DOI:
10.1039/C7SC05210C
(Perspective)
Chem. Sci., 2018,
9, 1408-1423
Porphyrinoids as a platform of stable radicals
Received
8th December 2017
, Accepted 5th January 2018
First published on 8th January 2018
Abstract
The non-innocent ligand nature of porphyrins was observed for compound I in enzymatic cycles of cytochrome P450. Such porphyrin radicals were first regarded as reactive intermediates in catabolism, but recent studies have revealed that porphyrinoids, including porphyrins, ring-contracted porphyrins, and ring-expanded porphyrins, display excellent radical-stabilizing abilities to the extent that radicals can be handled like usual closed-shell organic molecules. This review surveys four types of stable porphyrinoid radical and covers their synthetic methods and properties such as excellent redox properties, NIR absorption, and magnetic properties. The radical-stabilizing abilities of porphyrinoids stem from their unique macrocyclic conjugated systems with high electronic and structural flexibilities.
1. Introduction
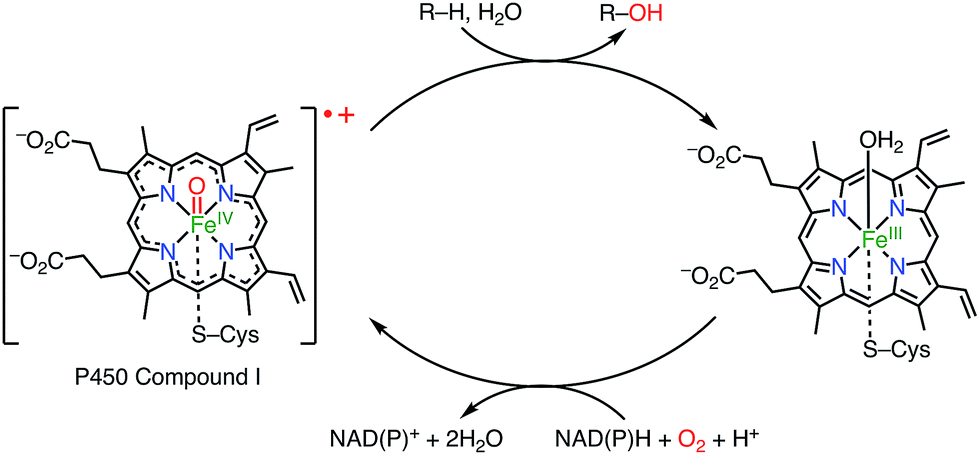
Porphyrins and related macrocycles1 play important roles in nature, as exemplified by the special pair of chlorophylls in the photosynthetic reaction center (RC), hemes, and cyanocobalamine (vitamin B12), to fulfill various biofunctions. Key features of these “pigments of life” are redox-active behaviors, coordination abilities, and light-harvesting properties. For example, the special pair of chlorophyll in the RC, upon photoexcitation, transfers an electron to an acceptor to form its radical cation, which triggers subsequent electron transfer events in photosynthesis known as the Z-scheme.2a Another example is cytochrome P450, which acts as a hydroxylation catalyst via the involvement of a radical cation intermediate (known as compound I; Fig. 1).2b Therefore, radicals and radical ions of porphyrins have been extensively investigated as models of intermediates in key biochemical processes. These radicals are usually short-lived and highly reactive. On the other hand, recent studies have revealed that some porphyrinoid radicals and radical ions are extremely stable.
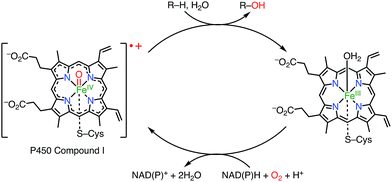 |
| | Fig. 1 Cytochrome P450 compound I. | |
Here, we review the exceptionally effective radical-stabilizing abilities of porphyrinoids, including porphyrins, ring-contracted porphyrins, ring-expanded porphyrins, and core-modified porphyrins. In early studies, radicals stabilized by porphyrinoids had been found serendipitously, but in recent studies it has been demonstrated that the installation of a porphyrinoid at an appropriate conjugative position can be a general strategy for producing a stable radical. Importantly, in most cases radicals have been stabilized by porphyrinoids to such an extent that they are tolerant of air, moisture, and silica gel column chromatography, so that they can be handled like usual organic molecules. In special cases, radicals can undergo chemical transformations for their further modification.
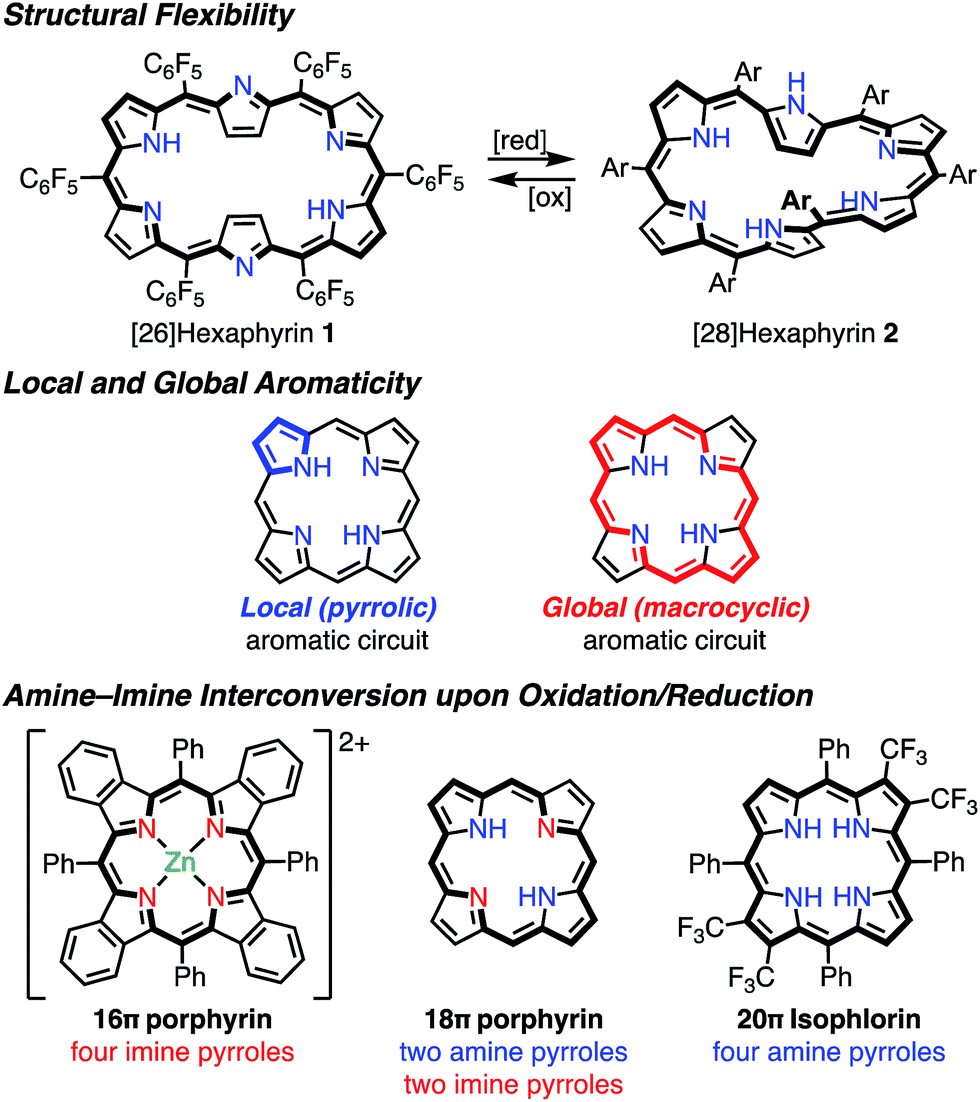
It has been shown that porphyrinoids, in particular expanded porphyrins, are a suitable platform for generating various electronic states such as Hückel and Möbius (anti)aromatic species.1f–i This ability has been attributed to their structural and electronic flexibilities. The structural flexibilities of expanded porphyrins allow facile conformational changes upon a change in the number of π-electrons. A representative example is hexaphyrin(1.1.1.1.1.1), which can exist as a planar 26π Hückel aromatic species 1 and a twisted 28π Möbius aromatic species 2, which are both stable compounds (Fig. 2). The electronic flexibilities of porphyrinoids are closely related to the facile amine–imine (pyrrole–pyrrolenine) interconversion of their pyrrole segments. For example, porphyrins consist of two amine-type and two imine-type pyrroles and can accommodate 16–20π electronic states via changes in the amine or imine forms of the pyrroles.3 Because the amine-type pyrroles can act as electron donors and the imine-type pyrroles can act as electron acceptors, porphyrins are subject to both oxidation and reduction. The stabilities of the resulting non- or anti-aromatic electronic states should be correlated with the macrocyclic aromatic stabilization energy.4 The total aromatic stabilization energies of porphyrinoids largely depend on the local aromaticity of the constituent pyrroles. Thus, the contribution of the macrocyclic aromatic stabilization energy is relatively small.4b Although the macrocyclic aromaticity is energetically low, aromatic porphyrinoids display an effective macrocyclic conjugated network as evidenced by strong diatropic ring currents, which are predominantly due to the global conjugation circuit. A theoretical interpretation of this specific balance between aromaticity and conjugation has been attempted.4a It is considered that these unique electronic and structural flexibilities generally play important roles in stabilizing radicals as well as aromatic and anti-aromatic species.
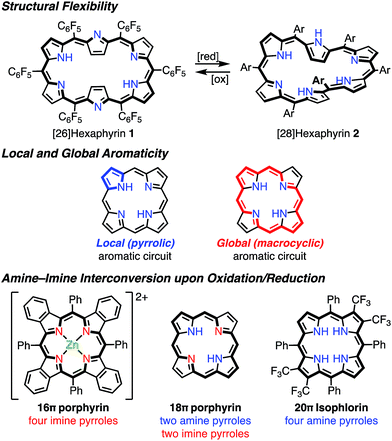 |
| | Fig. 2 Important ideas for understanding the structural and electronic properties of porphyrins. | |
Although there are many studies of porphyrinoids that are covalently linked to or coordinated with stable localized radicals (e.g. nitroxides and nitronyl nitroxides),5 this review will focus on organic radicals stabilized by spin delocalization over the π-electron systems of porphyrinoids.
2. Classification of porphyrinoid-based radicals
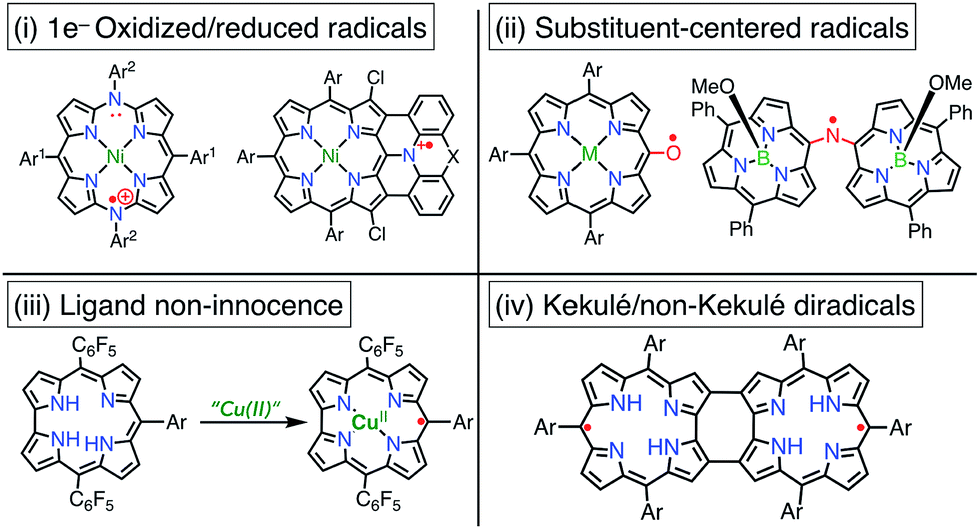
Porphyrinoid-based radicals can be classified into four types according to the origin of the radical, as shown in Fig. 3: (i) one-electron-oxidized/reduced radical ions (Chapter 3), (ii) substituent-centered neutral radicals (Chapter 4), (iii) non-innocent macrocyclic radicals of metal complexes (Chapter 5), and (iv) Kekulé/non-Kekulé-type diradicals (Chapter 6). Regardless of this classification, stable organic radicals are required to satisfy the following criteria: (1) effective delocalization of spin density for thermodynamic stabilization, (2) appropriate steric protection (kinetic stabilization) to avoid dimerization and undesired reactions, and (3) moderate redox potentials to prevent the facile reduction and oxidation of the radical itself. Therefore, most stable radicals are neutral species.
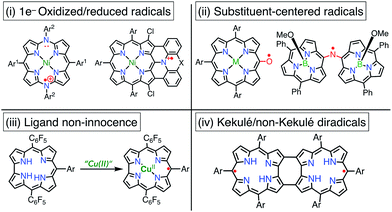 |
| | Fig. 3 Four types of porphyrinoid-based stable radicals. | |
2.1. Methodologies for characterization of porphyrinoid radicals
Because porphyrinoid radicals have unique open-shell electronic structures, they should be characterized very carefully. For example, delocalized radicals are NMR-silent owing to fast electronic relaxations. Because NMR spectroscopy is not effective for radicals, the structural determination of radicals largely relies on X-ray diffraction analysis. When X-ray analysis is impossible, NMR characterizations of 1e− oxidized or reduced compounds are also useful. EPR spectra with hyperfine couplings sometimes provide structural information, but porphyrinoid radicals frequently display isotropic signals around g = 2 without hyperfine structures. On the other hand, anisotropic EPR signals indicate the contribution of spin–orbit interactions with coordinated metals. Quantitative insights into spin states can be obtained by using VT-EPR, SQUID, and NMR studies. In the case of metalloporphyrinoids, the determination of the valence of the central metal is also important and is often provided by Mössbauer and X-ray photoelectron spectroscopies. The nature of the spin distribution can be estimated from hyperfine coupling constants in EPR spectra and via theoretical calculations.
3. One-electron-oxidized/reduced radical ions
Metalloporphyrins are easily oxidized or reduced to generate the corresponding radical cations or radical anions, which often occur not on the central metal but on the porphyrin macrocycle.6 These transformations can be accomplished by chemical reactions or electrochemical methods, but the radical ions thus generated are mostly reactive and thus unstable.7 However, in recent years, very stable porphyrinoid radical ions have been found. In this section, we discuss recent examples of stable radical ions of porphyrinoids.
3.1. Stable radical cations of porphyrinoids
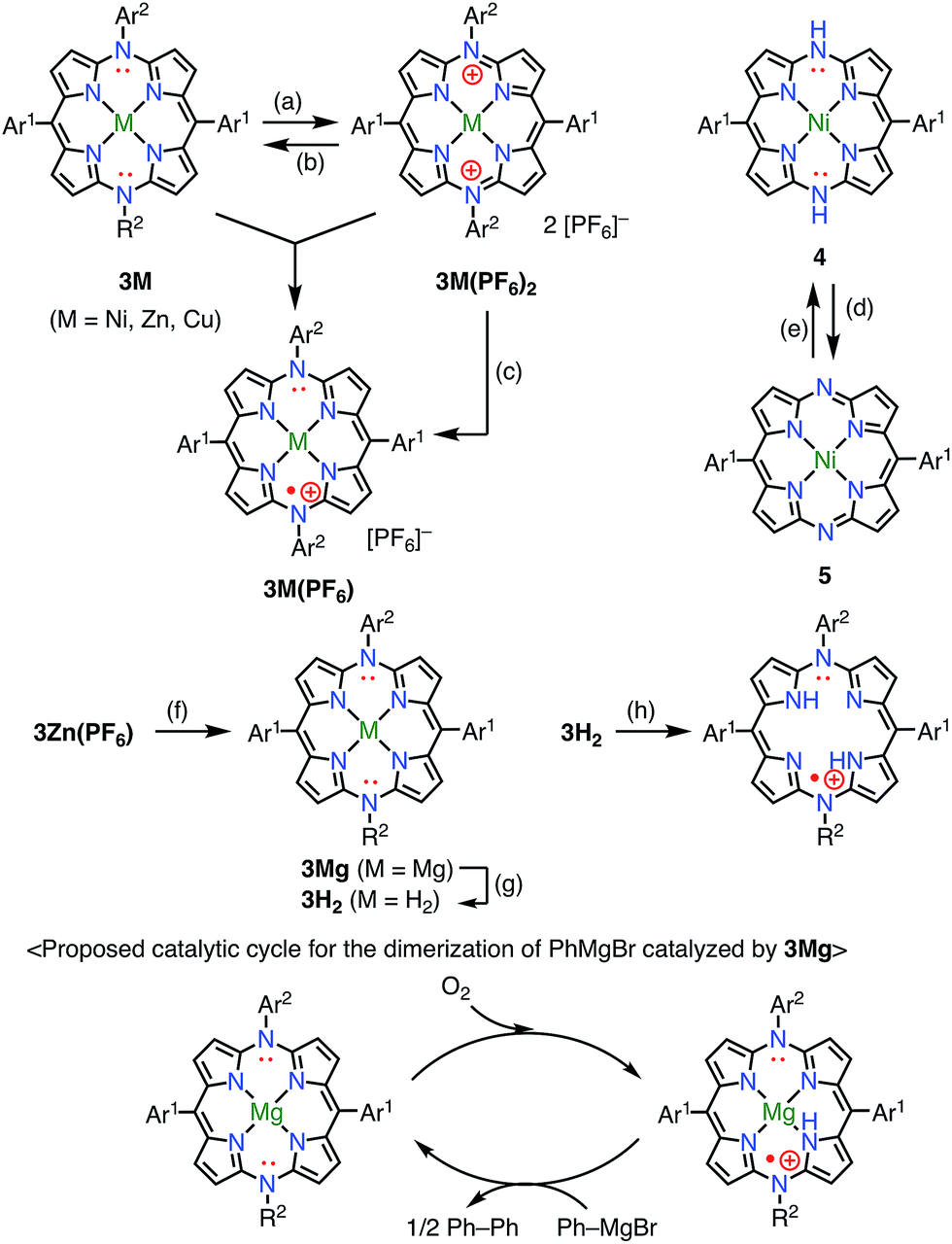
Diazaporphyrin is a macrocycle in which two methine-carbon of porphyrin are replaced by nitrogen atoms. Matano et al. reported multi-redox processes of the 20π anti-aromatic 5,15-diazaporphyrins 3M (M = Ni, Zn, Cu).8 Owing to their electron-rich nature, 3M exhibited multiple stable oxidation states. For example, two-electron oxidation of 3M with AgPF6 afforded 16π-electron dications 3M(PF6)2, and subsequent reduction with NaBH4 or CoCp2 gave 19π-electron monocations 3M(PF6) as stable compounds (Fig. 4). The free-base derivative 3H2(PF6) was also prepared via demetalation of the corresponding Mg(II) complex 3Mg followed by chemical 1e− oxidation. These radical cations were fairly stable under ambient conditions and were purified by silica gel column chromatography. The redox potentials of 3Ni(PF6) were determined by cyclic voltammetry to be Eox1 = 0.29 V (20π/19π) and Ered1 = −0.37 V (19π/18π) vs. Ag/Ag+ in CH2Cl2. Importantly, the mixing of 3Ni and 3Ni(PF6)2 also afforded the radical cation 3Ni(PF6) as evidenced by the EPR spectrum, which exhibited hyperfine coupling with the two meso-nitrogen atoms and four β-protons. The absorption spectrum of 3Ni(PF6) in toluene exhibited a characteristic band in the NIR region around 880 nm. 3Cu(PF6), 3Zn(PF6), and 3H2(PF6) also displayed electrochemical and optical properties that were quite similar to those of 3Ni(PF6). In the case of 3Cu(PF6), a magnetic interaction between the paramagnetic Cu(II) center and the π-radical is possible. In fact, neither EPR nor NMR signals were observed, which was presumably due to large zero-field splitting and the contribution of the triplet state, respectively. However, the assignment of the ground state is under discussion. Intriguingly, 3Mg was found to catalyze the dimerization of a phenyl Grignard reagent in the presence of air. NMR and EPR experiments suggested a mechanism involving a 20π/19π redox cycle of 3Mg, as shown in Fig. 4. Shinokubo et al. also reported the diazaporphyrin 4 without substituents on the meso-nitrogen atoms.9 In this case, a 19π species of the radical cation was not observed from the oxidation of the 20π species or the reduction of the 18π species 5, which was presumably due to facile disproportionation of the cationic intermediate.
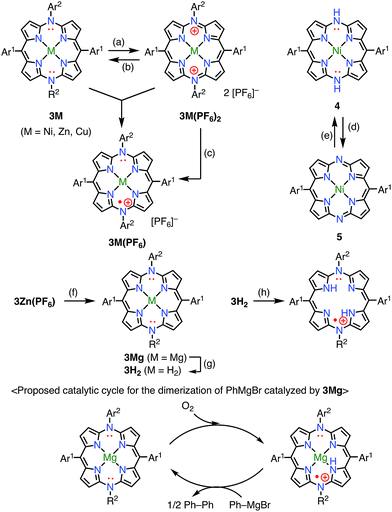 |
| | Fig. 4 Redox scheme of diazaporphyrins. (a) 2 equiv. AgPF6, CH2Cl2, rt, 2 min, 85% for 3Ni(PF6)2, 82% for 3Zn(PF6)2, 75% for 3Cu(PF6)2; (b) excess NaBH4, THF, rt, 8 h, 77% for 3Ni(PF6)2; (c) 1 equiv. NaBH4 or CoCp2, MeOH, 2 min, 80% for 3Ni(PF6), 85% for 3Zn(PF6), 75% for 3Cu(PF6); (d) N,N′-bis(trimethylsilyl)-1,4-dihydropyrazine, THF, rt, 48 h, then MeOH, 78%; (e) aerobic oxidation; (f) 61 equiv. PhMgBr, toluene/THF, rt, 10 min, not isolated; (g) trifluoroacetic acid, 70 °C, 7 h, 38% from 3Zn(PF6); (h) 1 equiv. AgPF6, CH2Cl2, rt, 2 min, 30%. Ar1 = Mesityl, Ar2 = 4-methoxyphenyl. | |
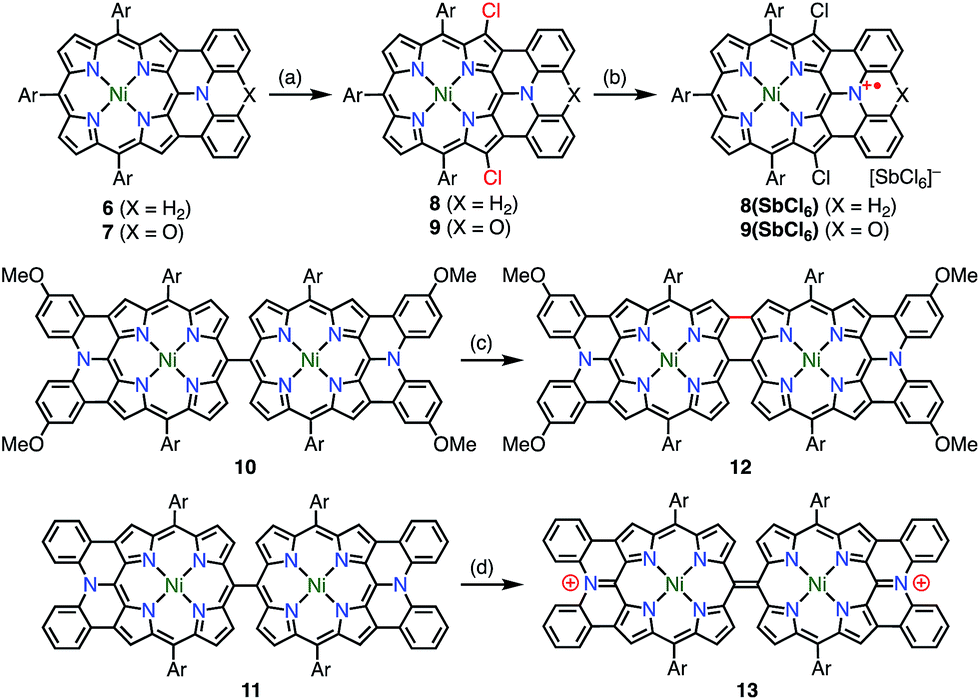
Osuka et al. synthesized the diphenylamine-fused porphyrins 6 and 7 (Fig. 5).10 The lone-pair electrons of the embedded nitrogen atom were well conjugated with the extended porphyrin conjugated network, which caused a decrease in oxidation potentials. Chemical 1e− oxidation gave the corresponding radical cations, both as unstable species. One of the degradation products was a β-chlorinated species, which indicated high spin density and reactivity at the β-position next to the fused moiety. To suppress this degradation pathway, 6 and 7 were chlorinated to give 8 and 9. Chemical oxidation of 8 and 9 gave 8(SbCl6) and 9(SbCl6), respectively, which were found to be extremely stable towards air, water, and silica gel column chromatography. As a result of this success, the diarylamine-fused porphyrin dimers 10 and 11 were prepared and their oxidation to the corresponding diradicals was attempted.11 Upon oxidation with magic blue, however, 10 and 11 were transformed into the partially fused dimer 12 and the closed-shell quinonoidal species 13, respectively, instead of open-shell diradicals.
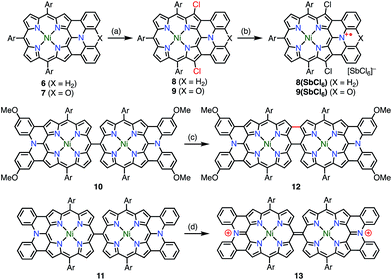 |
| | Fig. 5 Diarylamine-fused porphyrins. (a) 2.2 equiv. Palau'chlor, CHCl3, 0 °C to rt, 87% for 8, 77% for 9; (b) 1.1 equiv. magic blue, CH2Cl2, rt, 10 min, 79%; (c) 6.0 equiv. magic blue, CH2Cl2, rt, 15 min, then H2NNH2·H2O, 91%; (d) 2.2 equiv. magic blue, CH2Cl2, 10 min, 72%. Ar = 3,5-di-tert-butylphenyl. | |
3.2. Stabilizing radical anions is a challenge
Although the properties of 1e−-reduced porphyrin radical anions have been investigated,12 the isolation of stable radical anions of porphyrinoids has remained challenging. The reason is simply because porphyrinoids are usually electron-rich, and their radical anions are even more easily oxidized under aerobic conditions. Thus, the isolation of porphyrin radical anions has been difficult so far.
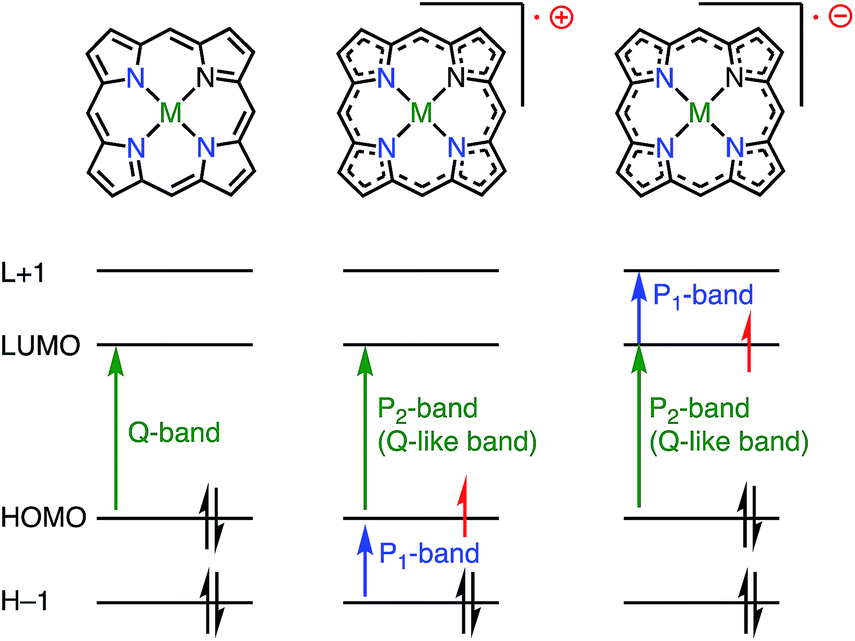
Whereas the SOMO orbital of radical cations reflects the characteristics of the HOMO of their neutral precursors, the SOMO orbital of radical anions represents the characteristics of the LUMO of their precursors. Therefore, the radical anions of porphyrinoids should have different electronic states from those of the radical cations, and it is also desired to investigate such radical anion species. Because most aromatic porphyrinoids have a [4n + 2]π-system, [4n + 1]π-radicals are easily accessible via the one-electron oxidation of neutral aromatic porphyrinoids, but [4n + 3]π-radicals are relatively difficult to generate. To the best of our knowledge, only two strategies for generating [4n + 3]π-radicals have been reported: (1) oxidative generation from stable [4n]π-species8,13,14 and (2) the incorporation of a positively charged center into the electron-deficient chromophore to balance the charge.15Fig. 6 summarizes the important orbitals and optical transitions of a neutral molecule, a radical cation, and a radical anion of porphyrin. Both radical anions and cations display similar P1 and P2 absorption bands. Although the origins of the P2 bands are similar for radical anions and radical cations, those of the P1 bands are different. Here, we use the terms P1 and P2 bands to indicate the lowest-energy transition (P1) and the following Q-like (P2) band.16,17
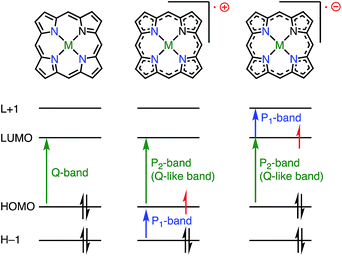 |
| | Fig. 6 Difference between radical cation and anion. | |
4. Substituent-centered radicals
4.1. Heme catabolism and meso-oxy radicals
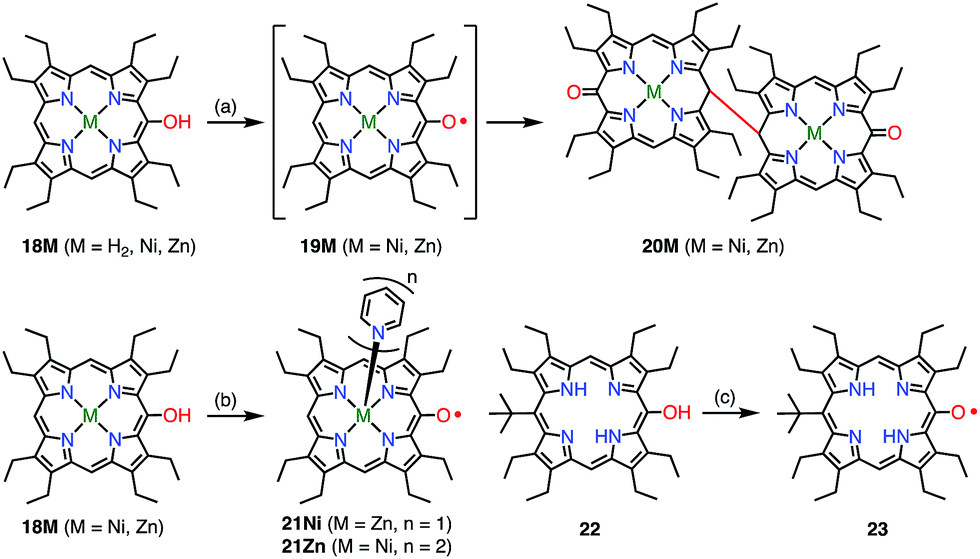
Heme is an iron complex of porphyrin that plays crucial roles in catalysis in heme proteins. However, once heme is released from the protein, it causes toxicity by generating reactive oxygen species. Therefore, the catabolism of heme is critically important in maintaining homeostasis. Because the first step of the catabolic process is known to be oxidation by heme oxygenase to give meso-oxyheme 15,18 its derivatives have attracted great interest (Fig. 7). The structure of 15 in heme oxygenase was determined to be 15A, but the reactivity that was observed toward molecular oxygen implied the contribution of the π-radical 15B.19 Although Bonnett et al. reported 1% paramagnetism for the free base β-octaethyl-meso-hydroxyporphyrin 18H2 in 1970,20 it was a long time until the first structural characterization of the paramagnetic species, i.e., the meso-oxy radical (oxophlorin radical), in 1992.21 Fuhrhop et al. investigated the reactivity of Zn(II) and Ni(II) complexes of the meso-oxy radical 19M (M = Ni, Zn) generated in situ and found rapid and irreversible dimerization at the 15-position.22
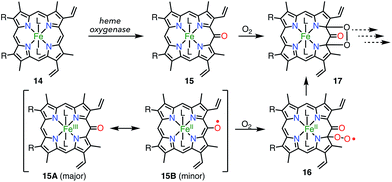 |
| | Fig. 7 Heme catabolism. | |
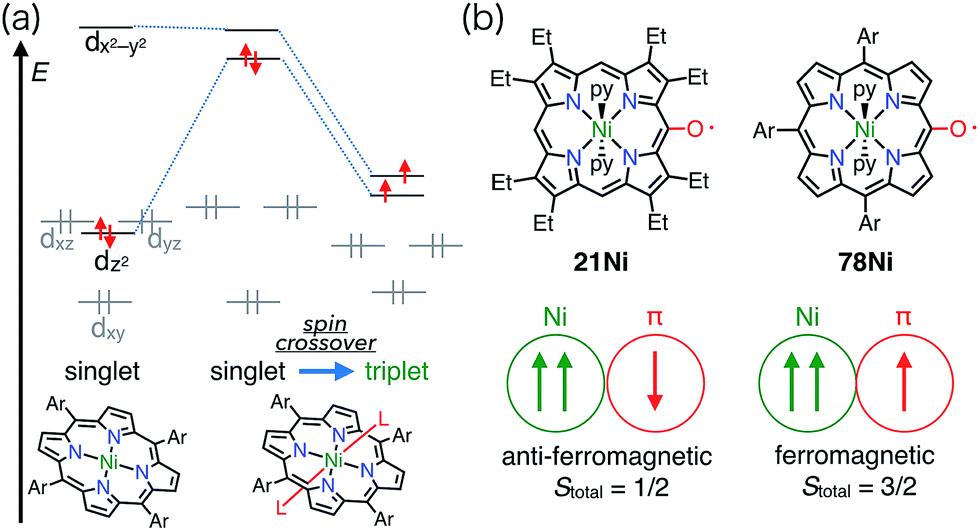
Balch et al. later investigated a series of β-octaethylporphyrin (OEP) meso-oxy radicals.23 They found that the oxidation of Zn(II) and Ni(II) meso-hydroxy OEP 18M in the presence of pyridine gave the meso-oxy radicals 21Zn and 21Ni as stable compounds (Fig. 8).21,24 X-ray diffraction analysis confirmed the axial coordination of the pyridine on the central metal. The coordinated pyridine was demonstrated to effectively stabilize the radical, because the removal of the pyridine led to the instantaneous dimerization of the radical. The radical 21Zn had a short Cmeso–O bond (1.268 Å) in comparison to that of 18Zn (1.392 Å), which is a common feature of meso-oxy radicals. In 21Ni, the octahedrally coordinated Ni(II) atom had a high-spin (S = 1) state as evidenced by an increase in the Ni–N distance, which arose from population of the dx2−y2 orbital. However, the EPR spectrum of 21Ni in pyridine displayed a sharp signal at g = 2.006, and its magnetic moment was determined to be 2.5(2) μB, which indicated the S = 1/2 character of the ground state. These results were interpreted in terms of an antiferromagnetic interaction between the porphyrin π-radical and the high-spin Ni(II) center.
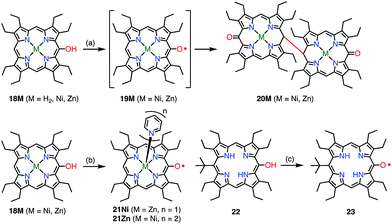 |
| | Fig. 8 Stable OEP meso-oxy radicals. (a) Electrochemical oxidation in CH2Cl2; (b) I2, pyridine, rt, 15 min, 48% for 21Ni; air, pyridine for 21Zn; (c) air, daylight, CH2Cl2, 90%. | |
Smith et al. synthesized the free-base meso-oxy radical 23 bearing a tert-butyl group at the 15-position. Steric protection at the 15-position, where the highest spin density was expected, led to enhanced stability.25 In fact, 23 displayed improved stability but underwent 30% degradation after two weeks in solution. By employing this strategy, the authors demonstrated an elegant way to construct meso–meso-linked oxophlorin oligomers by controlling radical–radical coupling.26
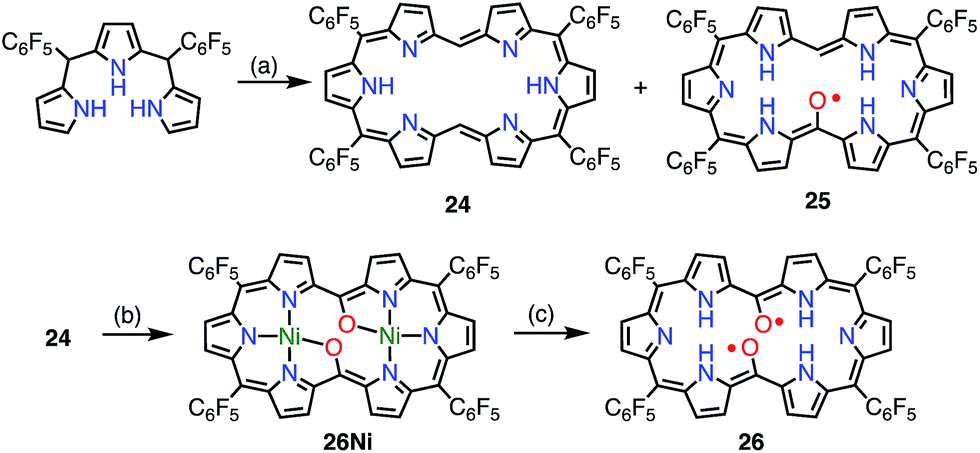
Osuka et al. attempted the synthesis of 15,30-bis(meso-free) [26]hexaphyrin(1.1.1.1.1.1) 24.13 The authors obtained 24 together with the meso-oxy radical 25via the aerobic oxidation of 24 (Fig. 9). X-ray diffraction analysis of 25 revealed a planar structure with a short Cmeso–O bond length of 1.281 Å, which indicated a highly delocalized spin distribution. As is typical for radical porphyrinoids, 25 displayed quite reversible oxidation and reduction waves with a small redox gap of 0.48 V. Furthermore, Osuka et al. found that the oxidation of 24 upon metalation with Ni(II) followed by demetalation gave the bis(meso-oxy) diradical 26.2726 was shown to be a stable non-Kekulé diradical, because SQUID magnetometry confirmed the singlet nature of its ground state with J/kB = −645 K, and CASSCF(2,2) calculations showed an open-shell singlet diradical ground state with a diradical index of 0.62. Reflecting the singlet nature of the ground state, the 1H NMR spectrum of 26 displayed signals due to β-protons at 8.0 and 9.6 ppm, as well as a signal due to the inner NH group at 4.5 ppm at −80 °C in CD2Cl2, which indicated a diatropic ring current in the 26π aromatic system. These signals became broader upon an increase in temperature owing to the population of the triplet diradical state. The aromaticity of 26 was further confirmed by theoretical calculations: an NICS(0) of −8.2 ppm and a net clockwise (diamagnetic) flow of current density in anisotropy of current-induced density (ACID) calculations. To the best of our knowledge, this is the first observation of ground-state open-shell aromaticity.28
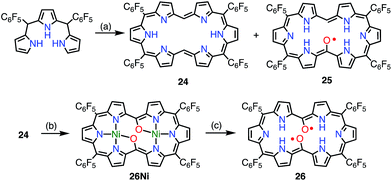 |
| | Fig. 9 Hexaphyrin mono- and bis(meso-oxy) radicals. (a) 22 equiv. HC(OMe)3, 17 equiv. MeSO3H, MS3A, CH2Cl2, 0 °C, 7 h in the dark, 17% for 24 and 15% for 25; (b) ca. 10 equiv. Ni(acac)2·nH2O, CH2Cl2/MeOH, rt, 1 h, 24%; (c) ca. 900 equiv. MeSO3H, CH2Cl2, rt, 30 min, 83%. | |
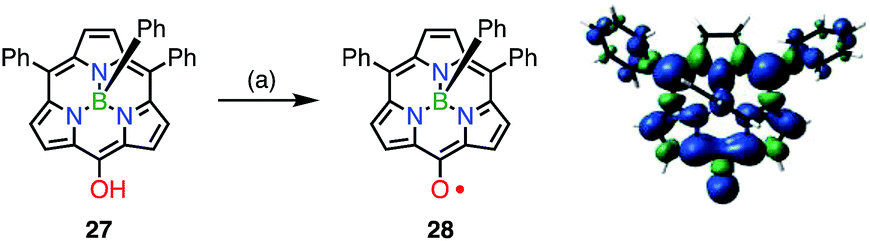
The observed high stabilities of 25 and 26 were considered to arise from effective spin delocalization over the large π-system of the hexaphyrin network. In 2015, however, the subporphyrin meso-oxy radical 28 was found to be very stable (Fig. 10).29meso-Hydroxysubporphyrin 27 was readily oxidized to 28 in the open air in solution, and a trace amount of 28 made the 1H NMR spectrum of 27 completely silent, which was presumably due to rapid hydrogen exchange. The oxidation of 27 with PbO2 gave the radical 28 in quantitative yields. An EPR study of 28 revealed hyperfine coupling with the protons of the meso-phenyl groups, as well as the subporphyrin core, which confirmed effective spin delocalization over the whole molecule except for the axial phenyl group. It is noteworthy that even a relatively small porphyrinoid such as 14π aromatic subporphyrin efficiently stabilizes the meso-oxy radical.
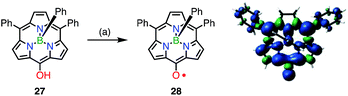 |
| | Fig. 10 Synthesis of the subporphyrin meso-oxy radical 28 and plot of spin density distribution of 28 (isovalue = 0.001). (a) 800 equiv. PbO2, CH2Cl2, rt, quant. | |
With the encouragement of these results, the 10,15,20-triarylporphyrin meso-oxy radicals 30M (M = H2, Ni, Zn) were synthesized by Osuka et al. (Fig. 11).30,31 In comparison with the OEP-based radicals, the stabilities of the 30M radicals were dramatically increased. Namely, the 30M radicals were stable in solution under ambient conditions and were purified by silica gel column chromatography. The Zn complex 30Zn was found to form the complementarily coordinated dimer 31 both in solution and in the solid state. The dimer 31 underwent quantitative dissociation into the monomer 30ZnPy on the addition of pyridine. The dimer exhibited a characteristic broad absorption band in the region of 1000–2000 nm, which was assigned to a symmetry-breaking charge transfer band. Although the optical and electrochemical properties were similar to those of the monomers regardless of the central metal, the magnetic properties of the solid states were strongly dependent on the metal. Namely, planar 30H2 and 30Zn formed efficiently overlapping π-dimers, which resulted in strong intermolecular antiferromagnetic interactions with J/kB = −790 K (30H2) and −720 K (30Zn). On the other hand, 30Ni has a saddle-like structure and forms an offset π-dimer with J/kB = −110 K. A strong intermolecular magnetic interaction via π–π stacking has also been revealed for porphyrin radical cations.32 Coordination with pyridine interrupted such π-stacking and thus allowed 30ZnPy to display an M–T curve that was typical of monoradical species. Arnold et al. also independently reported the facile aerobic oxidation of meso-hydroxyporphyrin 32 to furnish the radical 33.33 They showed that the lack of a 15-aryl group allowed the oxidative formation of the 15,15′-linked dimer 34.
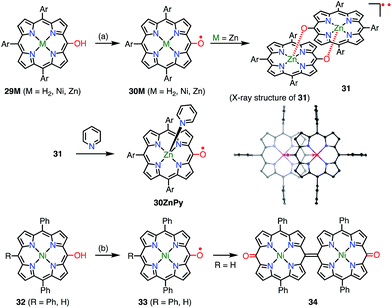 |
| | Fig. 11 10,15,20-Triarylporphyrin meso-oxy radicals. (a) 800 equiv. PbO2, CH2Cl2, rt, 5 min, 86% for 30H2, 91% for 30Ni, 89% for 31; (b) aerobic oxidation for 33, 7 equiv. DDQ, CH2Cl2, rt, 30 min, 26% for 34. Ar = 3,5-di-tert-butylphenyl. Hydrogen atoms and tert-butyl groups in the crystal structure of 31 were omitted for clarity. | |
4.2. Carbon-centered radicals
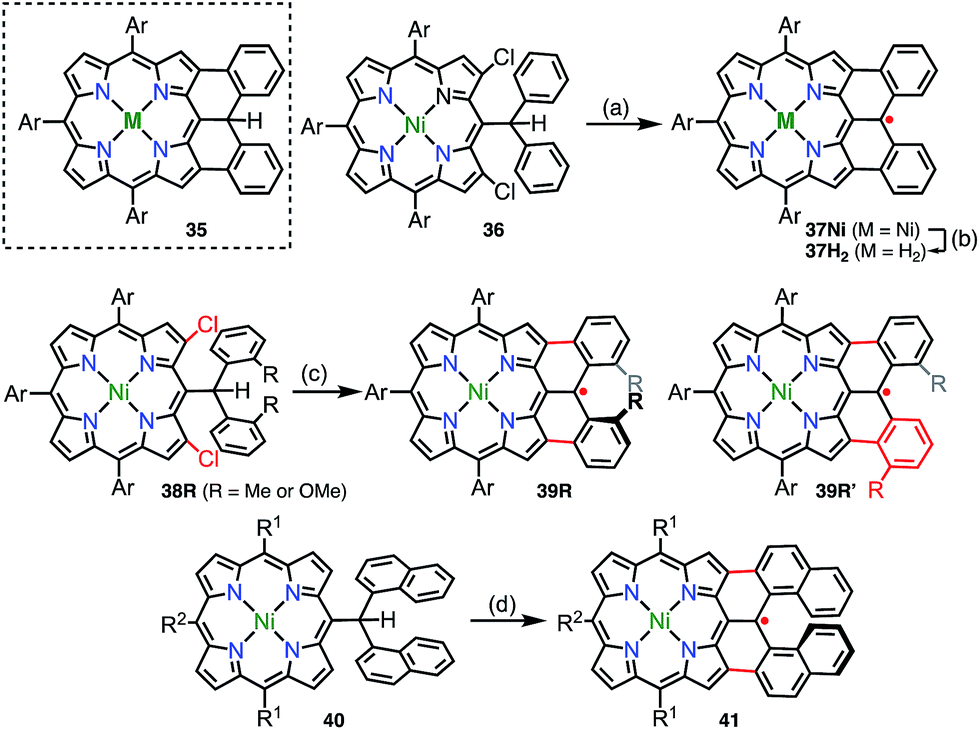
In addition to diphenylborane-34 and diphenylamine-fused porphyrins,35 the synthesis of the diphenylmethane-fused porphyrin 35 was attempted via an intramolecular C–H arylation reaction of 36.36 Surprisingly, the isolated product was not 35 but the neutral radical 37Nivia the loss of a hydrogen atom on the fused carbon atom (Fig. 12). The radical 37Ni was very stable under ambient conditions and did not react with hydrogen donors such as nBu3SnH and ascorbic acid. Taking advantage of the robust stability of 37Ni, its denickelation with sulfuric acid was accomplished to afford the free-base derivative 37H2 in a yield of 51%. The high stabilities of 37Ni and 37H2 arise from effective spin delocalization over the whole molecule and the structural rigidification.37 In a single crystal, both 37Ni and 37H2 form antiparallel π-stacked dimers with π–π distances of 3.51 and 3.56 Å for 37Ni and 3.56 and 3.61 Å for 37H2 together with an offset of 3.4–3.5 Å. The intermolecular exchange interaction in the solid state was determined to be J/kB = −391 K (37Ni) and −279 K (37H2), which reflected the slightly different π–π distances.
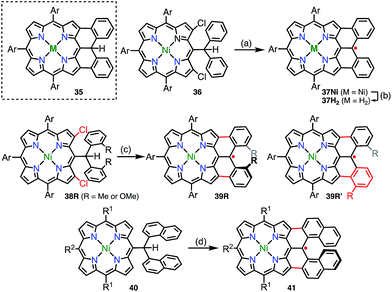 |
| | Fig. 12 Diarylmethyl-fused porphyrins. (a) 20 mol% Pd(OAc)2, 40 mol% PCy3·HBF4, 5 equiv. K2CO3, toluene, reflux, overnight, 72%; (b) H2SO4/CF3CO2H, 0 °C to rt, 45 min, 51%; (c) 20 mol% Pd(OAc)2, 40 mol% PCy3·HBF4, 5 equiv. K2CO3, toluene, reflux, overnight, 48% for 39Me/39Me′, 79% for 39OMe/39OMe′; (d) 10 equiv. DDQ, 10 equiv. Sc(OTf)3, toluene, 110 °C, 1 h, 19%. Ar = 3,5-di-tert-butylphenyl, R1 = mesityl, R2 = 4-methylphenyl. | |
Because the externally fused moiety of 37 can be regarded as a helicene-like structure, an extended series of 37 derivatives has been synthesized to obtain stable neutral helically chiral radicals.38 The bis(2-methylphenyl)methyl-, bis(2-methoxyphenyl)methyl-, and dinaphthylmethyl-fused porphyrins 39R (R = Me or OMe) and 41 were synthesized via Pd-catalyzed intramolecular fusion reactions (39R) and an oxidative fusion reaction (41). 39R and 41 are rare examples of stable radicals in which the spin is delocalized over a chiral π-system. Unfortunately, 39R and 39R′ were obtained as a mixture of regioisomers owing to an unexpected rearrangement (39Me/39Me′ = 32/68 and 39OMe/39OMe′ = 22/78), which was determined by analyzing 1e−-oxidized closed-shell cations [39R]+. The radical 41 was synthesized from 40 and was optically resolved using chiral HPLC. The circular dichroism spectra of the separated enantiomers exhibited weak but distinct mirror images up to 1400 nm. Because the lowest-energy band was assigned to the HOMO(β)–SOMO(β) transition, the circular dichroism observed in the NIR region indicated the spreading of the corresponding orbitals over the helical skeleton. In contrast to 37, the radical 41 did not form a π-stacked dimer in the solid state, and the intermolecular magnetic interaction was found to be slight (Weiss temperature of −2.7 K), owing to unfavorable steric interactions at the ends of the helices.
4.3.
meso-Aminyl radicals
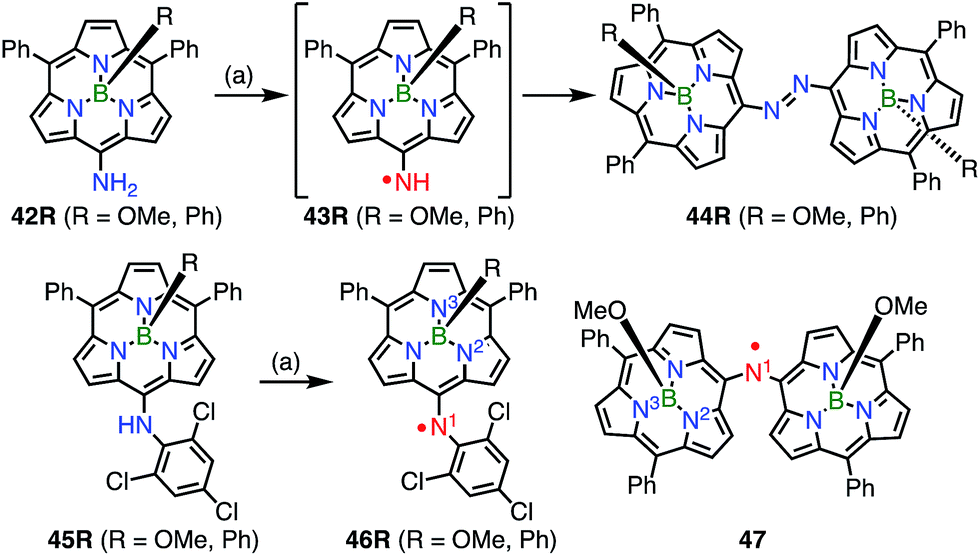
In contrast to oxygen- and carbon-centered radicals, aminyl radicals are unstable, and adjacent heteroatoms are usually required for their stabilization. Heteroatoms next to the aminyl center stabilize aminyl radicals via effective spin sharing as seen in the cases of hydrazinyl, oxyaminyl, and thioaminyl radicals.39 However, stable aminyl radicals without such adjacent heteroatoms are very rare, and the investigation of stable aminyl radicals without such resonance stabilization is a challenging issue. Osuka et al. attempted the oxidation of the meso-aminosubporphyrin 42R with PbO2 in the expectation that it might give the aminyl radical 43R, but the reaction gave the azosubporphyrin 44R, presumably via43R (Fig. 13).40 Thus, meso-(2,4,6-trichlorophenyl)subporphyrin 45 was prepared and oxidized to provide the subporphyrin meso-aminyl radical 46R in a high yield. Importantly, this radical was found to be fairly stable in spite of its neutral character without resonance stabilization.41 The bis(subporphyrinyl)aminyl radical 47 was also synthesized and found to be even more stable. Both the hyperfine coupling constants of the pyrrolic nitrogen atoms (N2 and N3 in Fig. 13) and DFT calculations indicated a higher spin density on the nitrogen atom in comparison with that in the corresponding oxygen-centered radicals, which was presumably because the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond energy is less than that of the CO bond. Whereas both radicals (46Ph and 46OMe) were quite stable, the spin delocalization over the subporphyrin core was more effective in 46Ph than in 46OMe, which may reflect the more electron-rich nature of 46Ph.
N bond energy is less than that of the CO bond. Whereas both radicals (46Ph and 46OMe) were quite stable, the spin delocalization over the subporphyrin core was more effective in 46Ph than in 46OMe, which may reflect the more electron-rich nature of 46Ph.
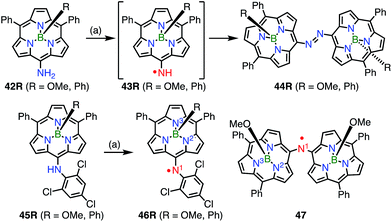 |
| | Fig. 13 Subporphyrin meso-aminyl radicals. (a) 200 equiv. PbO2, CH2Cl2, rt, 5–10 min, 83% for 44OMe, 95% for 44Ph, 88% for 46OMe, 87% for 46Ph. | |
5. Non-innocent macrocyclic radicals
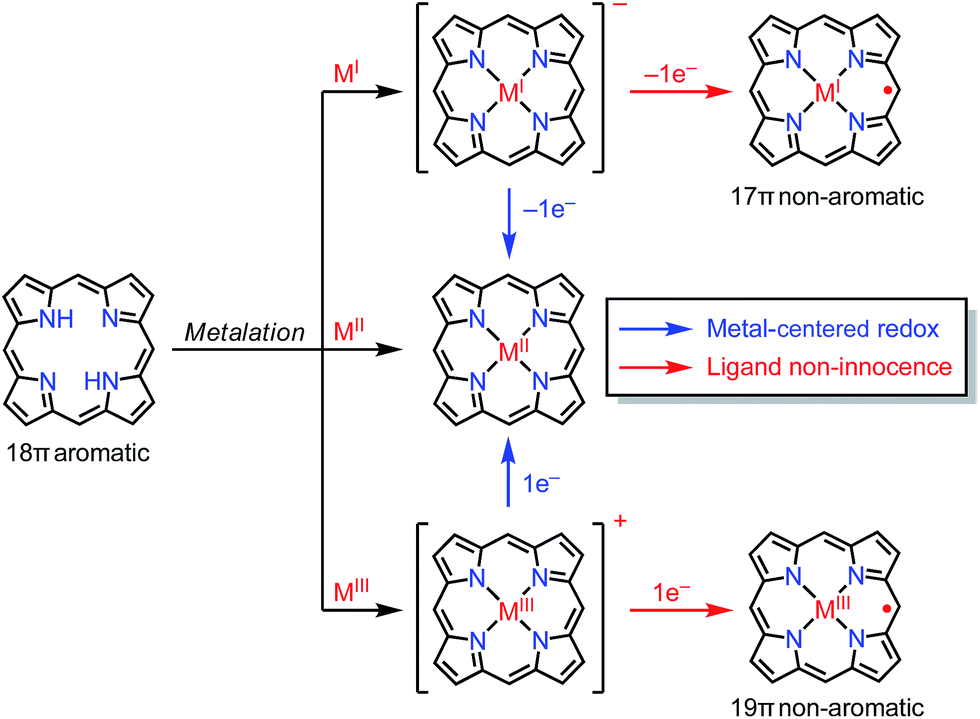
In the previous section, we discussed ligand-centered redox processes of M(II) metalloporphyrinoids that led to the formation of stable radicals owing to the non-innocent ligand42 character of porphyrinoids. In this section, we focus on other cases in which the redox-active character of porphyrinoid macrocycles enables the formation of stable radicals. Porphyrinoids can accommodate various metals. When the valence of the metal is mismatched with the cavities of the porphyrinoid and causes the complex to have a net charge, the resulting complexes sometimes tend to balance the charge by releasing or acquiring an electron (Fig. 14). These processes are facilitated by the fairly flexible π-networks of porphyrinoids.
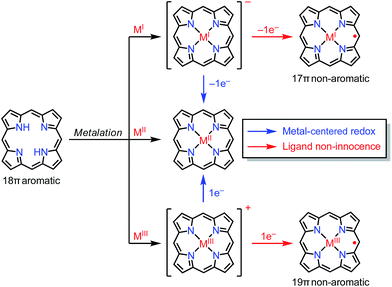 |
| | Fig. 14 General concept of ligand non-innocence in porphyrinoids. | |
5.1. Facile non-innocent behavior of porphyrinoids
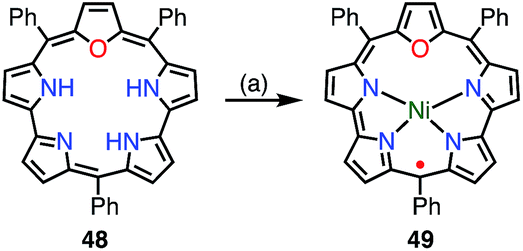
Chandrashekar et al. reported that the metalation with Ni(II) of oxasmaragdyrin 48, which is an expanded porphyrin bearing three pyrrolic protons, afforded a neutral π-radical via spontaneous oxidation of the macrocycle (Fig. 15).43 The Ni(II) complex 49 displayed a sharp EPR signal at g = 2.0031 with a linewidth of 11 G, which can be assigned to organic π-radical species. It should be noted that 49 also exhibited a slightly broader but distinct 1H NMR spectrum at 25 °C.
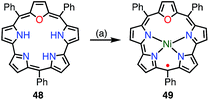 |
| | Fig. 15 Metalation with Ni(II) of oxasmaragdyrin. (a) 10 equiv. NiCl2, DMF, reflux, 10 h, 10%. | |
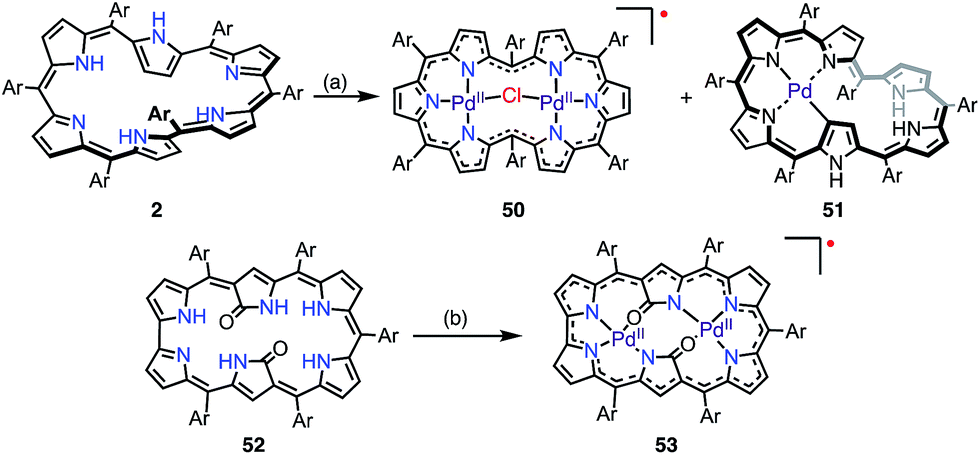
Osuka et al. reported that the metalation of [28]hexaphyrin with Pd(II) gave the bis-Pd(II) complex 50 together with the Möbius aromatic mono-Pd(II) complex 51 as a minor product (Fig. 16).14 In 50, [28]hexaphyrin acted as an inner 4NH ligand, but the bridging Cl− ion caused a valence mismatch. As a consequence, the hexaphyrin ligand released one electron to balance the charge to provide the radical 50. The EPR spectrum of 50 displayed a signal with an anisotropic g-factor of (2.0084, 1.9966, 1.9412), which indicated relatively strong spin–orbit coupling with the Pd(II) metal. The radical 50 is a rare example that has a delocalized spin over a non-planar conjugation circuit. Furuta et al. also reported a π-radical of a bis-Pd(II) complex of doubly N-confused hexaphyrin, i.e., 52, which had intriguing 2NH– and 3NH–NNNO cavities for metalation.44 The bis-Pd complex 53 displayed an isotropic EPR signal at g = 2.0021. Despite the bulky meso-pentafluorophenyl groups, 53 adopted a π-stacked structure in the solid state with an interplanar distance of 3.266 Å, which was shorter than the sum of the van der Waals radii. This close arrangement may arise from a moderate intermolecular antiferromagnetic interaction owing to the overlap of the SOMOs, which was proved by SQUID measurements (Weiss temperature of −42.9 K).
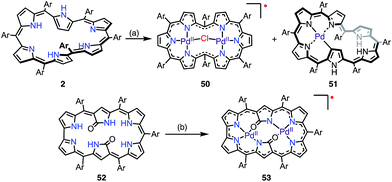 |
| | Fig. 16 Metalation of hexaphyrins with Pd. (a) 10 equiv. PdCl2, 10 equiv. NaOAc, CH2Cl2/MeOH, 45 °C, 24 h, 50% for 50, 4% for 51; (b) 10 equiv. Pd(OAc)2, CH2Cl2/MeOH, rt, 10 h, 75%. Ar = pentafluorophenyl. | |
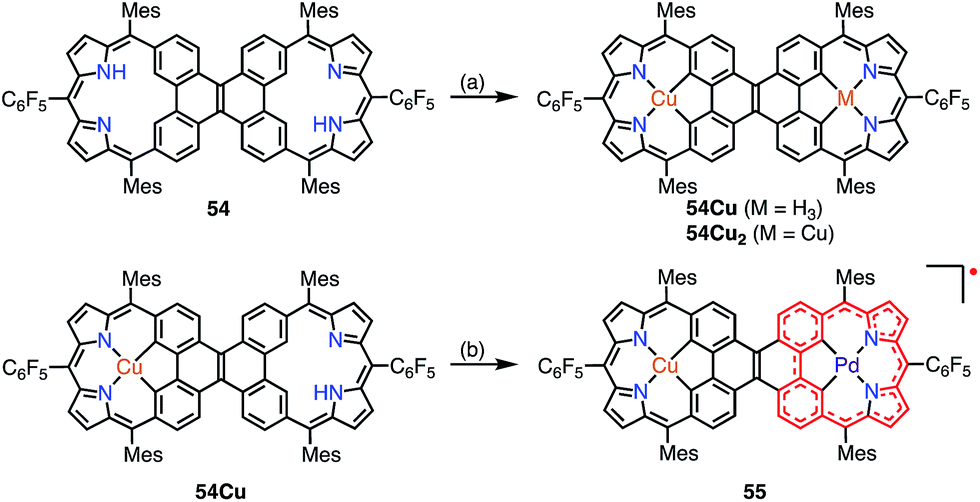
Sessler et al. recently reported the synthesis of the dicarbacorrole dimer 54, in which the monomers share a fused vinylene moiety in the center (Fig. 17).45 The macrocycle 54 has a symmetric structure with two trivalent CCNN cavities. The metalation of 54 with Cu afforded the mono- and bis-Cu(III) complexes 54Cu and 54Cu2. Interestingly, the complex 54Cu was further metalated with Pd(II) to give the neutral, stable π-radical 55via spontaneous oxidation. The spin density of 55 was delocalized only over the Pd-dicarbacorrole segment, which was confirmed by EPR spectroscopy and theoretical calculations. This is a rare example in which the spin density is not fully delocalized over a flat conjugated system.
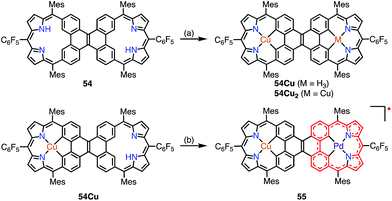 |
| | Fig. 17 Metalation of dicarbacorroles with Cu(III) and Pd(II). (a) Cu(OAc)2·H2O in CHCl3, reflux, 48 h; (b) Pd(PhCN)2Cl2 in PhCN, 180 °C, 2 h, 89%. | |
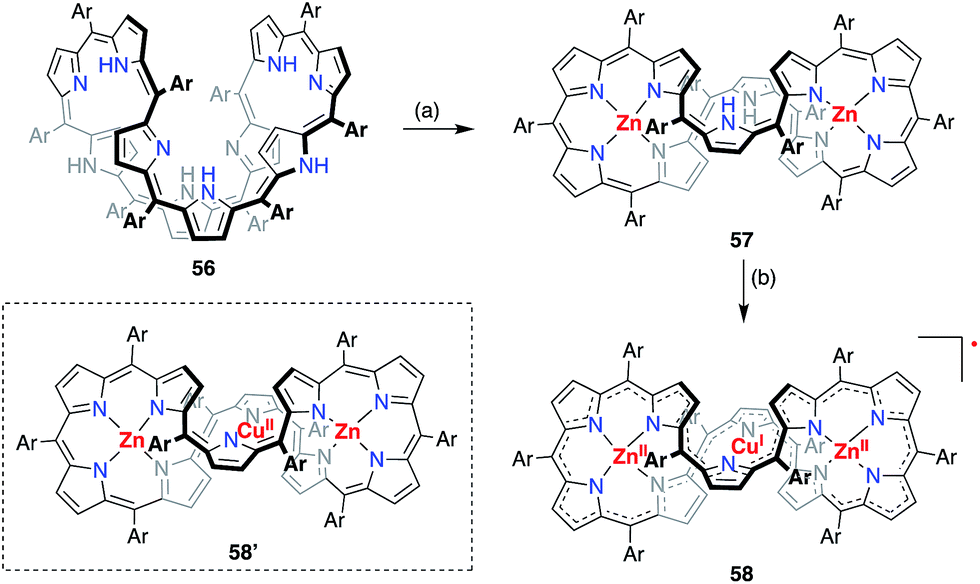
A valence mismatch between the ligand and metal is not necessary to induce ligand non-innocence. The [46]decaphyrin bis-Zn(II) complex 57 has a rigid structure and an inner cavity that is suitable for the coordination of a divalent metal in a linear manner. Osuka et al. examined the metalation of 57 with Cu(II) to obtain the unprecedented linearly coordinated Cu(II) complex 58′.46 However the resulting copper complex displayed a narrow EPR signal at g = 2.0177, which clearly indicated its purely organic π-radical character (Fig. 18). With the aid of theoretical calculations, the product was best interpreted as a Zn(II)–Cu(I)–Zn(II) complex of [45]decaphyrin, i.e., 58. These results indicated that almost instantaneous intramolecular electron transfer occurred from the decaphyrin ligand to the Cu(II) ion to generate the stable radical 58. Interestingly, the 45π non-aromatic radical 58 exhibited a broad absorption band in the NIR region that extended to 2100 nm.
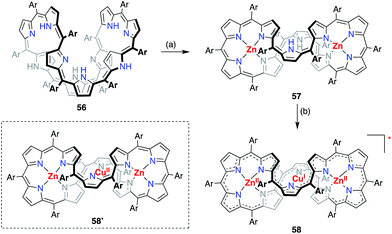 |
| | Fig. 18 Synthesis of the decaphyrin Zn(II)–Cu(I)–Zn(II) complex 58. (a) 5 equiv. Zn(OAc)2·2H2O, 5 equiv. NaOAc, MeOH, reflux, 2 h, 61%; (b) 10 equiv. Cu(OAc)2, 10 equiv. NaOAc, MeOH, rt, 4 h, 72%. Ar = pentafluorophenyl. | |
5.2. Tetraazaporphyrin phosphorus(V) complex
Kobayashi et al. reported the phosphorus(V) complex of tetraazaporphyrin 61 as an air-stable compound with a 19π electronic structure (Fig. 19).15 The complex 61 was synthesized by the insertion of phosphorus(V) into 59 followed by reduction with triethylamine (TEA). Solid samples of 61 were inert under ambient conditions for a year. The exceptional stability of 61 was ascribed to the electron-deficient nature of tetraazaporphyrins, in which the four meso-carbons of porphyrin are replaced by four nitrogen atoms. It was also suggested that charge neutralization by the central cationic phosphorus atom and steric protection by the peripheral bulky groups were both important. The radical 61 exhibited a very low electrochemical oxidation potential of −0.36 V vs. Fc/Fc+ in o-dichlorobenzene.
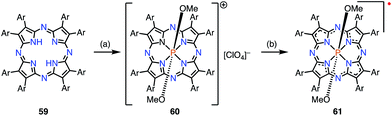 |
| | Fig. 19 Synthesis of 61. (a) POBr3 (200 equiv.), pyridine, 90 °C, 15 h, then CH2Cl2/MeOH, rt, 1 d. 60 was separated by SiO2 column chromatography (CH2Cl2/MeOH/CF3CO2H = 90/9.5/0.5), then NaClO4, CH2Cl2/MeCN, rt, 12 h, 56%; (b) 61 was separated by Al2O3 column chromatography (eluent: CH2Cl2/TEA = 99.5/0.5) without isolating 60, 44%. | |
5.3. Protonation-induced radical formation
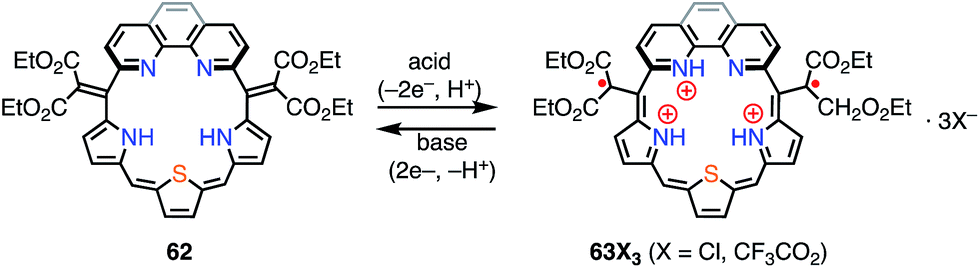
Protonation of the pyrrolic nitrogen atoms in the inner cavity strongly affected both the electronic and the structural properties of expanded porphyrinoids.47 For example, Naruta et al. synthesized the core-modified sapphyrin analogue 62, which contained two exo-methylene bonds (Fig. 20). Protonation of 62 induced facile 2e− oxidation to give the trication 63X3 (X = Cl−, CF3CO2−).48 The formation of 63X3 was reversible, depending on the acid concentration, as evidenced by titration experiments. 63X3 displayed a sharp EPR signal at g = 2.0041 and an NIR absorption band extending to 1400 nm, whereas the precursor 62 absorbed visible light up to 700 nm. X-ray diffraction analysis of 63X3 revealed a distorted structure, which indicated minimal spin delocalization through the phenanthroline bridge. The intramolecular spin–spin interaction was proved by a SQUID study to be antiferromagnetic with J/kB = −680 K. Besides these interesting redox behaviors upon protonation, 62 was also used as a fluorescent monitoring agent for sensing Mg2+ ions.49
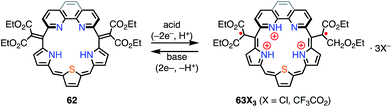 |
| | Fig. 20 Protonation behavior of the modified sapphyrin 62. | |
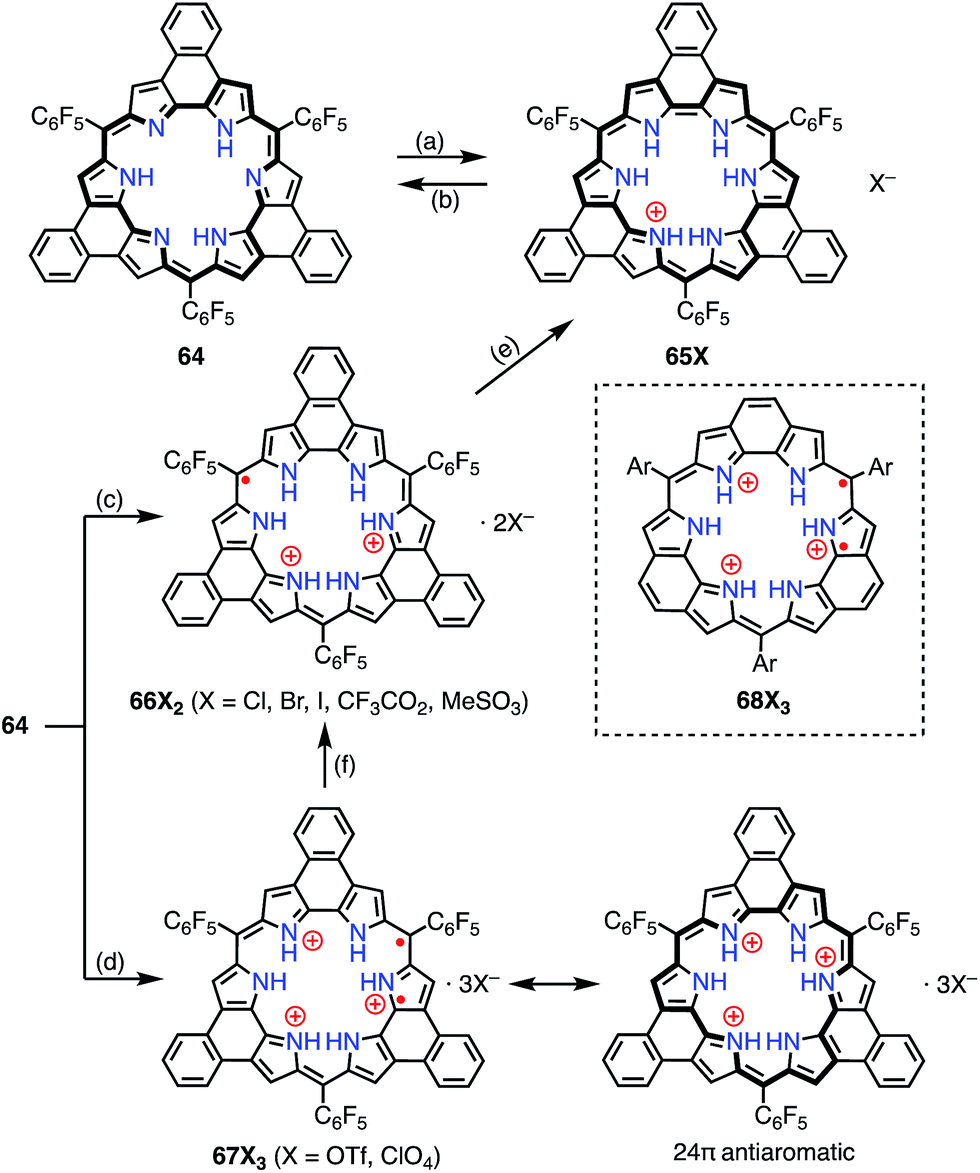
Sessler et al. reported the protonation behaviors of the o-phenylene-bridged annulated rosarin 64 (Fig. 21).50 The protonation of 64 with HX (X = Cl, Br, CF3CO2, MeSO3) afforded 66X2via a protonation-coupled electron transfer (PCET) process. The counterion has a great influence in this PCET process. For example, the protonation of 64 with HOTf or HClO4 only gave the tri-protonation product 67X3, but the subsequent addition of tetrabutylammonium salts (e.g., TBAX; X = Cl, Br) promoted the PCET process to give 66X2. When HI was used as an acid, the further reduction of 66X2 to 65X proceeded with decamethylferrocene.
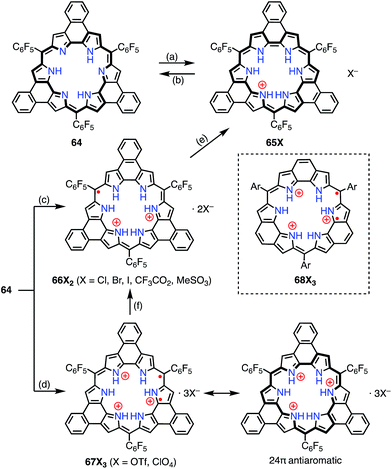 |
| | Fig. 21 Protonation-induced formation of the planar rosarin derivatives 64. (a) Saturated Na2S2O4 (aq), CH2Cl2, rt, 30 min, 87%; (b) MnO2 or triethylamine; (c) HX (X = Cl, Br, I, CF3CO2, MeSO3), CH2Cl2, rt; (d) HX (X = OTf, ClO4), CH2Cl2, rt; (e) Me10Fc, HI; (f) TBAX (X = Cl, Br, I), CH2Cl2, rt. | |
Intriguingly, Fukuzumi et al. revealed that 67X3 and the related rosarin derivatives 68X3 were diradicals with 24π anti-aromatic electronic structures.51 The intramolecular spin–spin interaction was found by VT-EPR measurements to be only weakly ferromagnetic (ΔEST ≈ +80 J mol−1 at 4 K for 68X3). Despite the weak magnetic interaction, the spin–spin distance was estimated to be short at 3.6 Å from the zero-field splitting value (D = 57.7 mT) on the assumption of the point dipole approximation. DFT calculations revealed the a1′′ and a2′′ symmetry of the SOMOs of the parent trication 68X3 These MOs are non-disjoint and should be responsible for the ferromagnetic interaction.
5.4. “Hidden” non-innocence
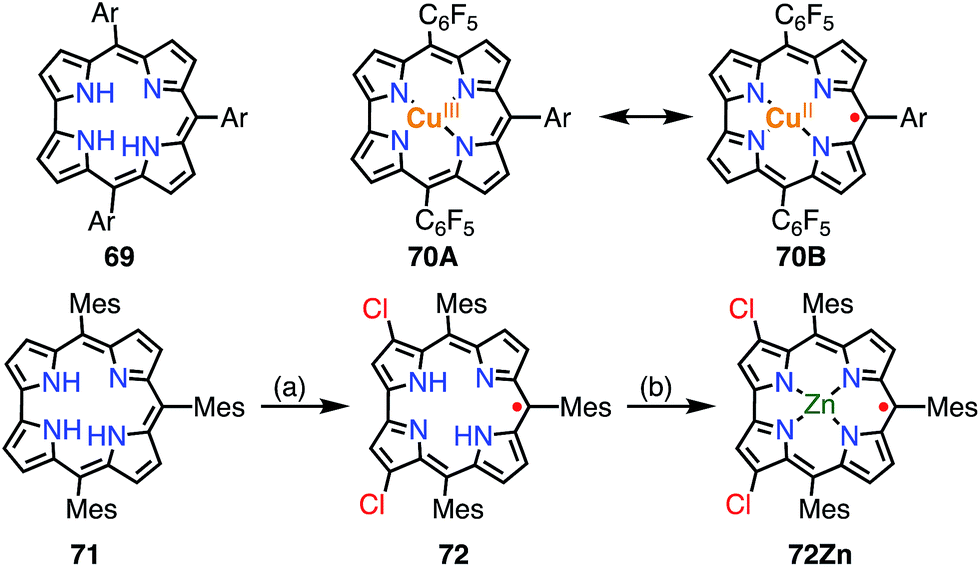
A corrole 69 is a one-methine-carbon-contracted porphyrin that has three inner pyrrolic NH protons and thus acts as a trivalent ligand (Fig. 22).52 In addition, corroles can incorporate metals in higher oxidation states such as Fe(V), Mn(VI), and Cr(VI). These complexes have often been used as mimics of biological intermediates.53 Fe(IV), Ni(II), and Cu(II) corroles are all known as ligand-centered radical species.54 Here, we review Cu(II) corroles in detail.
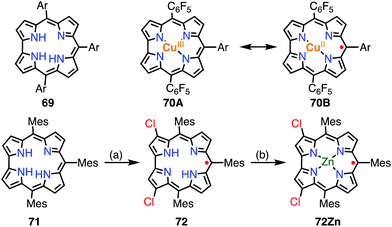 |
| | Fig. 22 Two plausible electronic structures of Cu(II) corrole (top) and corrole radicals. (a) WCl6, W(CO)6 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), PhCN, 200 °C, 8 h, 4–7%; (b) Zn(OAc)2·2H2O, pyridine, 50 °C, 2 h, 77%. Ar = 4-methoxycarbonylphenyl. :1), PhCN, 200 °C, 8 h, 4–7%; (b) Zn(OAc)2·2H2O, pyridine, 50 °C, 2 h, 77%. Ar = 4-methoxycarbonylphenyl. | |
Although the copper complex 70 was first synthesized in 1965,55 its electronic structure and the oxidation state of the Cu center have for a long time been under discussion.56 The observed diamagnetic character can be interpreted in terms of the Cu(III) corrole 70A or antiferromagnetically coupled Cu(II) and the corrole radical cation 70B. In 2010, Pierloot et al. suggested a non-innocent ligand character and a decreased singlet-triplet energy gap for 70 using ab initio calculations.57 In 2005, Kadish et al. and Sarkar et al. independently reported the existence of non-innocent ligand character in Cu(II) corroles by comparison with the corresponding silver corroles.58a,b The latter group also mentioned the existence of a mixed-orbital configuration in which the Cu corrole exists as an intermediate species between Cu(II) and Cu(III) complexes. In 2006, Lemon et al. further investigated the electronic structure of the copper corrole 70.58c An EPR study of 70 showed a doublet signal with hyperfine coupling with a 65Cu/63Cu nucleus (I = 3/2), which is characteristic of Cu(II) species. Further experimental support was given by X-ray photoelectron spectroscopy (XPS), which revealed shake-up satellite peaks in the Cu 2p region that were characteristic of Cu(II) ions. The magnetic interaction between the corrole π-radical and the Cu(II) center was found by VT-NMR measurements to be strongly antiferromagnetic (ΔEST = −13.9 ± 0.7 kJ mol−1). In this case, ligand-centered spin can only be observed when the paramagnetic character of the metal is eliminated, which is known as hidden non-innocence. The electronic structure of Cu corroles has been extensively discussed for decades from various experimental and theoretical aspects. However, further studies are required for a clear understanding of their electronic structure, in particular Cu(II)/Cu(III) mixed states and meso-substituent effects.
Recently, Bröring et al. synthesized the free corrole radical 72 and its Zn(II) complex 72Zn by the oxidation of the meso-triarylcorrole 71. Curiously, the oxidation of 71 with WCl6 and W(CO)6 gave compound 72 with concurrent β-chlorination, which indicated high spin densities on the β-carbons. Protection of these reactive β-positions is required for stabilization of the radicals.59 Both 72 and 72Zn were remarkably stable and exhibited split Soret-like bands around 400 nm and broad Q-like bands in the region of 500–800 nm.
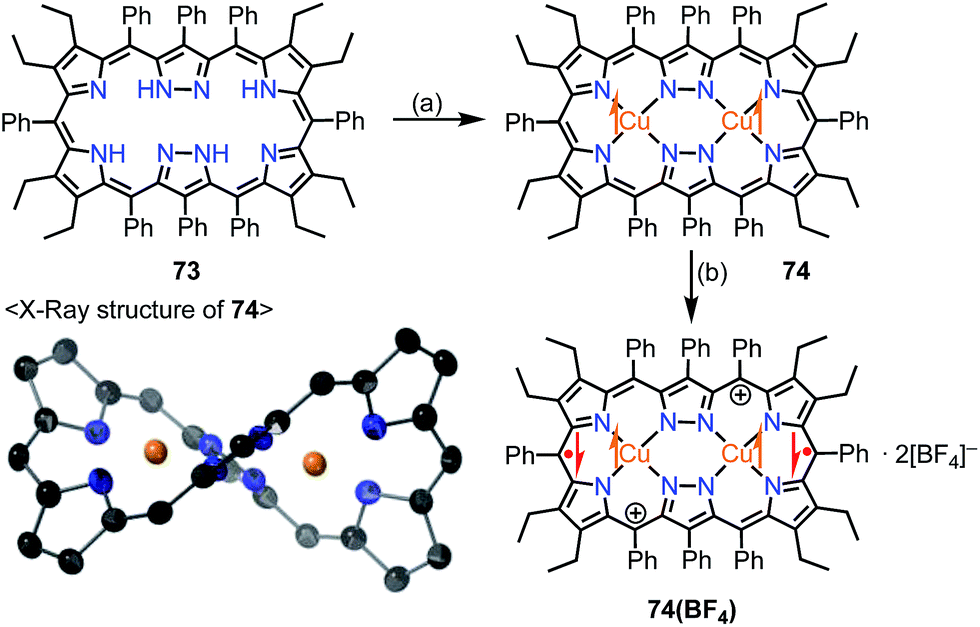
Brückner et al. recently reported the Siamese-twin porphyrin 73, which had a hexaphyrin-like skeleton with two pyrazole rings.60 The free base 73 had a flexible structure without macrocyclic aromaticity. The bis-Cu(II) complex 74 was shown to be twisted and exhibit a ferromagnetic interaction with J/kB = +23.5 K between the two S = 1/2 Cu(II) centers (Fig. 23). This magnetic interaction may arise from orbital orthogonality induced by the molecular twist. Two years later, Beer et al. investigated the oxidation of 74.61 Stepwise 1e− and 2e− oxidations of 74 were achieved with AgBF4. The monocation 74(BF4) was found to be a species with a total S = 1/2, in which the two Cu(II) centers remained in the same oxidation state but the ligand was oxidized to the radical cation. The observed doublet spin indicated an antiferromagnetic interaction between the monoradical [74]+ and the Cu(II) center. Moreover, the dication 74(BF4)2 was found to be a π-diradical, in which the two Cu(II) centers remained in the same oxidation state as determined by XAFS. X-ray diffraction analysis revealed the similar twisted structure of 74(BF4)2, which was feasible for ferromagnetic coupling of the two Cu(II) centers. However, 74(BF4)2 exhibited diamagnetic (S = 0) character. The authors suggested the ligand diradical character of [74]2+, which coupled strongly with the Cu(II) center to cancel out the net spin. The π-diradical character of [74]2+ may arise from the non-Kekulé nature of the dication owing to cross-conjugation between the pyrazole rings.
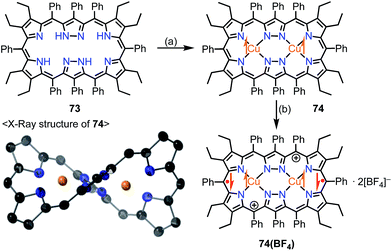 |
| | Fig. 23 Siamese-twin porphyrin and its Cu(II) complexes. (a) 4 equiv. Cu(OAc)2·H2O, CH2Cl2/MeOH, rt, overnight, 29%; (b) 2 equiv. AgBF4 in EtNO2, CH2Cl2, rt, 97%. | |
5.5. Magnetic interaction with the central metal
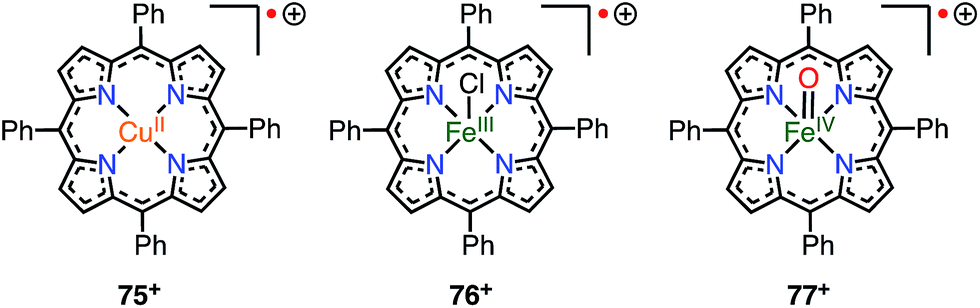
When a porphyrinoid π-radical has a paramagnetic metal in the central cavity, a magnetic interaction between the porphyrin radical ligand and the paramagnetic metal center is possible. Earlier, this was examined for the π-radical cation of meso-tetraphenyl Cu(II) porphyrin 75+, which was characterized as paramagnetic (S = 1 or S = 1/2, 1/2) in solution but diamagnetic (S = 0) in the solid state.62,63 Orthogonality between the au SOMO of the ligand and the dx2−y2 orbitals of Cu(II) has been assigned as responsible for the ferromagnetic interaction. Therefore, the ferromagnetic interaction was observed in solution owing to the planar structure and the resulting strict orthogonality, but a slight structural distortion in the solid state gave rise to an antiferromagnetic interaction. It should be noted that other metalloporphyrin radical ions such as 76+ and 77+ have also been investigated as model compounds of compound I (Fig. 24).64
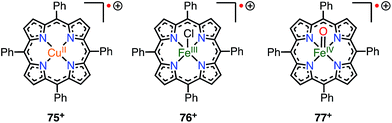 |
| | Fig. 24 Radical cations of metalloporphyrins. | |
Square-planar Ni(II) porphyrins are usually diamagnetic (low-spin, S = 0), but become paramagnetic (high-spin, S = 1) upon the coordination of an additional ligand (Fig. 25).65 The orbitals responsible for the spin of high-spin Ni(II) have σ-character, and the π-radicals should be orthogonal, which leads to a ferromagnetic interaction as in the case of Cu(II) porphyrins. Balch et al. reported that the β-octaethylporphyrin Ni(II) complex 21Ni existed as a doublet species (S = 1/2) as a consequence of an antiferromagnetic interaction between the high-spin Ni(II) atom and the π-radical. On the other hand, Osuka et al. reported a strong ferromagnetic interaction in the meso-pentafluorophenylporphyrin Ni(II) complex 78Ni.66 The authors suggested that the balance of magnetic interactions between the central metal and the ligand radical was likely to be sensitive, so that a very small structural distortion could alter the magnetic interaction.
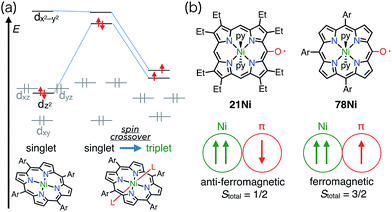 |
| | Fig. 25 (a) Coordination-induced spin crossover of Ni(II) center. (b) OEP and 10,15,20-triarylporphyrin meso-oxy radicals and their magnetic interaction with the Ni(II) center. Ar = pentafluorophenyl. | |
6. Kekulé/non-Kekulé radicals
Delocalized radicals can be divided into Kekulé and non-Kekulé molecules.67 In most Kekulé-type diradicals, the loss of bond-formation energy due to the presence of two unpaired electrons is compensated by a gain in aromatic stabilization energy.68 On the other hand, non-Kekulé radicals have structures that cannot be drawn as a closed-shell form.
6.1. Quinonoidal porphyrinoids
5,10- and 5,15-quinonoidal porphyrins, which can be drawn as diradicals with the retention of the macrocyclic aromatic conjugation circuit, are also promising candidates for obtaining stable diradical species. However, the macrocyclic aromatic stabilization energy of porphyrin seems relatively small in comparison with the bond dissociation energy.4 Consequently, the closed-shell quinonoidal electronic structure is more favorable, as seen in 34 and 79–82 (Fig. 26).33,69–71 These quinonoidal porphyrinoids exhibit unique optical and electrochemical properties owing to decreased HOMO–LUMO gaps and perturbed aromaticity.
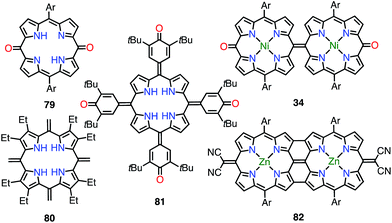 |
| | Fig. 26 Quinonoidal porphyrins. | |
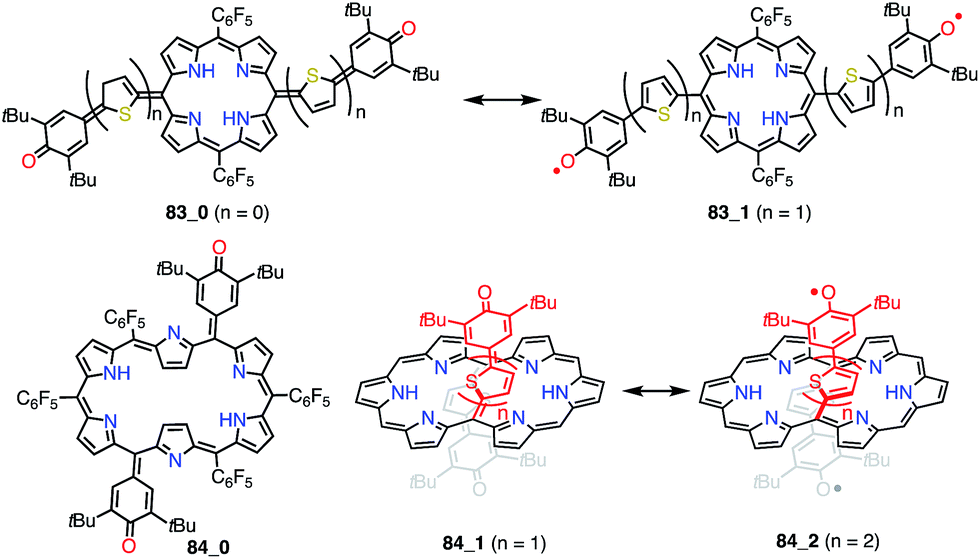
Osuka et al. prepared a series of 5,15-bis(3,5-di-tert-butyl-4-hydroxyphenyl-oligothienylene)-substituted porphyrins 83_n and [26]hexaphyrins 84_n (Fig. 27).72 Whereas 83_0, 84_0, and 84_1 exhibited sharp 1H NMR signals corresponding to a closed-shell quinonoidal structure, 83_1 and 84_2 remained silent or displayed broad signals even at low temperatures. Theoretical calculations at the UHF/6-31G*//B3LYP/6-31G* level revealed large diradical indices of 0.99 for 83_1 and 0.85 for 84_2. Compound 83_1 was sufficiently stable for isolation but sensitive to air and moisture. This chemical instability implied that the spin was less delocalized over the porphyrin core. On the other hand, 84_2 was highly stable, which was presumably due to effective spin delocalization over the hexaphyrin core, as indicated by DFT calculations. Judging from the exchange integral (J/kB = −33 K for 83_1 and J/kB = −940 K for 84_2), the interaction between the two phenoxy radicals was slight in 83_1 in comparison with that in 84_2, which also suggested that 83_1 was less conjugated. The strong antiferromagnetic interaction in 84_2 was presumably due to through-space spin–spin interactions, which are inaccessible for their porphyrin counterparts, as well as through-bond conjugation.
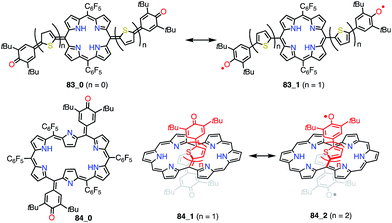 |
| | Fig. 27 Thienylquinonoidal porphyrins and hexaphyrins. The meso-C6F5 groups in 84_1 and 84_2 are omitted for clarity. | |
6.2. 2,18-Doubly linked corrole dimer
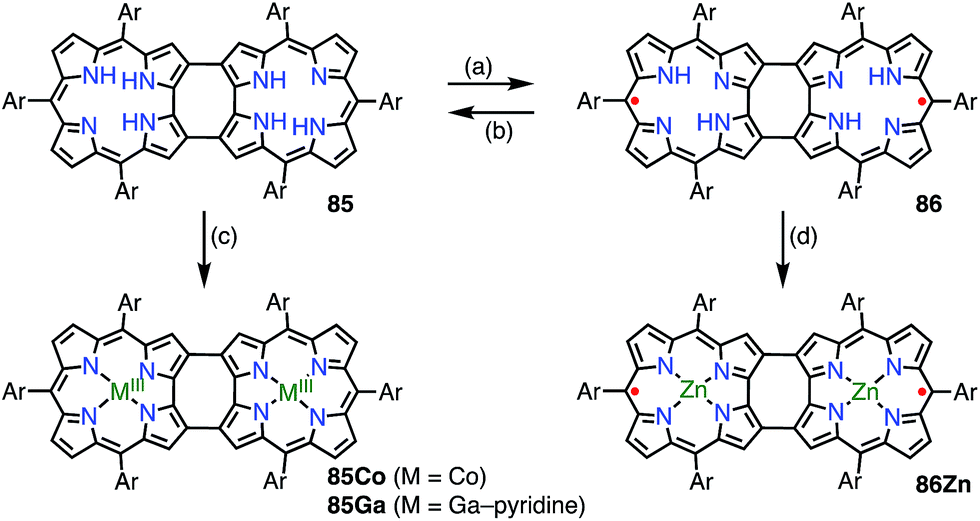
Shinokubo et al. synthesized the 2,18-doubly linked corrole dimer 85 with a cyclooctatetraene (COT) moiety in the center via oxidative fusion of a 2,2′-linked corrole dimer (Fig. 28).7385 was quantitatively oxidized with DDQ to give 86, and the metal complexes 85Co and 86Zn were also prepared. Whereas 85 and 85Co displayed absorption spectra that were typical of aromatic corroles, 86 and 86Zn exhibited largely perturbed spectra that extended to 1700 nm. 86Zn was NMR-silent but EPR-active and displayed a sharp signal at g = 2.0053. 86 and 86Zn were characterized as singlet diradicals. A SQUID study of 86Zn revealed an antiferromagnetic interaction with J/kB = −470 K.
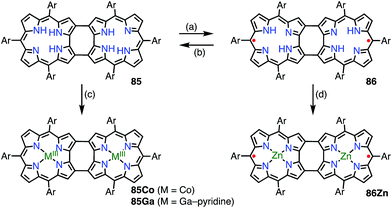 |
| | Fig. 28 2,18-Doubly linked corrole dimers. (a) 5 equiv. DDQ, toluene, rt, 2 h, 83%; (b) NaBH4, THF, rt, quant.; (c) 5 equiv. Co(OAc)2, 7.8 equiv. PPh3, EtOH, rt, 1.5 h, 78%; (d) sat. Zn(OAc)2 in EtOH, CHCl3, rt, 1 h, 54%. Ar = pentafluorophenyl. | |
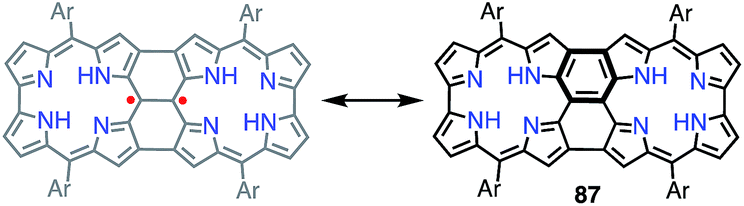
It is worth noting that the triply linked corrole dimer 87 also possessed a dimeric structure consisting of inner 2NH corroles, but 87 was a closed-shell molecule with a short C10–C10′ distance of 1.389 Å (Fig. 29).74a Recently, Gross et al. reported the synthesis and electronic structure of 85Ga.74b The authors showed that the HOMO–1 of 85Ga, which may correspond to the SOMO of the diradical 86, had a very low spin density on C2 and C18, but a large coefficient on C15. The small extent of the overlap of the SOMOs may be the origin of the diradical character of 86 and 86Zn.
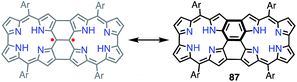 |
| | Fig. 29 Triply linked corrole dimer. Ar = pentafluorophenyl. | |
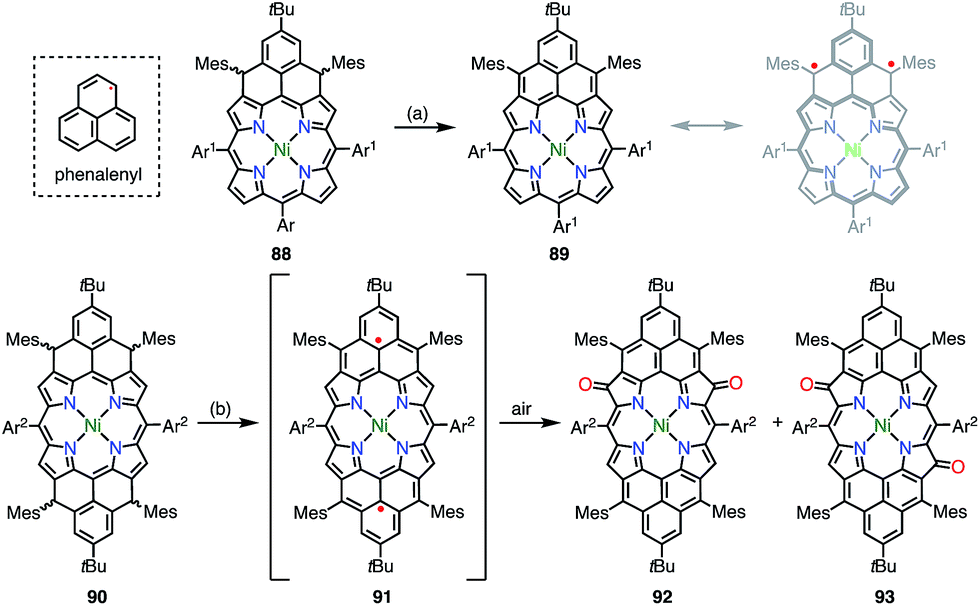
6.3. Phenalenyl-fused porphyrins
Phenalenyl is a very important stable organic π-radical and has been extensively studied from both experimental and theoretical viewpoints.75 Wu et al. prepared precursors of the mono- and bis-phenalenyl-fused porphyrins 88 and 90via an intramolecular Friedel–Crafts reaction.76 The authors concluded that the subsequent oxidation of 88 and 90 led to the formation of 89 and 91, respectively (Fig. 30). Compound 89 was characterized as a closed-shell chromophore with a sharp 1H NMR spectrum and a small diradical index of 0.06. Unfortunately, 91 was unstable under ambient conditions and gave the β-oxidized products 92 and 93. The EPR signal of 91 displayed clear hyperfine coupling with two sets of protons, which was consistent with the proposed structure. DFT calculations suggested that 91 was a ground-state triplet diradical with J/kB ≈ +1800 K, although no experimental evidence in support of the spin and oxidation state of 91 was reported. It is noteworthy that 91 formally bears two radicals on the sp2-carbon atoms attached to the meso-positions, but 91 cannot be drawn as a closed-shell structure owing to the externally fused conjugation circuit.
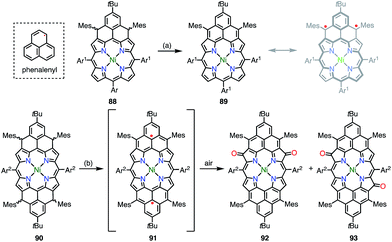 |
| | Fig. 30 Peripherally phenalenyl-fused porphyrins. Reagents and conditions: (a) 2 equiv. NIS, CH2Cl2, rt, 5 min, 49%; (b) 2 equiv. p-chloranil, CH2Cl2, rt, 10 min. 92 and 93 were obtained after exposure to air for 3.5 h, 20% for 92, 39% for 93. Ar1 = 4-tert-butylphenyl, Ar2 = 3,5-di-tert-butylphenyl. | |
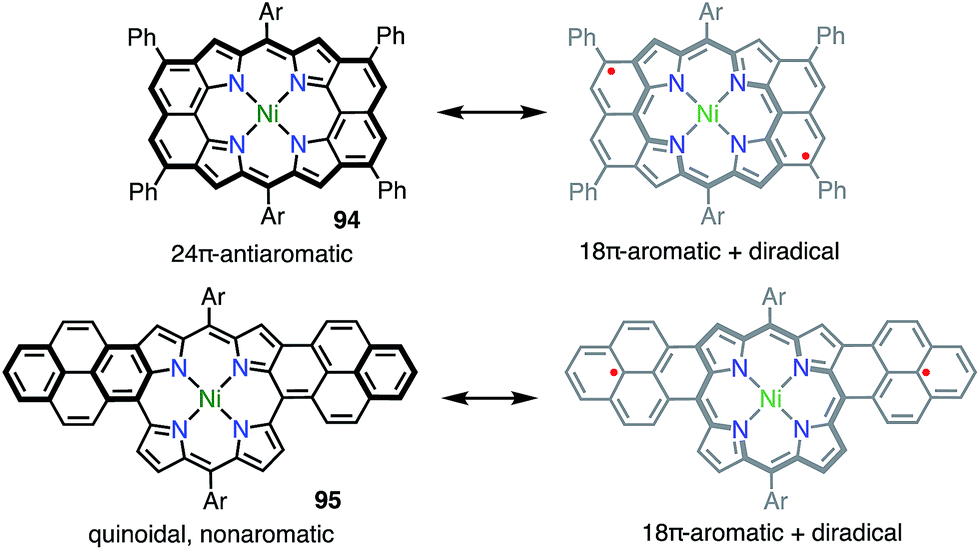
Osuka et al. also reported the similar peri-dinaphthoporphyrin 94,77 which comprises a partial structure of 91 and porphyrin tapes.7894 was characterized as a closed-shell anti-aromatic molecule (Fig. 31). Thompson et al. also prepared the bis-phenalenyl-fused porphyrin 95 by oxidative fusion followed by thermal dehydroaromatization.79 However, 95 was a closed-shell molecule with a calculated diradical index of <3%. The clear difference between 91, 94 and 95 is indicative that the design of the conjugation circuit is of great importance in producing a diradical.
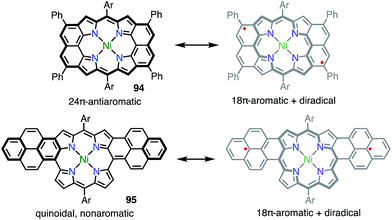 |
| | Fig. 31 Peripherally fused porphyrins. Ar = 3,5-di-tert-butylphenyl. | |
6.4. Thermally induced radical character
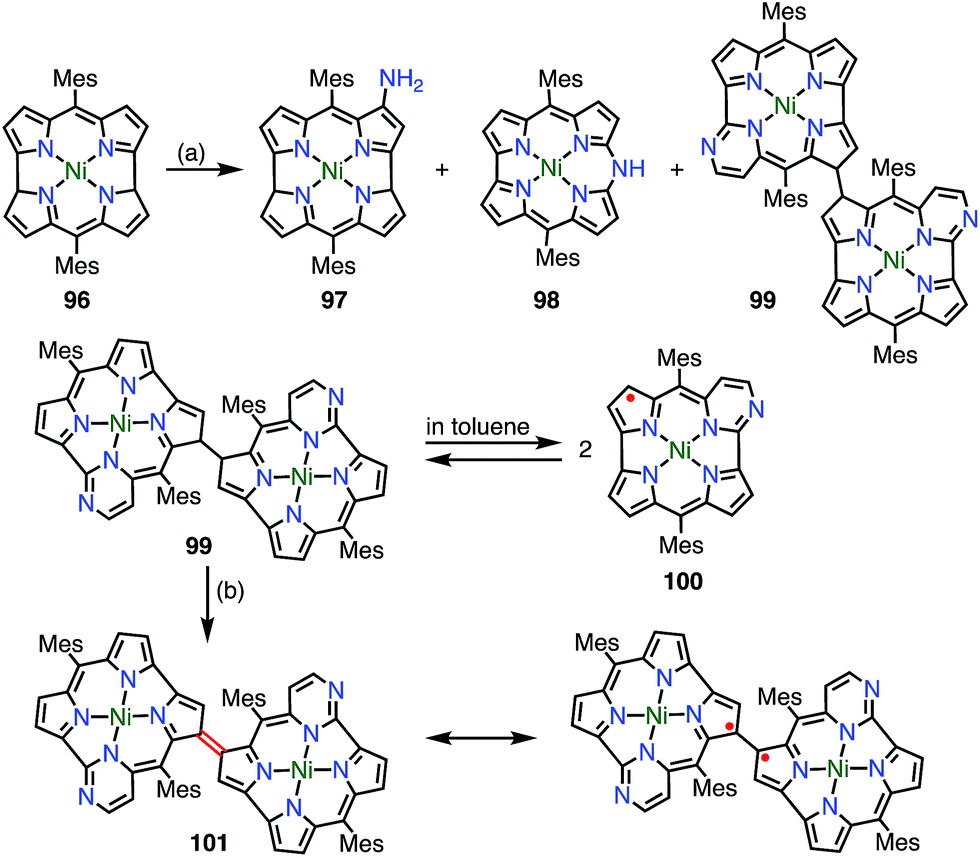
Chmielewski et al. reported the synthesis of the pyrimidinenorcorrole dimer 99via a unique ring expansion reaction of Ni(II)-norcorrole 96 (Fig. 32).8099 existed in equilibrium with its homolysis product 100 in toluene, which was proved by VT-EPR measurements and measurements of the temperature dependence of absorption. The population of 100 was estimated from VT-NMR measurements to be 12% at 300 K and almost 90% at 373 K. This process was reversible and reproducible in the open air, which indicated both the thermal stability and the chemical stability of 100. Surprisingly, the dimerization of 100 was completely regioselective, whereas the spin density of 100 was fairly delocalized, as evidenced by both the hyperfine coupling constants observed in the EPR spectrum and DFT calculations. Moreover, 99 was further oxidized in DMF (even degassed and under N2) to give 101. The oxidation of 99 to 101 also proceeded in toluene with the aid of a catalyst. X-ray diffraction analysis revealed a molecular twist around the C3–C3′ bond with a dihedral angle of 44° and a relatively short C–C distance of 1.437 Å. In the solid state, a small amount of a paramagnetic species was observed (6% on the assumption of a triplet species) by a SQUID study. The EPR signal of 101 in the solid state displayed a fine structure with a zero-field splitting parameter of D = 20 mT, which is typical of triplet species. A VT-NMR study of the spin-lattice relaxation time (T1) revealed that T1 was shorter at 300 K (0.04–0.5 s) in comparison with its value at 183 K (T1 > 1 s), which indicated thermally enhanced paramagnetic character. Rotation around the C3–C3′ bond was also observed in the form of averaging of the signals due to the meso-mesityl groups. The authors proposed that the population of the paramagnetic form was higher and/or chemical exchange between the forms was faster upon heating. These phenomena were further supported by quantum calculations that showed that the S–T gap was dependent on the dihedral angle.
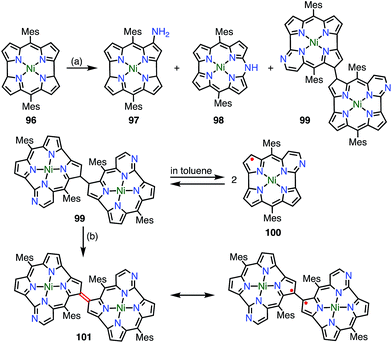 |
| | Fig. 32 Series of pyrimidinenorcorrole derivatives. (a) 12.5 equiv. 4-Amino-4H-1,2,4-triazole, 20 equiv. NaOH, THF, reflux, 2 h, 8.6% for 97, 28.8% for 98, 3.3% for 99, 32.2% for 100; (b) degassed DMF, N2, 80 °C, 30 min, 98%. | |
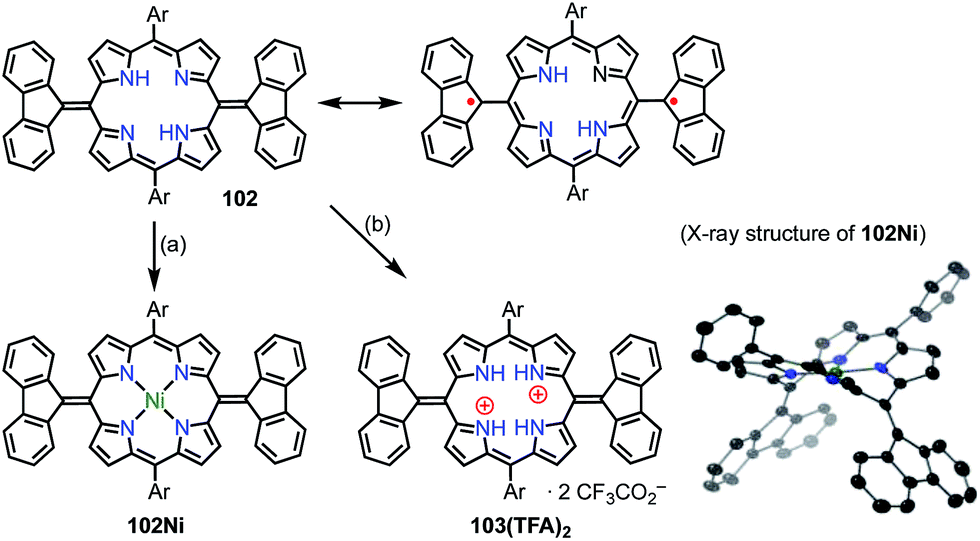
Wu et al. also proposed the thermally induced diradical characters of the 5,15-bis(9-fluorenylidene)porphyrin 102 and its Ni complex 102Ni (Fig. 33).81 Both 102 and 102Ni adopted quinonoidal structures in the single-crystal state with distortions due to steric hindrance by the fluorenyl groups. X-ray analysis revealed the highly bent structure of 102 due to steric repulsion between the β-proton and the fluorenyl group with a short Cmeso–Cfluorenyl bond length of 1.35–1.37 Å. A VT-NMR study detected signal broadening at high temperatures, which indicated the structural flexibility of 102 and 102Ni. 102Ni exhibited signal broadening at lower temperatures than 102, which has been interpreted in terms of the conformational flexibility of 102Ni, despite its metalation. A SQUID study of 102 and 102Ni revealed unusual open hysteresis loops for net magnetization.82 The estimated value of ΔEST was higher in 102 than in 102Ni, which was consistent with the VT-NMR results. From these results and DFT calculations, the authors proposed that the diradical character was triggered by a thermally induced “butterfly-like” structural change. Protonation also altered the diradical character of 102. For example, protonation of 102 gave 103(TFA)2 in quantitative yield, which exhibited sharp NMR signals even at high temperatures, which indicated closed-shell character.
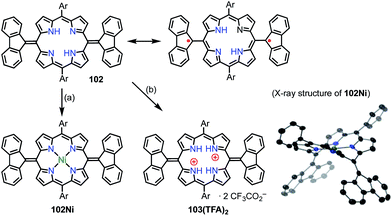 |
| | Fig. 33 5,15-Bis(9-fluorenyl)porphyrins and X-ray crystal structure of 102Ni. (a) 10 equiv. Ni(OAc)2·4H2O, toluene, 120 °C, 5 h, quant.; (b) 2 equiv. CF3CO2H, CH2Cl2, <10 min, quant. Ar = 3,5-di-tert-butylphenyl. Hydrogen atoms and tert-butyl groups in the crystal structure of 102Ni are omitted for clarity. | |
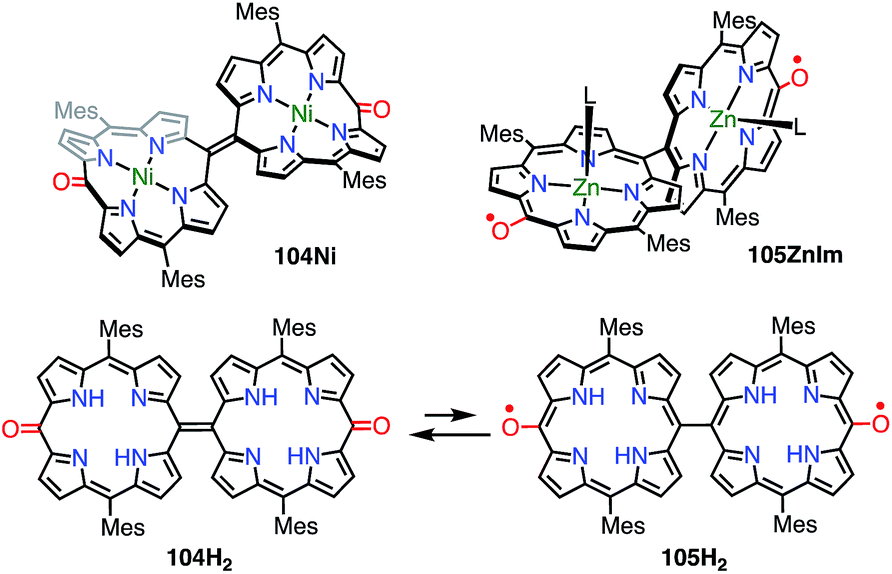
In the case of 102, structural flexibility induced the diradical character. Very recently, Osuka et al. reported a contrasting system in which diradical character was enhanced via structural rigidification.83 As shown above, the 5,5′-linked 15,15′-dioxo-Ni(II)-diporphyrin 34 was known to be a closed-shell quinonoid with a distorted structure. It was considered that the replacement of the central metal would lead to changes in the structure and thus magnetic interactions. In fact, the Ni(II) complex 104Ni was a closed-shell quinonoid similar to 34, but the Zn(II) complex 105ZnIm was found to be a stable diradical (Fig. 34). X-ray diffraction analysis of 105ZnIm showed a perpendicular structure consisting of two highly planar porphyrin segments with a dihedral angle of 71°. The C5–C5′ bond length (1.492 Å) clearly indicated single-bond character. These structural features indicated a diradical electronic state, which was supported by the silent 1H NMR spectrum, the active EPR spectrum, magnetism detected by SQUID experiments, a broad absorption tail extending to 1800 nm, and DFT calculations. Moreover, the free base 104H2 was found to exist as an equilibrium mixture of the closed-shell quinonoid 104H2 and the diradical 105H2 (104H2/105H2 = 81/19 in toluene at room temperature), as evidenced by VT-EPR and VT-NMR studies. The contribution of 105H2 increased at high temperatures.
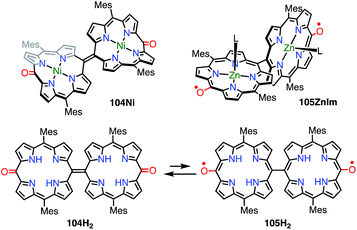 |
| | Fig. 34 5,5′-Linked 15,15′-dioxodiporphyrins. L = N-methylimidazole. | |
7. Other example
7.1. Pentathiapentaphyrin
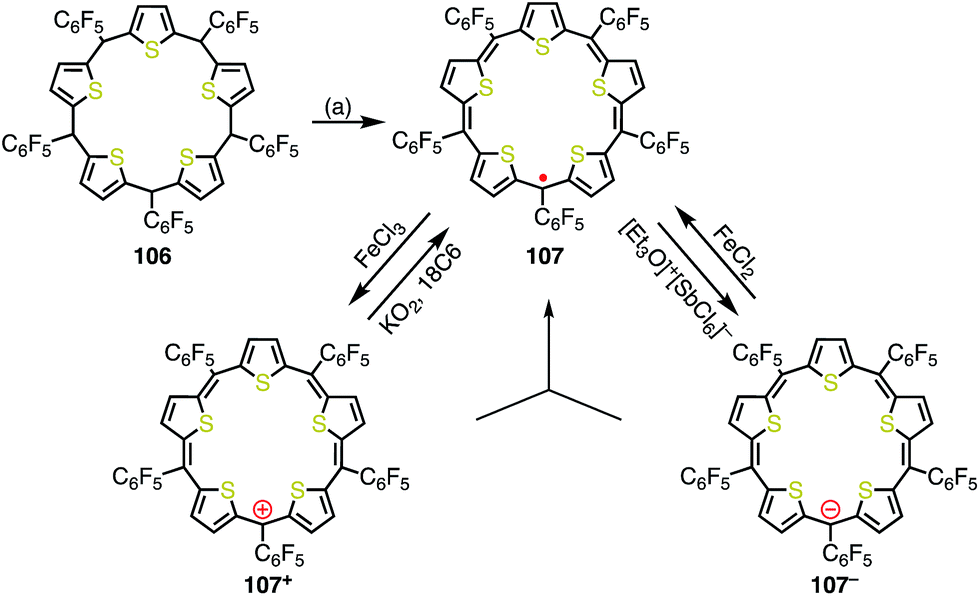
Anand et al. reported the synthesis of pentathiapentaphyrin(1.1.1.1.1) 107,84 which cannot be drawn as a neutral closed-shell form without an unpaired electron, in a similar way to the corrole radical 72 (Fig. 35). X-ray crystallographic analysis revealed its planar structure, which was suitable for spin delocalization. In fact, 107 displayed high stability towards air and moisture. The radical 107 could be reversibly oxidized/reduced to give the 24π anti-aromatic cation 107+ and the 26π aromatic anion 107−, respectively. The mixing of 107+ and 107− formed 107, which indicated the high tendency to comproportionation and thus the high stability of 107.
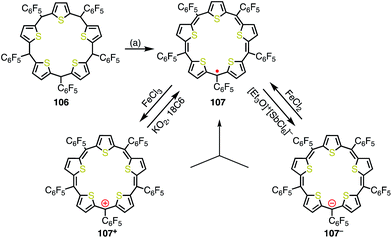 |
| | Fig. 35 Synthesis and redox behavior of pentathiapentaphyrin 107. (a) FeCl3, CH2Cl2, rt, 1 h, then triethylamine, rt, 38%. | |
8. Conclusion and perspectives
We reviewed porphyrinoid-based stable radicals mainly from the viewpoints of radical structure and stability. Early research focused on porphyrinoid-based radicals as important reaction intermediates in biochemistry. Recently, interest has been shifting to the investigation of functional materials based on stable radicals. The remarkable stability of porphyrinoid radicals is mainly due to effective spin delocalization (thermal stabilization). However, in some cases steric protection at positions where relatively high spin density is localized (kinetic stabilization) is necessary for chemical stability. The effective spin delocalization ability of porphyrinoids can be attributed to their unique structural and electronic flexibilities. Reflecting their delocalized π-radical characters, these radicals generally exhibit unique properties such as NIR absorption bands, magnetic properties, and small redox gaps.
There is ample room for further functionalization of stable porphyrinoids by taking advantage of their stability as well as properties such as light-harvesting functions, structural flexibility, and metal-coordinating ability. Owing to their chemical stability and unique properties, porphyrinoid-based radicals are promising candidates for future applications. The precise alignment of radicals in the solid and liquid-crystalline phases is required to achieve effective bulk properties, but general guidelines for the design of packing structures have not yet been established. Most of the stable radicals that have been synthesized so far are [4n + 1]π-species, and the stabilization of [4n + 3]π electronic radicals (and radical anions) still remains a challenging topic. The investigation of general strategies for stabilizing such electron-rich π-radicals will be important, because it will help in the creation of novel functional molecules. A recent study revealed the triplet Baird-type aromaticity of an internally dithienothiophene-bridged [34]octaphyrin dication.85 The excited-state dynamics of porphyrinoid-based radicals are also of great interest.
In addition, porphyrinoids may be promising for stabilizing radical-related unstable chemical species such as carbenes, nitrenes and other reactive species.86,87 Related studies are actively in progress in our laboratory, and we hope to report their results in the near future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank JSPS KAKENHI grant no. 25220802 and 16K13952 for supporting our stable radical chemistry. D. S. acknowledges a JSPS fellowship for young scientists.
Notes and references
-
(a)
Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing, Singapore, 2010, vol. 1–25 Search PubMed;
(b)
The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 1–10 Search PubMed;
(c) A. Osuka, E. Tsurumaki and T. Tanaka, Bull. Chem. Soc. Jpn., 2011, 84, 679 CrossRef CAS;
(d) C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192 CrossRef CAS PubMed;
(e) S. Shimizu, Chem. Rev., 2017, 117, 2730 CrossRef CAS PubMed;
(f) S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342 CrossRef CAS PubMed;
(g) A. Osuka and S. Saito, Chem. Commun., 2011, 47, 4330 RSC;
(h) T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584 CrossRef CAS PubMed;
(i) B. Szyszko, M. J. Białek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839 CrossRef CAS PubMed.
-
(a) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS PubMed;
(b) P. Hlavica, Eur. J. Biochem., 2004, 271, 4335 CrossRef CAS PubMed.
-
(a) Y. Yamamoto, Y. Hirata, M. Kodama, T. Yamaguchi, S. Matsukawa, K. Akiba, D. Hashizume, F. Iwasaki, A. Muranaka, M. Uchiyama, P. Chen, K. M. Kadish and N. Kobayashi, J. Am. Chem. Soc., 2010, 132, 12627 CrossRef CAS PubMed;
(b) S. Sugawara, Y. Hirata, S. Kojima, Y. Yamamoto, E. Miyazaki, K. Takimiya, S. Matsukawa, D. Hashizume, J. Mack, N. Kobayashi, Z. Fu, K. M. Kadish, Y. M. Sung, K. S. Kim and D. Kim, Chem.–Eur. J., 2012, 18, 3566 CrossRef CAS PubMed;
(c) M. Pohl, H. Schmisckler, J. Lex and E. Vogel, Angew. Chem., Int. Ed. Engl., 1991, 31, 1693 CrossRef;
(d) C. Liu, D.-M. Shen and Q.-Y. Chen, J. Am. Chem. Soc., 2007, 129, 5814 CrossRef CAS PubMed.
-
(a) J. Aihara, J. Phys. Chem. A, 2008, 112, 5305 CrossRef CAS PubMed;
(b) J. L. Wu, I. Fernandez and P. v. R. Schleyer, J. Am. Chem. Soc., 2013, 135, 315 CrossRef CAS PubMed;
(c) J. Aihara and H. Horibe, Org. Biomol. Chem., 2009, 7, 1939 RSC;
(d) M. Makino and J. Aihara, J. Phys. Chem. A, 2012, 116, 8074 CrossRef CAS PubMed.
-
(a) T. Asakura, J. S. Leigh Jr., H. R. Drott, T. Yonetani and B. Chance, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 861 CrossRef CAS PubMed;
(b) M. H. Rakowski, K. M. More, A. V. Kulikov, G. R. Eaton and S. S. Eaton, J. Am. Chem. Soc., 1995, 117, 2049 CrossRef.
- T. T. H. Tram, Y.-R. Chang, T. K. A. Hoang, M.-Y. Kuo and Y. O. Su, J. Phys. Chem. A, 2016, 120, 5511 Search PubMed.
- J. Rittle and M. T. Green, Science, 2010, 330, 933 CrossRef CAS PubMed.
-
(a) T. Satoh, M. Minoura, H. Nakano, K. Furukawa and Y. Matano, Angew. Chem., Int. Ed., 2016, 55, 2235 CrossRef CAS PubMed;
(b) K. Sudoh, T. Satoh, T. Amaya, K. Furukawa, M. Minoura, H. Nakano and Y. Matano, Chem.–Eur. J., 2017, 23, 16364 CrossRef CAS PubMed.
- A. Yamaji, H. Tsurugi, Y. Miyake, K. Mashima and H. Shinokubo, Chem.–Eur. J., 2016, 22, 3956 CrossRef CAS PubMed.
- N. Fukui, W. Cha, D. Shimizu, J. Oh, K. Furukawa, H. Yorimitsu, D. Kim and A. Osuka, Chem. Sci., 2017, 8, 189 RSC.
- N. Fukui, H. Yorimitsu and A. Osuka, Chem.–Eur. J., 2016, 22, 18476 CrossRef CAS PubMed.
- J. Mack and M. J. Stillman, J. Porphyrins Phthalocyanines, 2001, 5, 67 CrossRef CAS.
- T. Koide, G. Kashiwazaki, M. Suzuki, K. Furukawa, M.-C. Yoon, S. Cho, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 9661 CrossRef CAS PubMed.
- H. Rath, S. Tokuji, N. Aratani, K. Furukawa, J. M. Lim, D. Kim, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 1489 CrossRef CAS PubMed.
- T. Yoshida, W. Zhou, T. Furuyama, D. B. Leznoff and N. Kobayashi, J. Am. Chem. Soc., 2015, 137, 9258 CrossRef CAS PubMed.
- J. Rawson, P. J. Angiolillo and M. J. Therien, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13779 CrossRef CAS PubMed.
- M. D. Peeks, C. E. Tait, P. Neuhaus, G. M. Fischer, M. Hoffmann, R. Haver, A. Cnossen, J. R. Hammer, C. R. Timmel and H. L. Anderson, J. Am. Chem. Soc., 2017, 139, 10461 CrossRef CAS PubMed.
-
(a) T. Yoshida, M. Noguchi, G. Kikuchi and S. Sano, J. Biochem., 1981, 90, 125 CrossRef CAS PubMed;
(b) S. Sano, T. Sano, I. Morishima, Y. Shiro and Y. Maeda, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 531 CrossRef CAS PubMed;
(c) A. Wilks and P. R. O. de Montellano, J. Biol. Chem., 1993, 268, 22357 CAS;
(d) A. Gossauer, Chimia, 1994, 48, 352 CAS.
- K. M. Matera, S. Takahashi, H. Fujii, H. Zhou, K. Ishikawa, T. Yoshimura, D. L. Rousseau, T. Yoshida and M. Ikeda-Saito, J. Biol. Chem., 1996, 271, 6618 CrossRef CAS PubMed.
- R. Bonnett, M. J. Dimsdale and K. D. Sales, Chem. Commun., 1970, 962 RSC.
- A. L. Balch, B. C. Noll and E. P. Zovinka, J. Am. Chem. Soc., 1992, 114, 3380 CrossRef CAS.
- J.-H. Fuhrhop, S. Besecke, J. Subramanian, C. Mengersen and D. Riesner, J. Am. Chem. Soc., 1975, 97, 7141 CrossRef CAS.
- A. L. Balch, Coord. Chem. Rev., 2000, 200–202, 349 CrossRef CAS.
- A. L. Balch, B. C. Noll, S. L. Phillips, S. M. Reid and E. P. Zovinka, Inorg. Chem., 1993, 32, 4730 CrossRef CAS.
- R. G. Khoury, L. Jaquinod, A. M. Shachter, N. Y. Nelson and K. M. Smith, Chem. Commun., 1997, 215 RSC.
- R. G. Khoury, L. Jaquinod, R. Paolesse and K. M. Smith, Tetrahedron, 1999, 55, 6713 CrossRef CAS.
- T. Koide, K. Furukawa, H. Shinokubo, J.-Y. Shin, K. S. Kim, D. Kim and A. Osuka, J. Am. Chem. Soc., 2010, 132, 7246 CrossRef CAS PubMed.
- M. Ishida, J.-Y. Shin, J. M. Lim, B. S. Lee, M.-C. Yoon, T. Koide, J. L. Sessler, A. Osuka and D. Kim, J. Am. Chem. Soc., 2011, 133, 15533 CrossRef CAS PubMed.
- D. Shimizu, J. Oh, K. Furukawa, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 6613 CrossRef CAS PubMed.
- D. Shimizu, J. Oh, K. Furukawa, D. Kim and A. Osuka, J. Am. Chem. Soc., 2015, 137, 15584 CrossRef CAS PubMed.
- L. J. Esdaile, M. O. Senge and D. P. Arnold, Partial formation of similar meso-oxy radical had been suggested before, but isolation or characterization had not been achieved, Chem. Commun., 2006, 4192 RSC.
-
(a) H. Song, R. D. Orosz, C. A. Reed and R. Scheidt, Inorg. Chem., 1990, 29, 4274 CrossRef CAS;
(b) W. R. Scheidt, K. E. Brancato-Buentello, H. Song, K. V. Reddy and B. Cheng, Inorg. Chem., 1996, 35, 7500 CrossRef CAS;
(c) K. M. Barkigia, M. W. Renner and J. Fajer, J. Phys. Chem. B, 1997, 101, 8398 CrossRef CAS.
- L. J. Esdaile, L. Rintoul, M. S. Goh, K. Merahi, N. Parizel, R. M. Wellard, S. Choua and D. P. Arnold, Chem.–Eur. J., 2016, 22, 3403 Search PubMed.
- K. Fujimoto, J. Oh, H. Yorimitsu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 3196 CrossRef CAS PubMed.
- N. Fukui, W.-Y. Cha, S. Lee, S. Tokuji, D. Kim, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2013, 52, 9728 CrossRef CAS PubMed.
- K. Kato, W. Cha, J. Oh, K. Furukawa, H. Yorimitsu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 8711 CrossRef CAS PubMed.
- A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357 CrossRef CAS.
- K. Kato, K. Furukawa, T. Mori and A. Osuka, Chem.–Eur. J., 2018, 24, 572 CrossRef CAS PubMed.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321 CAS.
- D. Shimizu, S. K. Lee, D. Kim and A. Osuka, Chem.–Asian J., 2016, 11, 2946 CrossRef PubMed.
- D. Shimizu, K. Furukawa and A. Osuka, Angew. Chem., Int. Ed., 2017, 56, 7435 CrossRef CAS PubMed.
-
(a) P. J. Chirik, Inorg. Chem., 2011, 50, 9737 CrossRef CAS PubMed;
(b) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS.
- B. Sridevi, S. Jeyaprakash, R. Rao, T. K. Chandrashekar, U. Englich and K. Ruhlandt-Senge, Inorg. Chem., 2000, 39, 3669 CrossRef CAS PubMed.
- Y. Hisamune, K. Nishimura, K. Isakari, M. Ishida, S. Mori, S. Karasawa, T. Kato, S. Lee, D. Kim and H. Furuta, Angew. Chem., Int. Ed., 2015, 54, 7323 CrossRef CAS PubMed.
- X.-S. Ke, Y. Hong, P. Tu, Q. He, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2007, 139, 15232 CrossRef PubMed.
- Y. Tanaka, T. Yoneda, K. Furukawa, T. Koide, H. Mori, T. Tanaka, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 10908 CrossRef CAS PubMed.
- S. Zakavi, R. Omidyan and S. Talebzadeh, RSC Adv., 2016, 6, 82219 RSC.
- M. Ishida, S. Karasawa, H. Uno, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2010, 49, 5906 CrossRef CAS PubMed.
- M. Ishida, Y. Naruta and F. Tani, Angew. Chem., Int. Ed., 2010, 49, 91 CrossRef CAS PubMed.
- M. Ishida, S.-J. Kim, C. Preihs, K. Ohkubo, J. M. Lim, B. S. Lee, J. S. Park, V. M. Lynch, V. V. Roznyatovskiy, T. Sarma, P. K. Panda, C.-H. Lee, S. Fukuzumi, D. Kim and J. L. Sessler, Nat. Chem., 2013, 5, 15 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo, M. Ishida, C. Preihs, B. Chen, W. T. Borden, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2015, 137, 9780 CrossRef CAS PubMed.
- K. E. Thomas, A. B. Alemayehu, J. Conradie, C. M. Beavers and A. Ghosh, Acc. Chem. Res., 2012, 45, 1203 CrossRef CAS PubMed.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949 CrossRef CAS PubMed.
-
(a) E. Vogel, S. Will, A. S. Tilling, L. Neumann, J. Lex, E. Bill, A. X. Trautwein and K. Wieghardt, Angew. Chem., Int. Ed. Engl., 1994, 33, 731 CrossRef;
(b) S. Will, J. Lex, E. Vogel, H. Schmickler, J.-P. Gisselbrecht, C. Haubtmann, M. Bernard and M. Gross, Angew. Chem., Int. Ed. Engl., 1997, 36, 357 CrossRef CAS.
- A. W. Johnson and I. T. Kay, J. Chem. Soc., 1965, 1620 RSC.
-
(a) A. W. Johnson, Pure Appl. Chem., 1971, 28, 195 CrossRef CAS;
(b) N. S. Hush and J. M. Dyke, J. Inorg. Nucl. Chem., 1973, 35, 4341 CrossRef CAS;
(c) N. S. Hush and J. M. Dyke, J. Chem. Soc., Dalton Trans., 1974, 395 RSC;
(d) M. Bröring, F. Brégier, E. C. Tejero, C. Hell and M. C. Holthausen, Angew. Chem., Int. Ed., 2007, 46, 445 CrossRef PubMed.
- K. Pierloot, H. Zhao and S. Vancoillie, Inorg. Chem., 2010, 49, 10316 CrossRef CAS PubMed.
-
(a) K. E. Thomas, H. Vazquez-Lima, Y. Fang, Y. Song, K. J. Gagnon, C. M. Beavers, K. M. Kadish and A. Ghosh, Chem.–Eur. J., 2015, 21, 16839 CrossRef CAS PubMed;
(b) W. Sinha, M. G. Sommer, N. Deibel, F. Ehret, M. Bauer, B. Sarkar and S. Kar, Angew. Chem., Int. Ed., 2015, 54, 13769 CrossRef CAS PubMed;
(c) C. M. Lemon, M. Huynh, A. G. Maher, B. L. Anderson, E. D. Bloch, D. C. Powers and D. G. Nocera, Angew. Chem., Int. Ed., 2016, 55, 2176 CrossRef CAS PubMed.
- P. Schweyen, K. Brandhorst, R. Wicht, B. Wolfram and M. Bröring, Angew. Chem., Int. Ed., 2015, 54, 8213 CrossRef CAS PubMed.
- L. K. Frensch, K. Pröpper, M. John, S. Demeshko, C. Brückner and F. Meyer, Angew. Chem., Int. Ed., 2011, 50, 1420 CrossRef CAS PubMed.
- L. K. Blusch, K. E. Craigo, V. Martin-Diaconescu, A. B. McQuarters, E. Bill, S. Dechert, S. DeBeer, N. Lehnert and F. Meyer, J. Am. Chem. Soc., 2013, 135, 13892 CrossRef CAS PubMed.
-
(a) J. H. Fuhrhop and D. Mauzerall, J. Am. Chem. Soc., 1969, 91, 4174 CrossRef CAS PubMed;
(b) A. Wolberg and J. Manassen, J. Am. Chem. Soc., 1970, 92, 2982 CrossRef CAS PubMed;
(c) C. Mangersen, J. Subramanian and J.-H. Fuhrhop, Mol. Phys., 1976, 32, 893 CrossRef;
(d) D. Dolphin, Z. Muljiani, K. Rousseau, D. C. Borg, J. Fajer and R. H. Felton, Ann. N. Y. Acad. Sci., 1973, 206, 177 CrossRef CAS PubMed.
- W. F. Scholz, C. A. Reed, Y. J. Lee, W. R. Scheidt and G. Lang, J. Am. Chem. Soc., 1982, 104, 6791 CrossRef CAS.
-
(a) A. Wolberg and J. Manassen, J. Am. Chem. Soc., 1970, 92, 2982 CrossRef CAS PubMed;
(b) J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 1981, 103, 2884 CrossRef CAS;
(c) P. Gans, G. Buisson, E. Duée, J.-C. Marchon, B. S. Erler, W. F. Scholz and C. A. Reed, J. Am. Chem. Soc., 1986, 108, 1223 CrossRef CAS.
- S. Thies, H. Sell, C. Bornholdt, C. Schütt, F. Köhler, F. Tuczek and R. Herges, Chem.–Eur. J., 2012, 18, 16358 CrossRef CAS PubMed.
- C. Stähler, D. Shimizu, K. Yoshida, K. Furukawa, R. Herges and A. Osuka, Chem.–Eur. J., 2017, 23, 7217 CrossRef PubMed.
- M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed.
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC.
- C. Otto and E. Breitmaier, Liebigs Ann., 1991, 1347 CrossRef CAS.
- J. P. Hill, I. J. Hewitt, C. E. Anson, A. K. Powell, A. L. McCarty, P. A. Karr, M. E. Zandler and F. D'Souza, J. Org. Chem., 2004, 69, 5861 CrossRef CAS PubMed.
- I. M. Blake, A. K. Krivokapic, M. Katterle and H. L. Anderson, Chem. Commun., 2002, 1662 RSC.
- K. Naoda, D. Shimizu, J. O. Kim, K. Furukawa, D. Kim and A. Osuka, Chem.–Eur. J., 2017, 23, 8969 CrossRef CAS PubMed.
- S. Hiroto, K. Furukawa, H. Shinokubo and A. Osuka, J. Am. Chem. Soc., 2006, 128, 12380 CrossRef CAS PubMed.
-
(a) S. Ooi, T. Tanaka, K.-H. Park, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 6535 CrossRef CAS PubMed;
(b) S. Bhowmik, M. Kosa, A. Mizrahi, N. Fridman, M. Saphier, A. Stanger and Z. Gross, Inorg. Chem., 2017, 56, 2287 CrossRef CAS PubMed.
- T. Kubo, Chem. Rec., 2015, 15, 218 CrossRef CAS PubMed.
- W. Zeng, S. Lee, M. Son, M. Ishida, K. Furukawa, P. Hu, Z. Sun, D. Kim and J. Wu, Chem. Sci., 2015, 6, 2427 RSC.
- M. Umetani, K. Naoda, T. Tanaka, S.-K. Lee, J. Oh, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 6305 CrossRef CAS PubMed.
- A. Tsuda and A. Osuka, Science, 2001, 293, 5527 CrossRef PubMed.
- V. V. Diev, D. Femia, Q. Zhong, P. I. Djurovich, R. Haiges and M. E. Thompson, Chem. Commun., 2016, 52, 1949 RSC.
- B. Kin, T. Yoshida, X. Li, M. Stępień, H. Shinokubo and P. J. Chmielewski, Angew. Chem., Int. Ed., 2016, 55, 13142 CrossRef PubMed.
- H. Zhang, H. Plan, T. S. Herng, T. Y. Gopalakrishna, W. Zeng, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 13484 CrossRef CAS PubMed.
- Similar hysteresis behaviors were also observed for structurally flexible diradicals, but they showed closed hysteresis loop or phase transition behavior:
(a) T. Li, G. Tan, D. Shao, J. Li, Z. Zhang, Y. Song, Y. Sui, S. Chen, Y. Fang and X. Wang, J. Am. Chem. Soc., 2016, 138, 10092 CrossRef CAS PubMed;
(b) Y. Su, X. Wang, L. Wang, Z. Zhang, X. Wang, Y. Song and P. P. Power, Chem. Sci., 2016, 7, 6514 RSC.
-
Y. Jun-i, N. Fukui, K. Furukawa and A. Osuka, DOI:10.1002/chem.201705769.
- T. Y. Gopalakrishna, J. S. Reddy and V. G. Anand, Angew. Chem., Int. Ed., 2014, 53, 10984 CrossRef CAS PubMed.
- W.-Y. Cha, T. Kim, A. Ghosh, Z. Zhang, X.-S. Ke, R. Ali, V. M. Lynch, J. Jung, W. Kim, S. Lee, S. Fukuzumi, J. S. Park, J. L. Sessler, T. K. Chandrashekar and D. Kim, Nat. Chem., 2017, 9, 1243 CrossRef CAS PubMed.
- E. Iwamoto, K. Hirai and H. Tomioka, J. Am. Chem. Soc., 2003, 125, 14664 CrossRef CAS PubMed.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Atsuhiro
Osuka
and
Atsuhiro
Osuka