an average O⋯Se distance of 4.5 Å which is much larger than the sum of van der Waals radii of oxygen and selenium (3.4 Å)30 indicating the absence of the O⋯Se–Se/Se–C interaction. A general noticeable trend is that 2,6-disubstituted compounds containing CHO, CH2OH, and cyclic lactal functionalities tend to turn away the oxygen atom from the central carbon atom such that selenium is free from the steric clash with an oxygen atom. 77Se NMR spectroscopy is a powerful tool to probe the IChB in solution. Diselenides 9 and 17 showed 77Se NMR peaks in the considerably shielded region (376 ppm and 346 ppm respectively) rather than chemical shifts observed for the related 2-substituted diselenides 231 and 432 (468 ppm and 428 ppm respectively) with IChBs indicating the absence of IChBs in 9 and 17. Alternately these compounds have unique intramolecular C–H⋯Se interactions.25,28 This reflects the fact that, especially under a sterically crowded environment, it is energetically possible to dispose two small electropositive “H” atoms around the negative belt of the selenium atom than to place one/two larger “O” atoms with negative surface potential around it.25 These examples undoubtedly prove the fact that the O⋯Se–Se/Se–C interaction is highly sensitive to intramolecular steric crowding.
2.2. Adaptation of a conformation with weak IChBs
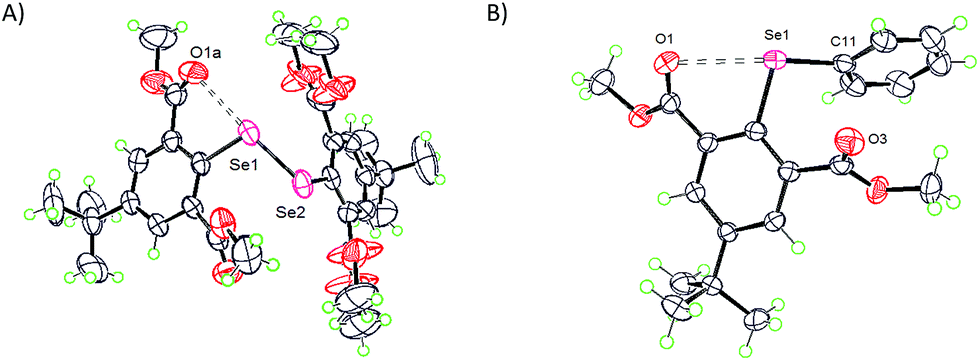
The ester group is a close relative to the formyl group regarding the carbonyl functionality, however, it has contrastingly different rotor behavior (vide infra). The formyl group in an aryl ring is a rigid rotor which tends to be strictly in the aryl plane with α and β either ≈180° or ≈0° which is essentially due to the resonance conjugation effect (vide infra). In contrast, the ester group is very adaptive and is a semi-rigid rotor which can adjust to the steric environment via tilting the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O π-framework slightly away from the aryl π-framework. This property not only allows the ester functionality to assume various α and β torsion angles, defined by the orientation of the oxygen atom of the carbonyl group concerning the C1 carbon atom, but also provides room for the survival of the O⋯Se–Se interaction even under a stressful environment.28 The compounds which cohere to this analogy are 10,281128 and 14.29 In one-half of diselenide 10, CO groups which are involved in the O⋯Se–Se interaction are located slightly (α = 23°) above the aryl plane and the CO of the ester group at the 6-position has moved significantly away from the aryl plane with β = 136° (Fig. 7A). Another half of 10 adopts entirely different α (159°) and β (137°) torsion angles. Compound 11 has similar structural features to compound 10.28 The α and β torsion angles observed for 14 are 27° and 49° respectively (Fig. 7B).29 The γ angle for all these compounds (10, 11 and 14) falls below 37° and CO⋯Se–Se/C (2.71–3.00 Å) distances observed are significantly lower than the van der Waals limit of the O⋯Se interaction (3.40 Å) indicating the presence of weak to moderately strong IChBs.
O π-framework slightly away from the aryl π-framework. This property not only allows the ester functionality to assume various α and β torsion angles, defined by the orientation of the oxygen atom of the carbonyl group concerning the C1 carbon atom, but also provides room for the survival of the O⋯Se–Se interaction even under a stressful environment.28 The compounds which cohere to this analogy are 10,281128 and 14.29 In one-half of diselenide 10, CO groups which are involved in the O⋯Se–Se interaction are located slightly (α = 23°) above the aryl plane and the CO of the ester group at the 6-position has moved significantly away from the aryl plane with β = 136° (Fig. 7A). Another half of 10 adopts entirely different α (159°) and β (137°) torsion angles. Compound 11 has similar structural features to compound 10.28 The α and β torsion angles observed for 14 are 27° and 49° respectively (Fig. 7B).29 The γ angle for all these compounds (10, 11 and 14) falls below 37° and CO⋯Se–Se/C (2.71–3.00 Å) distances observed are significantly lower than the van der Waals limit of the O⋯Se interaction (3.40 Å) indicating the presence of weak to moderately strong IChBs.
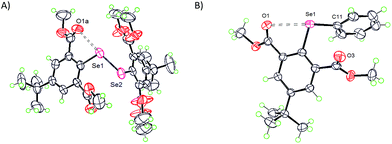 |
| | Fig. 7 X-ray structures showing the O⋯Se–Se interaction in 10 (A) and 14 (B). | |
It is noteworthy that compounds 10 and 11 possess both acyloxy (CO⋯Se–Se) and alkoxy (R–O⋯Se–Se) interactions and each half of the diselenide has a distinctly different spatial arrangement of atoms around each selenium.28 In a sterically less cumbersome diselenide 10, in comparison to 11, the former interaction is stronger than the latter. Nonetheless, the CO⋯Se–Se interaction found in 10 is much weaker than that observed in a similar 2-substituted system 3,33 indicating that the steric pressure still operates to reduce the effectiveness of the CO⋯Se–Se interaction in a sterically cumbersome environment. The CO⋯Se–Se distance observed for diselenide 10 is significantly shorter than the CO⋯Se–C(Ph) distance observed for compound 14 indicating that the Se–Se sigma hole is relatively more electron depleted than the Se–C sigma hole on selenium.
2.3. Twisting the skeletal aryl ring while maintaining IChBs
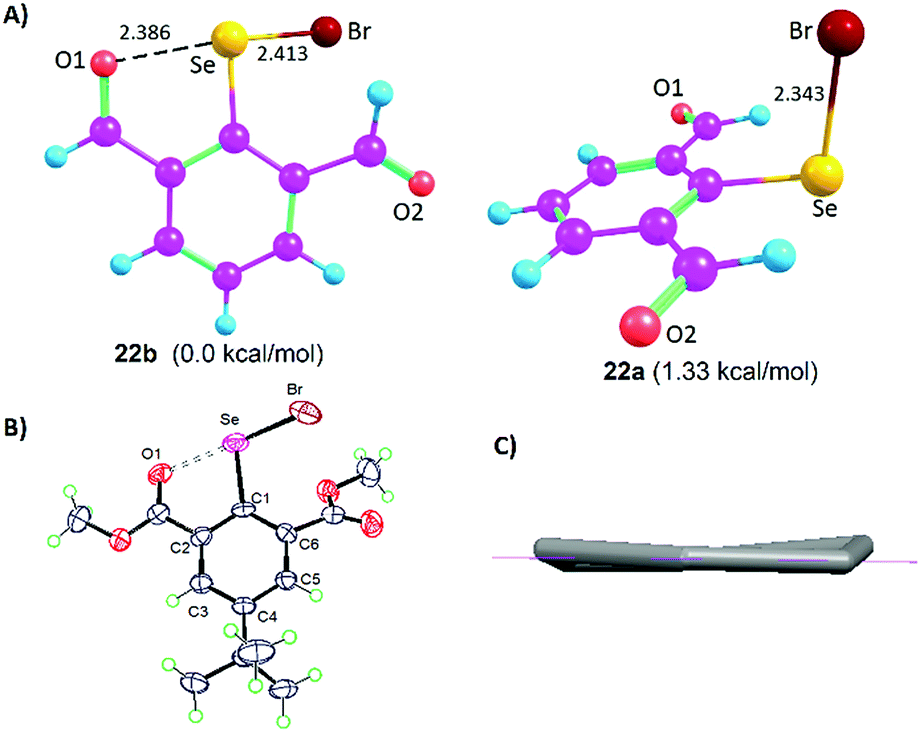
The Lewis base donors, OH, CHO, and COOMe are adaptive towards weak-to-moderately electron depleted sigma hole (Se–X, X= Se, C) donor functionalities. They use sidearm torsional motion as an adaptive strategy to either retract or establish weak IChBs with such sigma hole donors. Interestingly, these Lewis base donors tend to establish a strong IChB with a strong sigma hole donor (Se–Br). Such combination is overwhelmingly powerful to retain a strong IChB (O⋯Se–Br) with aryl ring skeletal deformation as an adaptation strategy.28 The oxidation of diselenide 9 leads to the formation of the corresponding selenenyl bromide intermediate 22 (Fig. 5).25 Despite the presence of the strong O⋯Se–Br interaction, our attempts to isolate selenenyl bromide 22 were unsuccessful. This instantaneously leads to the in situ formation of its cyclic selenenate ester derivative.25,34 The computationally optimized model systems (at the B3LYP-D2/6-31G(d) level), 22b and 22a conformers, showed that the former is 1.33 kcal mol−1 more stabilized than the latter (Fig. 8A). The energy barrier computed for the conversion of conformer 22b to 22a is considerably high (15.71 kcal mol−1). Therefore, it is probable that compound 22 would freeze in a 22b-like conformation with a strong O⋯Se–Br interaction (calculated O⋯Se distance 2.386 Å). This is an interesting example where the oxidation of diaryl 9 to arylselenenyl bromide 22 resulted in the switching of an “a-like” conformation in the former to a “b-like” conformation in latter. One would call this conformational switching either as a redox switch or more appropriately chalcogen bond donor dependent conformational switch. Despite its stabilization contribution, the over-dominating O⋯Se–Br interaction distorted the skeletal planarity of the aryl ring with non-zero dihedral angles for the carbon atoms in the benzene ring. This distortion helps the Br to move considerably away from the aryl plane (α = 3.6°, β = 169.2°, γ = −24.6°) and hence it keeps away from even the smallest hydrogen atom of the 6-formyl group. Although 22b is energetically more stable than 22a, the rigid rotor behavior of CHO (the tendency of α and β torsion angles to approach either 0° or 180°) in combination with the aryl ring distortion makes a 22b-like conformation very unstable.
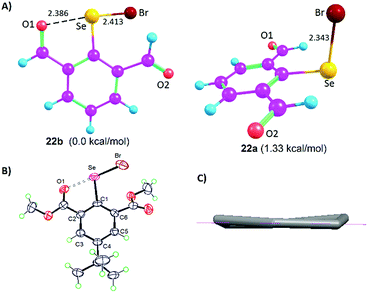 |
| | Fig. 8 (A) Computationally optimized conformers 22b and 22a. (B) X-ray structure of 16. (C) Non-planar aryl ring in 16. | |
This is in sharp contrast to the structural features that were observed for 2-formylphenylselenenyl bromide 8 where no aryl ring distortion was observed and it is stable under ambient conditions.24 The X-ray crystallographic studies on compound 16 provide a solid proof for the aryl ring distortion (Fig. 8B and C).28 The maximal distortion is observed around the C1 carbon atom with a C2–C1–C6–C5 torsion angle of 11°. It appears that compound 16 takes advantage of the stabilization resulting from the strong O⋯Se–Br interaction by slightly disturbing the planarity of the aryl ring. The semi-rigid rotor behavior of the ester group and its stericness (vide infra) are adaptively advantageous for the stabilization of 16 under ambient conditions.
2.4. Ionization of the E–X bond
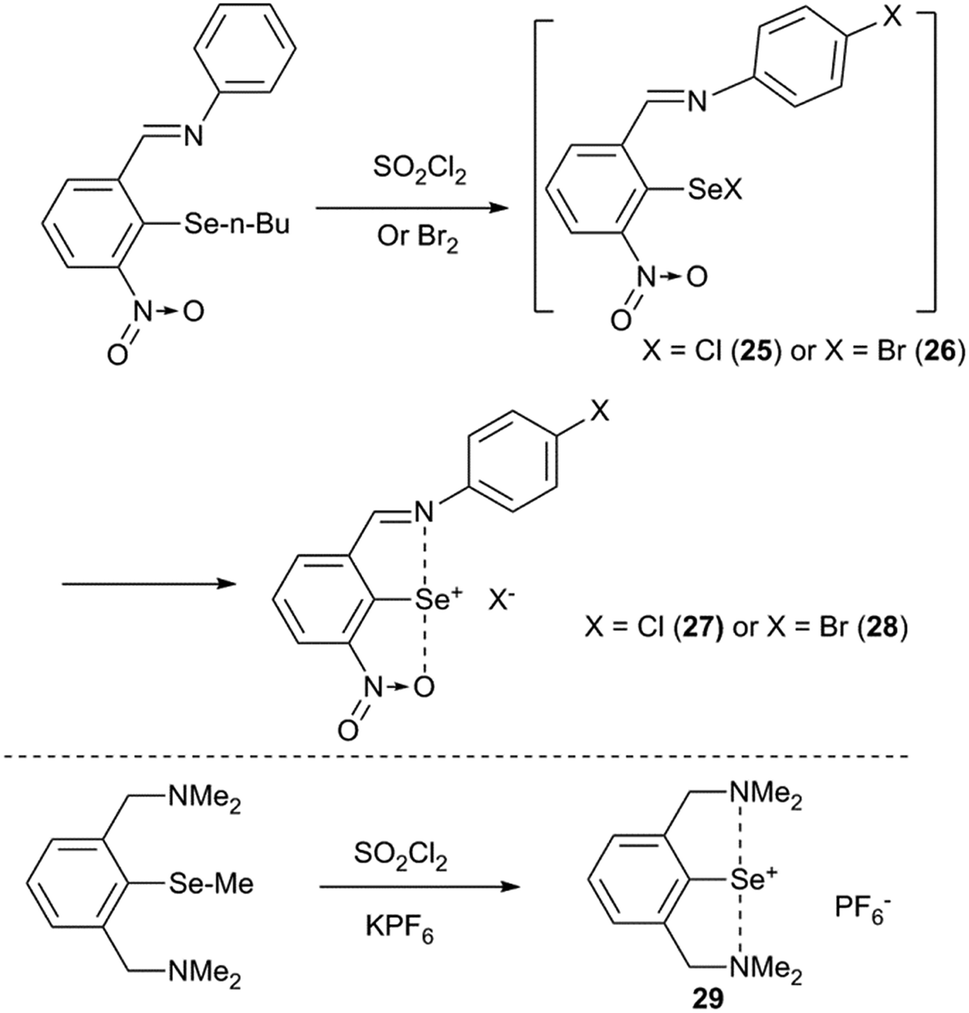
Any strained aryl ring is energetically unstable which would tend to planarize the aryl ring via a certain reaction pathway. One probable pathway to achieve this task is to ionize the compound via excommunication of the X substituent from the covalent radius limit of the selenium atom. For example, our attempts to isolate the covalent selenenyl halides (25 and 26) with the imino group as the donor and the nitro group as a second ortho-group resulted in the formation of the corresponding ionic selenenyl halides (27 and 28) (Fig. 9).35 Ionic selenenyl halides such as 27 and 28 assume a highly planar structure with an extremely strong pincer type N/O⋯Se⋯N interaction. In these compounds the torsion angles α ≈ β ≈ 0° are measured concerning the orientation of coordinated heteroatoms with respect to the C1 carbon atom. In contrast to 25 and 26, compound 7 with 2-nitro substitution was characterized as a covalent arylselenenyl bromide.36 Furukawa and co-workers reported the first structurally characterized 2,6-bis-CH2NMe2 stabilized cation associated with its counter anion PF6− (29). It was obtained via halogenating the corresponding methyl derivative and subsequent anion exchange.37 The same cation was very recently isolated with Cl− and Br− as counter ions.38
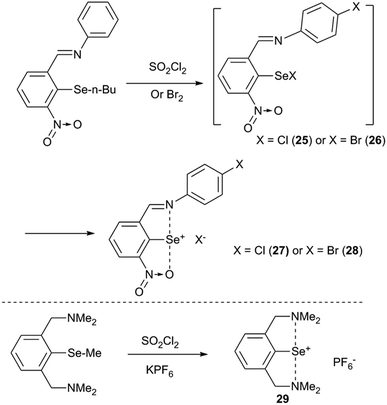 |
| | Fig. 9 Ionization of selenenyl halides. | |
The rationale behind the ionization of the Se–X bond in 25/26 is that the strong imine(N)···selenium atom interaction elongates the Se–Br/Cl bond along the interaction axis which leads to steric collision of halides with the nitro group.35 This, in turn, will either direct the Se–Br/Cl bond to move perpendicular to the aryl plane or to move in the aryl plane away from the selenium which leaves room for the nitro group to act as the second donor to participate in IChBs. This shows that strong IChBs in a sterically crowded environment lead to the ionization of selenenyl halides. The increased positive charge at the central chalcogen, resulting from ionization, enables it to accommodate two electronegative heteroatoms more effectively than its unionized form.
2.5. Intramolecular cyclization
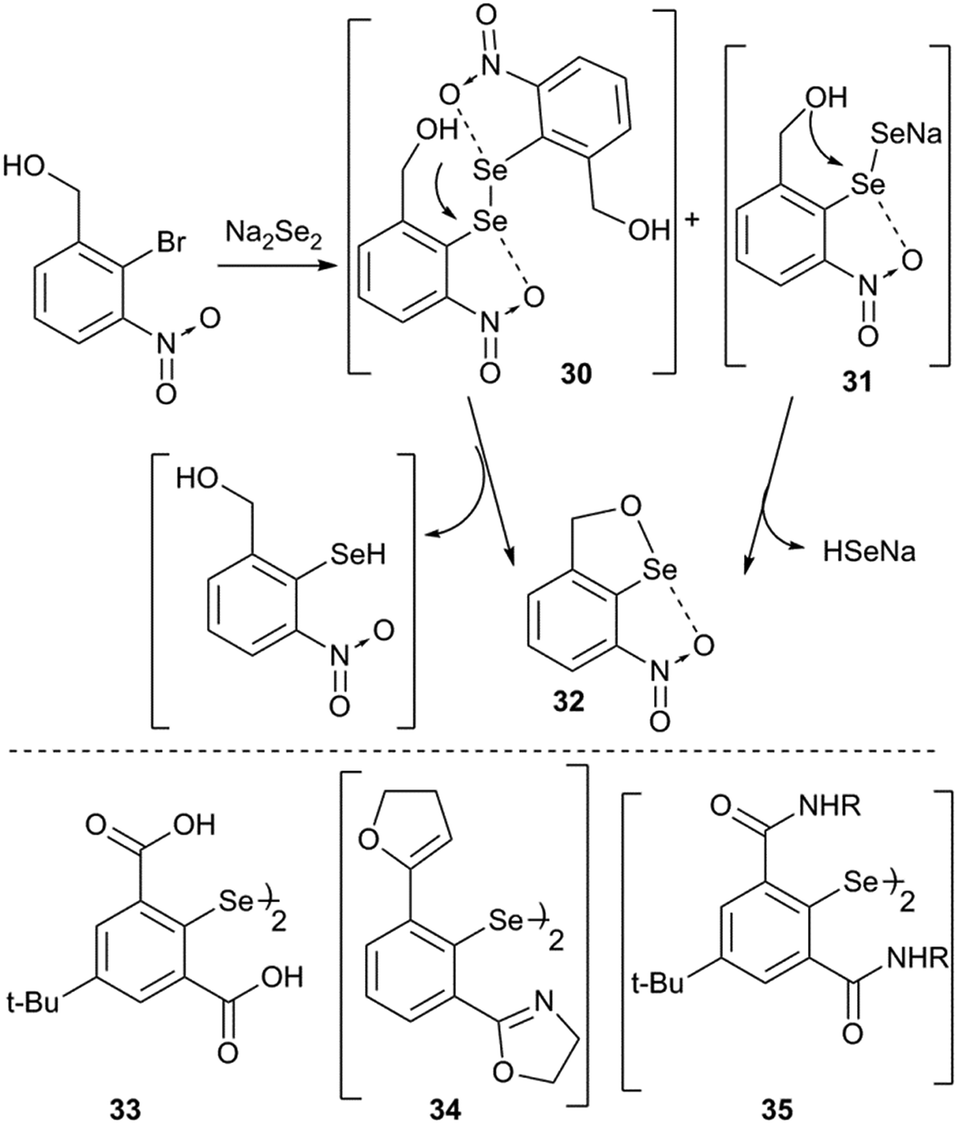
Intramolecular cyclization is an alternative adaptation strategy to the ionization process (Fig. 10). For instance, the occurrence of intramolecular cyclization of intermediates 30 and 31 could be due to the presence of the nitro group.36,39 IChBs force the outer selenium of the Se–Se bond to sterically collide with the ortho-hydroxymethyl group. In order to adjust to such a steric environment, the hydroxyl methyl group acts as an internal nucleophile to displace the peripheral selenium via the intramolecular addition–elimination reaction and provides 32 as the cyclized product.36 A facile intramolecular cyclization of 2,6-bis(carboxyl) substituted diaryl diselenide 33 in methanol medium, leading to the formation of a cyclic selenenate ester, has also been noticed.40 Attempted synthesis of 2,6-dioxazoline containing diselenide 34 by Mugesh and co-workers led to the formation of a cyclic selenenamide via unexpected hydrolysis of the oxazoline ring followed by a selenium centered intramolecular addition–elimination reaction.41 Expectedly, 2,6-bis-amide substitution facilitates (e.g.35) the formation of cyclic selenenamide derivatives via both lithiation42 and Na2Se2 routes43 under mild conditions, without any additional step. Selenenyl halides such as 2225 and 2336 are extremely unstable and undergo facile hydrolysis followed by intramolecular cyclization leading to the formation of their cyclic selenenate esters. Selenenyl bromide 16 is stable under ambient conditions.28 However, its hydrolysis and subsequent intramolecular cyclization are triggered in the presence of a mild base (triethyl amine) or passing through a silica gel column. It reacts very rapidly with strong diprotic nucleophiles such as H2S and primary amines leading to the formation of their cyclic compounds, thioselenenate ester and selenenamides respectively.44 Steric confinement of IChBs in combination with other common reaction pathways leads to the unprecedented synthesis of various heterocycles. The examples include pincer type dioxyselenurans29/telluranes,45 bicyclic46 and N-containing spirocyclic compounds.47 The chalcogen bonding aided formation of cyclic compounds, both aromatic and aliphatic compounds, continues to grow.48
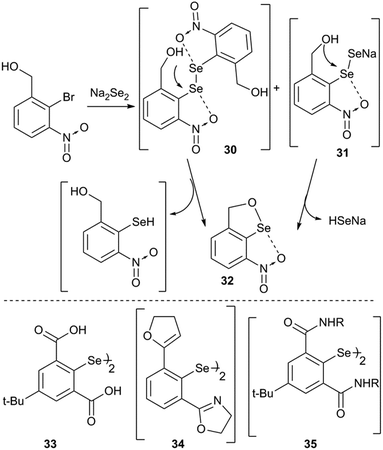 |
| | Fig. 10 Top panel shows the intramolecular addition–elimination reaction of diselenide motifs (30 and 31). The bottom panel shows the structures of other diaryldiselenide/diaryldiselenide intermediates. | |
3. Rotor mobility and conformational diversity
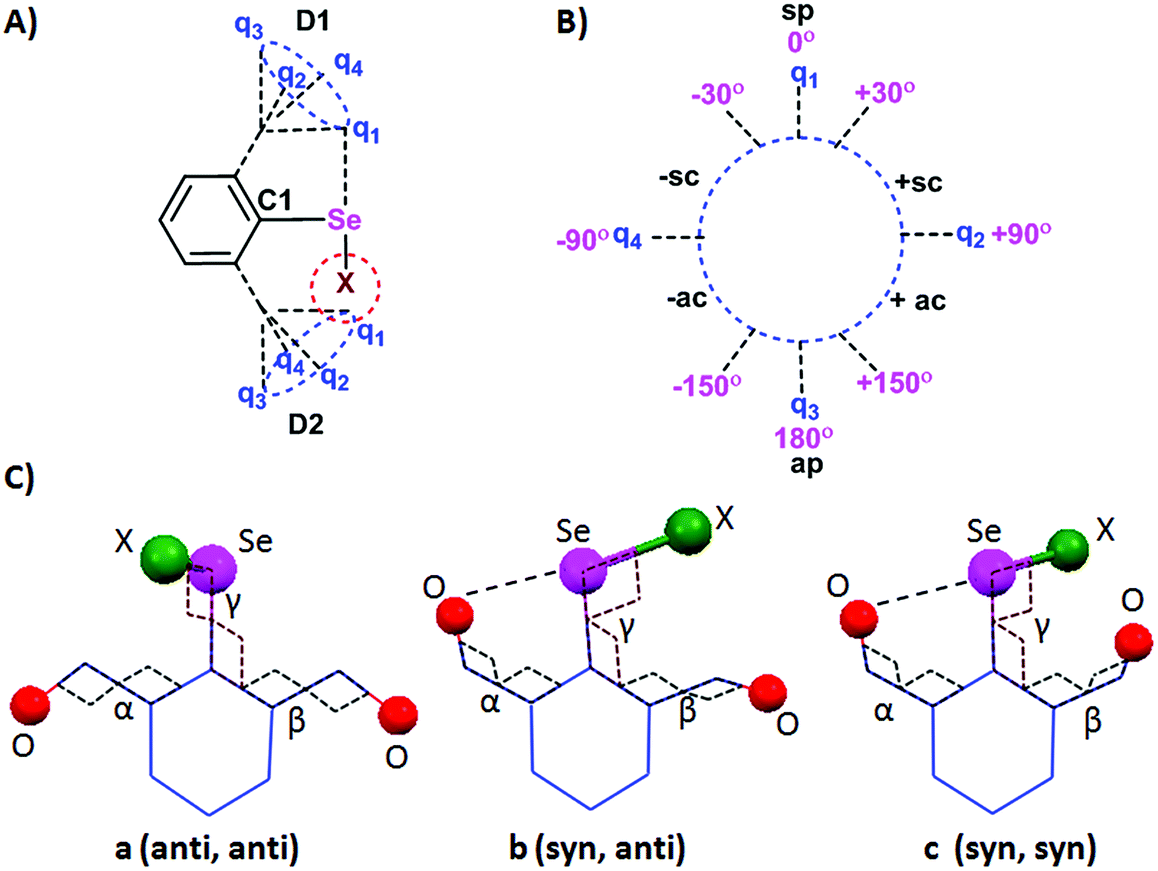
As we have discussed earlier, different sidearm rotors contribute differently to the observed structures and reactivity. Hence, a basic understanding of the rotor characteristic can serve as a means to utilize IChBs to construct functional molecular and supramolecular systems. This section provides an overview of conformational and structural aspects of 2,6-disubstituted compounds with the uses of definitions borrowed from organic chemistry.49 The relative orientations of C1 and D1, and C1 and D2 are expressed respectively as α and β torsion angles. The circle formed by the torsional motion of D1 or D2 around the C1 carbon of the aryl ring has been divided into quadrants by using the points q1, q2, q3, and q4 representing 0°, 90°, 180°, and −90° respectively (Fig. 11A and B). The torsional angle orientation of 0° ± 30° for C1 and D1 is referred to as the synperiplanar (sp) configuration or simply the syn configuration (Fig. 11B). The torsional angle orientation of 180° ± 30° for C1 and D1 is called as the antiperiplanar (ap) configuration or simply the anti-configuration. The intermediate torsion angle orientations are clinal orientations which could be either synclinal (sc, between 30 and 90°) or anticlinal (ac, between 90° and 150°). The same analogy also applies to C1-D2 torsional motions. Since 2,6-disubstituted systems have two sidearm torsional motions (Fig. 11A), the adaptive configurations appear as a pair. The three important configurations are a (anti, anti), b (syn, anti) and c (syn, syn) (Fig. 11C). The syn and anti-configurations in combination with synclinal (sc) and anticlinal (ac) configurations are also possible especially for rotationally flexible functionalities such as CH2OH and COOR. The other observed configurations for different molecular systems are provided in Table 1.
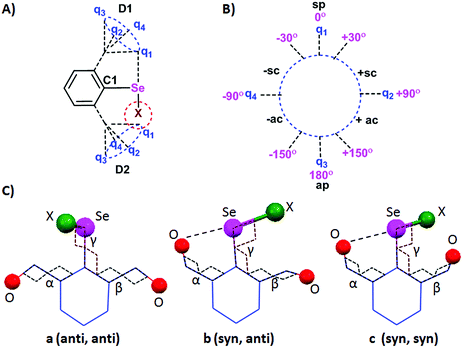 |
| | Fig. 11 (A) Rotational motion of D1 and D2. (B) Classification of the different torsion angles. (C) Three of the configurations of 2,6-disubstituted systems. | |
Table 1 List of observed configurations
| (anti, anti) |
(syn, anti) |
(syn, syn) |
(anti, SC) |
| *Computed structure. |
|
9, 12, 17 |
22b* |
Cyclic and ionized |
18
|
| (syn, AC) |
(SC, AC) |
(syn, SC) |
(AC, AC) |
|
10, 16 |
11
|
14
|
10, 11
|
Chemometric analysis such as principal component analysis (PCA) has been earlier used for the morphological classification of brucite particles.50 In principle, application of such methods to conformation classifications not only can further enhance our understanding of the emerging patterns of structure and reactivity under steric confinement but also has a predictive value. In the present case, the O⋯Se distances, and α, β, and γ torsion angles obtained for various 2-substituted and 2,6-substituted compounds have been used as the inputs for the PCA analysis.
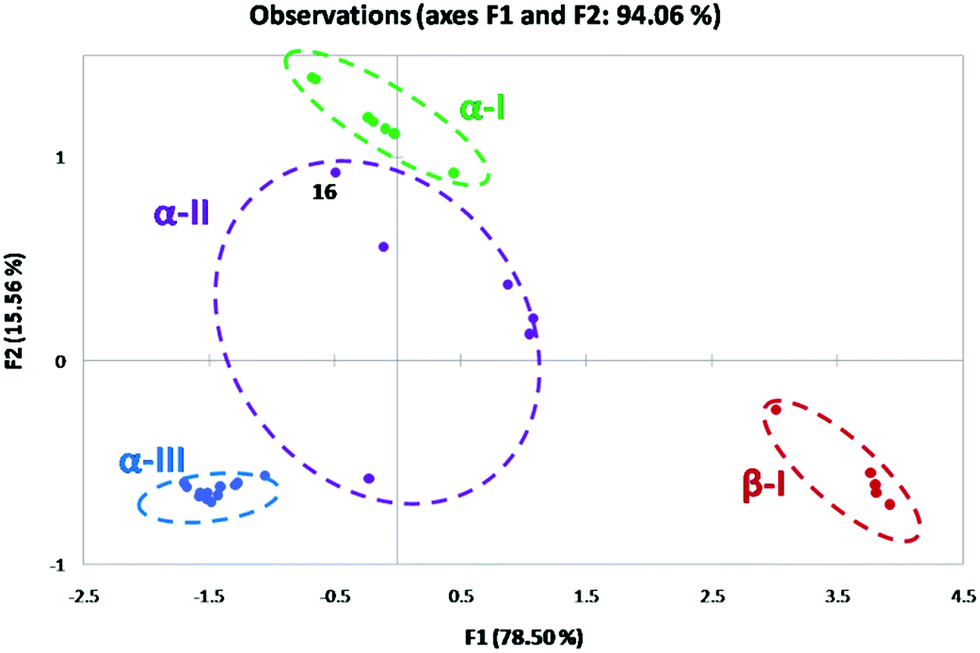
For 2-substituted systems, the α torsion angle approaches 0° (α → 0°) and the β torsion angle is approximated to be 180°. PCA reduces the multidimensionality of the data into a lower dimension. The corresponding conformational space produced in the PCA map for the 2-substituted system is referred to as the α-I space with a (syn, pseudo anti) configuration (Fig. 12). In principle, based on the observed geometry parameters, all the 2-substituted systems with the O or N-donor atom are expected to occupy the α-I space of the PCA map. 2,6-Disubstituted compounds with no IChBs fall in the β-I space where the α and β torsion angles approach 180° (α and β → 180°; (anti, anti)) and with no O⋯Se–X interaction. The conformations of compounds with semi rotor properties (ester group) occupy the α-II space with a large range of α and β torsion angles as evident from the spread of the α-II space. The possibility of IChBs and clinal orientations, due to the rotational mobility of the ester group, contributes to this large spread in the α-II space. Conformations of ionized compounds occupy the α-III space with α, β, and γ torsion angles approaching 0° (α, β, and γ → 0°; (syn, syn)-pincer type) where one of the heteroatoms of D2 serves as the X′ group after the displacement of X. This plot not only enables us to understand the emergence of distinct conformations upon subjecting the IChB to steric stress but also enables us to make some general understanding of the facts: (1) as the IChB becomes stronger, (α → 0°) and the β → 180° (e.g. arylselenenyl bromide 16), the associated points in the α-II conformational space closely approach the α-I conformational space (Fig. 12). (2) The conversion of compounds those occupy the β-I conformational space to the corresponding cyclic compound in the α-III space is most likely to accompany the formation of an intermediate species whose conformation may have a transient existence in the α-II conformational space. (3) 2,6-Tertiary amide systems (not known) have been anticipated to behave like 2,6-diester systems as the amide group is a semi-rigid rotor. As is obvious from the PCA map, the 2,6-bisformyl system adaptively takes on extreme conformations, (anti, anti) or (syn, anti), depending on the strength of the donors. For the formyl cases, it is intuitive to note that the strong sigma hole donor (Se–Br) favors (syn, anti) and weak sigma hole donors (Se–SeR or Se–Ph) favor the (anti, anti) conformation. In the case of the ester system, intermediate conformation preferences with considerable structural diversity have been encountered. This could be explained by having a look at the rotational motions of CHO, COOR and CONHR groups around the benzene plane.
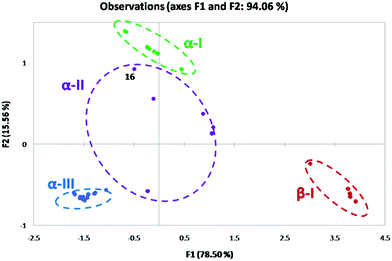 |
| | Fig. 12 Principal component analysis on the X-ray data. | |
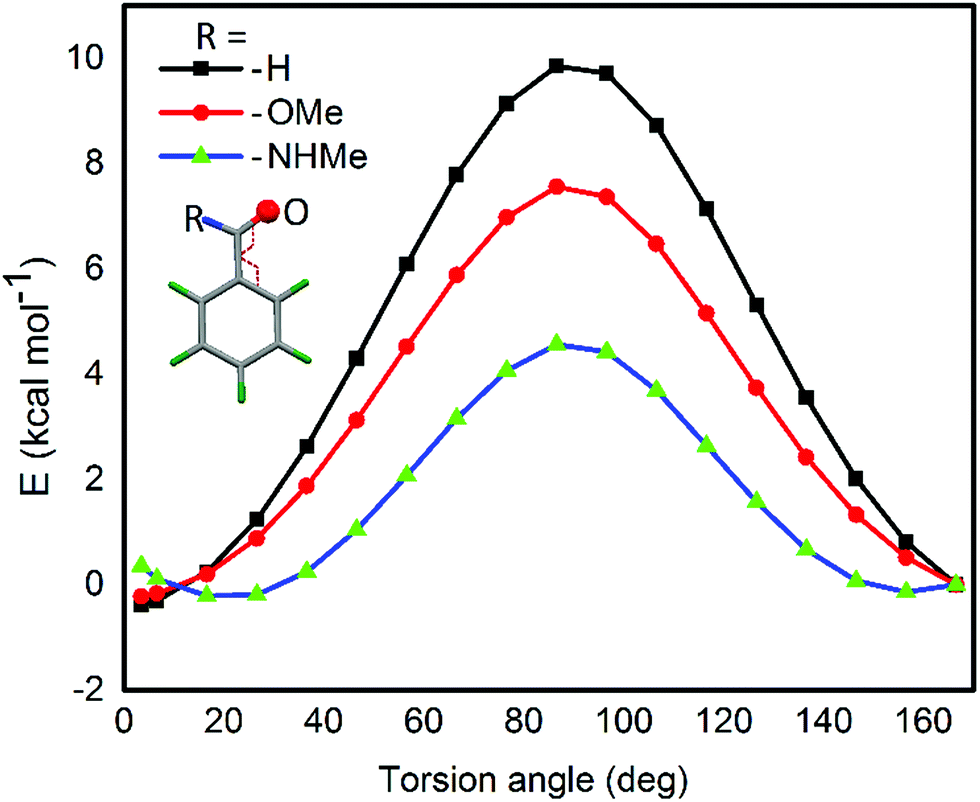
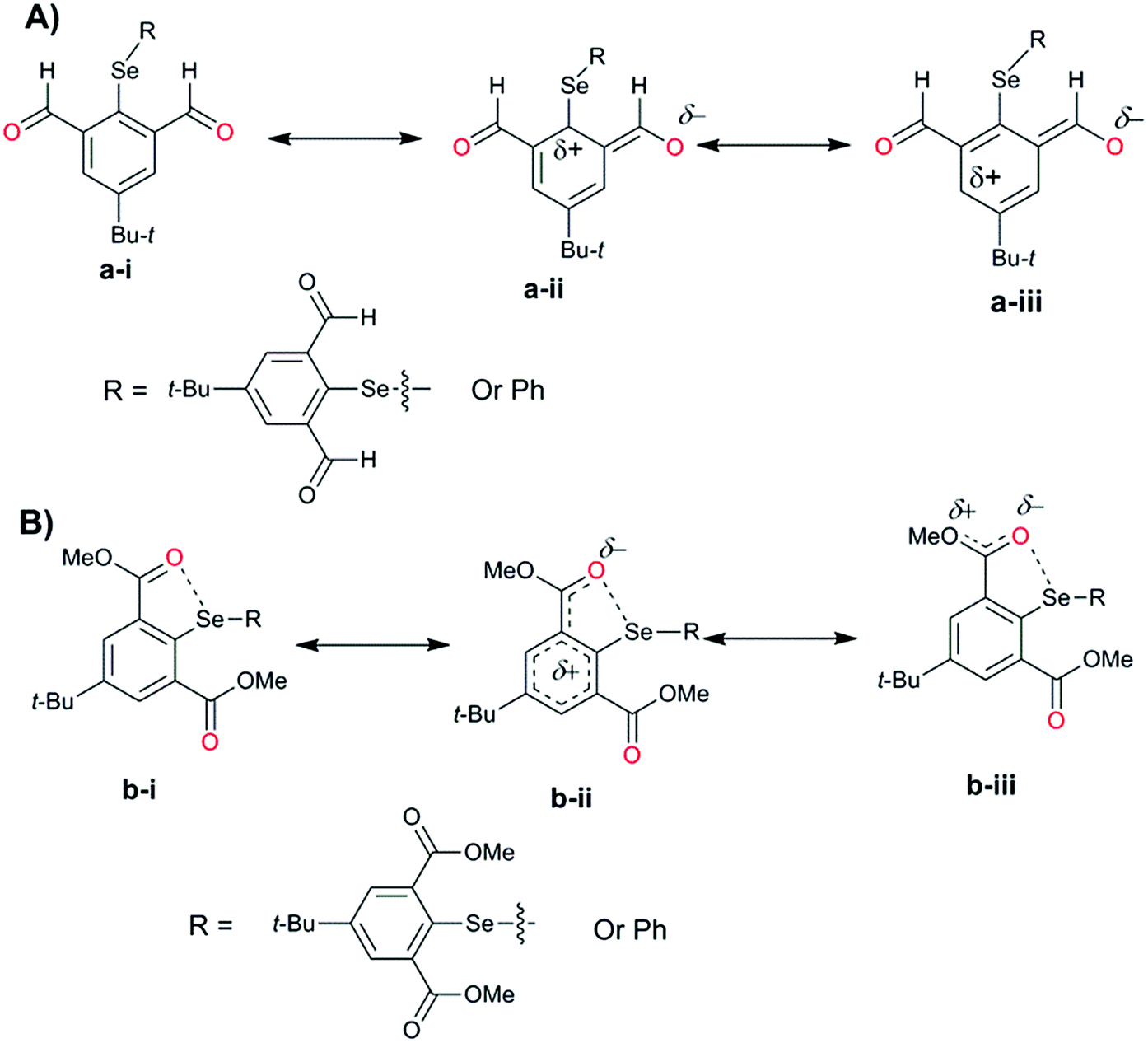
For the computational modeling of the rotational energy barrier, we have explored less complicated benzaldehyde, methylbenzoate, methylbenzamide systems. The variation of the torsional angle of Ph–COR (R = H, OMe, and NHMe) as a function of electronic energy reveals that the barrier height decreases in the order CHO > COOMe > CONHMe (Fig. 13). These trends are in agreement with the earlier experimental and computational results.51,52 This large energy barrier for the rotation of the CHO group around the aryl plane arises from the resonance delocalization of π-electrons between the formyl group and the aromatic ring (Fig. 14A(a-i–iii)). The reduced barrier height for COOMe rotation could be associated with the competing internal resonance delocalization of the π-electrons within the ester group via the contributing form b-iii (Fig. 14B) rather than b-ii. This facilitates a favorable intramolecular interaction of the oxygen of the ester functions with selenium and acts as a means for adjusting to the steric environment. The out-of-plane orientations of the ester groups observed in the X-ray structures of the diorganyl diselenides (10, 11) and monoselenide 14 indeed support the free rotation around the C–C bond of the aryl ring and ester group. More direct evidence comes from the X-ray structure of diselenide 10 in which, out of the four methyl ester groups, two were disordered due to the thermal motions of ester groups around the aryl plane. Due to the same reason, the barrier height of the amide rotation is further reduced as the N lone pair is more readily available for internal delocalization than the alkoxy O lone pair in ester systems. Hence, amide systems are also expected to display such adaptive rotor behaviors similar to the ester systems.
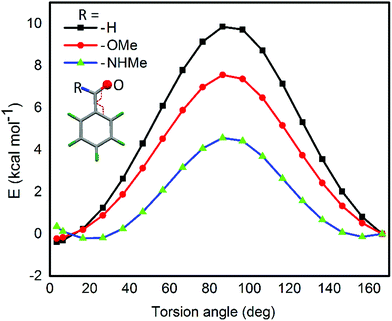 |
| | Fig. 13 Energy barriers for the rotational motion of carbonyl functions. | |
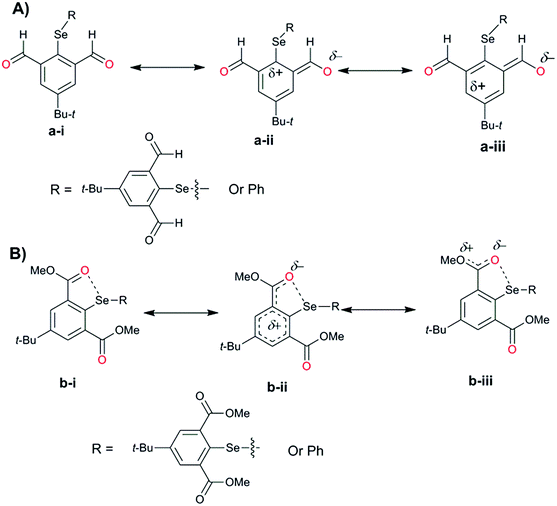 |
| | Fig. 14 Resonance contributing forms for (A) aldehyde and (B) ester derivatives. | |
4. The origin of molecular strain and its consequences
4.1. IChB induced aromatic ring strain
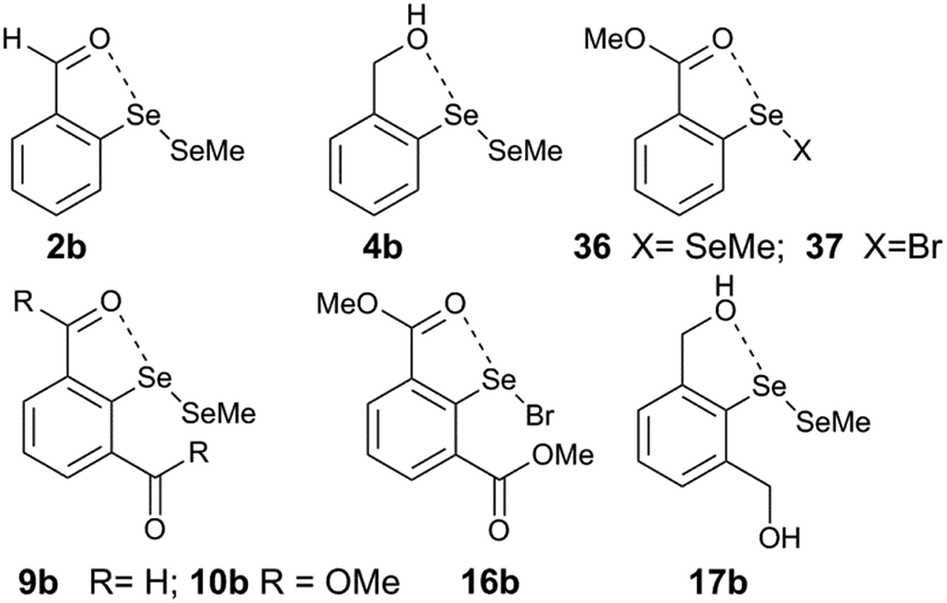
Computational analysis on model systems with the O⋯Se–X interaction (2b, 4b, 8, 9b, 10b, 17b, 16b, 22b, 36, and 37) enables us to understand further the self-correcting properties of IChB components under steric stress. The diselenide models 2b, 4b, 9b, 10b, 16b, and 22b are representative of compounds 2, 4, 9, 10, 16, and 22. For 2,6-disubstituted systems, b refers to the (syn, anti) conformation. The replacement of half of the diselenide motif with the SeMe group and the replacement of the “t-Bu” group in 2,6-disubstituted systems with “H” are often employed as approximations to reduce the computational time.25,28 The computationally optimized structures (at B3LYP-D2/6-31G(d)) of these systems show O⋯Se bond distances (Table 2) that are within the sum of van der Waals radii of O and Se (3.40 Å).
Table 2 The bond distances (O⋯Se), angles (O⋯Se–X), average torsion angles in the aryl ring (ψav), strain energy (Est), intramolecular chalcogen bond energy (EIChB), and net stabilization energies (Enet = EIChB + Est) were computed using the DFT-D method (B3LYP-D2/6-31G(d))
| Entry |
O⋯Se (Å) |
O⋯Se–X (°) |
ψ
av (°) |
E
st
|
E
IChB
|
E
net
|
|
2b
|
2.644 |
176.29 |
0.17 |
∼0 |
−3.35 |
−3.35 |
|
4b
|
2.925 |
173.48 |
0.91 |
∼0 |
−2.63 |
−2.63 |
|
8
|
2.33 |
178.13 |
0.02 |
∼0 |
−7.79 |
−7.79 |
|
36
|
2.611 |
176.26 |
0.18 |
∼0 |
−2.18 |
−2.18 |
|
37
|
2.373 |
177.93 |
0.01 |
∼0 |
−5.96 |
−5.96 |
|
9b
|
2.79 |
153.11 |
3.26 |
5.97 |
−3.65 |
2.32 |
|
10b
|
2.664 |
163.51 |
2.96 |
6.53 |
−2.31 |
4.23 |
|
16b
|
2.419 |
165.78 |
4.16 |
8.47 |
−7.30 |
1.07 |
|
17b
|
3.109 |
143.97 |
0.65 |
−2.07 |
−3.52 |
−5.59 |
|
22b
|
2.386 |
164.99 |
4.57 |
8.17 |
−9.51 |
−1.34 |
It is noteworthy that the 2-substituted systems show shorter chalcogen bonds with more linear O⋯Se–X angles than the corresponding 2,6-disubstituted systems indicating the weakening of IChBs in the latter due to the steric effect. An inspection of the average of the six torsion angles (ψav)28 of carbon atoms in the aromatic ring (Table 2) indicated that the ψav values for the optimized structures of the 2,6-disubstituted systems are significantly greater than that of the 2-substituted system. The exceptional case is 4b, which showed slightly higher ψav among the 2-substituted systems. This could be due to the IChB that forces the CH2 group to encounter steric interaction with the aryl π-electrons.
4.2. Rotor and sigma hole dependent strain energy
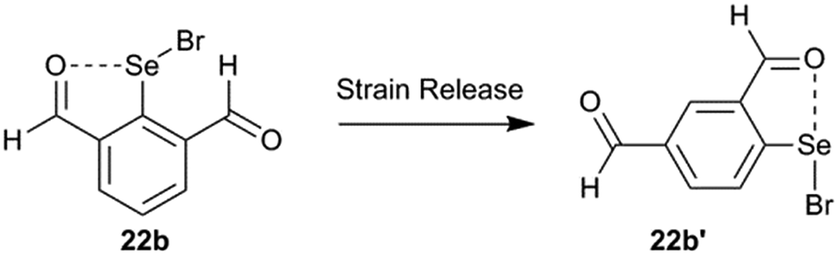
The strain energy contribution of the 6-H atom to destabilization of IChBs in the 2-substituted system is negligible and can be approximated to 0 kcal mol−1. The strain energy contribution of the 6-substituent to the destabilization of IChBs in the 2,6-disubstituted system is computed via interchanging the 4H and 6-substitution. For example, transforming the 2,6-diformyl system (22b) into its 2,4-diformyl isomer (22b′) would result in strain relief in the former (Fig. 15). In both systems (e.g. 2,4-diformyl and 2,6-diformyl systems), the meta-relationship between the substituents is maintained. Hence, this interchange is expected to conserve the electronic effects, and it only relaxes the molecule from a strained state to unstrained state. The energy difference between the two isomers (2,4 and 2,6-isomers) is referred to as the strain energy (Table 2). The magnitude of the strain energy depends on the nature of the rotor and the nature of the chalcogen bond donor (Table 2). Entry 17b with the flexible rotor –CH2OH showed a comparatively smaller distortion in the aryl ring than the 2,6-disubstituted entries with rigid and semi-rotor behavior. Consequently, this system has negative strain energy indicating that the free rotors are adaptive to the steric environment. Further, the strain energy calculations indicate that the entries with strong sigma hole donors (Se–Br, 16b and 22b) show comparatively larger strain energy than the entries with weak sigma hole donors (Se–Se, 9b and 10b). This correlates well with the large average aryl ring distortion torsion angle computed for the former.
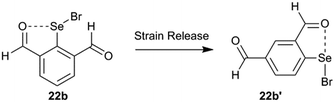 |
| | Fig. 15 An example of transforming the 2,6-disubstituted system into its 2,4-disubstituted isomer. | |
4.3. Intramolecular chalcogen bonding energy vs. strain energy
According to Politzer, the halogen/chalcogen bonding is mainly electrostatic that encompasses dispersion, induction and covalent forces.2a,22 The decomposition of their relative contributions to the chalcogen bonding is beyond the scope of this perspective. However, we have computed the intramolecular chalcogen bond energy (EIChB) using a similar approach adopted for the intermolecular case by Murray et al.,2a and the calculation of chalcogen bond energy (EChB) for intermolecular chalcogen bonds is very simple, which is the stabilization energy achieved through the complexation of the sigma hole donor and the Lewis acid donor (ΔE = Ecomplex − Esum of the energy of reactants).2a
Rather, it is cumbersome to calculate the intramolecular chalcogen bond energy (EIChB) where the sigma hole is masked intramolecularly with the Lewis base donor. We have attempted to obtain the EIChB using a homodesmic reaction. The homodesmic reaction, as shown for 8 (Fig. 16), has been previously used as a convenient means to obtain the chalcogen bond energy.18b The computed interaction energy for the chalcogen bond in the 2-substituted system (Table 2) is in good agreement with the previous reports on related systems. The interaction energy is normally higher for the system with chalcogens covalently bonded to halogens. Chalcogen bond stabilization is higher for selenenyl bromides than diselenides. In order to find the net stabilization energy conferred by IChBs in 2,6-disubstituted systems, we computed the EIChB for their strain relaxed isomers (2,4-isomers) (Fig. 16). The sum of strain energy and chalcogen bond energy (Enet = Est + EIChB) is the net stabilization energy (Enet). For the 2-substituted systems, the Est is negligible and the Enet is equal to the EIChB, whereas the steric function in 2,6-substituted systems makes a significant destabilization contribution to IChBs. The Enet calculated for the 2,6-disubstituted systems is >0 kcal mol−1 for the diselenides with a rigid rotor/semi-rigid rotor (9b and 10b; 2.32 and 4.23 kcal mol−1 respectively), and <0 kcal mol−1 for diselenides with flexible rotors (17b, −5.59 kcal mol−1). This can be used to rationalize the observed trends in the structures of diselenides to a certain extent. As a consequence of positive Enet, diselenide 9 with a rigid rotor compromises the IChB and diselenide with the semi-rigid rotor (10) maintains a weak IChB via making use of the semi-rotor property of the ester group. Although the CH2O(H)⋯Se–Se interaction is predicted to be favorable (−5.59 kcal mol−1), it is overpowered by other environmental effects in the solid and solution states.28,29,53 Also, the steric repulsion built within the pseudo-five-membered ring (–CHCH–CH2–O⋯Se–) formed by the O⋯Se–Se interaction in 4b and 17b might play a certain role in conformational preferences. It is interesting to note that Se–Br chalcogen bond donors favor the IChB under sterically demanding conditions (22b, −1.34 kcal mol−1). Selenyl bromide 16b is destabilized only to the extent of 1.07 kcal mol−1. However, this small stabilization energy (for 22b)/destabilization energy (for 16b) does not provide any reason to comprehend the observed reactivity of their real systems (16 and 22).
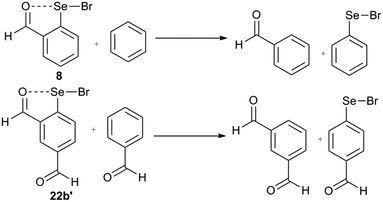 |
| | Fig. 16 The examples of homodesmic reactions for the 2-substituted (8) and 2,4-disubstituted systems (22b′). | |
4.4. Dispersion contribution to the IChB
The syntheses, X-ray structure determination, and computational studies on exceptionally stable diamondoid systems, yet sterically crowded, by Schreiner and co-workers54 have triggered a scientific revisit/inquiry into the long time underappreciated weak London dispersion interaction in organic compounds.55 These findings further substantiate the highly debated dispersion dependent origin of the higher stability of branched alkanes relative to linear alkanes.56 Along these lines, the contribution of dispersion to the chalcogen bonding has attracted considerable attention.57 While there has been tremendous effort made to understand the extent of dispersion contribution to the intermolecular chalcogen bonding,57 there are no straightforward methods available so far to study the extent of its contribution to the intramolecular interactions in general.58 The intramolecular version of symmetry adapted perturbation theory (ISAPT) is still at the early developmental phase.59 The computational studies, including symmetry adapted perturbation theory (SAPT) and dispersion corrected density functional theory (DFT-D) on intermolecular chalcogen bonding indicate the non-negligible dispersion contribution to the ChB.57 To figure out the extent of dispersion contribution to the chalcogen bonding in entries presented in Table 2, we went on to compare the Enet obtained for the systems using dispersion included (DFT-D) and not included DFT methods via homodesmic reactions (vide supra). Surprisingly, the Enet computed for the 2-substituted systems using the dispersion not included method (B3LYP/6-31G(d)) does not deviate much from the Enet computed using dispersion included DFT-D (both Grimme D2 and Grimme D3) methods with a correlation coefficient of 0.982 and 0.991 (respectively for DFT-D2 and DFT-D3, Fig. S3; ESI†). The only exception is 4b, which deviates slightly from the straight line. This might derive some stabilization from the dispersion interaction of the hydrogen atom of the CH2 group with the aryl π-cloud of the benzene ring. By considering 5–10% error contribution from Grimme D3 corrections, as known for intermolecular cases,57 the chalcogen bonding in 2-substituted systems derives negligible contribution from dispersion. This is in accordance with the experimental and computational studies on α-substituted thiophenes/selenophenes comprising formamide/thioformamide as Lewis acid donors.21 It has been shown that the experimental conformational free energy of the IChB stabilized system has a good correlation with the EIChB computed using the dispersion not included DFT method rather than with the EIChB computed using DFT-D methods implying a negligible contribution of dispersion force to IChBs.21 Further, in contrast to the 2-substituted systems, a slightly decreased level of correlation was obtained between the plot of Enet (DFT) and Enet (DFT-D) computed for 2,6-disubstituted systems (Grimme D2 and D3; 0.924 and 0.951 respectively, Fig. S4; ESI†). The decreased correlation coefficients obtained for the 2,6-disubstituted system are indicative of a dispersion contribution to the Enet in the 2,6-disubstituted system that must have an origin in the steric confinement. Further, the dispersion contribution to Enet is substantially larger (76 and 78%, respectively for 16b and 22b) for the systems with large aryl ring distortions than that of the systems with smaller aryl ring distortions. This dispersion contribution might contribute in part to the exceptional stability of the aryl ring strained (Ψav = 4.16°) arylselenenyl bromide 16. However, further experimental and computational studies are required to validate the role of dispersion contribution in the stability of the systems that originate from the steric function.
5. Structural/functional role of the steric confinement/proximal heteroatom
5.1. Kinetically trapped states
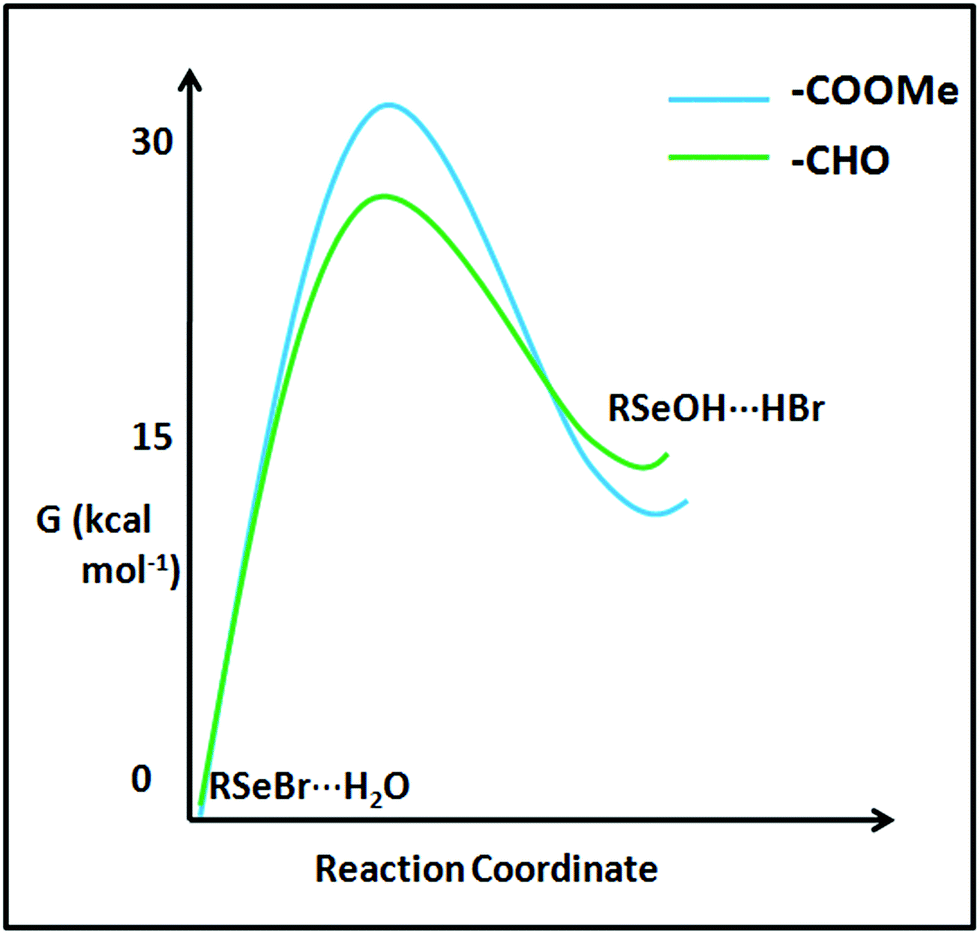
The stability difference between the selenenyl halide 22 (undergoing instantaneous hydrolysis) and 16 (considerably stable under ambient condition) can't be solely understood by looking at the ground state stabilization of the systems inferred from Enet. Destabilization of transition states of their hydrolysis paths may also play a role. The computational transition state analysis of the hydrolysis of selenenyl bromide model 16b reveals that the hydrolysis transition state of 16b is destabilized by about 5.15 kcal mol−1 compared to the hydrolysis transition state of 22b (Fig. 17). It appears that the bulky methoxy group in 16 acts as a kinetic trap to stabilize 16 under ambient conditions. This stability difference between 16 and 22 provides convincing evidence that the cyclization property is facile as long as there is no kinetic barrier. The kinetically trapped molecular systems can serve as reaction intermediates for the synthesis of chalcogen-containing heterocycles. Such trapped states of selenenyl bromides can be used as bench stable selenenylating agents. The use of such kinetically trapped states in the synthesis of heterocycles is well known,44 and their selenenylation reactions remain to be explored.60 In addition, such kinetically trapped states may emerge as potential reporting labels in clinical science and pharmacology. For instance, monitoring the cellular uptake of organic molecules, such as peptides and drugs, is crucial to understand the paths of physiological events and drug development processes. Gammelgaard and co-workers have recently shown the utility of SeMet (selenomethionine) labels to quantify the cellular peptide intake using Inductively-Coupled Plasma Mass Spectrometry (ICP-MS).61 In this experiment, 77Se+, 78Se+ and 82Se+ isotopes were monitored and 82Se+ was used for the quantitative determination of cellular peptide uptake by HeLa WT cells. We expect that the future research involving the development of methods for the synthesis of organochalcogen derivatives with sterically confined chalcogen bonds, yet kinetically stabilized species (e.g., 16 can easily react with the amino group of any molecule) would afford labels for mass spectrometry-based analytical tools. Provided if a 16 like moiety is tethered to a fluorescent reporter, it can report the presence of H2S. The kinetically trapped state may be used to build covalent frameworks. The covalent frameworks have exceedingly high porosity and find application in gas storage and catalysis.62
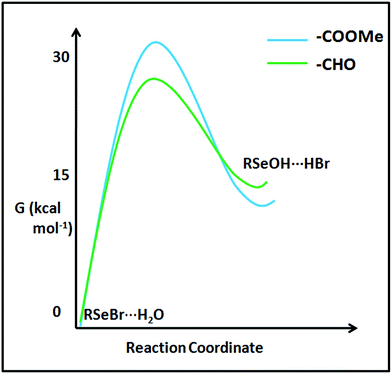 |
| | Fig. 17 Reaction coordinate computed for the hydrolysis of RSeBr (22b and 16b). | |
5.2. Stimulus-responsive sigma holes
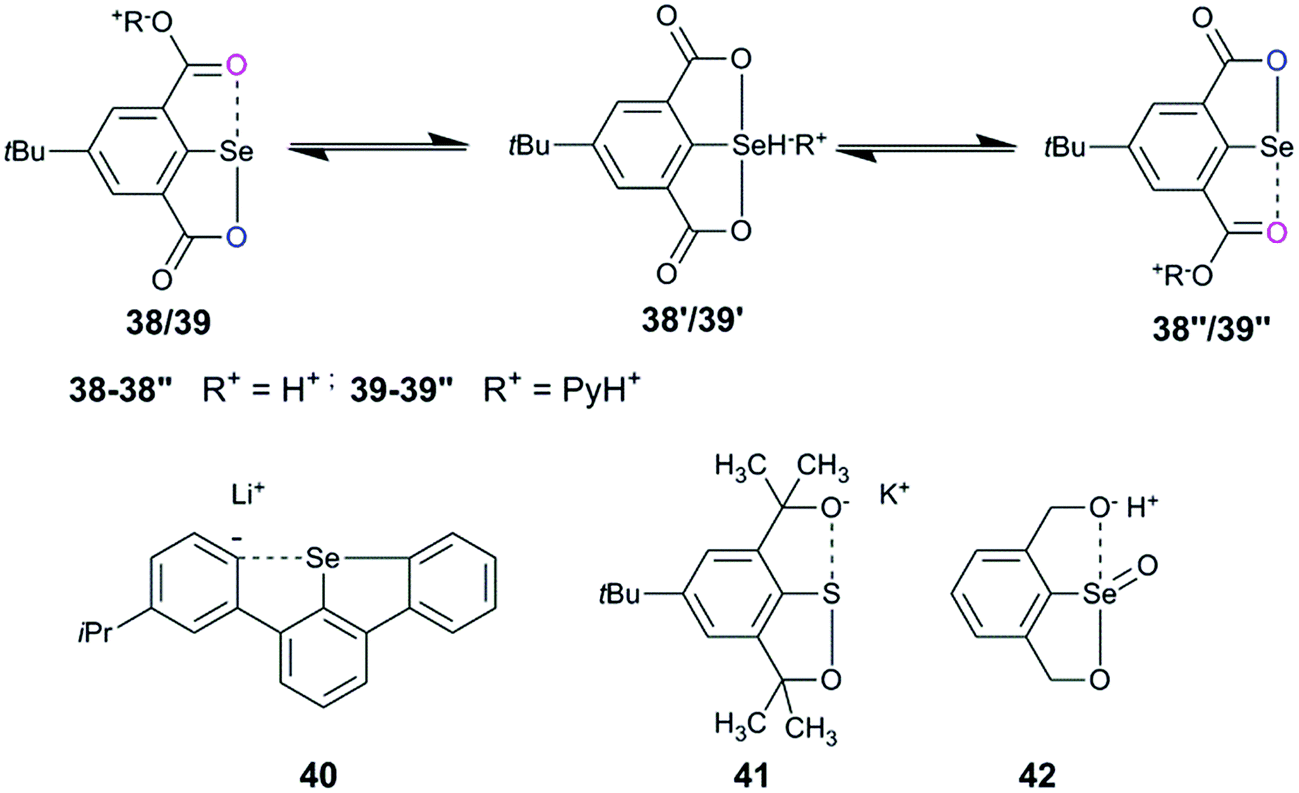
2,6-Disubstituted systems, such as 38/39, showed fluxional behavior under ambient conditions (Fig. 18).40 The cations such as H+ and PyH+ exchange between the two carboxylates via selenuranide intermediates (38′/39′). The fluxional behaviour observed in symmetrical selenocarboxylates (38/39) is further corroborated by the fluxional behaviour of symmetrical compounds (40–42) reported from Reich,63 Martin,64 and Back65 groups.
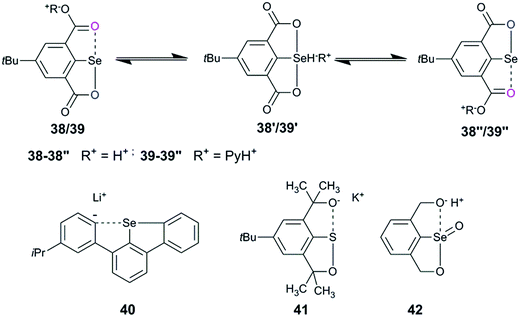 |
| | Fig. 18 Examples showing dynamic intramolecular exchange of cations. | |
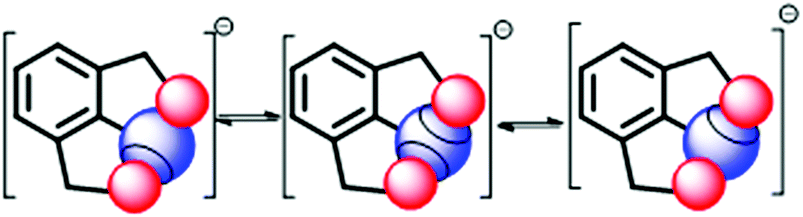
The fluxional behaviour in such systems can be accounted for via the stimulus-responsive (temperature dependent) exchange of sigma holes between two places of the chalcogen atom (Fig. 19). The exchange of the cation is rapid at RT. The selenuranide ion character is most prevalent below decoalescence temperature, which is evident from the formation of a species with an upfield shifted signal in 77Se NMR.40 In the selenuranide ion the O⋯Se bond characteristic appears to lie in between the weak and covalent interaction.40
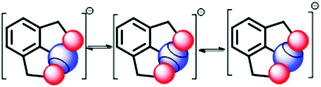 |
| | Fig. 19 Exchange of the sigma hole via the selenuranide intermediate. | |
5.3. Sigma hole sniffers and glutathione peroxidase mimics
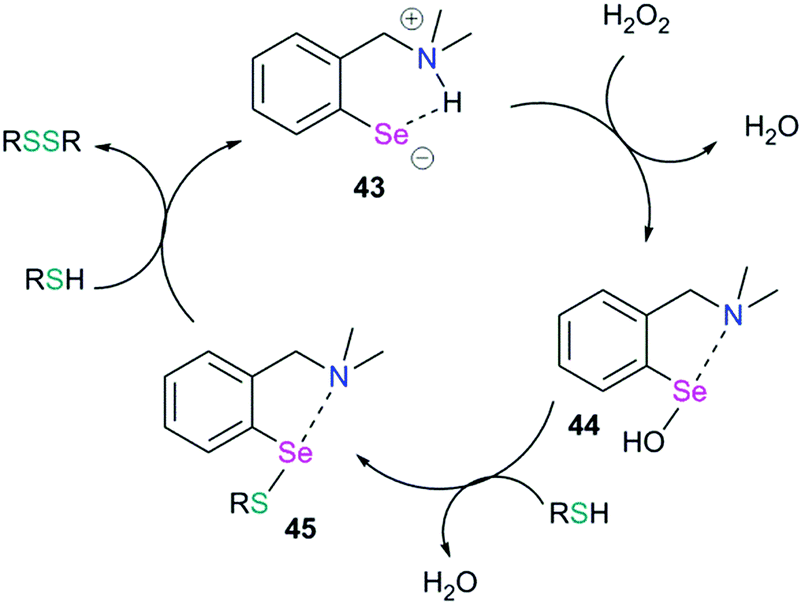
Chalcogen bonding plays an important role in glutathione peroxidase mimic catalytic cycles of arylchalcogen compounds.66 The first step in the catalytic cycle (Fig. 20) is the oxidation of selenol (RSeH, 43) by H2O2 which results in the formation of the selenenic acid intermediate (R–SeOH, 44). The prevalence of a strong IChB at the selenenic acid stage prevents its further oxidation. The selenenic acid has fleeting existence (in the 77Se NMR time scale) and reacts rapidly with the nucleophilic thiols to form the arylselenenyl sulfide intermediate (45).
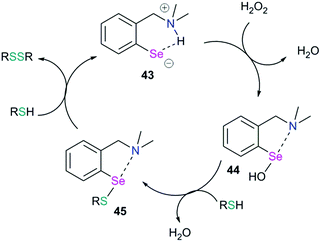 |
| | Fig. 20 Catalytic cycle showing the GPx-like mimic activity of an arylselenium system. | |
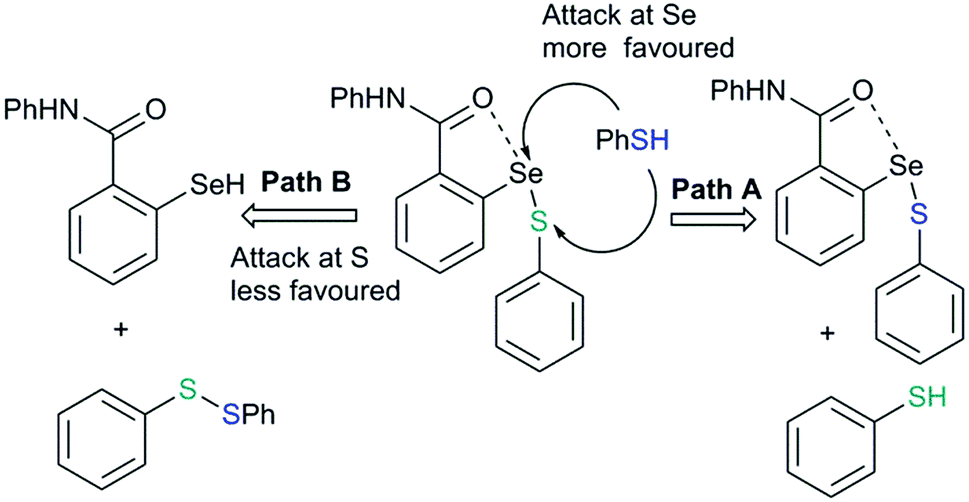
The reaction of this intermediate with an additional thiol molecule leading to the formation of selenol (43) is considered to be the rate-limiting step. Unfortunately, due to the primitive design of the synthetic models, this step is complicated by other competing processes. The presence of a strong IChB at this stage favors nucleophilic attack of the second thiol equivalent on the selenium centre which results in an unwanted thiol exchange reaction (Fig. 21, Path A).3d
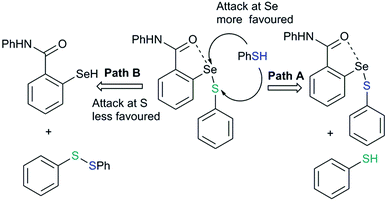 |
| | Fig. 21 The strong IChB at the selenenylsulfide stage facilitates the attack of thiol on sulfur rather than at selenium. | |
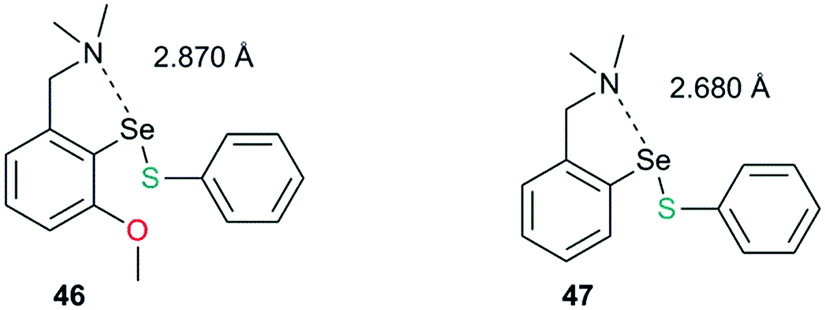
This strong interaction increases the electron density on the sulfur centre of the selenenylsulfide. As a consequence, the nucleophilic attack of the incoming thiol on sulfur is less favored (Fig. 21, Path B). There has been a speculation that adding more than one Lewis base heteroatom/steric function around the selenium center would weaken the D1⋯Se–SR interaction and will enhance GPx-like catalytic performance. Mugesh and co-workers have shown that the replacement of 6-H in benzylamine-type diselenides with an –OMe group indeed weakens the N⋯Se–SeR interaction at the selenenyl sulfide stage that facilitated the attack of incoming thiol on the sulfur center.67 The weakening of the N⋯Se interaction is inferred from the computational modeling. The 6-OMe substituted system (46) has a longer (weaker) N⋯Se distance than that calculated for the system without 6-OMe substitution (47).3d
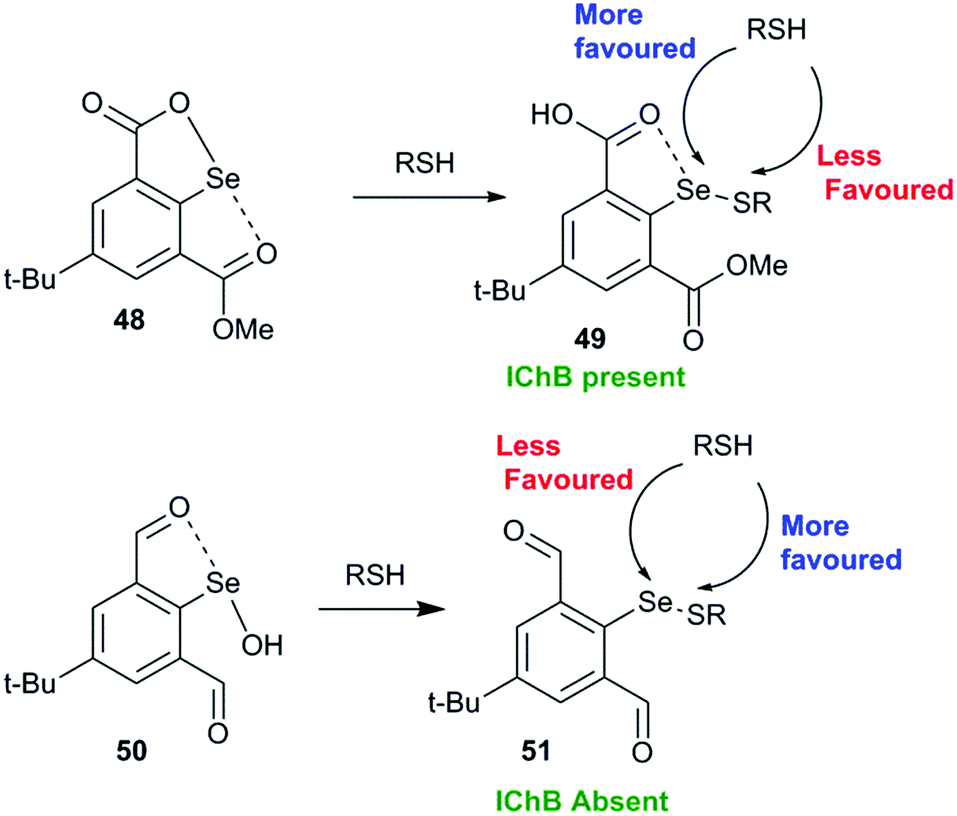
Another interesting study shows that it is advantageous to place the conformationally switchable side arm heteroatom donor around the catalytic chalcogen center (may be called as the sigma hole sniffer) that can undergo a conformational change depending upon the nature of the chalcogen sigma hole. The sniffer Lewis base in the sidearm must be able to exclusively form IChBs only with strong sigma hole donors (Se–OH) and not with the weak sigma hole donor (Se–SR) that will facilitate the second thiol attack “S” at the selenenyl sulfide stage. The 17 times enhanced peroxide decomposition activity of 9 (64.15 ± 2.26 μM min−1) with respect to the diselenide 10 3.86 ± 0.16 μM min−1) in the thiophenol assay can be explained with this paradigm.28 The poor activity of 10 is attributed to the semi-rigid rotor character of the methylester function which promotes the existence of IChBs in all intermediates in the catalytic cycle. As a consequence, the selenenyl sulfide derived from 10 maintains IChBs that promote thiol exchange and the oxidative intramolecular cyclization reaction. As an indirect proof, the selenenyl sulfide intermediate 49 derived from selenenate ester 48 (Fig. 22), a protected form of selenenic acid, showed a downfield 77Se NMR chemical shift (624 ppm) in comparison to the 77Se NMR chemical shift (512–565 ppm) of other arylselenenyl sulfides that show the O/N⋯Se–SR IChB.68 The high catalytic activity of 9 was rationalized on the basis of computational studies, which show that arylselenenyl sulfide 51 derived from 9 having no IChB (anti, anti) conformation is more stable than its conformer having IChBs (syn, anti). However, an opposite trend is observed for selenenic acid 50 derived from 9.28 The absence of IChBs in 51 favours the attack of the second thiol on the sulfur atom. This conformational switching is analogous to the 9→22 conformational switch. The sidearm dependent switchable chalcogen bond is a new paradigm in GPx-like mimics and has to be further studied for better understanding.
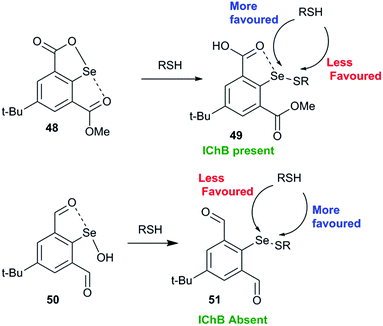 |
| | Fig. 22 Steric confinement with adaptive rotor characteristics prevents IChB and favours the nucleophilic attack on sulfur. | |
6. Conclusions and outlook
While weak non-covalent and steric effects have been traditionally treated as a subject of the organic chemistry scholarship, the amazing conformational diversity encountered in 2,6-disubstituted organochalcogen compounds confronts us with the fact that such interactions are ubiquitous and instrumental in defining the chemistry of main group p-block element containing organometallic/metalloid compounds. The adoption of conformational formalism, borrowed from organic chemistry, to describe the structures of main group organometallic/metalloid compounds would certainly add up new entries into conformational chemistry. Indeed, the conformational chemistry of main group compounds and the role of weak interactions as conformational control are not well established regarding both experiments and theory. We hope that sigma hole dependent switching of conformational states with the flexible side arm function is of great importance in future research. Along these lines, the concept of sigma hole sniffer in the GPx-like activity of the organoselenium system is a new paradigm which has to be studied in detail that in turn will expand the application of IChBs in the development of other functional molecular systems. We have shown in many cases that the heterocycle formation is a dominant process when the central chalcogen is bonded to more electronegative elements. In many circumstances, this process occurs via elusive intermediates in an uncontrolled manner. The methods of kinetically trapping the intermediates with IChBs and steric functions could be useful in developing bench stable selenium reagents for selenenylation reactions and developing methods for the controlled way of synthesizing chalcogen-containing heterocycles. In addition to this, such kinetically trapped organoselenium compounds can serve as mass-spectrometric labels. Kinetically trapped states may emerge as a tool for the controlled synthesis of covalent frameworks and construction of novel multivalent Lewis acid receptors for anions. We believe that, with detailed experimental and theoretical studies, the structural and functional role of sterically confined IChB components would attract interest in supramolecular chemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
H. B. S. gratefully acknowledges the Department of Science and Technology (DST), New Delhi for the J. C. Bose Fellowship. K. S. is thankful to DST, New Delhi for awarding the INSPIRE Faculty Grant. We acknowledge our colleagues Dr V. P. Singh and Dr S. S. Zade, and people from other research groups who have contributed to this research area. Their names are mentioned in the references.
Notes and references
-
(a) G. E. Garett, G. L. Gibson, R. N. Stratus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126–4133 CrossRef PubMed;
(b) L. Brammer, Faraday Discuss., 2017, 203, 485–507 RSC;
(c) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
-
(a) J. S. Murray, P. Lane, T. Clark and P. Politzer, J. Mol. Model., 2007, 13, 1033–1038 CrossRef PubMed;
(b) H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef PubMed;
(c) W. Wang, B. Ji and Y. Zhang, J. Phys. Chem. A, 2009, 113, 8132–8135 CrossRef PubMed;
(d) J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 1–53 CrossRef;
(e) R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef PubMed.
-
(a) S. R. Wilson, P. A. Zucker, R.-R. C. Huang and A. Spector, J. Am. Chem. Soc., 1989, 111, 5936–5937 CrossRef;
(b) G. Mugesh, W.–W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125–2179 CrossRef PubMed;
(c) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255–6285 CrossRef PubMed;
(d) K. P. Bhabak and G. Mugesh, Acc. Chem. Res., 2010, 43, 1408–1418 CrossRef PubMed.
-
(a) M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, F. Marini, L. Bagnoli and A. Temperini, Chem.–Eur. J., 2002, 8, 1118–1124 CrossRef PubMed;
(b) L. Uehlin, G. Fragale and T. Wirth, Chem.–Eur. J., 2002, 8, 1125–1133 CrossRef PubMed;
(c)
Organoselenium Chemistry: A Practical Approach, ed. T. G. Back, Oxford University Press, Oxford, 1999 Search PubMed;
(d)
Organoselenium Chemistry: Synthesis and Reactions, ed. T. Wirth, Wiley-VCH, Weinheim, 2011 Search PubMed.
-
(a) P. Wonner, L. Vogel, F. Kniep and S. M. Huber, Chem.–Eur. J., 2017, 23, 16972–16975 CrossRef PubMed;
(b) P. Wonner, L. Vogel, M. Düser, L. Gomes, F. Kniep, B. Mallick, D. B. Werz and S. M. Huber, Angew. Chem., Int. Ed., 2017, 56, 12009–12012 CrossRef PubMed;
(c) S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812–815 CrossRef PubMed.
-
(a) H. Zhao and F. Gabbaï, Nat. Chem., 2010, 2, 984–990 CrossRef PubMed;
(b) G. E. Garrett, E. I. Carrera, D. S. Seferos and M. S. Taylor, Chem. Commun., 2016, 52, 9881–9884 RSC;
(c) N. A. Semenov, A. V. Lonchakov, N. A. Pushkarevsky, E. A. Suturina, V. V. Korolev, E. Lork, V. G. Vasiliev, S. N. Konchenko, J. Beckmann, N. P. Gritsan and A. V. Zibarev, Organometallics, 2014, 33, 4302–4314 CrossRef;
(d) J. Y. C. Lim, I. Marques, A. L. Thompson, K. E. Christensen, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 3122–3133 CrossRef PubMed.
-
(a) A. F. Cozzolino, P. S. Whitfield and I. Vargas-Baca, J. Am. Chem. Soc., 2010, 132, 17265–17270 CrossRef PubMed;
(b) T. Suzuki, H. Fujii, Y. Yamashita, C. Kabuto, S. Tanaka, M. Harasawa, T. Mukai and T. Miyashi, J. Am. Chem. Soc., 1992, 114, 3034–3043 CrossRef;
(c) A. F. Cozzolino, N. E. Gruhn, D. L. Lichtenberger and I. Vargas-Baca, Inorg. Chem., 2008, 47, 6220–6226 CrossRef PubMed;
(d) A. F. Cozzolino, I. Vargas-Baca, S. Mansour and A. H. Mahmoudkhani, J. Am. Chem. Soc., 2005, 40, 4966–4971 Search PubMed;
(e) A. F. Cozzolino, J. F. Britten and I. Vargas-Baca, Cryst. Growth Des., 2006, 6, 181–186 CrossRef;
(f) T. Chivers, X. L. Gao and M. Parvez, Inorg. Chem., 1996, 35, 9–15 CrossRef PubMed.
-
(a) T. Niksch, H. Görls, M. Friedrich, R. Oilunkaniemi, R. Laitinen and W. Weigand, Eur. J. Inorg. Chem., 2010, 74–94 CrossRef;
(b) S. P. Thomas, K. Satheeshkumar, G. Mugesh and T. N. Guru Row, Chem.–Eur. J., 2015, 21, 6793–6800 CrossRef PubMed.
- L. Chen, J. Xiang, Y. Zhao and Q. Yan, J. Am. Chem. Soc., 2018, 140, 7079–7082 CrossRef PubMed.
- P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kübel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299 CrossRef PubMed.
-
(a) J. Beckmann, J. Bolsinger and A. DuthieK, Chem.–Eur. J., 2011, 17, 930–940 CrossRef PubMed;
(b) K. Srivastava, S. Sharma, H. B. Singh, U. P. Singh and R. J. Butcher, Chem. Commun., 2010, 46, 1130–1132 RSC.
-
(a) K. Lekin, S. M. Winter, L. E. Downie, X. Bao, J. S. Tse, S. Desgreniers, R. A. Secco, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2010, 132, 16212–16224 CrossRef PubMed;
(b) M. Risto, R. W. Reed, C. M. Robertson, R. Oilunkaniemi, R. S. Laitinen and R. T. Oakley, Chem. Commun., 2008, 3278–3280 RSC;
(c) L. S. Konstantinova, I. V. Baranovsky, E. A. Pritchina, M. S. Mikhailov, I. Y. Bagryanskaya, N. A. Semenov, I. G. Irtegova, G. E. Salnikov, K. A. Lyssenko, N. P. Gritsan, A. V. Zibarev and O. A. Rakitin, Chem.–Eur. J., 2017, 23, 17037–17047 CrossRef PubMed.
-
(a) D. H. R. Barton, M. B. Hall, Z. Lin, S. I. Perekh and J. Riebenspies, J. Am. Chem. Soc., 1993, 115, 5056–5059 CrossRef;
(b) M. Iwaoka and S. Tomoda, J. Org. Chem., 1995, 60, 5299–5302 CrossRef;
(c) A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357–4416 CrossRef PubMed;
(d)
T. Chivers and R. S. Laitinen, in Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. A. Devillanova, RSC Publishing, Cambridge UK, 2007, ch. 4, p. 223 Search PubMed.
- S. Zhang, X. Wang, Y. Su, Y. Qiu, Z. Zhang and X. Wang, Nat. Commun., 2014, 5, 4127 CrossRef PubMed.
-
(a) T. Chivers and R. S. Laitinen, Dalton Trans., 2017, 46, 1357–1367 RSC;
(b) A. J. Karhu, O. J. Pakkanen, J. M. Rautiainen, R. Oilunkaniemi, T. Chivers and R. S. Laitinen, Dalton Trans., 2016, 45, 6210–6221 RSC.
- N. W. Alcock, Adv. Inorg. Chem. Radiochem., 1972, 15, 1–58 CrossRef.
-
(a) T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2015, 44, 1725–1739 RSC;
(b)
T. Chivers, A Guide to Chalcogen-Nitrogen Chemistry, World Scientific, London, 2005 CrossRef;
(c) F. De Vleeschouwer, M. Denayer, B. Pinter, P. Geerlings and F. De Proft, J. Comput. Chem., 2018, 39, 557–572 CrossRef PubMed;
(d) V. Oliveira, D. Cremer and E. Kraka, J. Phys. Chem. A, 2017, 121, 6845–6862 CrossRef PubMed.
-
(a) M. Iwaoka, H. Komatsu, T. Katsuda and S. Tomoda, J. Am. Chem. Soc., 2004, 126, 5309–5317 CrossRef PubMed;
(b) D. Roy and R. B. Sunoj, J. Phys. Chem. A, 2006, 110, 5942–5947 CrossRef PubMed;
(c) B. K. Sharma and G. Mugesh, ChemPhysChem, 2009, 10, 3013–3020 CrossRef PubMed.
-
(a) S. Tsuzuki and N. Sato, J. Phys. Chem., B, 2013, 117, 6849–6855 CrossRef PubMed;
(b) M. E. Brezgunova, J. Lieffrig, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, J. G. Ángyán, M. Fourmigué and E. Espinosa, Cryst. Growth Des., 2013, 13, 3283–3289 CrossRef;
(c) M. S. Pavan, A. K. Jana, S. Natarajan and T. N. Guru Row, J. Phys. Chem., B, 2015, 119, 11382–11390 CrossRef PubMed.
-
N. Furukawa and S. Sato, Chemistry of hypervalent compounds, ed. K.-y. Akiba, Wiley-VCH, New York, 1999, ch. 8, p. 241 Search PubMed.
- D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef PubMed.
-
(a) P. Politzer, J. S. Murray and M. C. Concha, J. Mol. Model., 2008, 14, 659–665 CrossRef PubMed;
(b) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
-
(a) G. Mugesh, A. Panda, H. B. Singh, R. J. Butcher and S. Kumar, Chem.–Eur. J., 1999, 5, 1411–1421 CrossRef;
(b) K. Kandasamy, H. B. Singh and R. J. Butcher, New J. Chem., 2004, 28, 640–645 RSC.
- M. Baiwir, M. G. Llabres, O. Dideberg, L. Dupont and J. L. Piette, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2188–2191 CrossRef.
- S. S. Zade, S. Panda, H. B. Singh, R. B. Sunoj and R. J. Butcher, J. Org. Chem., 2005, 70, 3693–3704 CrossRef PubMed.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef PubMed.
- C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2956–2965 CrossRef.
- K. Selvakumar, H. B. Singh and R. J. Butcher, Chem.–Eur. J., 2010, 16, 10576–10591 CrossRef PubMed.
- K. Selvakumar, H. B. Singh, N. Goel, U. P. Singh and R. J. Butcher, Dalton Trans., 2011, 40, 9858–9867 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef.
- T. Chakraborty, K. Srivastava, S. Panda, H. B. Singh and R. J. Butcher, Inorg. Chim. Acta, 2010, 363, 2905–2911 CrossRef.
- S. K. Tripathi, U. Patel, D. Roy, R. B. Sunoj, H. B. Singh, G. Wolmershäuser and R. J. Butcher, J. Org. Chem., 2005, 70, 9237–9247 CrossRef PubMed.
- K. Selvakumar, P. Shah, H. B. Singh and R. J. Butcher, Chem.–Eur. J., 2011, 17, 12741–12755 CrossRef PubMed.
- S. S. Zade, H. B. Singh and R. J. Butcher, Angew. Chem., Int. Ed., 2004, 43, 4513–4515 CrossRef PubMed.
- V. P. Singh, H. B. Singh and R. J. Butcher, Eur. J. Inorg. Chem., 2010, 637–647 CrossRef.
- V. P. Singh, H. B. Singh and R. J. Butcher, Chem.–Asian. J., 2011, 6, 1431–1442 CrossRef PubMed.
- H. Fujihara, H. Mima and N. Furukawa, J. Am. Chem. Soc., 1995, 117, 10153–10154 CrossRef.
- A. Pop, A. Silvestru, E. J. J. Pérez, M. Arca, V. Lippolis and C. Silvestru, Dalton Trans., 2014, 43, 2221–2233 RSC.
- T. Wirth and G. Fragale, Chem.–Eur. J., 1997, 3, 1894–1902 CrossRef.
- K. Selvakumar, H. B. Singh, N. Goel, U. P. Singh and R. J. Butcher, Chem.–Eur. J., 2012, 18, 1444–1457 CrossRef PubMed.
- G. Roy and G. Mugesh, J. Am. Chem. Soc., 2005, 127, 15207–15217 CrossRef PubMed.
- B. Kersting and M. Delion, Z. Naturforsch. B, 1999, 54, 1042–1047 Search PubMed.
- S. S. Zade, S. Panda, S. K. Tripathi, H. B. Singh and G. Wolmershäuser, Eur. J. Org. Chem., 2004, 3857–3867 CrossRef.
- K. Selvakumar, H. B. Singh and R. J. Butcher, Tetrahedron Lett., 2011, 52, 6831–6834 CrossRef.
- K. Selvakumar, H. B. Singh, N. Goel and U. P. Singh, Organometallics, 2011, 30, 3892–3896 CrossRef.
- V. P. Singh, H. B. Singh and R. J. Butcher, Eur. J. Org. Chem., 2011, 5485–5497 CrossRef.
- V. P. Singh, H. B. Singh and R. J. Butcher, Chem. Commun., 2011, 47, 7221–7223 RSC.
- P. R. Prasad, K. Selvakumar, H. B. Singh, V. I. Minkin and R. J. Butcher, J. Org. Chem., 2016, 81, 3214–3226 CrossRef PubMed.
-
(a) D. H. R. Barton and R. C. Cookson, Q. Rev., Chem. Soc., 1956, 10, 44–82 RSC;
(b) V. Dragojlovic, ChemTexts, 2015, 1, 14 CrossRef.
- C. R. S. Matos, M. J. Xavier, L. S. Barreto, N. B. Costa Jr and I. F. Gimenez, Anal. Chem., 2007, 79, 2091–2095 CrossRef PubMed.
- D. M. Pawar, K. K. Wilson and E. A. Noe, J. Org. Chem., 2000, 65, 1552–1553 CrossRef PubMed.
- R. Vargas, J. Garza, D. Dixon and B. P. Hay, J. Phys. Chem. A, 2001, 105, 774–778 CrossRef.
- K. Selvakumar, V. P. Singh, P. Shah and H. B. Singh, Main Group Chem., 2011, 10, 141–152 Search PubMed.
- P. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Nature, 2011, 477, 308–311 CrossRef PubMed.
- H. Schwertfeger, A. A. Fokin and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 1022–1036 CrossRef PubMed.
-
(a) S. Grimme, Angew. Chem., Int. Ed., 2006, 45, 4460–4464 CrossRef PubMed;
(b) M. D. Wodrich, C. S. Wannere, Y. Mo, P. D. Jarowski, K. N. Houk and P. v. R. Schleyer, Chem.–Eur. J., 2007, 13, 7731–7744 CrossRef PubMed.
-
(a) C. Bleiholder, D. B. Werz, H. Köppel and R. Gleiter, J. Am. Chem. Soc., 2006, 128, 2666–2674 CrossRef PubMed;
(b) A. F. Cozzolino, P. J. W. Elder, L. M. Lee and I. Vargas-Baca, Can. J. Chem., 2013, 91, 338–347 CrossRef;
(c) R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef PubMed.
-
(a) J. F. Gonthier and C. Corminboeuf, Chimia, 2014, 68, 221–226 CrossRef PubMed;
(b) A. Wuttke and R. A. Mata, J. Comput. Chem., 2017, 38, 15–23 CrossRef PubMed.
- R. M. Parrish, J. F. Gonthier, C. Corminboeuf and C. David Sherrill, J. Chem. Phys., 2015, 143, 051103 CrossRef PubMed.
-
(a) G. Fragale, M. Neuburger and T. Wirth, Chem. Commun., 1998, 1867–1868 RSC;
(b) M. Spichty, G. Fragale and T. Wirth, J. Am. Chem. Soc., 2000, 122, 10914–10916 CrossRef.
- B. Gammelgaard and B. P. Jensen, J. Anal. At. Spectrom., 2007, 22, 235–249 RSC.
-
(a) Y. Jin, Y. Zhu and W. Zhang, CrystEngComm, 2013, 15, 1484–1499 RSC;
(b) L.-H. Li, X.-L. Feng, X.-H. Cui, Y. X. Ma, S.-Y. Ding and W. Wang, J. Am. Chem. Soc., 2017, 139, 6042–6045 CrossRef PubMed.
- H. J. Reich, M. J. Bevan, B. O. Gudmundsson and G. L. Puckett, Angew. Chem., Int. Ed., 2002, 41, 3436–3439 CrossRef PubMed.
- P. H. W. Lau and J. C. Martin, J. Am. Chem. Soc., 1978, 100, 7077–7079 CrossRef.
- N. M. R. McNeil, M. C. Matz and T. G. Back, J. Org. Chem., 2013, 78, 10369–10382 CrossRef PubMed.
- K. P. Bhabak and G. Mugesh, Chem.–Eur. J., 2008, 14, 8640–8651 CrossRef PubMed.
- D. Bhowmick, S. Srivastava, P. D'Silva and G. Mugesh, Angew. Chem., Int. Ed., 2015, 54, 8449–8453 CrossRef PubMed.
-
(a) K. P. Bhabak and G. Mugesh, Chem.–Eur. J., 2009, 15, 9846–9854 CrossRef PubMed;
(b) K. P. Bhabak and G. Mugesh, Chem.–Asian J., 2009, 4, 974–983 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and coordinates. See DOI: 10.1039/c8sc01943f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Harkesh B.
Singh
*a and
Harkesh B.
Singh