
Regioselective C–H alkenylation of imidazoles and its application to the synthesis of unsymmetrically substituted benzimidazoles†
Hyeongwoo
Kim‡
,
Ye Ji
Hwang‡
,
Inhyuk
Han
and
Jung Min
Joo
 *
*
Department of Chemistry and Chemistry Institute of Functional Materials, Pusan National University, Busan 46241, Republic of Korea. E-mail: jmjoo@pusan.ac.kr
First published on 9th May 2018
Abstract
A palladium-catalyzed C–H alkenylation of imidazoles has been developed. High C5 selectivity was achieved for C2-unsubstituted and C2-substituted imidazoles using oxygen and copper(II) acetate, respectively, as oxidants. The obtained products were applied to benzannulation through a sequence involving transposition of N-alkyl groups to give C4-alkenyl imidazoles, alkenylation, thermal 6π-electrocyclization, and oxidation, affording unsymmetrically substituted benzimidazoles.
Imidazoles and their benzannulated forms, benzimidazoles, are an important class of heterocycles, which are found in a number of commercial drugs, biologically active compounds, N-heterocyclic carbene ligands, and ionic liquids.1 Thus, selective syntheses of (benz)imidazoles have been essential to provide a wide range of derivatives containing a chosen functional group at a desired position. Particularly, parent (NH)-(benz)imidazoles are in tautomeric equilibrium, being formally symmetric, and approaches to access unsymmetrically substituted (benz)imidazoles have been of great interest in organic synthesis. However, most of the available methods rely on the formation of C–N bonds, which presents difficulties in the preparation of more sterically hindered regioisomers (Fig. 1A).2,3 The synthesis of unsymmetrically substituted benzimidazoles also depends on C–N bond forming processes, such as the installation of primary amines onto multi-substituted aromatic rings and subsequent ring closure, making it challenging to increase structural diversity in the aromatic ring (Fig. 1B).4 Alternatively, the regioselective formation of C–C bonds via direct C–H functionalization of N-substituted imidazoles could provide a systematic access to functionalized (benz)imidazoles.5 In fact, notable progress has been made in the installation of aryl groups to afford C5-arylated imidazoles.6,7 However, C–H alkenylation, also known as the Fujiwara–Moritani reaction, has been much less successful than arylation.8–10 Despite the importance of alkenyl imidazoles in drug discovery and natural product synthesis, either as chemical motifs or as latent functional groups, the regioselective C5-alkenylation of N-alkylimidazoles using readily available olefins has not been reported to the best of our knowledge.11 Furthermore, the installed alkenyl groups offer a great opportunity to build unsymmetrically substituted benzimidazoles; however, this possibility has been underexplored.12
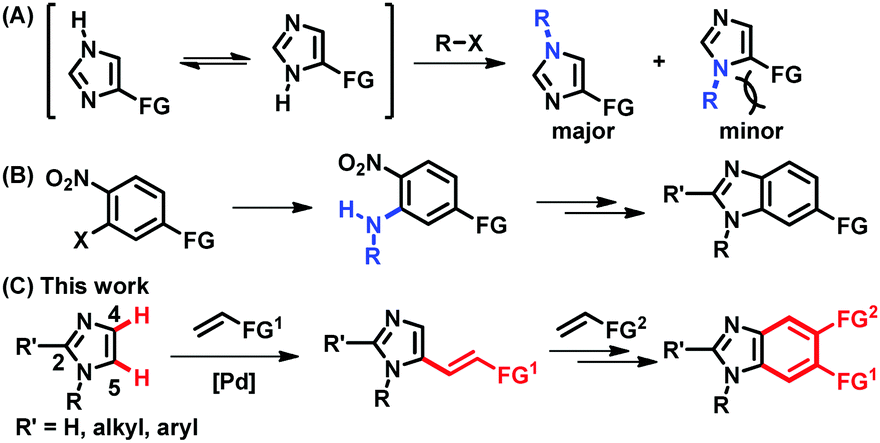 | ||
| Fig. 1 (A) (NH)-Imidazoles in tautomeric equilibrium leading to regioisomeric mixtures of N-substituted products. (B) Synthesis of benzimidazoles from multi-substituted arenes via C–N bond formation. (C) Synthesis of C5-alkenyl imidazoles and functionalized benzimidazoles via regioselective C–C bond forming reactions. | ||
The underdevelopment of imidazole C–H alkenylation is attributed to the unique reactivity of this heterocyclic core. Compared to the other two-nitrogen-containing five-membered heterocycle, pyrazole, imidazole is significantly basic, undergoing protonation at the nitrogen atom in the presence of acids and outcompeting nitrogen-based ligands that are frequently used in aerobic oxidation reactions.13,14 Instead of searching for ligands that could compete with imidazoles, we envisioned that an imidazole substrate itself could be a competent ligand to facilitate dehydrogenative alkenylation of imidazoles.15 More specifically, we hypothesized that imidazole-ligated Pd(II) carboxylate complexes should enable palladation at the more nucleophilic C5 position in preference to the C2 position of imidazoles, which requires strong bases and/or Lewis acidic metal additives.16,17 Herein, we report the regioselective C5-alkenylation of imidazoles and its application to the construction of unsymmetrically substituted benzimidazoles (Fig. 1C).
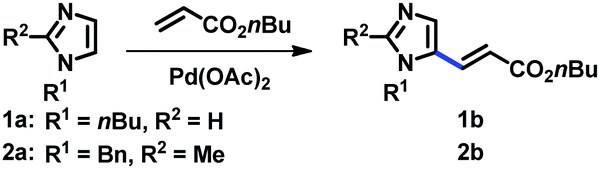
In order to develop a general protocol for the alkenylation of imidazoles, the optimization process was performed with a 1-alkylimidazole and a 1,2-dialkylimidazole in parallel (Table 1, see the ESI† for details). Simple imidazole 1a was reacted with two equivalents of olefins, whereas the amount of olefins was increased to three equivalents for C2-substituted imidazole 2a. Employing the C2-alkenylation conditions reported by Ong's group did not provide the corresponding C5-alkenylated imidazoles (entries 1 and 2).9a Only when we replaced the Ag salt by oxygen, we observed some selectivity towards the C5 position, and the reaction was more efficient in DMA than in 1,4-dioxane (entries 3 and 4). The presence of carboxylate salts was critical for the conversion, and potassium pivalate was superior to potassium acetate (entries 5 and 6).18 The kinetic isotope effect (KIE) was measured under the conditions of entry 6 (kH/kD = 2.76; see the ESI†), suggesting that the cleavage of the C–H bond at C5 was rate-limiting. In addition, it is notable that these aerobic conditions were not compatible with related 1,3-azoles (not shown), i.e. thiazoles and oxazoles, indicating the distinctive ligand effect of imidazole. Cesium pivalate was as competent as potassium pivalate, however, the same efficiency was not achieved with a combination of potassium carbonate and pivalic acid (entries 7 and 8, respectively). Aerobic conditions using air instead of oxygen reduced the conversion of both substrates (entry 9). To increase the conversion of C2-substituted imidazole 2a, where the regioselectivity issue is alleviated, the addition of Cu salts was investigated. As anticipated, the alkenylation product 2b was formed in high yields in the presence of Cu(OAc)2, which was slightly more efficient than its hydrate form (entries 11 and 10, respectively). The addition of pyridine as a ligand did not increase the conversion, presumably due to the strong ligand effect of imidazoles (entry 12). Heating the reaction mixture at 120 °C promoted the alkenylation at both C4 and C5 positions, resulting in a decreased yield of 2b (entry 13). It should be noted that under the Cu conditions, the selectivity was generally low for simple imidazole 1a and a significant amount of the C2-alkenylation product was formed. For example, the reaction of 1a using the conditions described in entry 11 gave 38% yield of the corresponding C2-alkenylation product along with 12% of C5-alkenylation product 1b. This result is consistent with the fact that copper salts activate the C2 position of imidazoles, an effect that has been observed since the pioneering reports on the C–H arylation of imidazoles.17 Therefore, the conditions described in entries 6 and 11 were used for the C5-alkenylation of C2-unsubstituted and C2-substituted imidazoles, respectively.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive | Oxidant | Solvent | Temp. (°C) | Yield (%) | |
| 1b | 2b | |||||
| a Reaction conditions: imidazole (1.0 mmol), n-butyl acrylate (2.0 and 3.0 mmol for 1a and 2a, respectively), Pd(OAc)2 (0.10 mmol), additive (2.0 mmol), oxidant (2.0 mmol or O2 balloon), solvent (0.33 M), 120 °C, 24 h. b GC yield. c 1H NMR yield. d Reaction conditions: imidazole (1.0 mmol), n-butyl acrylate (5.0 mmol), Pd(TFA)2 (0.10 mmol), 1,10-phenanthroline (0.15 mmol), AgTFA (2.0 mmol), toluene (0.50 M), 130 °C (ref. 9a). e PivOH (0.50 mmol) was used. | ||||||
| 1d | 1,10-Phenanthroline | AgTFA | Toluene | 130 | <5 | <1 |
| 2 | — | AgTFA | 1,4-Dioxane | 100 | <5 | <1 |
| 3 | — | O2 (1 atm) | 1,4-Dioxane | 120 | 11 | 7 |
| 4 | — | O2 (1 atm) | DMA | 120 | 32 | 50 |
| 5 | KOAc | O2 (1 atm) | DMA | 120 | 57 | 61 |
| 6 | KOPiv | O2 (1 atm) | DMA | 120 | 67 | 67 |
| 7 | CsOPiv | O2 (1 atm) | DMA | 120 | 64 | 64 |
| 8e | K2CO3/PivOH | O2 (1 atm) | DMA | 120 | 54 | 40 |
| 9 | KOPiv | Air | DMA | 120 | 52 | 50 |
| 10 | — | Cu(OAc)2·H2O | 1,4-Dioxane | 100 | 20 | 68 |
| 11 | — | Cu(OAc)2 | 1,4-Dioxane | 100 | 12 | 76 |
| 12 | Pyridine (20 mol%) | Cu(OAc)2 | 1,4-Dioxane | 100 | 14 | 72 |
| 13 | — | Cu(OAc)2 | 1,4-Dioxane | 120 | 6 | 62 |
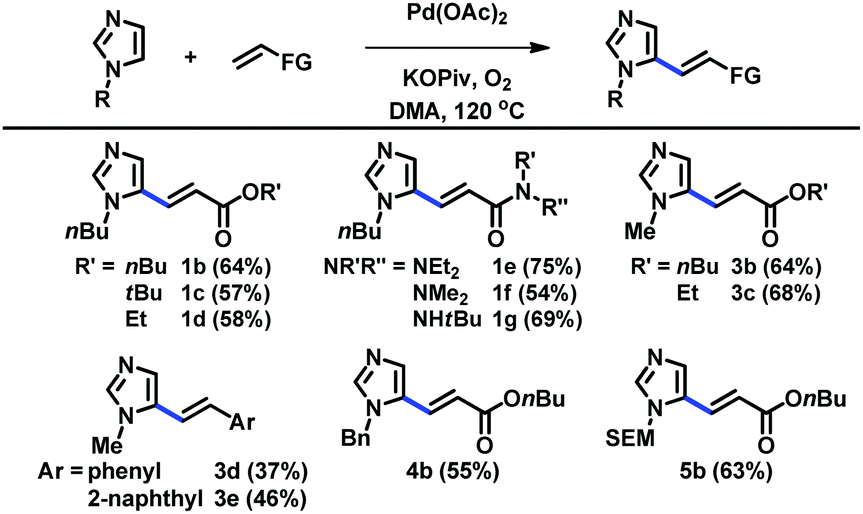
The scope of C5-alkenylation of C2-unsubstituted imidazoles was examined using the catalytic system derived from Pd(OAc)2, KOPiv, O2, and DMA (Table 2). Reactions with acrylates and acrylamides gave good yields of C5-alkenylation products (1b–1g, 3b, and 3c). Styrene derivatives were also used to afford the corresponding products (3d and 3e). Furthermore, imidazoles substituted with different alkyl groups at the N1 nitrogen were well tolerated (4b and 5b). C5-Alkenyl imidazoles have previously been accessible via the Horner–Wadsworth–Emmons and Heck reactions of the corresponding C5-prefunctionalized imidazoles, which, however, are difficult to access.11 Thus, our strategy based on the direct C–H alkenylation of readily available imidazoles offers a highly useful alternative access to a range of these structural motifs.
| Reaction conditions: imidazole (1.0 mmol), alkene (2.0 mmol), Pd(OAc)2 (0.10 mmol), KOPiv (2.0 mmol), O2 balloon (1.0 atm), DMA (3.0 mL), 120 °C, 24 h. |
|---|
|
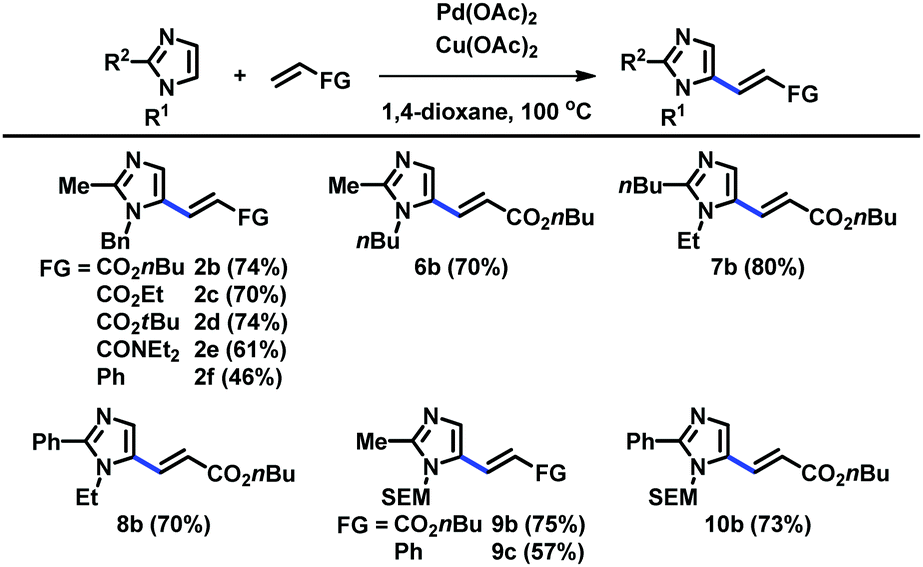
When the C2 position was substituted with alkyl and aryl groups, the Pd-catalyzed and Cu-mediated alkenylation worked well with a variety of alkenes, including acrylates, acrylamides, and styrene (Table 3). Substituents at the N1 and C2 positions were varied to readily provide a range of C5-alkenylated imidazoles.
| Reaction conditions: imidazole (0.50 mmol), alkene (1.5 mmol), Pd(OAc)2 (0.050 mmol), Cu(OAc)2 (1.0 mmol), 1,4-dioxane (1.5 mL), 100 °C, 15 h. |
|---|
|
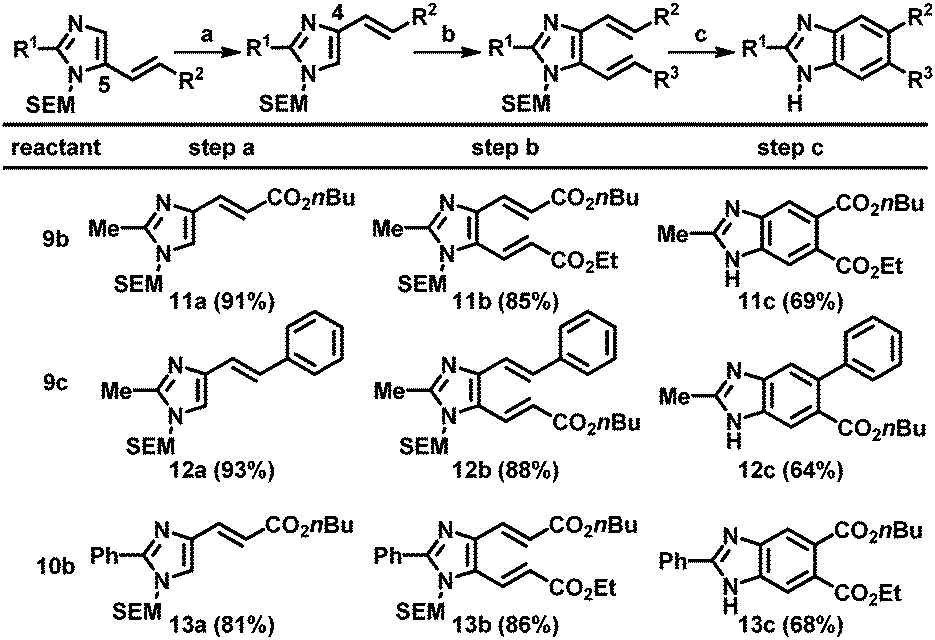
Next, the resulting C5-alkenylated imidazoles were subjected to additional alkenylation. Despite various attempts, the yield of C4-alkenylation of C5-alkenylated imidazoles remained modest (<40%). Thus, an alternative strategy based on the SEM-group switch, which had previously been demonstrated for the preparation of C4,5-diarylated imidazoles, was adopted.2b,16 Heating the C5-alkenyl imidazoles in the presence of a catalytic amount of 2-(trimethylsilyl)ethoxymethyl chloride (SEM-Cl) afforded the corresponding C4-alkenyl imidazoles in good-to-high yields (Table 4, step a). For imidazoles bearing a substituent at the C2 position, the alkenylation at the C5 position using Pd(OAc)2 and Cu(OAc)2 was applied to smoothly generate the corresponding dialkenylated products (11b, 12b, and 13b; step b). For the subsequent ring closure, the oxidative conditions developed by Langer's group gave a mixture of products, resulting from undesired pathways, including SEM-switch, removal of the SEM group, and most significantly, incomplete oxidation.12 Other oxidizing reagents that could enable thermal 6π-electrocyclization and oxidation were tested; the desired benzimidazoles were ultimately formed employing catalytic amounts of DDQ and NaNO2 under an oxygen atmosphere (step c).19 However, high temperatures (200 °C) were required and thus, the scrambling of the SEM group could not be eliminated. Hence, SEM deprotection was performed to obtain the final (NH)-free benzimidazoles (11c, 12c, and 13c).
| Step a: imidazole (1.0 equiv.), SEM-Cl (0.050 equiv.), CH3CN (0.10 M), 100 °C, 24 h. Step b: imidazole (1.0 equiv.), alkene (3.0 equiv.), Pd(OAc)2 (0.10 equiv.), Cu(OAc)2 (2.0 equiv.), 1,4-dioxane (0.33 M), 100 °C, 15 h. Step c: DDQ (0.10 equiv.), NaNO2 (0.10 equiv.), Ph2O (0.10 M), 180 °C, 36 h; 3 N HCl, 80 °C. |
|---|
|
This strategy using sequential alkenylation, switch of the N-substituent, and cyclization was useful for the regioselective formation of N-methylbenzimidazoles. For example, the alkenylation product 5b was subjected to trimethyloxonium tetrafluoroborate, and the obtained product was subsequently treated with TFA at ambient temperature, giving 1-methyl-4-alkenyl imidazole 14a (Scheme 1). Remarkably, the next alkenylation of imidazole 14a bearing an electron-withdrawing group at the C4 position took place preferentially at the C5 position, giving 14b. Ring closure led to the formation of benzimidazole 14c, which has different substituents at the C5 and C6 positions and fixed regiochemistry at the N1 position.
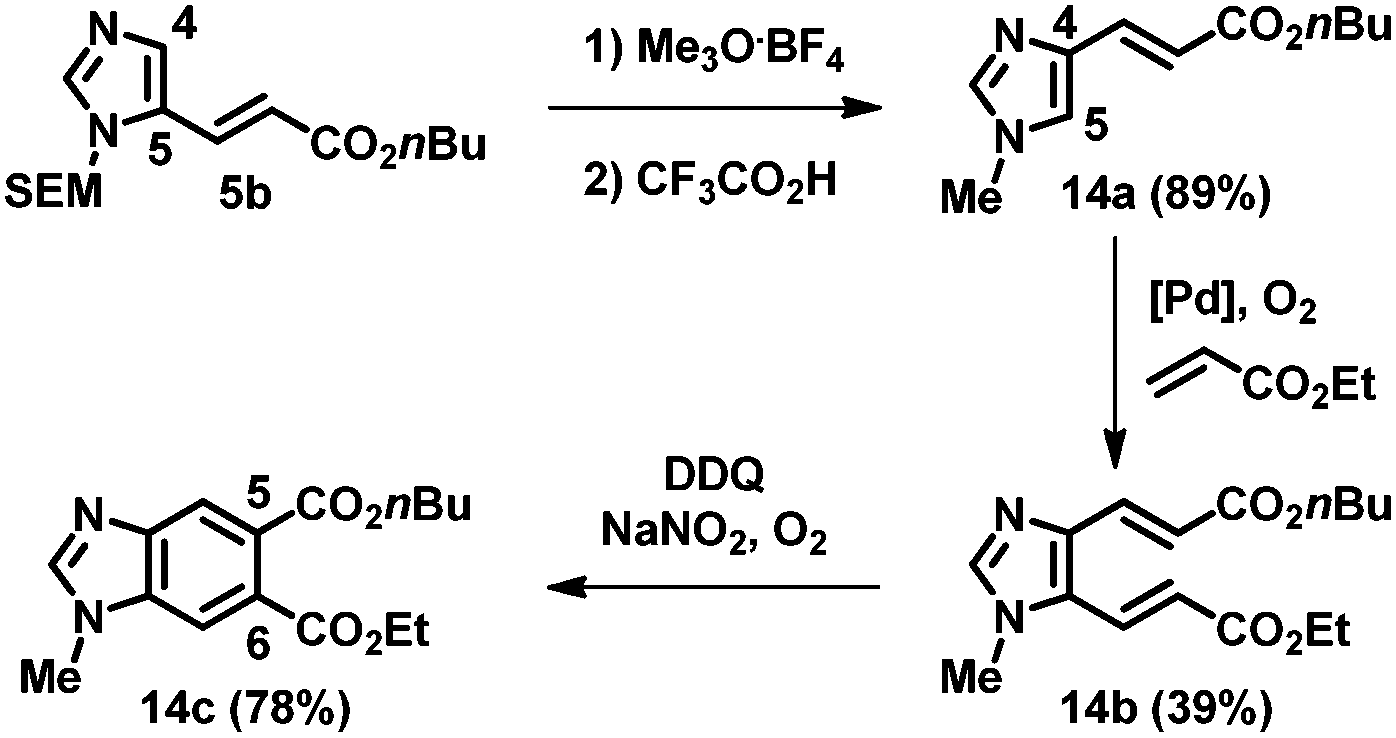 | ||
| Scheme 1 Benzannulation of a C2-unsubstituted imidazole using N-methylation. | ||
In conclusion, we have demonstrated Pd-catalyzed C–H alkenylation reactions of imidazoles at the C5 position. Imidazole substrates served as effective ligands in the palladium-catalyzed system, allowing the oxidative coupling to olefins. A variety of alkenylated imidazoles were easily prepared from readily available simple imidazoles and olefins in a single step. Furthermore, the resulting imidazoles were converted to unsymmetrically substituted benzimidazoles in a systematic way. In addition to the importance in the synthesis of imidazole and benzimidazole derivatives, these results suggest the potential role of imidazoles as ligands in dehydrogenative C–C bond forming reactions, which are currently being investigated in our laboratory and will be reported in due course.
This research was supported by the National Research Foundation of Korea (NRF-2016R1D1A1B03930762).
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Xi, Q. Huang and L. Liu, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 4, pp. 143–364 Search PubMed.
- (a) J. A. Joule and K. Mills, Heterocycl. Chem., Wiley-Blackwell, New York, 5th edn, 2010, pp. 462–463 Search PubMed; (b) Y. He, Y. Chen, H. Du, L. A. Schmid and C. J. Lovely, Tetrahedron Lett., 2004, 45, 5529 CrossRef.
- For recent examples of regioselective N-alkylation of (benz)imidazoles to afford the more sterically hindered regioisomers, see: (a) E. Van Den Berge and R. Robiette, J. Org. Chem., 2013, 78, 12220 CrossRef PubMed; (b) S. Chen, R. F. Graceffa and A. A. Boezio, Org. Lett., 2016, 18, 16 CrossRef PubMed.
- (a) P. Picconi, C. Hind, S. Jamshidi, K. Nahar, M. Clifford, M. E. Wand, J. M. Sutton and K. M. Rahman, J. Med. Chem., 2017, 60, 6045 CrossRef PubMed; (b) K. Kuroda, S. Tsuyumine and T. Kodama, Org. Process Res. Dev., 2016, 20, 1053 CrossRef; (c) C. N. Kuzniewski, J. Gertsch, M. Wartmann and K.-H. Altmann, Org. Lett., 2008, 10, 1183 CrossRef PubMed.
- For reviews on the rapid synthesis of complex molecules via C–H bond functionalization, see: (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef PubMed; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef PubMed; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef PubMed; (d) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef PubMed; (e) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef PubMed; (f) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef PubMed.
- For reviews, see: (a) C. B. Bheeter, L. Chen, J.-F. Soulé and H. Doucet, Catal. Sci. Technol., 2016, 6, 2005 RSC; (b) F. Bellina and R. Rossi, Adv. Synth. Catal., 2010, 352, 1223 CrossRef.
- For C5-arylation of imidazoles, see: (a) F. Bellina, S. Cauteruccio, L. Mannina, R. Rossi and S. Viel, J. Org. Chem., 2005, 70, 3997 CrossRef PubMed; (b) B. B. Touré, B. S. Lane and D. Sames, Org. Lett., 2006, 8, 1979 CrossRef PubMed; (c) F. Bellina, S. Cauteruccio, A. Di Fiore, C. Marchetti and R. Rossi, Tetrahedron, 2008, 64, 6060 CrossRef; (d) B. Liégault, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826 CrossRef PubMed; (e) J. Roger and H. Doucet, Tetrahedron, 2009, 65, 9772 CrossRef; (f) F. Shibahara, E. Yamaguchi and T. Murai, J. Org. Chem., 2011, 76, 2680 CrossRef PubMed; (g) A. Mahindra, N. Bagra and R. Jain, J. Org. Chem., 2013, 78, 10954 CrossRef PubMed; (h) P. V. Kumar, W.-S. Lin, J.-S. Shen, D. Nandi and H. M. Lee, Organometallics, 2011, 30, 5160 CrossRef; (i) M. Baghbanzadeh, C. Pilger and C. O. Kappe, J. Org. Chem., 2011, 76, 8138 CrossRef PubMed; (j) F. Shibahara, T. Yamauchi, E. Yamaguchi and T. Murai, J. Org. Chem., 2012, 77, 8815 CrossRef PubMed; (k) F. Bellina, M. Lessi and C. Manzini, Eur. J. Org. Chem., 2013, 5621 CrossRef; (l) S. Guo and H. V. Huynh, Organometallics, 2014, 33, 2004 CrossRef; (m) X.-X. He, Y. Li, B.-B. Ma, Z. Ke and F.-S. Liu, Organometallics, 2016, 35, 2655 CrossRef; (n) J.-Y. Lee, J.-S. Shen, R.-J. Tzeng, I.-C. Lu, J.-H. Lii, C.-H. Hu and H. M. Lee, Dalton Trans., 2016, 45, 10375 RSC; (o) J.-S. Ouyang, Y.-F. Li, D.-S. Shen, Z. Ke and F.-S. Liu, Dalton Trans., 2016, 45, 14919 RSC; (p) L.-Q. Hu, R.-L. Deng, Y.-F. Li, C.-J. Zeng, D.-S. Shen and F.-S. Liu, Organometallics, 2018, 37, 214 CrossRef . Also, see ref. 16 and 17a.
- (a) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef PubMed; (b) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138 CrossRef PubMed.
- (a) A single example of C2-selective alkenylation of N-methylimidazole was reported. W.-C. Lee, T.-H. Wang and T.-G. Ong, Chem. Commun., 2014, 50, 3671 RSC; (b) For a directing-group-assisted C5-alkenylation of 2-substituted imidazoles, see: H. Zhao, J. Xu, C. Chen, X. Xu, Y. Pan, Z. Zhang, H. Li and L. Xu, Adv. Synth. Catal., 2018, 360, 985 CrossRef.
- For Ni-catalyzed alkenylation of imidazoles using alkynes, see: K. S. Kanyiva, F. Löbermann, Y. Nakao and T. Hiyama, Tetrahedron Lett., 2009, 50, 3463 CrossRef.
- (a) J.-G. Boiteau, N. Rodeville, C. Martin, S. Tabet, C. Moureou, F. Muller, J.-C. Jetha and I. Cardinaud, Org. Process Res. Dev., 2015, 19, 646 CrossRef; (b) X. Wang and C. Chen, Tetrahedron, 2015, 71, 3690 CrossRef PubMed; (c) A. Al-Mourabit, M. A. Zancanella, S. Tilvi and D. Romo, Nat. Prod. Rep., 2011, 28, 1229 RSC; (d) Y. Todoroki, K. Naiki, H. Aoyama, M. Shirakura, K. Ueno, M. Mizutani and N. Hirai, Bioorg. Med. Chem. Lett., 2010, 20, 5506 CrossRef PubMed; (e) E. A. B. Kantchev, G.-R. Peh, C. Zhang and J. Y. Ying, Org. Lett., 2008, 10, 3949 CrossRef PubMed . Note that only limited types of 1-alkyl-5-haloimidazoles are commercially available. Also, see ref. 2b.
- For the Heck reaction of dibromoimidazoles and cyclization for the synthesis of benzimidazoles having the same substituents on the aromatic ring, see: S.-M. T. Toguem and P. Langer, Synlett, 2010, 1779 Search PubMed.
- For alkenylation of pyrazoles, see: (a) H. T. Kim, H. Ha, G. Kang, O. S. Kim, H. Ryu, A. K. Biswas, S. M. Lim, M.-H. Baik and J. M. Joo, Angew. Chem., Int. Ed., 2017, 56, 16262 CrossRef PubMed; (b) S. J. Han, H. T. Kim and J. M. Joo, J. Org. Chem., 2016, 81, 689 CrossRef PubMed.
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636 CrossRef PubMed.
- (a) L. A. Perego, L. Grimaud and F. Bellina, Adv. Synth. Catal., 2016, 358, 597 CrossRef; (b) M. S. Szulmanowicz, W. Zawartka, A. Gniewek and A. M. Trzeciak, Inorg. Chim. Acta, 2010, 363, 4346 CrossRef; (c) C. J. Mathews, P. J. Smith and T. Welton, J. Mol. Catal. A: Chem., 2003, 206, 77 CrossRef.
- For C2-arylation of imidazoles using a strong base, see: J. M. Joo, B. B. Touré and D. Sames, J. Org. Chem., 2010, 75, 4911 CrossRef PubMed.
- For C2-arylation of imidazoles using Lewis acidic metal additives, see: (a) P.-A. Sommai, S. Tetsuya, K. Yoshiki, M. Masahiro and N. Masakatsu, Bull. Chem. Soc. Jpn., 1998, 71, 467 CrossRef; (b) F. Bellina, C. Calandri, S. Cauteruccio and R. Rossi, Tetrahedron, 2007, 63, 1970 CrossRef; (c) F. Bellina, S. Cauteruccio and R. Rossi, Eur. J. Org. Chem., 2006, 1379 CrossRef; (d) F. Bellina, S. Cauteruccio, L. Mannina, R. Rossi and S. Viel, Eur. J. Org. Chem., 2006, 693 CrossRef . For a computational study, see: ; (e) S. I. Gorelsky, Organometallics, 2012, 31, 794 CrossRef.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef PubMed.
- W. Zhang, H. Ma, L. Zhou, Z. Sun, Z. Du, H. Miao and J. Xu, Molecules, 2008, 13, 3236 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, and crystallographic data. See DOI: 10.1039/c8cc02405g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |